
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA newly designed softoxometalate [BMIm]2[DMIm][α-PW12O40]@hydrocalumite that controls the chain length of polyacrylic acid in the presence of light†
Santu Das and
Soumyajit Roy*
Eco-Friendly Applied Materials Laboratory (EFAML), Materials Science Centre, Department of Chemical Sciences, Mohanpur Campus, Indian Institute of Science Education & Research, Kolkata, 741246 West Bengal, India. E-mail: s.roy@iiserkol.ac.in
First published on 30th March 2016
Abstract
An immediate need in polymer chemistry is to control the chain length of a polymer. Hence it is necessary to develop a strategy that controls the chain length of a polymer. In our present work we have tried to control the chain length of acrylic acid polymer by polymerizing it using a soft-oxometalate (SOM) based catalyst. This soft-oxometalate has a porous material hydrocalumite core and on its pores, a polyoxometalate [BMIm]2[DMMm][α-PW12O40] (1) (where BMIm = 1-butyl 3-methylimidazole; DMIm = 1,3-dimethylimidazole). The soft-oxometalate, [BMIm]2[DMIm][α-PW12O40]@hydrocalumite (SOM 2) was synthesized by sonication within an hour. Using this SOM 2 as an initiator catalyst we obtained poly-acrylic acid of MW 1.2 kDa. While using AIBN@hydrocalumite 3 as an initiator catalyst, the MW of PAA is obtained at around 150 kDa. This difference is attributed to the more favourable interaction of ionic POM with inorganic hydrocalumite, as compared to the less favourable interaction of organic AIBN with inorganic hydrocalumite. More favourable interaction of POM 1 with hydrocalumite leads to higher radical generation and hence shorter polymer chains as compared to that in the case of AIBN in 3, where longer chains are generated due to the low abundance of radicals at the pores of hydrocalumites. SOM 2 was characterized by X-ray diffraction (XRD), horizontally attenuated total reflection infrared spectroscopy (HATR-IR), scanning electron microscopy (SEM), electron dispersive scattering technique (EDS) and dynamic light scattering (DLS).
Introduction
The ability to control macromolecular architecture is an increasingly important area of polymer chemistry. In synthetic polymer chemistry an important goal is to develop a controlled radical polymerization process that can yield polymers with controlled and well-defined chain lengths. In the present work we have developed a new softoxometalate based on a porous frame-work hydrocalumite and a polyoxometalate [BMIm]2[DMMm][α-PW12O40] (1) (where BMIm – 1-butyl 3-methylimidazole, DMIm – 1,3-dimethylimidazole) which polymerizes acrylic acid in a controlled manner with light. The architecture of the catalyst, determined by an array of techniques, is such that it yields polyacrylic acid of same molecular weight.Polyacrylic acid (PAA) is a very useful compound in chemistry. It is used in pharmaceuticals, paints and cosmetics industries as an emulsifying, thickening, dispersing and suspending agent.1–5 Also polyacrylic acid and its derivatives are very useful in disposable diapers.6,7 It is also a versatile agent in many applications, viz. in the removal of heavy and radioactive metallic ions, controlled release of reagents,8 artificial gardening etc.9–14 Many such applications require a PAA of controlled chain length. Therefore there is a need to synthesize PAA with controlled molecular weight and thus controlled chain length.15,16 Here we address this need.
There are several routes to address this need. For instance different types of homopolymers and copolymers with complex architectures have become easily accessible via the route of controlled free radical polymerization (CRP).17–19 Different techniques have been developed such as nitroxide-mediated polymerization (NMP),20–22 reversible transfer techniques [reversible addition fragmentation chain transfer (RAFT)23–26 and macromolecular design via the interchange of xanthates,27 MADIX9 (ref. 27)], and atom transfer radical polymerization (ATRP).28–31 Among these free-radical polymerization processes are well known and are very useful for industrial applications.32–35 All the above methods have been applied to control the polymerization of acrylic acid by different groups.15,16 For instance first attempt of controlling nitroxide mediated radical polymerization was done by Charleux group.16 Later research has been developed using different ratio of H2O2 as a catalyst.36 Also different catalytic system has been developed for controlling the chain length of polyacrylic acid.37–40
However there is another constraint. Not all of the above routes are green. It is now becoming increasingly important in polymer industries to develop green synthetic methods of polymerization, and photo-polymerization is one step forward in that direction.41–44 Hence the need of the hour is a greener photochemical route for synthesizing PAA of controlled chain length.
In this paper we have developed a new catalyst based on softoxometalate (SOM)45–51 which converts acrylic acid to mono disperse polyacrylic acid in the presence of light. We have already shown in our earlier work that POM of type phosphotungstate generates radical in presence of UV-light which can act as a catalyst for radical polymerization method.52 We have also shown that our synthesized [BMIm]2[DMIm][α-PW12O40] (1), a polyoxometalate based catalyst,52 can perform photochemical radical as well as cationic polymerization. So, now question arises can we synthesize monodisperse polyacrylic acid using this catalyst by the modification of its surface? To address this question we have used softoxometalate (SOM) based catalyst materials.53–55 In our present work we have synthesized SOM based on 1. Our group already reported that SOM can be synthesized from 1 which forms vesicle like superstructure in water dispersion.53 Using this superstructure as photocatalyst we could synthesize polymeric spheres. But now we want to control the chain length of polyacrylic acid, using a two component SOM and an electroactive template which will facilitate the catalytic process. With this end in view, we have chosen hydrocalumite as a template material with well defined pores and we have loaded our catalyst in those pores of hydrocalumite (Fig. 1). The composite material [BMIm]2[DMIm][α-PW12O40]@hydrocalumite (SOM 2) is characterized by infrared spectroscopy, powder X-ray diffraction, scanning electron microscopy, elemental dispersive scattering and dynamic light scattering technique, and used this material as a photocatalyst for controlled polymerization of acrylic acid.
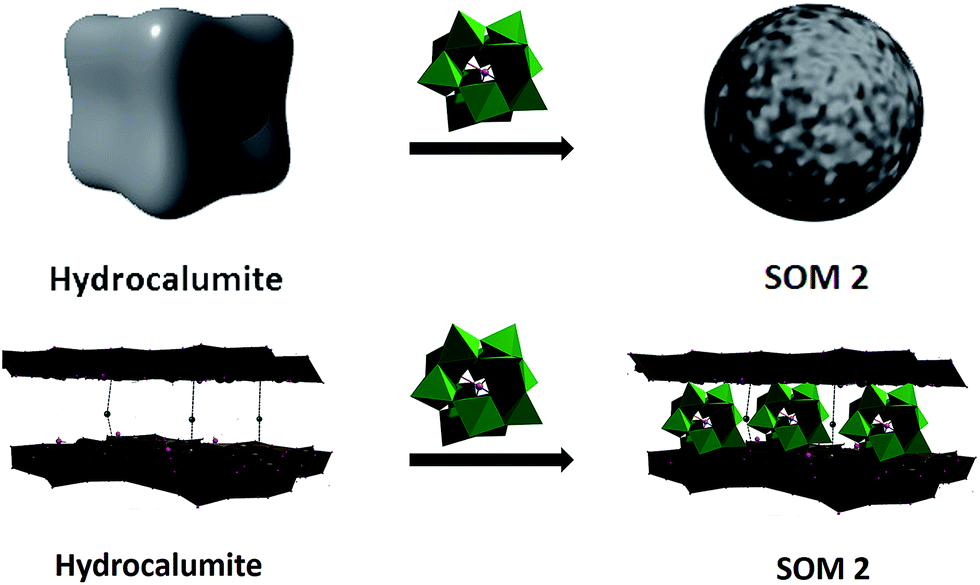 | ||
| Fig. 1 Schematic representation of reaction of hydrocalumite with [BMIm]2[DMIm][α-PW12O40]. Dice schematically represents hydrocalumite in dispersion and sphere represents hydrocalumite@[BMIm]2[DMIm][α-PW12O40] composite at dispersion. Such transformations are observed experimentally in our system. | ||
Hydrocalumite is a porous material, it is an ore of calcium and aluminum, and it can also be synthesized artificially. Hydrocalumite is used as a catalyst for different purposes.56–58 General formula of hydrocalumite shown by Campos-Molina et al. is Ca2Al(OH)6Cl·2H2O.57 Its structure is closely analogous to that of hydrotalcite.59–61 It is a Ca–Cl–Al hydroxide layer. Due to the branching between two layers it forms a porous network. Porous frame-work is important as a template. On doping other materials on hydrocalumite, doped materials will go to the porous framework of hydrocalumite (Fig. 1). In this paper we have doped hydrocalumite with [BMIm]2[DMIm][α-PW12O40] (1), and 1 goes to the pores of hydrocalumite. Such a doping results in macroscopic topological transformation in hydrocalumite particles upon loading with POMs ([BMIm]2[DMIm][α-PW12O40]) forming spherical SOMs. Hence the loading causes a marked topological transformation from cube to sphere of composite SOMs. This composite SOM has been characterized by different techniques which have been explored in details in next part of paper and we further used it as a photocatalyst for polymerization of acrylic acid.
Result and discussion
Synthesis and characterization of hydrocalumite
Hydrocalumite has been synthesized using a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of CaCl2 and AlCl3, which was dissolved in water. Layered hydroxide framework of hydrocalumite was prepared by slow addition of 2 M NaOH solution into the reaction mixture with vigorous stirring for a while and further stirring it for 18 h at 65 °C. A white precipitate of hydrocalumite was obtained. In molecular level it forms a layer lattice type hydroxide; two layers are linked through Cl− bridge. SEM images of hydrocalumite reveals its porous surface which can be used as a host for POMs (Fig. 2a). EDS-spectrum reveals the presence of aluminum, calcium and chlorine in hydrocalumite framework. Detailed structural characterization was obtained from X-ray diffraction of hydrocalumite which shows that it forms a hexagonal lattice structure (Fig. 3a). We have got three characteristic peaks from X-ray diffraction analysis, which is observed due to the presence of (003), (006), (110) Miller planes in hexagonal hydrocalumite which are characteristic for hexagonal hydrocalumite.56 The lattice parameter of hydrocalumite was also similar to the literature with a = 5.749, c = 23.492 (Fig. 3a). Further we have characterized it using HATR-IR spectroscopy it shows peak at 1480, and 876 cm−1 (Fig. 3b). We have dispersed hydrocalumite in water by sonication method. It forms a stable dispersion in water (Fig. S1a†) with dice type morphology in dispersion (Fig. 2c). We have characterized the dispersion using DLS method (Fig. S3†). Hydrodynamic radius of hydrocalumite was around 150 nm which was also similar to the radius obtained from SEM image of hydrocalumite. HATR-IR spectroscopy of the dispersion reveals that hydrocalumite is stable in dispersion over time.
:1 mixture of CaCl2 and AlCl3, which was dissolved in water. Layered hydroxide framework of hydrocalumite was prepared by slow addition of 2 M NaOH solution into the reaction mixture with vigorous stirring for a while and further stirring it for 18 h at 65 °C. A white precipitate of hydrocalumite was obtained. In molecular level it forms a layer lattice type hydroxide; two layers are linked through Cl− bridge. SEM images of hydrocalumite reveals its porous surface which can be used as a host for POMs (Fig. 2a). EDS-spectrum reveals the presence of aluminum, calcium and chlorine in hydrocalumite framework. Detailed structural characterization was obtained from X-ray diffraction of hydrocalumite which shows that it forms a hexagonal lattice structure (Fig. 3a). We have got three characteristic peaks from X-ray diffraction analysis, which is observed due to the presence of (003), (006), (110) Miller planes in hexagonal hydrocalumite which are characteristic for hexagonal hydrocalumite.56 The lattice parameter of hydrocalumite was also similar to the literature with a = 5.749, c = 23.492 (Fig. 3a). Further we have characterized it using HATR-IR spectroscopy it shows peak at 1480, and 876 cm−1 (Fig. 3b). We have dispersed hydrocalumite in water by sonication method. It forms a stable dispersion in water (Fig. S1a†) with dice type morphology in dispersion (Fig. 2c). We have characterized the dispersion using DLS method (Fig. S3†). Hydrodynamic radius of hydrocalumite was around 150 nm which was also similar to the radius obtained from SEM image of hydrocalumite. HATR-IR spectroscopy of the dispersion reveals that hydrocalumite is stable in dispersion over time.
 | ||
| Fig. 2 SEM image of (a) hydrocalumite in solid state, (b) SOM 2 in solid state (c) hydrocalumite in dispersion, (d) SOM 2 in dispersion. | ||
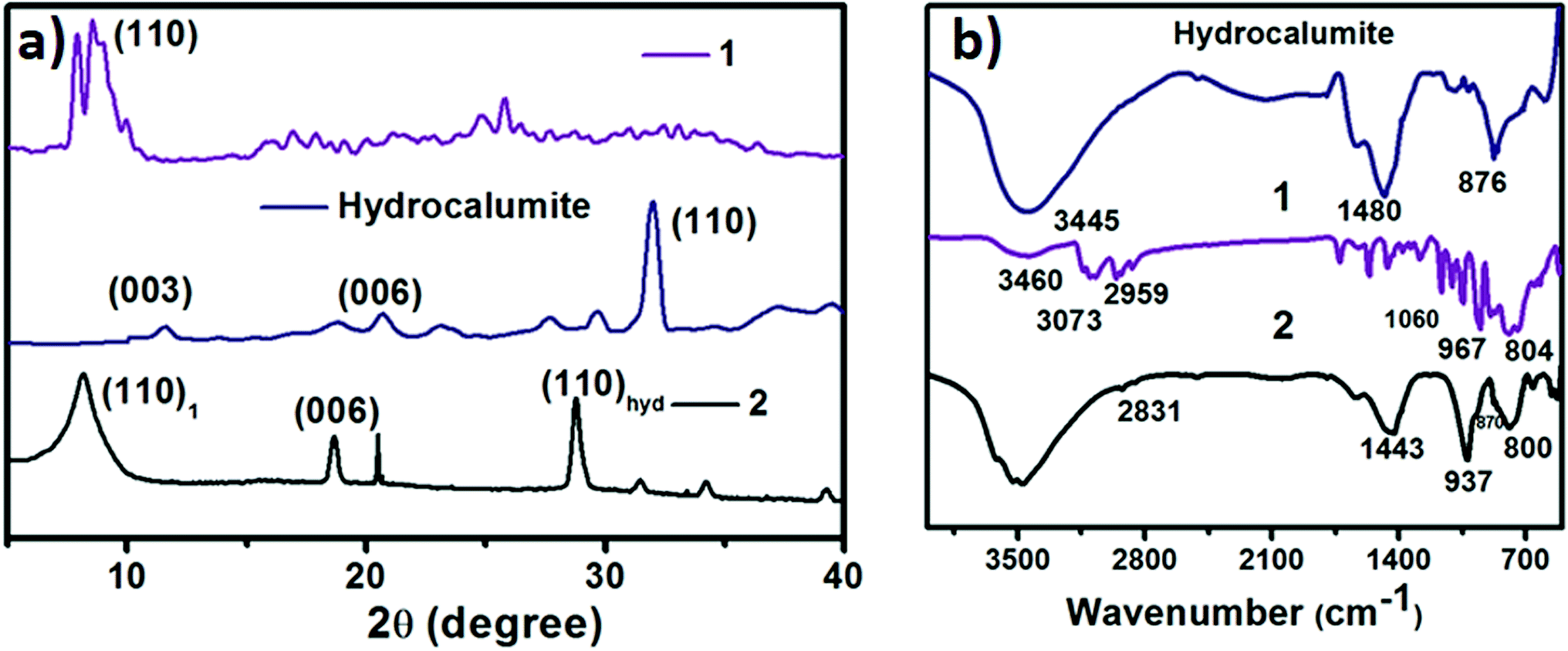 | ||
| Fig. 3 (a) X-ray diffraction of hydrocalumite (blue line), 1 (violet line) and SOM 2 (black line). (b) HATR-IR spectrum of hydrocalumite (blue line), 1 (violet line) and SOM 2 (black line). | ||
Synthesis and characterization of [BMIm]2[DMIm][PW12O40] (1)
1 was synthesized following literature procedure52 and was further characterized by FT-IR spectroscopy (Fig. 3b), powder X-ray diffraction analysis (Fig. 3a) we get a sharp peak at 10.190° which corresponds to (110) Miller plane. The inter planar distance was observed 8.6921 Å. 1 forms vesicle like superstructure in DMSO-water mixture.53 The hydrodynamic radius of this vesicle can be controlled by changing the polarity of system.53 We now synthesize a new composite material based on hydrocalumite and 1, and to do so we have added 1 (DMSO solution) in a dilute dispersion of hydrocalumite.Synthesis of hydrocalumite@[BMIm]2[DMIm][PW12O40] composite (SOM 2) and its characterization
Hydrocalumite is a porous material. It has fixed pores. To load 1 on the pores of hydrocalumite we slowly add 1 (DMSO solution) to a dispersion of hydrocalumite in water. Further we sonicate the reaction mixture for an hour. After sonication the composite material is formed in water (Fig. S1b†). Initially it forms a homogeneous dispersion, but after few hours (∼12 h) on standing, the composite, forms a gel like material that indicates the formation of [BMIm]2[DMIm][α-PW12O40]@hydrocalumite (SOM 2) composite (Fig. S1c†). Upon sonication of this gel, a homogeneous dispersion is obtained. SOM 2 has been characterized in dispersion as well as in solid state. The morphology of SOM 2 in dispersion is obtained from SEM image (Fig. 2d). SEM image of composite material is different from that of hydrocalumite. It forms symmetrical spherical superstructure in dispersion (Fig. 2d). It further indicates that upon loading of 1 on the surface of hydrocalumite, which has cuboidal shape, turns to spheroidal shaped SOM 2, the new composite material in dispersion. We now investigate the loading of 1 on hydrocalumite by SEM and X-ray diffraction.Pure hydrocalumite surface appears grayish. Upon loading of 1, due to enriched electron density on the surface of hydrocalumite, the surface lightens up. It looks brighter implying successful loading of electron rich 1 on hydrocalumite, to make SOM 2. This composite SOM 2 is further characterized by X-ray diffraction analysis. X-ray diffraction pattern gives characteristic information about SOM 2. From the X-ray diffraction pattern of SOM 2 it is observed that the both hydrocalumite and 1 are present in the SOM 2. Three peaks are observed at 10.855°, 18.67°, and 28.79° (Fig. 3a). Among these three peaks first peak is obtained due to Bragg reflection from (110) plane of 1, and other two peaks are obtained due to Bragg reflections from (006) and (110) planes of hydrocalumite. All the three peaks in composite SOM 2 have shifted from their original positions obtained in their pure state. This shifting in peaks indicate the formation of new composite material in reaction. Thus SOM 2 is not a physical mixture of hydrocalumite and 1 but it is a composite of the two materials. Further detailed structural information is also obtained from X-ray diffraction analysis. As in the reaction system hydrocalumite is always taken in excess and also during synthesis of SOM 2, 1 is added on hydrocalumite slowly therefore it is natural that the composite material contains unit cell of hydrocalumite. Also from X-ray diffraction pattern it is observed that the X-ray diffraction pattern of SOM 2 is quite similar with hydrocalumite X-ray diffraction pattern. Therefore composite material SOM 2 is composed of hexagonal unit cell with lattice spacing of 4.81 Å between (006) planes and 3.197 Å between (110) planes of hydrocalumite. The lattice parameter of newly synthesized composite material SOM 2 has been obtained from least square method, a = 6.3969 Å, and c = 28.904 Å which is different from that of hydrocalumite. Here POM 1 may be incorporated into the lattice of hydrocalumite to finally form a hexagonal lattice.
Further formation of composite material SOM 2 is characterized from HATR-IR spectroscopy (Fig. 3b). We do not get any detailed structural information from HATR-IR spectrum due to the low concentration of SOM 2 in dispersion. From the IR spectrum two peaks are obtained-one at 937 cm−1 which corresponds to W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Ot stretching and second at 800 cm−1 that corresponds to W–O–W stretching. Other two peaks are obtained at 1443 and 870 cm−1 respectively, which correspond to the presence of hydrocalumite in SOM 2. All the peaks are well shifted from the IR spectra of pure materials (i.e. hydrocalumite and 1), which further reveals the formation of new composite material instead of a physical mixture of hydrocalumite and 1. Furthermore hydrodynamic radius of SOM 2 has been monitored by dynamic light scattering method which shows greater hydrodynamic radius compared to hydrodynamic radius of hydrocalumite. This is also attributed to the formation of composite material in which we believe that, deposition of 1 on the surface of hydrocalumite leads to an increase in hydrodynamic radius of SOM 2 (Fig. S3†). Further we have measured the surface area of starting hydrocalumite as well as composite SOM 2. The surface area of hydrocalumite is 330 cm2 g−1 while that of SOM 2 is 300 cm2 g−1, which indicates that loading of 1 partly blocks the pores of hydrocalumite therefore the surface area of hydrocalumite decreases after formation of composite with 1. From all the above characterization techniques we can conclude that we have synthesized a new composite material SOM 2 by simple sonication method. We now use this material as a photocatalyst in the polymerization of acrylic acid under UV-light (λ = 373 nm, energy density = 19.2 mW cm−2).
Ot stretching and second at 800 cm−1 that corresponds to W–O–W stretching. Other two peaks are obtained at 1443 and 870 cm−1 respectively, which correspond to the presence of hydrocalumite in SOM 2. All the peaks are well shifted from the IR spectra of pure materials (i.e. hydrocalumite and 1), which further reveals the formation of new composite material instead of a physical mixture of hydrocalumite and 1. Furthermore hydrodynamic radius of SOM 2 has been monitored by dynamic light scattering method which shows greater hydrodynamic radius compared to hydrodynamic radius of hydrocalumite. This is also attributed to the formation of composite material in which we believe that, deposition of 1 on the surface of hydrocalumite leads to an increase in hydrodynamic radius of SOM 2 (Fig. S3†). Further we have measured the surface area of starting hydrocalumite as well as composite SOM 2. The surface area of hydrocalumite is 330 cm2 g−1 while that of SOM 2 is 300 cm2 g−1, which indicates that loading of 1 partly blocks the pores of hydrocalumite therefore the surface area of hydrocalumite decreases after formation of composite with 1. From all the above characterization techniques we can conclude that we have synthesized a new composite material SOM 2 by simple sonication method. We now use this material as a photocatalyst in the polymerization of acrylic acid under UV-light (λ = 373 nm, energy density = 19.2 mW cm−2).
Polymerization of acrylic acid
Polymerization reaction of acrylic acid is performed under UV light. In each case 5 mL dispersion of SOM 2 is taken in quartz tube and 500 μL of acrylic acid is mixed & kept in UV photo reactor (λ = 373 nm, energy density = 19 mW cm−2) with irradiation time of 12 h. The polymerization reaction is terminated by adding acid solution. Polymerization of acrylic acid is confirmed from GPC. Molecular weight (mass average molecular weight) of polyacrylic acid (PAA) obtained from polymerization reaction is 1.2 kDa. In this process catalyst acts as co-initiator. We have also prepared another composite catalyst material with hydrocalumite and AIBN (3). As we already know that AIBN is used as a commercial polymerization catalyst.62–64 The reaction follows radical pathway and when the same reaction is carried out as above for 2 hours, PAA with molecular weight of 150 kDa is obtained.Effect of catalyst loading on the Mw of the polymer
Now we are interested to observe the effect of catalyst loading on the Mw of PAA. From the catalyst loading study (Fig. 3a) it is observed that for all different catalyst loading Mw of all PAA are in the same range of around 1.2 kDa (Fig. 4a) while that for 3 is around 150 kDa (Fig. 4a). Thus we conclude that for a given catalyst, chain length of PAA formed depends not on the catalyst loading in hydrocalumite-catalyst composite but it depends on the nature of catalyst or more precisely the pore size of the hydrocalumite. This is so because only pore size of hydrocalumite has remain unchanged in all the reactions for a given catalyst, while other parameters have varied. Hence for a particular catalyst at different loading of initiator catalyst in hydrocalumite, amount of initiator catalyst present on the pore is same; therefore we get constant chain length of PAA. But chain length of PAA is different for different initiator catalyst-hydrocalumite composite. This is explained by considering the amount of initiator catalyst present in the pores of hydrocalumite. As for 1 it interacts with the surface of hydrocalumite strongly through non bonded ionic interaction, thus large amount 1 is present in the pores of hydrocalumite, but in case of AIBN as it is an organic molecule its interaction with the porous surface of hydrocalumite is less therefore less amount of AIBN is held by the hydrocalumite pores. Thus SOM 2 generates large number of radicals in the reaction that leads to generation of small chain length polyacrylic acid, where as in case of 3 the situation is just the reverse. In case of 3 less number of radicals are generated therefore we get long chain length polyacrylic acid, due to lesser loading of initiator catalyst AIBN on hydrocalumite pores.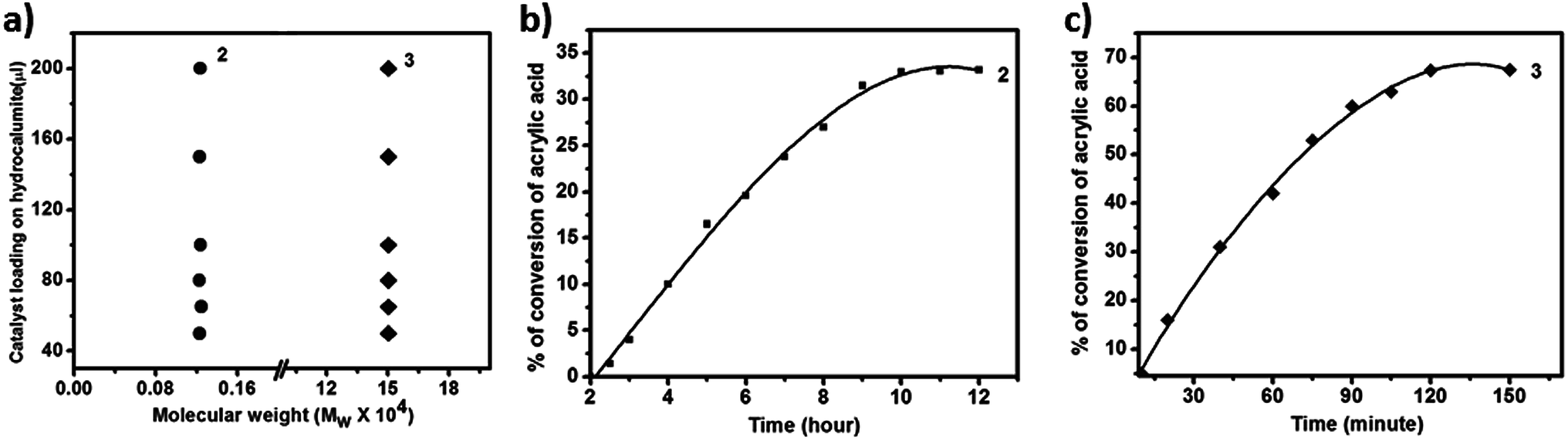 | ||
| Fig. 4 (a) Mass average molecular weight distribution of polyacrylic acid with different loading of catalyst on hydrocalumite. (b) Time dependent photo polymerization of acrylic acid using 2 as a catalyst. (c) Time dependent photo polymerization of acrylic acid using 3 as a catalyst. | ||
Kinetics study of polymerization reaction of acrylic acid
We have also performed time dependent polymerization experiment with both SOM 2 (Fig. 4b) and 3 (Fig. 4c). Time dependent study reveals that it follows general heterogeneous catalyst kinetics.65 Initially with increasing time polymer concentration increases rapidly. However at the later stages the rate of increment of polymerization reaction decrease with time and finally it has almost ceased (Fig. 4b and c). From time dependent study it is observed that 3 undergoes rapid polymerization compared to 2, this may be due to the higher activity of AIBN as compared to 1. In case of SOM 2 the extent of conversion of acrylic acid is around 33% while in case of 3 extent of conversion is around 67%. Furthermore we have studied the stability of the catalyst during the course of photo polymerization reaction.We have performed the HATR-IR spectroscopy of SOM 2 before and after reaction, and it is observed that the catalyst is stable throughout the course of the reaction (Fig. 5a). As SOM 2 is stable throughout the reaction, therefore we can reuse the catalyst for further reactions. We have performed polymerization reaction with this catalyst up to 5 cycles (Fig. 5b). The catalyst is stable up to 5 cycles (Fig. 5a). In all cases extent of polymerization of acrylic acid is almost same and molar mass obtained in each case is also the same. Hence we can say we have developed a new softoxometalate based polymerization catalyst which is stable and can control the chain length of polyacrylic acid.
 | ||
| Fig. 5 (a) HATR-IR spectroscopy of SOM 2 before reaction and after reaction (after different catalytic cycle). (b) Percentage of acrylic acid conversion at different catalytic cycle using SOM 2 as a catalyst. | ||
Now we are in a position to describe the pathway of the polymerization reaction. Here we show a possible reaction path way (Fig. 6a). In the composite catalyst system 1 acts as active photocatalyst, hydrocalumite does not absorb any light, and hence photochemical activity of SOM 2 solely depends on 1 (Fig. 6c). Initially SOM 2 absorbs light and goes to the excited state SOM 2*. A H˙ radical is transferred from imidazolium group to PW12 unit. This leads to the reduction of PW12 unit and generation of imidazolium radical cation. This imidazolium radical cation further initiates the radical/cationic polymerization process. The reduced PW12 unit is again oxidized by air when we keep the sample in open air. To prove that the reaction follows radical path way we have performed EPR spectroscopy at intermediate stages of the reaction (Fig. 6b) which gives a peak in the spectrum and it indicates presence of radical in the reaction mixture. Further proof that the reaction proceeds through a radical pathway and has been proved by using catechol in the reaction system, in presence of catechol quenching of radicals, inhibit polymerization reaction altogether which indicates the reaction follows radical pathway (Fig. 6d).
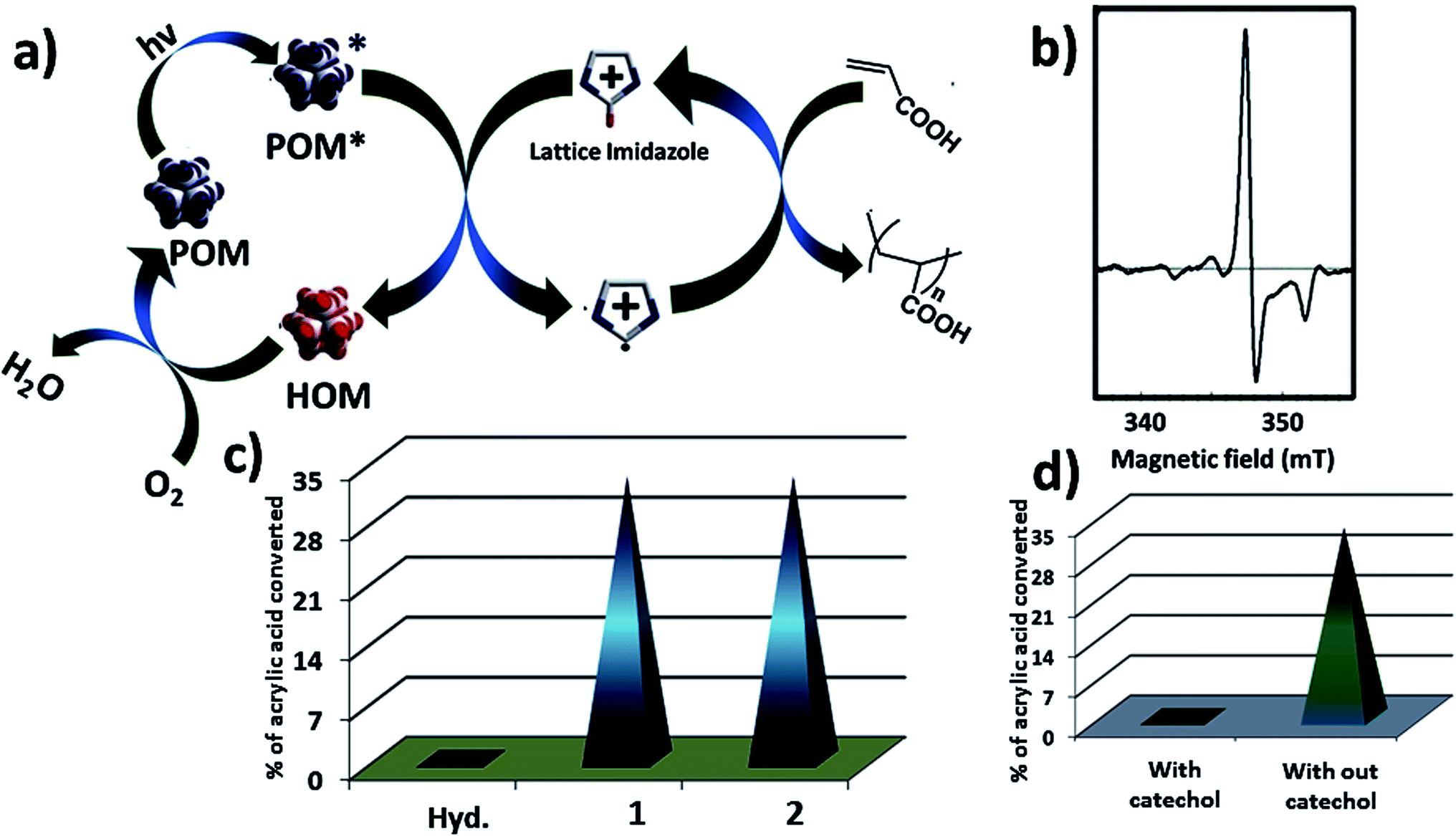 | ||
| Fig. 6 (a) Possible reaction pathway of photo polymerization using SOM 2 as a catalyst. (b) EPR spectrum of SOM 2 after 15 minute of reaction showing radical generates in reaction. (c) Photo polymerization of acrylic acid using hydrocalumite, 1 and SOM 2 as a photo catalyst. (d) Photo polymerization in presence of catechol and in absence of catechol. | ||
Conclusion
To summarize here we have introduced a new designed softoxometalate, which is formed from two component materials, viz., a porous template (i.e. hydrocalumite) and a polyoxometalate i.e. 1. We have prepared it just by simple sonication. The polyoxometalate 1 goes to the pores of hydrocalumite and forms the softoxometalate in water. This softoxometalate forms stable dispersion in water, and it is characterized by X-ray diffraction, SEM-EDS, HATR-IR, Raman spectroscopy. The composite SOM 2 is used as a photo catalyst in the polymerization of acrylic acid. It can control the chain length of polyacrylic acid. The chain length of polymer depends on the interaction of POMs with the pores of hydrocalumite. More favorable is the interaction higher is the loading of POM 1 on hydrocalumite. Higher the loading more radicals are generated and hence lower is the Mw of PAA produced. In case of SOM 2 (with POM 1) as catalyst, the PAA Mw is 1.2 KDa for all catalyst loading. However as we change POM 1, with AIBN, as AIBN has unfavorable interaction with hydrocalumite and hence lesser loading of AIBN take place with hydrocalumite composite (3). For all loading with AIBN based catalyst the Mw of PAA is obtained to be 150 kDa, since the radical concentration in this case is low. In short we have exploited the interaction of an oxometalate in a SOM, based on hydrocalumite to control polymer chain length. This method may be explored to all layer double hydroxide for controlling polymer chain lengths, in cationic as well as in radical polymerization.Experimental procedure
Material and reagents
All the materials were purchased from commercially available sources and used without further purification. All the glasswares were first boiled in acid bath then washed with water and finally cleaned with acetone. They were properly dried in hot air oven over-night. We have used doubly distilled deionized water in all the experiments.Synthesis of hydrocalumite and dispersed it in water
Hydrocalumite was synthesized by following the typical method, 2 M NaOH solution was added dropwise to a mixed aqueous solution of calcium chloride (2 mM) and aluminium chloride (1 mM) at 65 °C under vigorous stirring. The concentration of metal ion was adjusted to 0.66 mol L−1, and finally reaction pH was adjusted at 11.5. The resulting precipitate was kept at 65 °C for 18 h. After that the reaction mixture was cooled at room temperature and filtered through glass flirt. Further the precipitate was washed with 200 mL of doubly distilled water and dried under vacuum. Further it was dried at 100 °C for one day. Solid hydrocalumite was obtained and used for catalysis.Preparation of dispersion of hydrocalumite
20 mg of hydrocalumite was mixed in 5 mL of doubly distilled water and further sonicated at room temperature for an hour; a homogeneous dispersion of hydrocalumite was obtained.Synthesis of [BMIm]2[DMIm][α-PW12O40] (1)
The ionic liquid BMImCl (8.725 g, 50 mmol) was added slowly to a solution of phosphotungstic acid (1.44 g, 5 mM) with vigorous stirring at 30 °C. A white dispersion was formed in a minute, then the reaction mixture was kept at 30 °C for one day to complete the reaction. The reaction mixture was cooled at room temperature and filtered through glass flirt. The precipitate was washed with doubly distilled water for several times and the precipitate was re-dissolved in acetonitrile. White crystals appeared after a week which were dried under vacuum.Synthesis of [BMIm]2[DMIm][α-PW12O40]@hydrocalumite composite (SOM 2)
To prepare 2 we have first prepared a dispersion of hydrocalumite (2 mg mL−1). Further 20 mg of 1 was dissolved in 2 mL DMSO. Now 1 was added (different proportion) in a 5 mL dispersion of hydrocalumite (2 mg mL−1) and further 5 mL water was added. Then the reaction mixture was sonicated for an hour. The composite material 2 was formed as a homogeneous dispersion in water. The dispersion kept for a day formed gel at the bottom of the reaction container which further indicated the formation of composite material, if we have further sonicated this gel it again formed stable dispersion in water (Table 1).| Sl. No. | Hydrocalumite (4 mg mL−1) | [BMIm]2[DMIm][α-PW12O40] (10 mg mL−1) |
|---|---|---|
| 1 | 5 mL | 50 μL |
| 2 | 5 mL | 65 μL |
| 3 | 5 mL | 80 μL |
| 4 | 5 mL | 100 μL |
| 5 | 5 mL | 200 μL |
General procedure of sonication
10 mL sample is taken in a sealed vial, and placed it in an ultrasonicator chamber which contained 200 mL of water. We have sonicated the sample in it for one hour.General procedure of photo polymerization of acrylic acid
In a quartz tube, 5 mL dispersion of SOM 2 has taken in a quartz tube. 0.5 mL acrylic acid was added to the catalyst and shaken well to mix it properly. The reaction mixture was sealed and bubbled with nitrogen for half an hour. Then the reaction mixture was kept in the photo reactor (energy density = 0.19 mW cm−2, λ = 373 nm) for 12 h. After 12 h reaction was terminated by adding dilute acid solution. Further the solvent was evaporated in vacuum. A solid residue of polyacrylic acid and SOM 2 was present in the reaction chamber. Polyacrylic acid was dissolved in water and it was separated from SOM 2 via centrifugation. Solid SOM 2 was present in the bottom of centrifuged tube which was further washed five times with water to remove polyacrylic acid completely (removal of PAA was confirmed from FT-IR spectroscopy). Further we have dispersed the catalyst in water again and performed the polymerization of acrylic acid in the next cycle.Characterization techniques
Acknowledgements
SR gratefully acknowledges the grants from IISER-Kolkata, India, DST-fast track and BRNS-DAE grant.References
- J. E. Mark, Polymer Data Handbook, Oxford University Press, New York, 1999 Search PubMed.
- K. J. Lissant, US Pat., US3974116 A, 1976.
- R. L. Ibsen, A. Mathews, T. C. Chadwick and X. Yu, US Pat., US6458340 B1, 2002.
- J. W. Johnson, R. D. Prottas and C. H. Strolle, US Pat., US5051464 A, 1991.
- H. Omidian and K. Park, J. Drug Delivery Sci. Technol., 2008, 18, 83–93 CrossRef CAS.
- Y. Irie, K. Kajikawa, H. Takahashi and T. Fujiwara, US Pat., US5264495 A, 1993.
- A. Pourjavadi, M. Ayyari and M. Amini-Fazl, Eur. Polym. J., 2008, 44, 1209–1216 CrossRef CAS.
- S. Biswas, E. Mani, A. Mondal, A. Tiwari and S. Roy, Soft Matter, 2016, 12, 1989–1997 RSC.
- P. Cañizares, Á. Pérez and R. Camarillo, Desalination, 2002, 144, 279–285 CrossRef.
- S. Kiatkamjornwong, W. Chomsaksakul and M. Sonsuk, Radiat. Phys. Chem., 2000, 59, 413–427 CrossRef CAS.
- S. Francis, M. Kumar and L. Varshney, Radiat. Phys. Chem., 2004, 69, 481–486 CrossRef CAS.
- A. Bhattacharya, Prog. Polym. Sci., 2000, 25, 371–401 CrossRef CAS.
- D. M. Devine, S. M. Devery, J. G. Lyons, L. M. Geever, J. E. Kennedy and C. L. Higginbotham, Int. J. Pharm., 2006, 326, 50–59 CrossRef CAS PubMed.
- G.-B. Cai, G.-X. Zhao, X.-K. Wang and S.-H. Yu, J. Phys. Chem. C, 2010, 114, 12948–12954 CAS.
- C. Ladavière, N. Dörr and J. P. Claverie, Macromolecules, 2001, 34, 5370–5372 CrossRef.
- L. Couvreur, C. Lefay, J. Belleney, B. Charleux, O. Guerret and S. Magnet, Macromolecules, 2003, 36, 8260–8267 CrossRef CAS.
- K. Matyjaszewski and T. P. Davis, Handbook of radical polymerization, Wiley Online Library, 2002 Search PubMed.
- C. J. Hawker, J. Am. Chem. Soc., 1994, 116, 11185–11186 CrossRef CAS.
- B. P. Fors and C. J. Hawker, Angew. Chem., Int. Ed., 2012, 51, 8850–8853 CrossRef CAS PubMed.
- D. Bertin, D. Gigmes, S. R. Marque and P. Tordo, Chem. Soc. Rev., 2011, 40, 2189–2198 RSC.
- J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013, 38, 63–235 CrossRef CAS.
- D. Gigmes and S. R. Marque, Nitroxide-Mediated Polymerization and its Applications, Wiley Online Library, 2012 Search PubMed.
- M. Semsarilar and S. Perrier, Nat. Chem., 2010, 2, 811–820 CrossRef CAS PubMed.
- G. Moad, E. Rizzardo and S. H. Thang, Polym. Int., 2011, 60, 9–25 CrossRef CAS.
- S. Xu, J. Li and L. Chen, J. Mater. Chem., 2011, 21, 4346–4351 RSC.
- D. J. Keddie, Chem. Soc. Rev., 2014, 43, 496–505 RSC.
- S. Perrier and P. Takolpuckdee, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5347–5393 CrossRef CAS.
- D. A. Shipp, J.-L. Wang and K. Matyjaszewski, Macromolecules, 1998, 31, 8005–8008 CrossRef CAS.
- J. Xia, S. G. Gaynor and K. Matyjaszewski, Macromolecules, 1998, 31, 5958–5959 CrossRef CAS.
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef CAS.
- A. J. Magenau, N. C. Strandwitz, A. Gennaro and K. Matyjaszewski, Science, 2011, 332, 81–84 CrossRef CAS PubMed.
- L. Kan, Z. Xu and C. Gao, Macromolecules, 2010, 44, 444–452 CrossRef.
- O. Eksik, M. A. Tasdelen, A. T. Erciyes and Y. Yagci, Compos. Interfaces, 2010, 17, 357–369 CrossRef CAS.
- G. Zhang, I. Y. Song, K. H. Ahn, T. Park and W. Choi, Macromolecules, 2011, 44, 7594–7599 CrossRef CAS.
- D. S. Chu, J. G. Schellinger, J. Shi, A. J. Convertine, P. S. Stayton and S. H. Pun, Acc. Chem. Res., 2012, 45, 1089–1099 CrossRef CAS PubMed.
- A. Asati, S. Santra, C. Kaittanis, S. Nath and J. M. Perez, Angew. Chem., Int. Ed., 2009, 48, 2308–2312 CrossRef CAS PubMed.
- S. Fréal-Saison, M. Save, C. Bui, B. Charleux and S. Magnet, Macromolecules, 2006, 39, 8632–8638 CrossRef.
- P. E. Millard, L. Barner, M. H. Stenzel, T. P. Davis, C. Barner-Kowollik and A. H. Müller, Macromol. Rapid Commun., 2006, 27, 821–828 CrossRef CAS.
- H.-Y. Yu, Z.-K. Xu, Q. Yang, M.-X. Hu and S.-Y. Wang, J. Membr. Sci., 2006, 281, 658–665 CrossRef CAS.
- S. Muthukrishnan, E. H. Pan, M. H. Stenzel, C. Barner-Kowollik, T. P. Davis, D. Lewis and L. Barner, Macromolecules, 2007, 40, 2978–2980 CrossRef CAS.
- T. Wan, X. Wang, Y. Yuan and W. He, J. Appl. Polym. Sci., 2006, 102, 2875–2881 CrossRef CAS.
- K. D. Jandt and R. W. Mills, Dent. Mater., 2013, 29, 605–617 CrossRef CAS PubMed.
- P. Kissel, D. J. Murray, W. J. Wulftange, V. J. Catalano and B. T. King, Nat. Chem., 2014, 6, 774–778 CrossRef CAS PubMed.
- M.-A. Tehfe, F. Dumur, B. Graff, D. Gigmes, J.-P. Fouassier and J. Lalevée, Macromolecules, 2013, 46, 3332–3341 CrossRef CAS.
- S. Roy, CrystEngComm, 2014, 16, 4667–4676 RSC.
- S. Das, S. Kumar, A. Mallick and S. Roy, J. Mol. Eng. Mater., 2015, 3, 1540008 CrossRef CAS.
- S. Roy, Comments Inorg. Chem., 2011, 32, 113–126 CrossRef CAS.
- S. Das, P. Thomas and S. Roy, Eur. J. Inorg. Chem., 2014, 2014, 4551–4557 CrossRef CAS.
- S. Das, A. Misra and S. Roy, New J. Chem., 2015, 40, 994 RSC.
- S. Roy, H. J. Meeldijk, A. V. Petukhov, M. Versluijs and F. Soulimani, Dalton Trans., 2008, 2861–2865 RSC.
- S. Roy, M. C. Mourad and M. T. Rijneveld-Ockers, Langmuir, 2007, 23, 399–401 CrossRef CAS PubMed.
- D. Chen, A. Sahasrabudhe, P. Wang, A. Dasgupta, R. Yuan and S. Roy, Dalton Trans., 2013, 42, 10587–10596 RSC.
- K. Das and S. Roy, Chem.–Asian J., 2015, 10, 1884–1891 CrossRef CAS PubMed.
- A. Mallick, D. Lai and S. Roy, New J. Chem., 2016, 40, 1057 RSC.
- P. Thomas, C. Pei, B. Roy, S. Ghosh, S. Das, A. Banerjee, T. Ben, S. Qiu and S. Roy, J. Mater. Chem. A, 2015, 3, 1431–1441 CAS.
- Y. Kuwahara, K. Tsuji, T. Ohmichi, T. Kamegawa, K. Mori and H. Yamashita, Catal. Sci. Technol., 2012, 2, 1842–1851 CAS.
- M. a. J. Campos-Molina, J. Santamaría-González, J. Mérida-Robles, R. n. Moreno-Tost, M. C. Albuquerque, S. Bruque-Gámez, E. Rodríguez-Castellón, A. Jiménez-López and P. Maireles-Torres, Energy Fuels, 2009, 24, 979–984 CrossRef.
- Y. Kuwahara, K. Tsuji, T. Ohmichi, T. Kamegawa, K. Mori and H. Yamashita, ChemSusChem, 2012, 5, 1523–1532 CrossRef CAS PubMed.
- F. Cavani, F. Trifirò and A. Vaccari, Catal. Today, 1991, 11, 173–301 CrossRef CAS.
- D. G. Cantrell, L. J. Gillie, A. F. Lee and K. Wilson, Appl. Catal., A, 2005, 287, 183–190 CrossRef CAS.
- M. Di Serio, M. Ledda, M. Cozzolino, G. Minutillo, R. Tesser and E. Santacesaria, Ind. Eng. Chem. Res., 2006, 45, 3009–3014 CrossRef CAS.
- Q.-L. Zhao, E.-H. Liu, G.-X. Wang, Z.-H. Hou, X.-H. Zhan, L.-c. Liu and H. Wu, J. Polym. Res., 2014, 21, 1–6 CAS.
- Q. Zhang, J. M. Galvan-Miyoshi, F. Pezzotti and W. Ming, Eur. Polym. J., 2013, 49, 2327–2333 CrossRef CAS.
- G. Gody, T. Maschmeyer, P. B. Zetterlund and S. Perrier, Nat. Commun., 2013, 4 DOI:10.1038/ncomms3505.
- J. J. Stracke and R. G. Finke, ACS Catal., 2014, 4, 909–933 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra02685k |
| This journal is © The Royal Society of Chemistry 2016 |