
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enzyme-responsive polyion complex (PIC) nanoparticles for the targeted delivery of antimicrobial polymers†
Ignacio
Insua
a,
Evangelos
Liamas
b,
Zhenyu
Zhang
b,
Anna F. A.
Peacock
a,
Anne Marie
Krachler
cd and
Francisco
Fernandez-Trillo
*ad
aSchool of Chemistry, University of Birmingham, B15 2TT Birmingham, UK
bSchool of Chemical Engineering, University of Birmingham, B15 2TT Birmingham, UK
cSchool of Biosciences, University of Birmingham, B15 2TT Birmingham, UK
dInstitute of Microbiology and Infection, University of Birmingham, B15 2TT Birmingham, UK. E-mail: f.fernandez-trillo@bham.ac.uk
First published on 16th March 2016
Abstract
Here we present new enzyme-responsive polyion complex (PIC) nanoparticles prepared from antimicrobial poly(ethylene imine) and an anionic enzyme-responsive peptide targeting Pseudomonas aeruginosa's elastase. The synthetic conditions used to prepare these nanomaterials allowed us to optimise particle size and charge, and their stability under physiological conditions. We demonstrate that these enzyme responsive PIC nanoparticles are selectively degraded in the presence of P. aeruginosa elastase without being affected by other endogenous elastases. This enzyme-responsive PIC particle can exert an elastase-specific antimicrobial effect against P. aeruginosa without affecting non-pathogenic strains of these bacteria. These targeted enzyme-responsive PIC nanoparticles constitute a novel platform for the delivery of antimicrobial peptides and polymers, and can be a powerful tool in the current race against antimicrobial resistance.
Introduction
There is an increasing concern with the emergence of microbial strains that can resist the action of current antimicrobials.1–3 As evidenced over the last decades, the limited pipeline of new antibiotics cannot cope with this increasing number of resistant microbes,4–6 and new antimicrobial strategies are needed that go beyond the development of new drugs.7,8 One such strategy is the re-evaluation of drug candidates, which have been discarded due to toxicity and side effects. This is the case for antimicrobial peptides,9–11 that are amongst the few last-resort treatments against multidrug resistant strains.12,13Despite being powerful antimicrobial agents, the widespread use of antimicrobial peptides in the clinic is limited by their inactivation by serum proteases, toxicity to eukaryotic cells and high production cost.9–11 The positively-charged amphiphilic structure of antimicrobial peptides is responsible for damaging bacterial membranes and cytosol components, resulting in a fast and multi-target antimicrobial action with broad spectra, but also in toxicity to the host.9–11 Polymeric mimics of these peptides have been developed as an affordable alternative with increased protease stability and selectivity for prokaryotic cells. However, the clinical application of these polymer mimics is compromised by poor bioavailability, limited diffusion in tissues, polyelectrolyte complexation with negatively charged microbial peptidoglycans and extracellular polymers, and insufficient toxicological characterisation.14,15 Further strategies to improve the therapeutic profile of antimicrobial peptides and mimics include masking of the positive charge.16 However this often results in a reduced antimicrobial activity as a consequence of slow or incomplete cleavage of the masking group.
Less explored has been the use of controlled (and ideally targeted) delivery when optimising antimicrobial therapeutic profile.17,18 However, this is a very attractive alternative to improve the medical profile of current antimicrobials: strategies that can deliver antimicrobials at the site of infection can minimise off-target effects such as toxicity or the emergence of reservoirs of microbial resistance genes in non-pathogenic strains such as commensal bacteria.5
Towards this end, here we present a new targeted enzyme-responsive nanoparticle that selectively delivers antimicrobial polymers in the presence of Pseudomonas aeruginosa, whose infections are very hard to treat.19 In our system, the toxicity of cationic antimicrobial poly(ethylene imine) (PEI) was minimised through polyelectrolyte complexation with an anionic peptide, to form well-defined polyion complex (PIC) nanoparticles (Scheme 1). We demonstrate that our synthetic approach is highly modular, letting us tune particle size and charge, and the stability of the PIC nanoparticles under physiological conditions. By introducing a P. aeruginosa's elastase (LasB) cleavable sequence into our anionic peptide, PIC nanoparticles could be degraded in the presence of this P. aeruginosa's virulence factor,20,21 and more importantly, remained unchanged when treated with human leucocyte elastase (HLE), a human elastase secreted at the site of infection. Finally, we demonstrate that these enzyme-responsive PIC nanoparticles have a targeted antimicrobial effect against P. aeruginosa and that this antimicrobial effect is not observed for P. aeruginosa strains that do not produce the triggering LasB elastase.
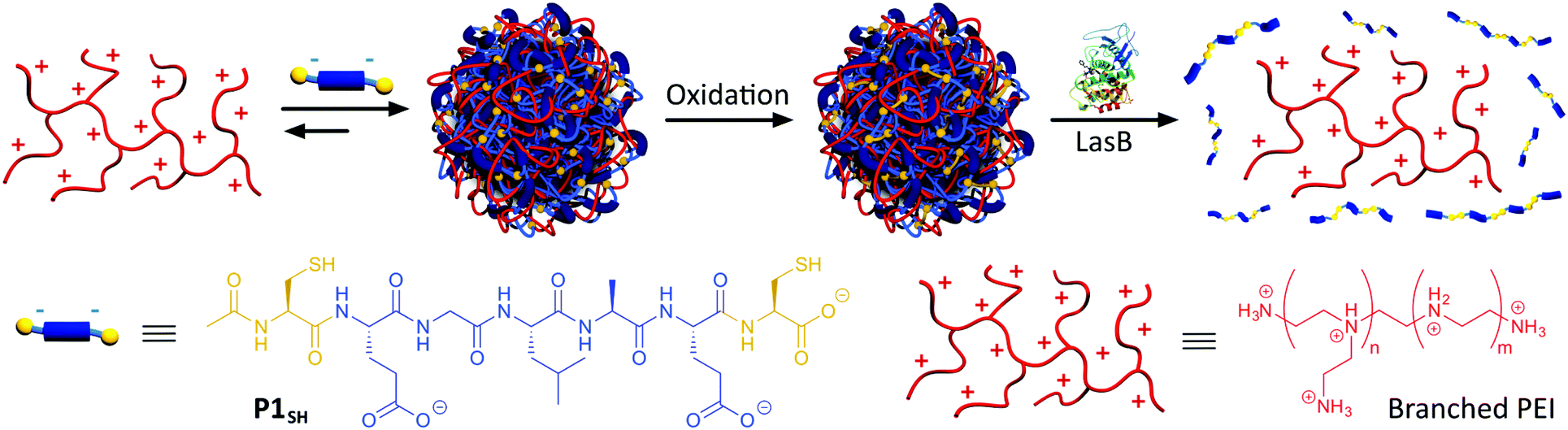 | ||
| Scheme 1 Assembly and oxidative cross-linking of PIC nanoparticles from P1SH (Ac-C-E-GLA-E-C-OH) and antimicrobial branched PEI. Degradation of PIC nanoparticles by LasB and subsequent PEI release. | ||
Experimental
Materials
Enzymes (Pseudomonas aeruginosa Elastase (LasB): EC 3.4.24.26 and Human Leucocyte Elastase (HLE): EC 3.4.21.37) were purchased from Merck Millipore. Branched poly(ethylene imine) 25 kDa average molecular weight (B-PEI25), 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES), Luria Bertani (LB) broth (Miller) and Dulbecco's modified eagle medium (DMEM) were bought from Sigma-Aldrich®. Fluorescamine and dimethylsulphoxide (DMSO) were purchased from Acros Organics™. Ethylenediaminetetraacetic acid (EDTA) was purchased from Alfa Aesar®. Nylon 0.45 μm syringe filters were purchased from Camlab. P. aeruginosa strains were kindly donated by Suzanne M. J. Fleiszig. We studied PAO1V as a pathogenic strain and a deletion strain of lasA and lasB genes of PAO1V was used as LasB-negative control (ΔlasAB).22 Live/Dead® BacLight™ viability kit was purchased from Thermo Fisher Scientific Inc.Instrumentation
Dynamic light scattering (DLS) and ζ-potential measurements were carried out in a Zetasizer Nano ZS (Malvern Instruments Ltd) stabilised at 37 °C. DLS was read at 173° (backscattering) for 60 seconds in triplicate and ζ-potentials were recorded 30 times at 140 V. A FLUOstar Omega (BMGLabtech Gmbh) microplate reader was used to measure fluorescamine reactions. Atomic force microscopy (AFM) measurements were carried out in 5 mM HEPES buffer at pH 7.4 with intermittent contact mode, using a NanoWizard® II (JPK Instruments Ltd) fitted with SiNi cantilevers (BudgetSensors®) with a spring constant of 0.27 N m−1. PIC particle size was measured from AFM images with ImageJ software (version 1.48v). FACS was performed on an Attune® Acoustic Focusing Cytometer (Applied Biosystems™) measuring 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 events per sample. BacLight™ fluorescent dyes were excited at 488 nm and emission was read at 515–545 and 640 nm for green and red probes, respectively.
000 events per sample. BacLight™ fluorescent dyes were excited at 488 nm and emission was read at 515–545 and 640 nm for green and red probes, respectively.
Enzymatic degradation – fluorescamine assay
Stock solutions of peptide (1 mM) or succinyl casein (0.5 mg mL−1) were prepared in 2.5 mM Na2B4O7 buffer at pH 8.0 with 10 mM CaCl2 and 10% DMSO. 125 μL of these substrate solutions were added to a 96-well black-walled microplate and mixed with 125 μL of the same buffer without DMSO, containing 0.6 μg of enzyme (LasB or HLE). Solutions of enzymes and substrates alone were prepared as blanks. Every sample was prepared in triplicate. The microplate was incubated at 37 °C for 4 hours under orbital shaking. After 4 hours, 50 μL of 0.1 M ethylenediaminetetraacetic acid (EDTA) in water at pH 8.0 were added to each well to quench all enzymatic activity.23 Then, each sample was mixed in a 1:1 volume with a 1 mM solution of fluorescamine24 in methanol. The microplate was incubated at 37 °C under orbital shaking for 30 minutes. After this time, fluorescence was measured exciting at 355 nm and reading the emission at 460 ± 10 nm.
Preparation of PIC nanoparticles
For nanoparticles prepared at a 1:0.3 N:COOH ratio (defined as the ratio between amines in PEI and carboxylic acids in the peptides), stock solutions of B-PEI25 (2.5 mM in amines) and Ac-C-E-GLA-E-C-OH (P1SH) (0.25 mM) in 5 mM 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES) buffer at pH 7.4 were prepared. Then, both solutions were filtered and mixed drop-wise under stirring. The reaction mixture was stirred at room temperature for 24 hours open to air to allow thiol oxidation with atmospheric oxygen. PIC nanoparticles prepared at different N:COOH ratios were obtained by changing the concentration of peptide stock solution and mixing with 2.5 mM B-PEI25 following the same protocol (Table S2†). After 24 hours, samples were analysed directly by DLS and ζ-potential without prior filtration.
Enzymatic degradation of PIC nanoparticles
1 mL of PIC nanoparticles prepared at 1:0.3 N:COOH ratio was incubated at 37 °C with 50 μL (5 μg) of LasB in 2.5 mM Na2B4O7 buffer at pH 8.0. As controls, PIC nanoparticles were incubated under the same conditions with HLE (5 μg) or Na2B4O7 buffer to assess the enzymatic specificity and effect of salts in the buffer, respectively. Each sample was prepared in triplicate from three different batches of PIC nanoparticles. DLS data was acquired over time (1–4 hours). The number of amines was monitored after 4 hours of incubation as described before (fluorescamine assay).
Antimicrobial activity of PIC nanoparticles
1 mL aliquots of P. aeruginosa PAO1V and ΔlasAB cultures in LB broth (OD600 = 1.0) were centrifuged and resuspended in 1 mL of DMEM. These samples were incubated at 37 °C for 6 hours under orbital shaking to allow LasB production. After this time, samples were centrifuged and half of the supernatant replaced with the same volume of 5 mM HEPES buffer at pH 7.4 (live control), PIC nanoparticles (prepared at a 1:0.3 N:COOH ratio as described above) and 1.25 mM B-PEI25 in 5 mM HEPES buffer at pH 7.4. A dead control was also prepared by replacing all the supernatant after centrifugation with 70% v/v aqueous 2-propanol. Samples were resuspended and incubated at 37 °C under orbital shaking. Each sample was prepared in triplicate. Every hour, 200 μL of each sample were taken and the rest was kept shaking at 37 °C. These 200 μL aliquots were incubated in the dark for 10 minutes with 1 μL of a 10-fold diluted 1:1 mixture of BacLight™ probes in DMEM. Dead control samples had to be centrifuged and resuspended in assay buffer before staining with fluorescent dyes. Then, samples were analysed by FACS setting gates for dead bacteria in green versus red emission dot plots from the live and dead controls.
Results and discussion
Peptide synthesis and enzymatic degradation
Our goal was to develop a delivery method that did not require chemical modification of the antimicrobial. PIC particles25 are ideal for this purpose because the cationic antimicrobial peptides or mimics thereof can be encapsulated within a particle formed upon polyelectrolyte complexation with negatively charged materials. This strategy of complexing oppositely charged materials has been extensively used to deliver other “precious” therapeutics, in particular nucleic acids.26In our case, PIC nanoparticles would be prepared by the combination of a tailor-made anionic peptide and the positively charged backbone of the antimicrobial (Scheme 1). In addition, we wanted our system to be selectively degraded in the presence of pathogenic bacteria, but not in their absence: for example, in the presence of commensal bacteria or non-pathogenic strains, unable to produce virulence factors. As such, our system would be pathogen specific and enable a targeted delivery of the antimicrobial. Here we chose P. aeruginosa as a model pathogen: P. aeruginosa is an opportunistic pathogen commonly associated with hospital-acquired infections, often colonising wounds and immunocompromised patients.19 Interestingly, amongst the virulence factors that P. aeruginosa secretes to mediate infection, LasB,20,21 a zinc-metalloprotease, constituted an excellent target to develop our system. LasB has elastolytic activity and a preference to cleave sites with small hydrophobic residues.27,28 Thus, we designed our anionic peptides to contain a short LasB-cleavable sequence (e.g. GLA,27Scheme 1) flanked by two glutamic acids, responsible for providing overall negative charge to our peptide. We postulated that multivalency would be lost upon LasB mediated cleavage of the peptide, resulting in degradation of the particle and delivery of the antimicrobial (Scheme 1).
Two peptides were prepared at this stage: P1, containing the Gly-Leu-Ala sequence27 and P2, containing a tetraalanine site,28 both sequences having been reported to be cleaved by LasB. The N-terminus of both peptides was capped as a neutral acetamide to mask the cationic amine functionality. All peptides were prepared by solid-phase synthesis in good to excellent yields and high purity without any chromatographic purification (see ESI: section 3, Fig. S1–S6† for experimental details and characterisation).
A fluorogenic assay was then used to evaluate enzymatic degradation of these peptides.24 Peptides were incubated with LasB, treated with fluorescamine and the increase in emission at 460 nm correlated with the amount of amines produced upon enzymatic hydrolysis of the peptides. In each assay, degradation of succinyl casein by the evaluated elastases was monitored, to confirm that the enzymes had not lost their activity upon storage.29 Additionally, the values obtained from fluorescence measurements were normalised to that produced by a model peptide (H2N-LA-E-OH (P3), Fig. S7†), which should be produced upon enzymatic hydrolysis of P1.27 In our hands, the GLA sequence in P1 was degraded to a higher extent than the tetraalanine in P2, showing an increase in amine content of 60% after four hours (Fig. 1). More importantly, when these peptides were incubated with HLE, a human elastase secreted by the immune system during inflammation, little to no degradation was observed for both peptides. This demonstrated that both of these peptides were excellent candidates to develop LasB-responsive nanoparticles, which would only be degraded by LasB, even if endogenous elastases were present in the media. All subsequent experiments were performed with P1 because it is degraded to a greater extent by LasB.
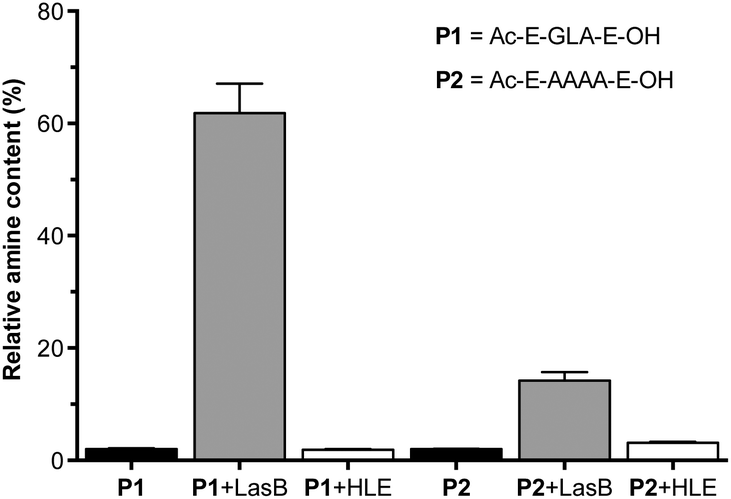 | ||
| Fig. 1 Relative amine content in samples of LasB responsive anionic peptides evaluated in this work. Relative amine content was calculated from fluorescamine conjugates formed following incubation with enzymes for 4 hours and normalised to the fluorescence observed with a model degradation peptide (P3, H2N-LA-E-OH) (Fig. S7†). Peptide blanks (i.e. without enzyme) in black. n = 3. | ||
PEI as a model antimicrobial polymer against P. aeruginosa
Having established the peptide sequence to be employed in the synthesis of our enzyme-responsive PIC nanoparticles, we then turned our attention to the choice of antimicrobial. PEI has been reported as an efficient antimicrobial against both Gram positive and Gram negative bacteria.30,31 Its antimicrobial activity can be tuned as a function of its degree of branching and molecular weight. Moreover, PEI has been extensively applied as a starting material for the preparation of polyelectrolyte complexes.25 These features, and its commercial availability, made it an ideal candidate with which we could optimise the preparation of our PIC nanoparticles.However, the antimicrobial activity of PEI towards P. aeruginosa had not been reported. Thus, five commercial PEIs of various molecular weights and branching were evaluated for their antimicrobial activity against P. aeruginosa. A pathogenic P. aeruginosa strain (PAO1V) was cultured overnight in the absence or presence of different concentrations of PEI and viability checked using a LIVE/DEAD® staining assay (Fig. S8†). This assay reports on membrane integrity and thus is especially suited to evaluate the activity of antimicrobial peptides and mimics. As expected, activity depended on the amount of PEI used. Aggregates could be visibly observed on the sides of the cuvettes for the larger polymers at the highest concentration tested (0.1 mg mL−1), compromising the characterisation of the system. These aggregates may form as a result of the clustering of bacteria by the cationic polymers,32,33 or these polymers being trapped by debris from dead bacteria. Increasing the molecular weight resulted in an increase in the percentage of dead cells. Moreover, the results at 0.01 mg mL−1 suggested that branching had a beneficial effect on antimicrobial activity. Therefore, B-PEI25 was selected as the candidate PEI to prepare our enzyme-responsive PIC nanoparticles.
PIC nanoparticle synthesis
Enzyme-responsive PIC nanoparticles were prepared by mixing stock solutions of the peptide (in 5 mM HEPES buffer at pH 7.4) with a 2.5 mM stock solution of B-PEI25 prepared in the same buffer. Initial attempts to make PIC nanoparticles with P1 did not result in the formation of stable aggregates. Thus, we decided to introduce two cysteine residues in our peptides, to allow for chemical oxidation during the nanoparticle formation and cross-linking of the peptides. This strategy has been employed in the synthesis of other PIC complexes, such as nucleic acid polyplexes.34–36 PIC nanoparticles were successfully made with this modified peptide (P1SH), by mixing peptide and B-PEI25 stock solutions and allowing the nanoparticles to oxidise overnight under stirring and open to air (Scheme 1). The kinetics of thiol cross-linking was studied by monitoring the decrease of accessible free thiols during nanoparticle formation using Ellman's reagent.37 Complete depletion of thiols was observed after nine hours for P1SH in the presence of B-PEI25, whereas only 13% of initial thiols contained in P1SH had oxidised after ten hours in its absence (Fig. S9†). This result is in agreement with the literature, where it is proposed that electrostatic complexation of polyions increases the local concentration of thiols and thus increases oxidation rates.38Different mixtures of B-PEI25 to P1SH (expressed as N:COOH ratio) were evaluated to determine the optimal ratio to prepare PIC nanoparticles (Fig. 2, Table S2†). Of all the N:COOH ratios tested, only those between 1:0.8 and 1:0.3 formed stable PIC nanoparticles. This phenomenon could be due to the lack of enough peptide below 1:0.3 to condense B-PEI25 into particles, and the generation of neutral and unstable coacervates at equimolar mixtures of positive and negative charge, in agreement with the decrease in ζ-potential observed with increasing peptide content (Fig. 2b). Interestingly, we were not able to form PIC nanoparticles in the presence of an excess of COOH groups (i.e. peptide). We believe the nucleation of these PIC nanoparticles is driven by complexation of the anionic peptides onto the B-PEI25 backbone. Once the positive charges on the surface of the growing nanoparticles are neutralised, complexation of further peptide is hindered and thus prevents the formation of negatively charged particles.
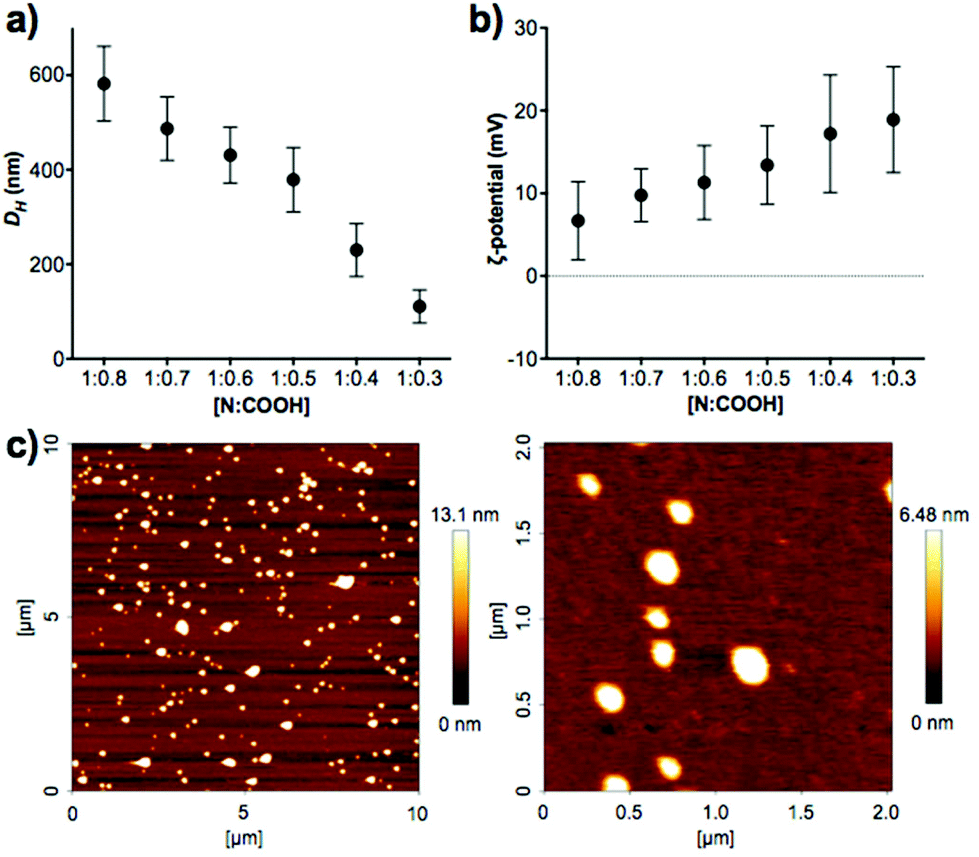 | ||
| Fig. 2 (a) Hydrodynamic diameter (DH) and (b) ζ-potential of P1SH PIC nanoparticles prepared at different N:COOH ratios. (c) AFM characterisation of PIC nanoparticles prepared at a 1:0.3 N:COOH ratio. D = 127 ± 42 nm, n = 48. | ||
A clear trend was found in particle size, with smaller particles being formed with decreasing amounts of peptide (Fig. 2a). AFM measurements were carried out to confirm the size and shape of these nanomaterials. PIC nanoparticles prepared at a 1:0.3 N:COOH ratio were selected for AFM analysis due to their narrow size distribution in DLS characterisation. AFM images showed the presence of spherical nanoparticles of 127 ± 42 nm in diameter (Fig. 2c), in agreement with the 111 ± 35 nm hydrodynamic diameter measured by DLS.
Physiological stability of PIC nanoparticles
PIC aggregates are disrupted in the presence of other electrolytes, which can shield and weaken the electrostatic forces holding the aggregates together, and potentially lead to the rupture of the particles.25 In this regard, the high contents of salts in the body could compromise the efficacy of PIC nanoparticles in biological settings. With this in mind, the stability of PIC nanoparticles under physiological conditions was assessed by diluting them into a final concentration of 154 mM NaCl and incubating them at 37 °C.39 The size of PIC nanoparticles incubated under these conditions was monitored by DLS over four hours (Fig. 3). All PIC nanoparticles tested swelled under experimental conditions, as expected from the addition of small Na+ and Cl− ions that can permeate these soft nanoparticles and shield the interactions between the polyions. A trend in the extent of this swelling was observed and, in general, decreasing the amount of P1SH (i.e. COOH component) resulted in nanoparticles with increasing swelling. Interestingly, only the nanoparticles prepared at a 1:0.3 N:COOH ratio remained stable in size after the initial swelling for the whole duration of the experiment, with all of the other nanoparticles eventually reducing their size after two hours of incubation. PIC nanoparticles prepared at high N:COOH ratios were the most unstable and no aggregates could be detected for most of these systems following incubation for at least two hours (Fig. 3, hollow symbols).
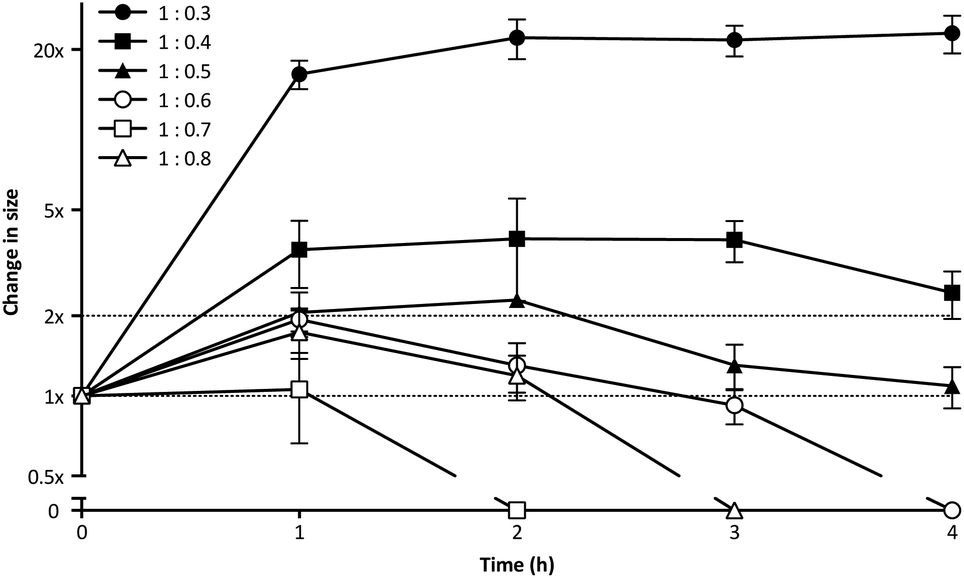 | ||
| Fig. 3 Relative change in size for P1SH PIC nanoparticles prepared at different N:COOH ratios and incubated under simulated physiological conditions (154 mM NaCl, pH 7.4, 37 °C). Particle size was normalised to that of a control in the absence of NaCl (1×). n = 3. | ||
DLS autocorrelation function curves (Fig. S10†), size distributions (Fig. S11†) and count plots (Fig. S12†) strongly support these observations: while PIC nanoparticles prepared with higher concentrations of peptide (N:COOH ratios 1:0.8–1:0.6) were unable to scatter light following incubation with NaCl, in some cases for as little as two hours, those prepared with a lower proportion of peptide (N:COOH ratios 1:0.5–1:0.3) did produce autocorrelation curves that could be fitted by the DLS software. However, only those prepared at a 1:0.3 N:COOH ratio were stable enough to scatter light over a prolonged period of time, as observed in the detection counts. Consequently, PIC nanoparticles prepared at this N:COOH ratio of 1:0.3 were selected for biological studies due to their higher stability.
A similar effect was observed when particles were incubated with other biologically relevant cations, such as Ca2+ (Fig. S13†). Multivalent cations have a high affinity for polyanionic peptides and thus our PIC particles swelled when exposed to CaCl2, as evidenced by the shift in their DLS autocorrelation curves towards longer relaxation times. This swelling was bigger with bigger amounts of CaCl2 added and resulted in uneven light scattering profiles and multimodal size distributions at the highest concentrations tested.
Enzymatic degradation of PIC nanoparticles
The enzyme-triggered disassembly of these (1:0.3)-PIC nanoparticles was evaluated first by DLS, monitoring the amount of light scattered by three different batches of these nanomaterials when exposed to LasB at 37 °C (Fig. 4, hollow circles). Even after only one hour of incubation, PIC nanoparticles treated with LasB scattered 25% less light, and this scattering kept decreasing for the duration of the experiment, reaching 40% of the initial scattering intensity after four hours.
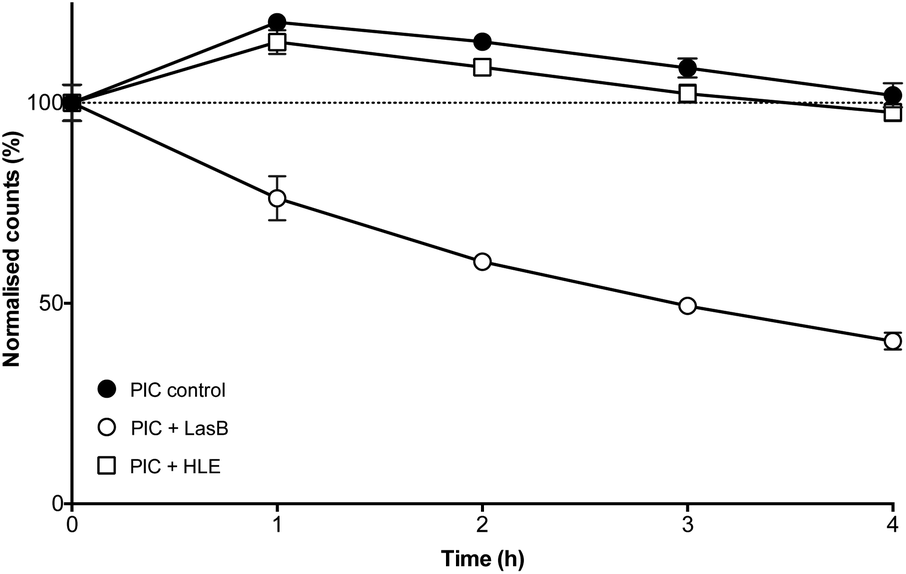 | ||
| Fig. 4 Normalised detection counts (%) for P1SH PIC nanoparticles prepared at a 1:0.3 N:COOH ratio in the absence (●) and presence of LasB (○) and HLE (□). Data normalised to the initial counts for each of the individual experiments. n = 3. | ||
As a comparison, incubation with HLE caused nanoparticles to scatter 15% more light after one hour incubation and remained close to 100% of the initial scattering for the duration of the experiment (Fig. 4, hollow squares). This initial increase in scattering intensity may be due to the swelling of the PIC nanoparticles after addition of the enzyme buffer (2.5 mM Na2B4O7, pH 8.0), in a similar fashion to the swelling observed under physiological conditions (Fig. 3). A similar swelling was observed when PIC nanoparticles were incubated with this buffer in the absence of any of the enzymes (Fig. 4, dark circles). No significant difference was observed when this scattering was compared to that of HLE, proving that the human enzyme was not having an effect on the stability of these PIC nanoparticles. A similar effect could be observed in the DLS size distributions (Fig. S14†) with broader distributions and smaller sizes being observed upon treatment with LasB, as opposed to the almost invariable profile for HLE and the buffer controls.
The ability of LasB to degrade these PIC nanoparticles was also assessed, as before, monitoring the formation of amines upon enzymatic treatment. When PIC nanoparticles were reacted with fluorescamine after four hours of incubation with LasB or HLE, no significant difference in amine content was observed for those treated with HLE when compared to PIC nanoparticles incubated in the absence of any enzyme (Fig. S15†). However, an increase in fluorescence emission (up to 30%) was observed for the samples incubated with LasB. This increase in the amount of amines, together with the DLS data, confirms the selective cleavage of P1SH within the nanoparticle and consequent disassembly of these PIC nanoparticles when exposed to the bacterial elastase.
Antimicrobial activity of PIC nanoparticles
Having established the selectivity of the degradation of these PIC nanoparticles in the presence of P. aeruginosa LasB, the antimicrobial activity of these enzyme-responsive nanoparticles was assessed. P. aeruginosa PAO1V, a pathogenic strain that secretes LasB, was incubated with PIC nanoparticles (1:0.3 N:COOH ratio) or B-PEI25, aliquots were taken every hour for four hours, and stained using the LIVE/DEAD® viability kit. As expected, the antimicrobial activity for the PIC nanoparticles was significantly reduced when compared to B-PEI25, with the nanoparticles exhibiting, after two hour incubation, less than 10% of the activity of B-PEI25 (Fig. 5), thus confirming the potential of these PIC nanoparticles to reduce the toxicity of antimicrobial polymers. This antimicrobial activity was slowly recovered upon incubation with the bacteria, and up to 25% of the antimicrobial activity of B-PEI25 could be observed for PIC nanoparticles after four hours of incubation, before the assay conditions become too harsh to have viable controls of P. aeruginosa. When the same experiment was performed with a LasB-deletion strain (ΔlasAB), which does not produce this elastase, a similar reduction in toxicity could be observed with the PIC nanoparticles (Fig. 5, white bars), despite the slightly higher susceptibility of this strain towards the antimicrobial polymer (Fig. S16 and S17†). More importantly, the antimicrobial activity for the PIC nanoparticles against this ΔlasAB mutant remained constantly low over the duration of the experiment, demonstrating that this strain was unable to degrade the nanoparticle and thus release the antimicrobial polymer.
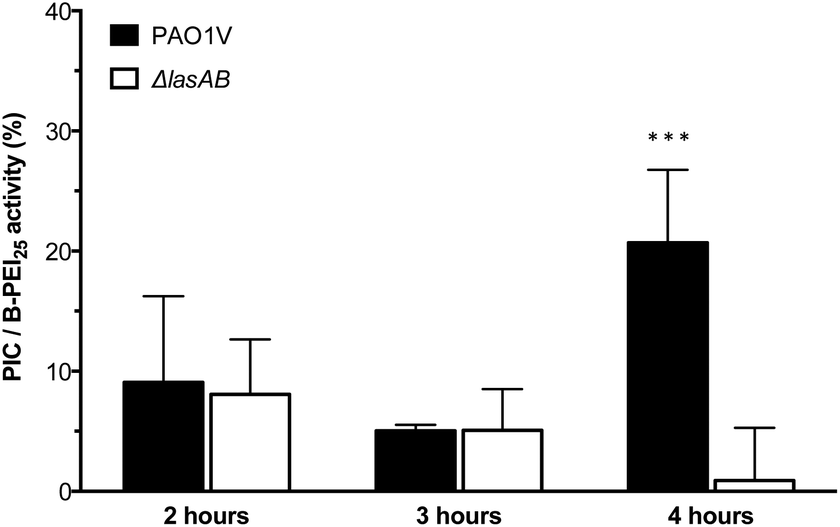 | ||
| Fig. 5 Normalised antimicrobial activity over time of P1SH PIC nanoparticles prepared at a 1:0.3 N:COOH ratio. Activity has been normalised by dividing the relative antimicrobial activity (Fig. S17†) from PIC nanoparticles by that of B-PEI25. *** p < 0.001 between PAO1V and ΔlasAB (CI = 99.9%) after 4 hours. n = 3. | ||
Conclusions
Here, we have developed an easy methodology for the preparation, under aqueous conditions, of PIC nanoparticles from an enzyme-responsive anionic peptide and a cationic antimicrobial polymer. The size of these nanoparticles could be tuned between 100 and 600 nm by simply adjusting the N:COOH ratio. We have demonstrated that disulfide mediated cross-linking and N:COOH ratios were fundamental to impart these PIC nanoparticles with stability under physiological conditions. Furthermore, we have shown that both the starting peptide and the formed PIC nanoparticles can be selectively degraded in the presence of LasB elastase, a virulence factor secreted by the opportunistic pathogen P. aeruginosa, while no degradation was observed in the presence of HLE, an elastase secreted by human leucocytes. Finally, we demonstrate that the toxicity of B-PEI25 was significantly reduced within these nanoparticles. Antimicrobial activity was slowly recovered when these PIC nanoparticles were incubated with a pathogenic P. aeruginosa strain, while remaining non-toxic in the presence of a strain that cannot produce the elastase.
We believe these enzyme-responsive PIC nanoparticles have a great versatility and potential in the current fight against antimicrobial resistance, and our efforts to optimise antimicrobial activity, while maintaining the low toxicity against non-targeted cells we observe here, will be reported in due course.
Author contributions
All authors contributed to the experimental set-up and discussed the results. II and FFT designed the nanoparticle synthesis and characterisation; and II, FFT and AMK designed the microbiological assays. EL and ZZ designed and performed the AFM experiments. II carried out all other experiments. FFT and AMK secured funding. II and FFT analysed the data and wrote the paper, with all other authors contributing to the final version of the manuscript.Acknowledgements
FFT thanks the University of Birmingham for the John Evans Fellowship. AMK thanks the University of Birmingham for a Birmingham Fellowship. FFT and AMK thank the Wellcome Trust (177ISSFPP) for funding. II thanks the University of Birmingham for a PhD studentship. Some equipment used was obtained through Birmingham Science City: Innovative Uses for Advanced Materials in the Modern World (West Midlands Centre for Advanced Materials Project 2) and Birmingham Science City Translational Medicine: Experimental Medicine Network of Excellence project, with support from Advantage West Midlands (AWM) and part funded by European Regional Development fund (ERDF). The authors thank Dr Marie-Christine Jones and Dr Hannene Ali-Boucetta (School of Pharmacy, UoB) for the access to the DLS, and Nicolas Perez for useful discussions and help with the microbiology assays. The authors thank Prof. Suzanne M. J. Fleiszig for the kind donation of P. aeruginosa strains.Notes and references
- European Antimicrobial Resistance Surveillance Network, Antimicrobial resistance surveillance in Europe, European Centre for Disease Prevention and Control, 2013 Search PubMed.
- World Health Organization, Antimicrobial resistance: global report on surveillance, World Health Organization, 2014 Search PubMed.
- K. Hede, Nature, 2014, 509, S2–S3 CrossRef PubMed.
- M. A. Cooper and D. Shlaes, Nature, 2011, 472, 32 CrossRef CAS PubMed.
- K. Bush, P. Courvalin, G. Dantas, J. Davies, B. Eisenstein, P. Huovinen, G. A. Jacoby, R. Kishony, B. N. Kreiswirth, E. Kutter, S. A. Lerner, S. Levy, K. Lewis, O. Lomovskaya, J. H. Miller, S. Mobashery, L. J. V. Piddock, S. Projan, C. M. Thomas, A. Tomasz, P. M. Tulkens, T. R. Walsh, J. D. Watson, J. Witkowski, W. Witte, G. Wright, P. Yeh and H. I. Zgurskaya, Nat. Rev. Microbiol., 2011, 9, 894–896 CrossRef CAS PubMed.
- J. O'Neill, The Wellcome Trust, HM Government, 2015 Search PubMed.
- C. Nathan, Sci. Transl. Med., 2012, 4, 140sr2 Search PubMed.
- B. Spellberg, J. G. Bartlett and D. N. Gilbert, N. Engl. J. Med., 2013, 368, 299–302 CrossRef CAS PubMed.
- R. E. W. Hancock and H.-G. Sahl, Nat. Biotechnol., 2006, 24, 1551–1557 CrossRef CAS PubMed.
- C. D. Fjell, J. A. Hiss, R. E. W. Hancock and G. Schneider, Nat. Rev. Drug Discovery, 2012, 11, 37–51 CAS.
- J. L. Fox, Nat. Biotechnol., 2013, 31, 379–382 CrossRef CAS PubMed.
- M. E. Evans, D. J. Feola and R. P. Rapp, Ann. Pharmacother., 1999, 33, 960–967 CrossRef CAS.
- E. A. Azzopardi, E. L. Ferguson and D. W. Thomas, J. Crit. Care, 2013, 28, 219 CrossRef PubMed.
- K. Kuroda and G. A. Caputo, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2013, 5, 49–66 CrossRef CAS PubMed.
- A. Jain, L. S. Duvvuri, S. Farah, N. Beyth, A. J. Domb and W. Khan, Adv. Healthcare Mater., 2014, 3, 1969–1985 CrossRef CAS PubMed.
- J. Bergen, J. Li, C. R. Rayner and R. L. Nation, Antimicrob. Agents Chemother., 2006, 50, 1953–1958 CrossRef PubMed.
- M.-H. Xiong, Y. Bao, X.-Z. Yang, Y.-H. Zhu and J. Wang, Adv. Drug Delivery Rev., 2014, 78, 63–76 CrossRef CAS PubMed.
- V. W. L. Ng, J. M. W. Chan, H. Sardon, R. J. Ono, J. M. García, Y. Y. Yang and J. L. Hedrick, Adv. Drug Delivery Rev., 2014, 78, 46–62 CrossRef CAS PubMed.
- E. B. M. Breidenstein, C. de la Fuente-Núñez and R. E. W. Hancock, Trends Microbiol., 2011, 19, 419–426 CrossRef CAS PubMed.
- B. Wretlind and O. R. Pavlovskis, Rev. Infect. Dis., 1983, 5, S998–S1004 CrossRef CAS PubMed.
- E. Kessler and D. E. Ohman, in Handbook of Proteolytic Enzymes - (Third Edition) - ScienceDirect, ed. N. D. Rawlings and G. Salvesen, Academic Press, London, 3rd edn, 2013, vol. 1, pp. 582–592 Search PubMed.
- B. A. Cowell, S. S. Twining, J. A. Hobden, M. S. F. Kwong and S. M. J. Fleiszig, Microbiology, 2003, 149, 2291–2299 CrossRef CAS PubMed.
- C. T. Supuran and A. Mastrolorenzo, Curr. Enzyme Inhib., 2011, 7, 2–23 CrossRef CAS.
- S. Udenfriend, S. Stein, P. Böhlen, W. Dairman, W. Leimgruber and M. Weigele, Science, 1972, 178, 871–872 CAS.
- H. Yoon, E. J. Dell, J. L. Freyer, L. M. Campos and W.-D. Jang, Polymer, 2014, 55, 453–464 CrossRef CAS.
- U. Lächelt and E. Wagner, Chem. Rev., 2015, 115, 11043–11078 CrossRef PubMed.
- K. Morihara and H. Tsuzuki, Arch. Biochem. Biophys., 1971, 146, 291–296 CrossRef CAS PubMed.
- J. M. Saulnier, A. M. Rayssiguie, M. C. Duclos and J. M. Wallach, Biochem. Soc. Trans., 1990, 18, 900–901 CrossRef CAS PubMed.
- T. Hatakeyama, H. Kohzaki and N. Yamasaki, Anal. Biochem., 1992, 204, 181–184 CrossRef CAS PubMed.
- K. A. Gibney, I. Sovadinova, A. I. Lopez, M. Urban, Z. Ridgway, G. A. Caputo and K. Kuroda, Macromol. Biosci., 2012, 12, 1279–1289 CrossRef CAS PubMed.
- T. He and V. Chan, J. Biomed. Mater. Res., Part A, 2010, 95A, 454–464 CrossRef CAS PubMed.
- L. T. Lui, X. Xue, C. Sui, A. Brown, D. I. Pritchard, N. Halliday, K. Winzer, S. M. Howdle, F. Fernandez-Trillo, N. Krasnogor and C. Alexander, Nat. Chem., 2013, 5, 1058–1065 CrossRef CAS PubMed.
- I. Louzao, C. Sui, K. Winzer, F. Fernandez-Trillo and C. Alexander, Eur. J. Pharm. Biopharm., 2015, 95, 47–62 CrossRef CAS PubMed.
- D. L. MacKenzie, K. Y. Kwok and K. G. Rice, J. Biol. Chem., 2000, 275, 9970–9977 CrossRef.
- R. Nasanit, P. Iqbal, M. Soliman, N. Spencer, S. Allen, M. C. Davies, S. S. Briggs, L. W. Seymour, J. A. Preece and C. Alexander, Mol. BioSyst., 2008, 4, 741–745 RSC.
- M. Soliman, S. Allen, M. C. Davies and C. Alexander, Chem. Commun., 2010, 46, 5421–5433 RSC.
- P. W. Riddles, R. L. Blakeley and B. Zerner, Anal. Biochem., 1979, 94, 75–81 CrossRef CAS PubMed.
- T. Blessing, J. S. Remy and J. P. Behr, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 1427–1431 CrossRef CAS.
- B. M. Koeppen and B. A. Stanton, in Renal Physiology, Elsevier, 2013, pp. 1–14 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Peptide synthesis, further details of PIC nanoparticle characterisation and full data from microbiology assays. See DOI: 10.1039/c6py00146g |
| This journal is © The Royal Society of Chemistry 2016 |