
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Preparation and characterisation of an 57Fe enriched haemoglobin spike material for species-specific isotope dilution mass spectrometry†
C.
Brauckmann
*a,
C.
Frank
a,
D.
Schulze
a,
P.
Kaiser
b,
R.
Stosch
a and
C.
Swart
a
aPhysikalisch-Technische Bundesanstalt, Bundesallee 100, 38116 Braunschweig, Germany. E-mail: christine.brauckmann@ptb.de
bINSTAND e. V., Ubierstraße 20, 40223 Düsseldorf, Germany
First published on 10th March 2016
Abstract
Haemoglobin (HGB) and its various isoforms are powerful markers for diagnostic and treatment control. For the first time a species-specific isotope dilution mass spectrometry (IDMS) method for the quantification of the whole protein without peptide digestion was developed in this study. The required species-specific HGB spike material was prepared by substituting the entire protohaem containing natFe for the same group enriched with 57Fe. The spike material was characterised regarding isotopic enrichment and structural modification compared to natural HGB. Two different separation methods using a MonoQ® ion exchange chromatography column and a size exclusion column (SEC), respectively, have been developed for the separation of HGB from the matrix and their results for the quantification using IDMS were compared. For this, a high-performance liquid chromatography (HPLC) system was linked to an inductively coupled plasma mass spectrometer (ICP-MS) and HGB was quantified via its iron content. The IRMM/IFCC-467 and JCCRM 912-2 reference materials were used as control samples and for comparison of both separation techniques (MonoQ® and SEC). The HGB values obtained for both methods were found to be identical within the expanded uncertainty.
Introduction
Haemoglobin (HGB) is the most important protein in vertebrates for oxygen transport and is responsible for the red colour of blood. Anaemia, one of the most widespread diseases worldwide, is defined by the cut off concentrations of HGB1–3 with concentrations of 14–18 g dL−1 for men and 12–16 g dL−1 for women considered as normal in whole blood.4 Many factors can affect the HGB concentration, for example the altitude where the person lives. Due to its tetrameric structure, HGB has many different variants. The most common form is HBA0 consisting of two α- and two β-chains and four protoporphyrin IX (PPIX) complexed with Fe(II). HBA1c, the β-N-terminal glycated form of HBA0, is an important biomarker for diagnosis and quality management of diabetes mellitus. As a longitudinal parameter, HBA1c is monitored for therapeutic control over a long period.5 In the case of long-term biomarkers, the reproducibility and reliability of the values in consecutive measurements over a long time is essential. For this reason, several approaches exist for the quantification of both total HGB and HBA1c. Therefore, measurement systems with metrological traceability to the International System of Units (SI) are of great importance to establish comparability of values over time and space. For HBA1c the lack of metrological traceability in the current reference measurement procedure was already recognized by Kaiser et al. and an isotope dilution tandem mass spectrometer method with calibration based on SI traceable peptide standards was established.6,7In this work, we will focus mainly on the determination of total HGB. In 1978 the International Committee for Standardization in Haematology (ICSH) recommended a reference method for total HGB which is based on the photometric determination of cyanmethaemoglobin (HiCN method).8 However, its results are not traceable to the SI because the calibration is based on a reference preparation which is certified by the WHO using the HiCN method but not linked to a pure protein standard.9,10 Furthermore, the HiCN method requires highly toxic potassium cyanide to be used for derivatisation and stabilisation. Another spectral absorbance method, which uses less toxic substances, is the alkaline haematin detergent (AHD)-method. This method focuses on the determination of protohaem which is converted into a stable AHD complex.11,12 To establish this method as a reliable and comparable routine method, it is desirable to have a primary reference method that is independent of the AHD-method for validation and comparison and which ensures the traceability to the SI.
Therefore, the aim of this study is the development of primary reference measurement procedures with results traceable to the SI. These are essential for the improvement of the comparability of the methods currently used in clinical laboratories and, thus, the diagnosis of widespread diseases. As the iron containing protohaem group occurs in all HGB forms, quantification of the iron content allows the determination of total HGB. Del Castillo Busto et al. have already published a species-unspecific or post-column isotope dilution mass spectrometry (SU-IDMS) approach for HGB applying high performance liquid chromatography coupled to inductively coupled plasma mass spectrometry (HPLC-ICP-MS).13 However, a species-specific IDMS (SS-IDMS) approach using HPLC-ICP-MS is published here for the first time. The advantage of SS-IDMS is that the spike material is added to the sample as early as possible and from this moment on a stable isotope ratio is established which is not affected by contamination or sample loss during sample preparation and separation. Therefore, SS-IDMS achieves a higher level of accuracy and reliability in the quantification of total HGB than SU-IDMS. Furthermore, important parameters for SU-IDMS such as column recovery and injection volume need not be known in the case of SS-IDMS. Hence, a species-specific spike material was produced and characterised carefully regarding the isotope enrichment and protein conformation and applied for double and triple IDMS experiments.14 The calibration strategy for double and triple IDMS approaches is based on a so-called back-spike blend containing the pure analyte and a species-specific spike material in a matrix and a concentration as close to the sample as possible (exact matching method).15 The double IDMS approach can be explained as single IDMS of the sample and reversed IDMS for characterisation of the spike material within one step.16 The advantage of this approach is that the concentration of the spike need not be known exactly and, furthermore, the influence of many sources of uncertainties, for example mass bias of the MS, non-linearity or death time of the detector, is greatly diminished.16 The triple IDMS strategy is based on the double IDMS approach with all its advantages using an additional spike-reference blend. This results in the cancelation of the isotope ratio of the spike material from the equation as one single IDMS and two reversed IDMS samples are applied.14,15 For all approaches, a well characterised reference material is required that forms the reference point for IDMS and enables the traceability of the results to the SI. However, such a material is not commercially available in the case of HGB.
Experimental
Chemicals
Ultrapure water (18 MΩ cm) was prepared using a Millipore Elix 5 UV combined with a Milli-Q Element A10 water purification system (Millipore, Merck, Darmstadt, Germany), and used for the preparation of all solutions.Ammonium acetate (NH4Ac) (bioultra), tris(hydroxymethyl)methylamine (Tris) (bioultra), potassium cyanide (KCN) (bioultra), ferrous stabilised human haemoglobin A0 (FSH-HGB) and PPIX (≥95%) were purchased from Sigma-Aldrich (St Louis, USA). Acetone (suprasolve), hydrochloric acid 30% (HCl) (suprapur), disodium hydrogen phosphate (Na2HPO4) (normapur), sodium dihydrogen phosphate (NaH2PO4) (normapur), sodium hydrogen carbonate (NaHCO3) (normapur), disodium carbonate (Na2CO3) (normapur), ethanol (EtOH) (suprasolve), formic acid (FA) (suprapur) and acetic acid (HAc) (suprapur) were obtained from Merck (Darmstadt, Germany). The 57Fe spike used was from Trace Science International Corp. (Ontario, Canada) with a certified isotopic abundance of 54Fe 0.00%, 56Fe 0.57%, 57Fe 95.93% and 58Fe 3.50%. Solutions of isotopically enriched Fe were prepared by dissolving the pure metal in concentrated nitric acid (HNO3, 65%, subboiled, Merck, Darmstadt, Germany) and diluted gravimetrically to a stock solution of 1000 mg kg−1. The IRMM/IFCC-467 reference material “haemoglobin isolated from whole blood” was obtained from IRMM (Geel, Belgium) and the JCCRM 912-2 “certified reference material for total haemoglobin measurements” was from Reference Material Institute for Clinical Chemistry Standard (ReCCS) (Kanagawa, Japan). Human erythrocytes (ERY) were obtained from the German Blood Bank (Braunschweig, Germany).
Instrumentations
Fe isotopes were detected with a quadrupole ICP-MS (Agilent 7700cx) equipped with a Scott-type spray chamber, a PFA-ST 700 μL Micro Flow nebulizer (700 μL min−1, AHF, Tübingen, Germany), a shield torch with an injector (i.d. 1.00 mm), an octopole collision/reaction cell, and Ni sampler and skimmer cones. The parameters of the lens system were optimised for sensitivity, oxide rate, and relative standard deviation on a daily basis. For the hyphenation, the HPLC was coupled to the ICP-MS via a PEEK capillary (i.d. 0.13 mm, ERC GmbH). The applied instrumental parameters for the different methods are listed in Table 1. Within the HPLC method a column equilibration step (5 min washing step under starting conditions) is included before each sample injection.
| Ion exchange chromatography (MonoQ®) | |
| Column | MonoQ® GL 5/50 from GE Healthcare (Uppsala, Sweden) |
| Mobile phase A | 12.5 mmol L−1 Tris at pH = 7.8 |
| Mobile phase B | 12.5 mmol L−1 Tris + 125 mmol L−1 NH4Ac at pH = 7.8 |
| Gradient | 0 min → 0% B, 25 min → 100% B, 30 min → 100% B |
| Column oven temp | 30 °C |
| Flow rate | 0.5 mL min−1 |
| Injection volume | 15 μL |
| DAD wavelength | 254 nm, 415 nm |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|
| Size exclusion chromatography (SEC) | |
| Column | Zenix® SEC-150 Gel Column, 30 cm × 4.6 mm, 3 μm particle size, PEEK from Supelco (Sigma-Aldrich, St. Louis, USA) |
| Mobile phase | 12.5 mmol L−1 Tris + 125 mmol L−1 NH4Ac at pH = 7.8 |
| Gradient | Isocratic |
| Column oven temp | 30 °C |
| Flow rate | 0.35 mL min−1 |
| Injection volume | 15 μL |
| DAD wavelength | 254 nm, 415 nm |
|
|
| ICP-MS | |
| RF power | 1500 W |
| Nebulizer | PFA-ST 700 μL Micro Flow nebulizer |
| Spray chamber temp | 2 °C |
| Plasma gas | Ar, 15 L min−1 |
| Reaction gas | H2, 6 mL min−1 |
| m/z monitored | 53, 54, 56, 57, 58, 60 |
| HPLC-ESI-MS | |
|---|---|
| Column | Jupiter300-C4 (Phenomenex, Torrance, USA) |
| Column oven temp | 40 °C |
| Flow rate | 0.2 mL min−1 |
| Eluent A | 0.1% FA in H2O |
| Eluent B | 0.1% FA in CH3CN |
| Gradient | 5 min → 5% B, 55 min → 65% B, 65 min →65% B |
| Mass range | m/z 100–m/z 3000 |
| Polarity | Positive |
For measurements of protohaem and PPIX, one drop (0.3 μL) of each solution was deposited on a 200 nm gold coated silicon wafer and allowed to dry under ambient conditions. The acquisition time was 200 s. To perform the HGB measurements, two drops were deposited on top of each other (0.3 μL each for preconcentration of the solution) and measured with an acquisition time of 1200 s.
HGB spike preparation
:1. All masses were air buoyancy corrected. 12.5 mmol kg−1 Tris at pH = 7.8 was used for dissolution and dilution. The FSH-HGB reference stock solution was prepared freshly on the day of measurement. 8 mg of commercially available FSH-HGB were dissolved in 1 g Tris buffer. All solutions for the preparation of the blends in IDMS were adjusted to a concentration of about 4 mg g−1 HGB.
Three different blends were prepared from the sample (x), reference (z) and spike (y) solutions:
| bx: HGB sample + HGB spike |
| bz1, bz2: HGB reference + HGB spike |
Two different blends (bz1 and bz2) with an isotope ratio of 57Fe/56Fe close to one are required for the triple IDMS approach. For proof of principle, the IRMM/IFCC-467 reference material was used as the HGB sample in the blend bx. The volume of the blends bx, bz1 and bz2 was 60 μL. For the HPLC-ICP-MS measurements the blends were diluted with 1 mL of Tris buffer and homogenised. Furthermore, 50 μL of a 50 mmol kg−1 KCN solution were added. The samples were stored in an HPLC auto-sampler at 4 °C and were measured using a nested sample sequence as is commonly used in bracketing sequences with two calibration blend standards (bz1 and bz2). The samples were analysed with two different HPLC methods: firstly with the MonoQ® column and secondly with the SEC column.
Additionally, the JCCRM 912-2 reference material was measured as an unknown sample following the same sample preparation procedures as with IRMM/IFCC-467.
Results and discussion
Apo-HGB
The production of apo-HGB according to the protocol described in the Experimental section was controlled by means of HPLC-UV-Vis-ICP-MS. In the case of apo-HGB, the tetrameric structure of HGB is destroyed and the monomeric α- and β-chains are released. The iron containing protohaem groups are no longer bound by the monomeric chains and can be removed during purification.23Due to the instability of the apo-protein, HPLC-UV-Vis-ICP-MS measurements were conducted without further dilution steps immediately after purification. The chromatograms obtained by ICP-MS (a) and UV-Vis (b) are shown in Fig. 1. As both detection techniques are coupled online, a shift in the retention time of about 0.2 min can be observed due to the distance of the two detectors.
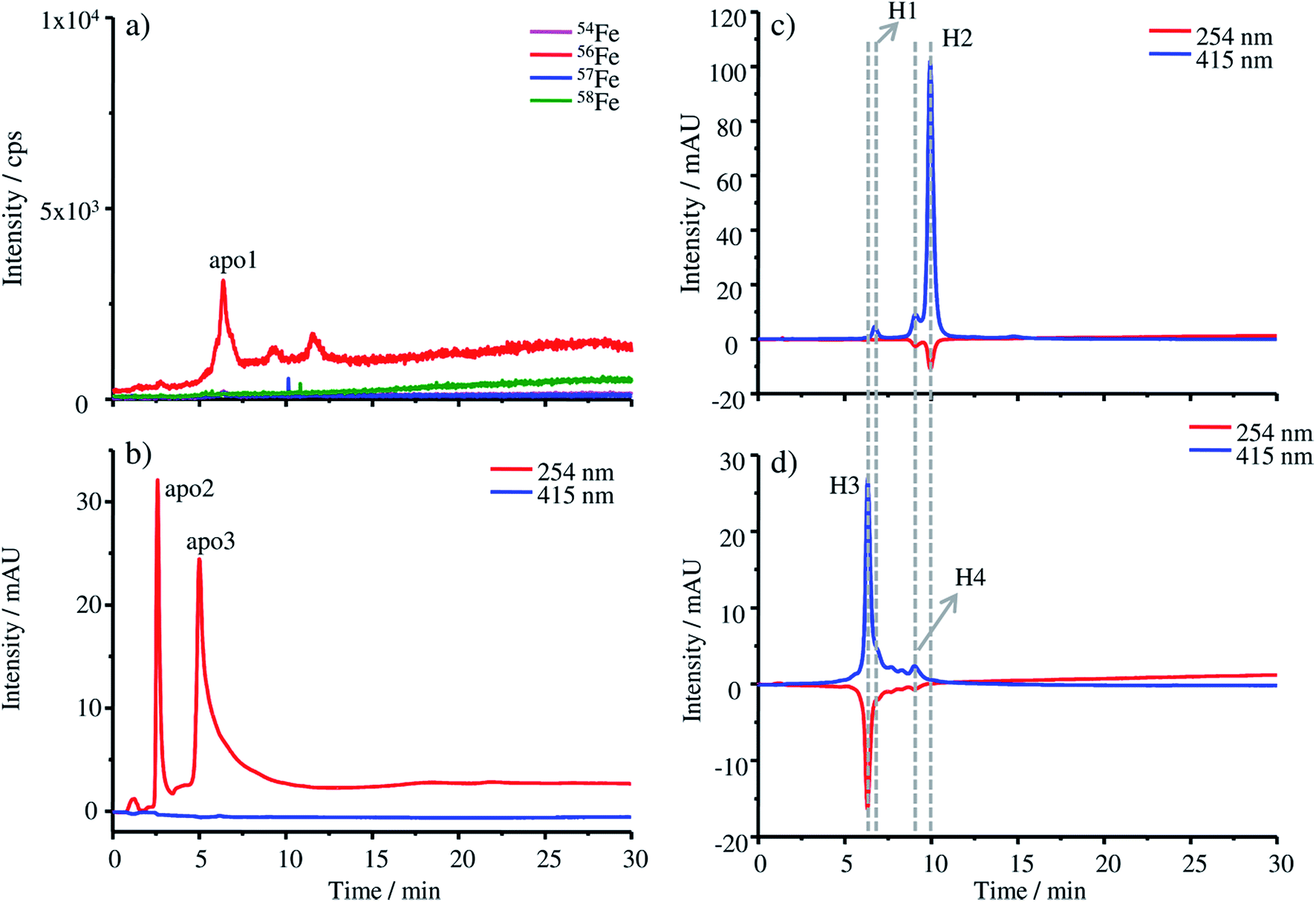 | ||
| Fig. 1 (a) HPLC-ICP-MS and (b) HPLC-UV-Vis chromatograms of apo-HGB produced by removing the protohaem group from natural HGB, (c) HPLC-UV-Vis chromatogram of natural FSH-HGB and (d) HPLC-UV-Vis chromatogram of the resulting 57Fe enriched HGB spike material. | ||
In Fig. 1a only a low iron signal of the most abundant isotope 56Fe is detected at 6.38 min (apo1), which might be a dimeric form of HGB and two further very low peaks at 9.27 min and 11.58 min, which might be caused by small iron impurities are also detected. In the case of the UV-Vis chromatograms at a wavelength of 254 nm, two peaks at 2.59 min (apo2) and 5.00 min (apo3) can be detected. The fact that these peaks cannot be detected by ICP-MS indicates that they originate from iron-free protein chains. At 415 nm, the absorption wavelength characteristic of intact HGB, no signal can be observed indicating the complete denaturation of the protein.
For comparison, the absorption of the starting material FSH-HGB and the HGB spike material at 415 nm is shown in the HPLC-UV-Vis chromatogram in Fig. 1c and d. A solution containing 0.3 mg g−1 HGB dissolved in Tris buffer at a pH of 7.8 was used for these measurements. Next to the main peak H2 at 9.97 min, a further peak H1 appears at 6.74 min. Both peaks, H1 and H2, have a shoulder at 6.21 min and 9.10 min. However, neither H1 nor H2 is detected in the chromatograms of apo-HGB, leading to the conclusion that the protohaem free α- and β-chains were successfully prepared.
57Fe enriched protohaem
Raman spectroscopy was used for monitoring the molecular vibrations during the HGB spike preparation in two different steps of the synthesis. A first check was carried out after completing the formation of the 57Fe enriched protohaem, resulting in the spectra shown in Fig. 2a.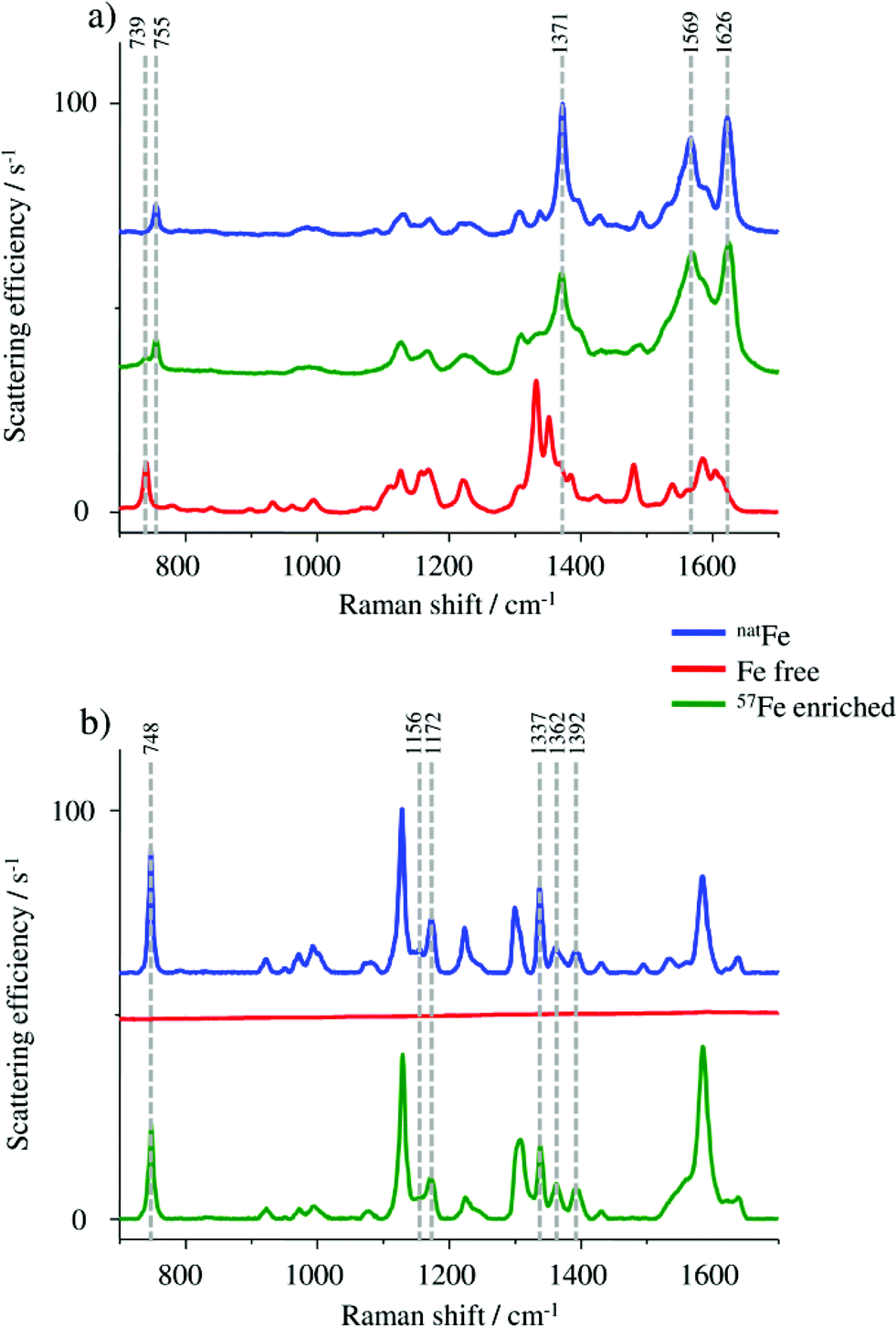 | ||
| Fig. 2 (a) Raman spectra of haemin chloride containing natFe as a control (blue), 57Fe enriched protohaem (green) and the starting material PPIX (red). (b) Raman spectra of FSH-HGB including natFe (blue), the apo-HGB without iron (red) and the 57Fe enriched HGB spike material (green). | ||
In particular, the band at 755 cm−1 in the spectrum of the 57Fe enriched protohaem indicates the successful reaction to the desired product. The bands at 1371 cm−1, 1569 cm−1, and 1626 cm−1 confirm this result, correlating well with the spectrum of the haemin chloride used as the control sample. A comparison of the product spectrum and the PPIX spectrum also indicates the presence of some unconverted PPIX in the prepared material. One obvious indication is the presence of the band at 739 cm−1 but also the regions from 1300 cm−1 to 1400 cm−1 and from 1530 cm−1 to 1650 cm−1 show some influence of the typical PPIX signature.24–26 As the remaining PPIX cannot be incorporated into the protein chains; it does not disturb the reconstruction of HGB. For this reason no further purification was performed.
HGB spike material
The HGB spike material was produced by combining apo-HGB with the 57Fe enriched protohaem. At the applied pH value of about 8 the typically tetrameric structure of HGB is expected, which is also the major HGB form in humans. The HGB spike material was characterised by HPLC-ESI-MS, Raman spectroscopy and HPLC-UV-Vis-ICP-MS.Due to the acidic separation conditions during HPLC-ESI-MS measurements, the HGB structure is decomposed into its constituents (protohaem group and α- and β-chains).13,27,28 Therefore, the protohaem group and α- and β-chains of HGB are separated on the column and are detected at different retention times by ESI-MS.
The deconvoluted mass spectra of the α- and β-chain of (a) the starting material HBA0 and (b) the HGB spike are shown in Fig. 3.
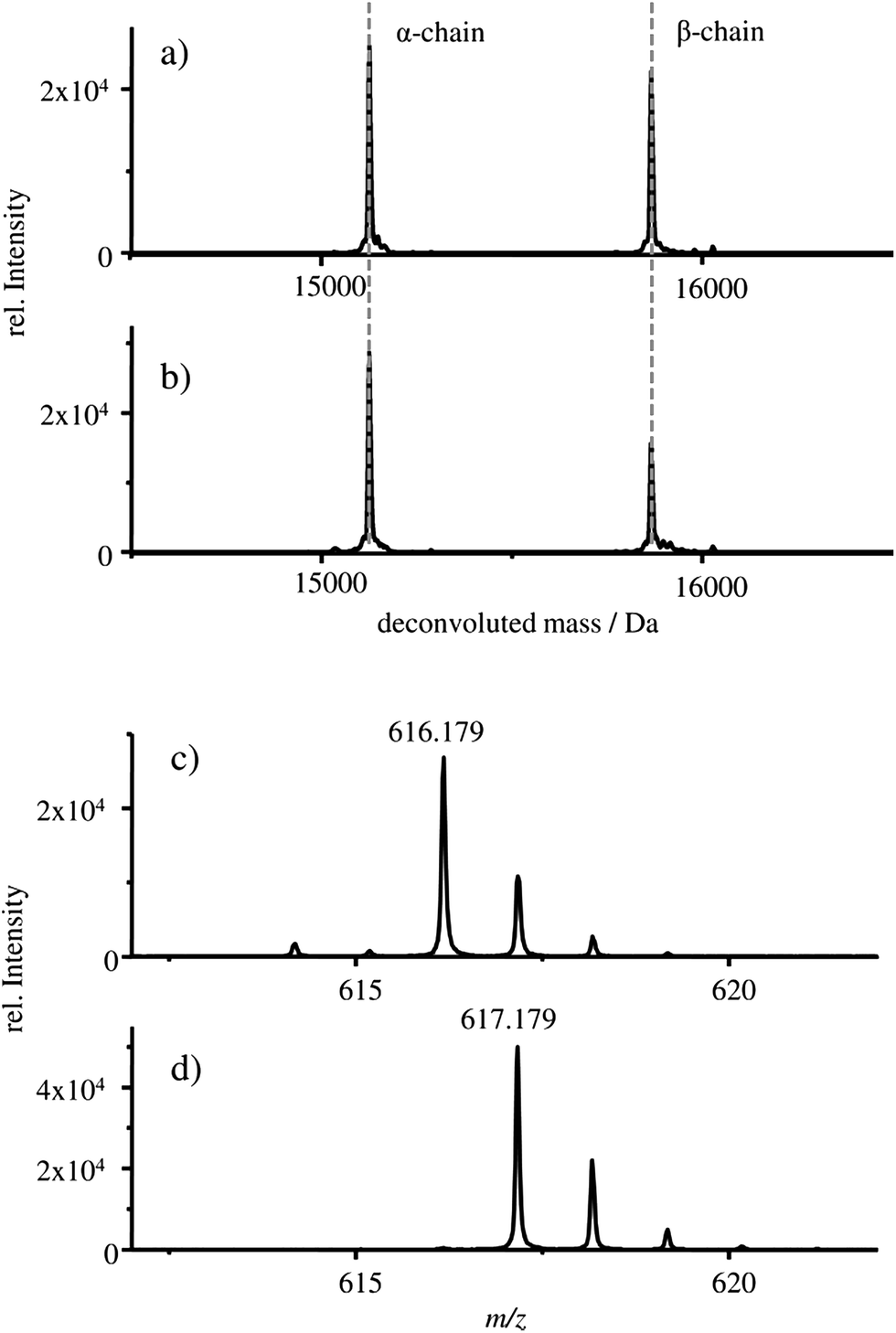 | ||
| Fig. 3 Deconvoluted mass spectrum of samples: (a) FSH-HGB and (b) HGB spike material. The deconvoluted mass spectra of the α- and β-chain are shown, extracted from the respective TIC at 28.0 min to 30.0 min. Mass spectra of the samples: (c) FSH-HGB and (d) HGB spike material, extracted from the respective TIC at 31.7 min to 32.4 min. In these mass spectra the protohaem group with natFe (c) and 57Fe (d) are shown. | ||
In both cases, a mass of 15125 Da is detected for the α-chain and 15866 Da for the β-chains which correlates well with the published masses of 15126 Da and 15867 Da for the α- and β-chains of HGB, respectively.13,27–30 Therefore, it can be concluded that intact α- and β-chains are implemented in the reconstructed HGB spike material.
Besides the protein chains, the protohaem group is also detected. The mass spectra of the protohaem groups of the sample FSH-HGB and the HGB spike material are shown in Fig. 3c and d, respectively. The most abundant isotope of the protohaem group containing natural iron (Fig. 3c) has a m/z of 616.179 ([C34H32N4O4Fe(III)]+, calculated m/z = 616.177, deviation = 3.2 ppm) and the protohaem group with 57Fe enriched iron is shown in Fig. 3d. In this case, the most abundant isotope has a m/z of 617.179 ([C34H32N4O457Fe(III)]+, calculated m/z = 617.180, deviation = 1.6 ppm). The exchange of natural iron against 57Fe enriched iron within the protohaem group can be observed in the mass deviation (Δm = 0.999 Da) and the change of the isotopic pattern. Since the Fe(II) oxidation state is not stable under the applied conditions, for both protohaem groups the same oxidation state Fe(III) is expected.31 The results obtained from HPLC-ESI-MS analysis demonstrate that all parts of HGB can be identified in the spike material and, furthermore, the 57Fe enriched protohaem group can be clearly distinguished from the natFe protohaem.
In the next step, the HGB spike material was analysed by Raman spectroscopy. By means of this method it is possible to investigate if the natFe protohaem was removed from the FSH-HGB and if the 57Fe-enriched HGB spike material is reconstructed correctly. However, natFe and 57Fe containing HGB cannot be distinguished. Therefore, Raman spectra were collected from the FSH-HGB starting material, the intermediate product apo-HGB, and the 57Fe-HGB spike material (Fig. 2b). The bands in the HGB spectra are predominantly caused by protohaem vibrations which are complex in HGB. For instance, the band at 748 cm−1 can be assigned to the pyridine ring breathing mode for the implemented protohaem while the bands at 1156 cm−1, 1172 cm−1, 1337 cm−1, 1362 cm−1, and 1392 cm−1 originate from pyridine half- and quarter-ring vibrations.24 After removing the natFe protohaem from the HGB, this characteristic signature has completely disappeared. The characteristic absorption bands are also detected in the reconstructed 57Fe enriched HGB spike material. Therefore, it can be concluded that the HGB spike material was successfully reconstructed. Although the Raman spectra obtained from FSH-HGB and HGB spike are in good agreement and it can be concluded that the spike and starting material have the same structure, the enrichment of the 57Fe isotope still requires validation.
Therefore, a suitable HPLC-ICP-MS method was developed in order to compare the produced HGB spike material with natural FSH-HGB. For this purpose, the MonoQ® column, a strong ion exchange column, is applied. The pH value of the mobile phase was adjusted to pH = 7.8 taking into account that the tetrameric form of HGB is only stable at a neutral pH. A dissociation of protohaem is expected when the αβαβ tetrameric HGB structure is degraded due to basic or acidic conditions (pH > 6 or pH < 9) and dimeric (αβ) or monomeric (α, β) structures of HGB are formed.23,32,33
The chromatograms of the FSH-HGB and HGB spike material obtained with HPLC-ICP-MS are shown in Fig. 4a and b.
 | ||
| Fig. 4 MonoQ®-ICP-MS chromatograms of (a) pure FSH-HGB, (b) HGB spike material, (c) FSH-HGB derivatised with KCN and (d) HGB spike material derivatised with KCN. The iron isotopes 54Fe, 56Fe, 57Fe and 58Fe are monitored for all samples. | ||
The most abundant peaks are detected for the 57Fe isotope in the case of the HGB spike material compared to 56Fe in natural HGB. For the HGB spike material the observed retention time of the main peak H3 is 6.45 min and 9.45 min in the case of the peak H4. Compared to FSH-HGB shown in Fig. 4a the main peak of FSH-HGB H2 and the smaller peak H1 cannot be observed for the HGB spike material, although, the shoulder peak of H1 and H2 match with the retention time of H3 and H4. This small shift in retention times might be caused by different ligands which are complexed by the protohaem group. However, the main peaks of the HGB spike material and FSH-HGB are different which indicates that different isoforms of HGB have been formed in the spike material compared to FSH-HGB. These results lead to the assumption that H1 and H3 are caused by methaemoglobin (met-HGB) whereas H2 and H4 are caused by HGB. The fact that FSH-HGB contains less than 15% met-HGB according to the product speciation sheet supports this conclusion (peak H1 in Fig. 4a). Furthermore, in the case of the spike material, 57Fe(III) was used for the production of the protohaem, which means that mainly met-HGB was produced. Thus, FSH-HGB contains predominantly HGB with complexed Fe(II), whereas the spike material consists of met-HGB with incorporated Fe(III). According to Bunn et al.34 met-HGB has an isoelectric point (IEP) of about 0.2 pH units higher than that of HGB. This explains the observed shift in the retention time as the retention time of analytes decreases with increasing IEP on the applied MonoQ® strong anion exchange column.
The successful application of IDMS using the MonoQ® requires both the natural HGB and HGB spike material to be in the same oxidation state. Therefore, two approaches are possible: either the reduction of met-HGB to HGB or the oxidation of HGB to met-HGB. Experiments with ascorbic acid as the reducing agent and sodium nitride as the oxidizing agent did not yield quantitative transformation and formed no stable products as already described in the literature.35 Therefore, KCN, a highly reactive agent, was investigated for the conversion of the analyte (Fig. 4c and d). After the treatment with KCN, the retention time of the main peaks in both chromatograms is at 9.50 min (C1). The shift of the retention time of natural FSH-HGB (H2 at 10.17 min) is less than the shift observed for the spike material because the formed cyanmethaemoglobin and natural HGB have a similar IEP.34 As both, the main peak of the spike material (H3) at 6.45 min and the peak H2 of FSH-HGB at 6.92 min, are shifted to the same retention time, it can be assumed that the difference in retention times previously observed is caused by the different oxidation states of Fe and not by incorrect reconstitution of the proteins. Small amounts of impurities appearing in the HGB spike material (peaks at 6.45 min C2 and at 8.07 min C3) require further investigations.
Furthermore, the successful exchange of 56Fe with 57Fe can be clearly seen when comparing the isotopic abundance of Fe in FSH-HGB and HGB spike. A mixture of HGB spike and FSH-HGB can be adjusted to the isotope ratio of 57Fe/56Fe = 1, which is required for exact matching IDMS.
Besides the separation with the MonoQ®, SEC-ICP-MS was used to compare the size of FSH-HGB and the produced HGB spike (Fig. 5).
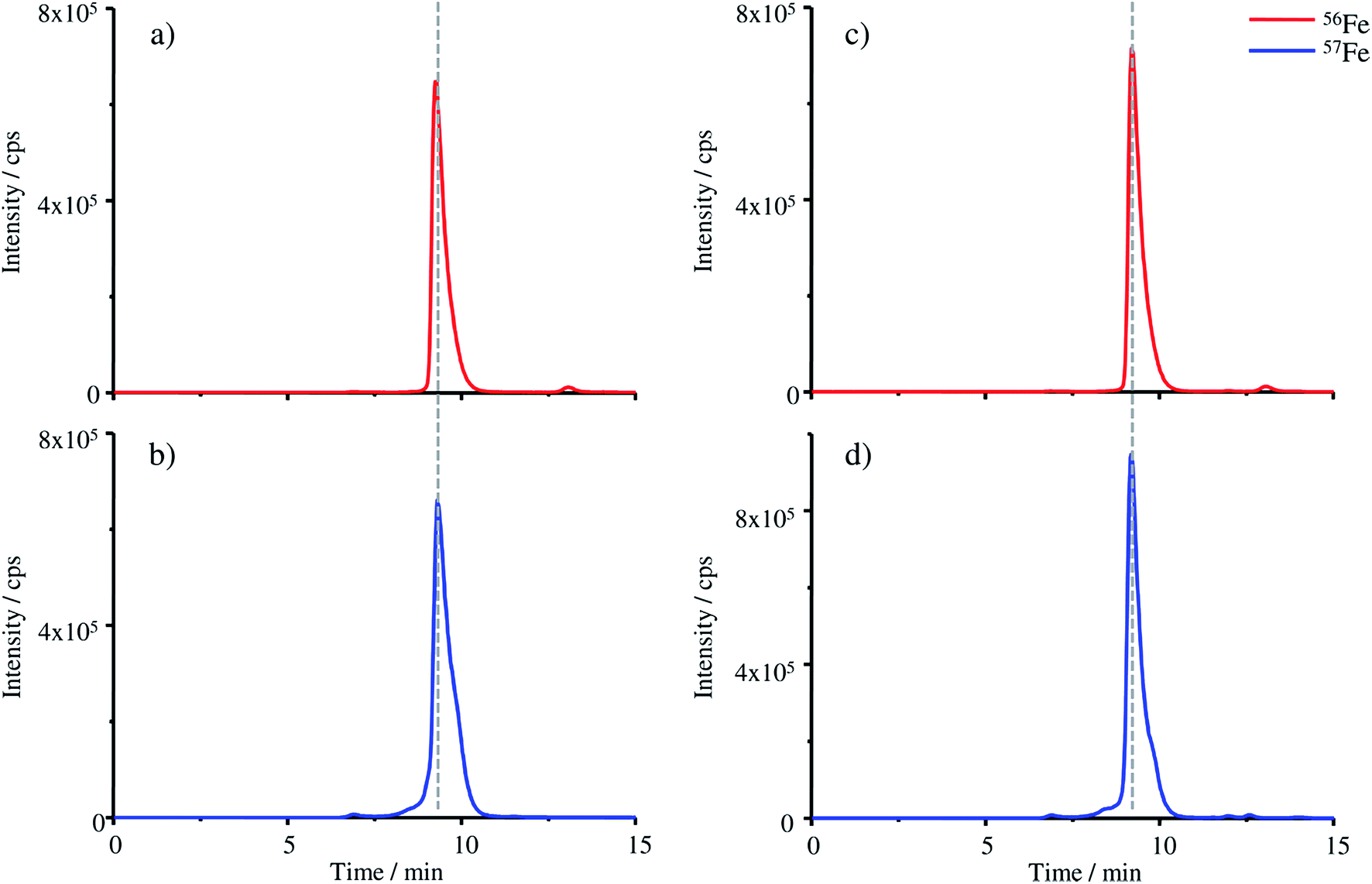 | ||
| Fig. 5 SEC-ICP-MS chromatograms of (a) pure FSH-HGB, (b) HGB spike material, (c) FSH-HGB derivatised with KCN and (d) HGB spike material derivatised with KCN. The iron isotope 56Fe is monitored for the FSH-HGB containing samples and 57Fe for HGB spike material samples. | ||
Four different samples were analysed with the SEC column: FSH-HGB (a), the HGB spike material (b), FSH-HGB derivatised with KCN (c) and the HGB spike material derivatised with KCN (d). FSH-HGB has a retention time of 9.27 min and after the reaction with KCN the peak appears at 9.21 min. The HGB spike material is detected at 9.31 min and after KCN treatment the peak shifts also to a retention time of 9.21 min. The small variation of the retention times of the untreated samples might be caused by the different oxidation states of iron which was already observed with the MonoQ® column. After derivatisation with KCN the spike material as well as FSH-HGB show the same retention time. However, as the shift in the retention time is only small, it can be considered to evaluate the peaks even without prior treatment of the samples with KCN, thus eliminating this toxic compound from the procedure. The results illustrated in Fig. 5 demonstrate that the reconstruction of the HGB spike material was successful and the typical tetrameric form of HGB has been reconstituted.
Stability of the HGB spike
The stability of the HGB spike was investigated in Tris buffer as well as in blends with FSH-HGB and haemolysed erythrocyte concentrate. Two different aliquots (batch 8.1 and 8.2) were compared since batch 8.1 was dissolved directly after production and batch 8.2 was stored as solid at −20 °C for 5 months. The results of the stability investigation are shown in Fig. 6.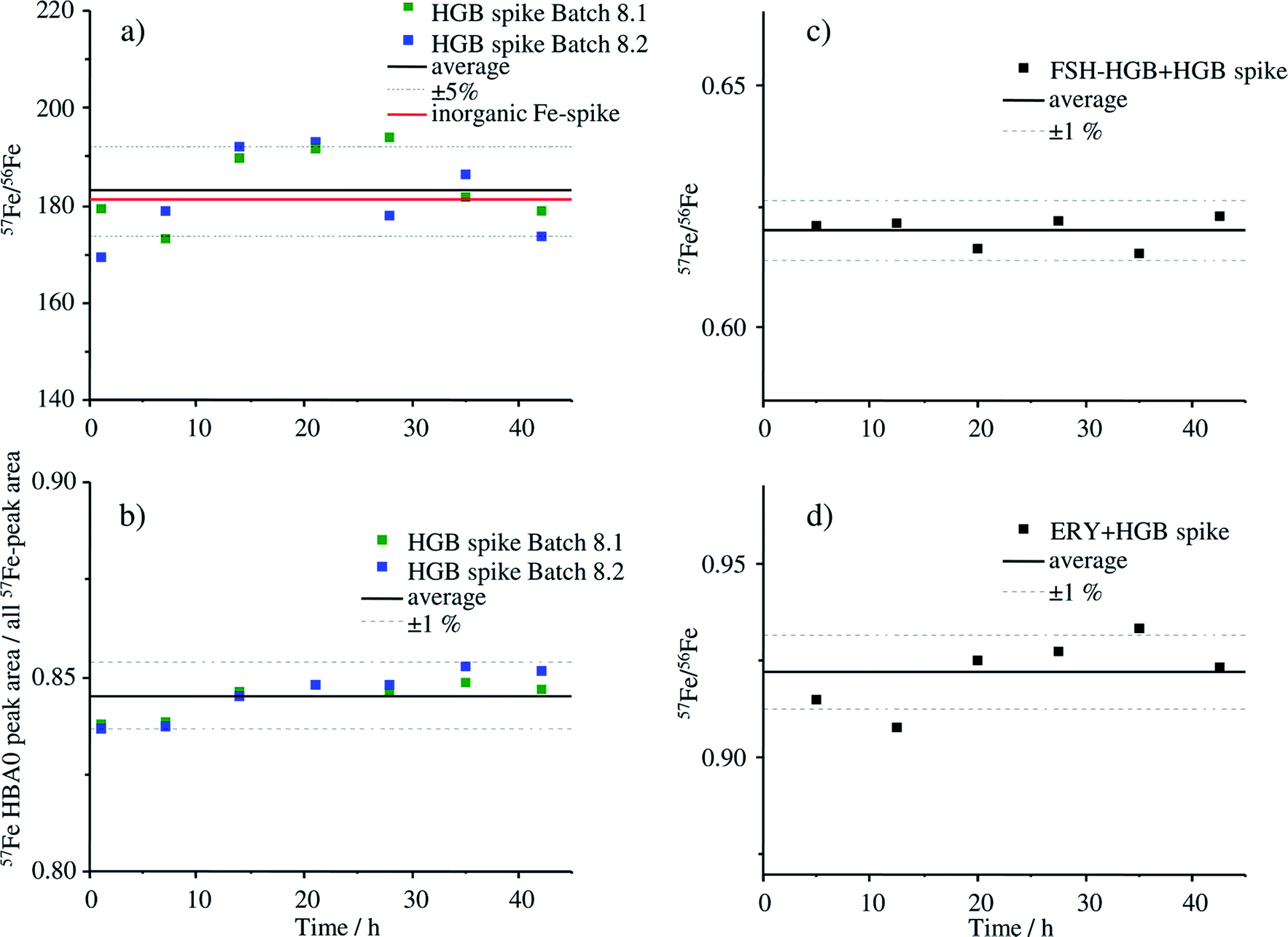 | ||
| Fig. 6 Stability investigation of the HGB spike of two different aliquots from the same batch (batch 8) using MonoQ®-ICP-MS. Batch 8.1 was dissolved in 12.5 mol Tris-buffer pH 7.8 and stored at −20 °C for 5 months, batch 8.2 was dissolved on the day of the measurement. For the stability investigation in real samples, the HGB spike material of batch 8.2 was used. (a) Isotope ratio of 57Fe/56Fe of the HGB spike of batch 8.1 and batch 8.2 over a time period of 42 h, (b) the ratio of the peak areas of HBA0/sum of all Fe peaks of batch 8.1 and batch 8.2 over a time period of 42 h, (c) isotope ratio of 57Fe/56Fe of a blend containing HGB spike and FSH-HGB, (d) isotope ratio of 57Fe/56Fe of a blend containing HGB spike and haemolysed ERY. | ||
The stability of the HGB spike dissolved in Tris buffer is shown in Fig. 6a and b. The isotope ratio 57Fe/56Fe is stable over the analysis time of 42 h and, furthermore, there is no difference between the freshly dissolved aliquot of batch 8.2 and of batch 8.1, which was dissolved 5 months before and stored at −20 °C. The relative standard deviation (RSD) of 4.5% is caused by the high background signal at m/z 56 which is caused by a small unresolved ArO+ interference. The ratio 58Fe/57Fe with much lower RSDs is presented in Fig. ESI-1† for comparison. The degradation of the HGB spike was investigated by means of comparing the peak areas of HBA0 and the sum of all detected Fe peaks that appear in the chromatogram of MonoQ®-ICP-MS over a time period of 42 h applying the fresh and HGB spike stored for 5 months in a freezer, batch 8.2 and batch 8.1, respectively. The corresponding chromatograms after 1 h and 42 h are shown in Fig. ESI-2.† The RSD of below 1% indicates that no Fe containing degradation products can be detected which would be the case if there was any decomposition of the protein. Therefore, we can conclude that over the analysis time of 42 h at 4 °C and also when stored in Tris buffer at −20 °C for five months the HGB spike is stable. The stability of the HGB spike was tested in blends with FSH-HGB and erythrocyte haemolysate and is shown in Fig. 6c and d, respectively. In both cases a stable 57Fe/56Fe isotope ratio can be monitored over 42 h and a RSD of about 1% was observed. These results demonstrate that the stability of the produced HGB spike is suitable for species-specific IDMS quantification.
HGB spike applied in species-specific IDMS
In the last step, the prepared HGB spike material was applied for species-specific IDMS using HPLC-ICP-MS. For the characterisation of the HGB spike material, two different HPLC separation methods (MonoQ® column and SEC column) were developed and used for the IDMS experiment. For the double and triple IDMS approach, the sample (x), the 57Fe enriched HGB spike material (y) and a reference material (z) are required. As the reference, the FSH-HGB obtained from Sigma-Aldrich was characterised regarding its purity. As the sample, the reference material IRMM/IFCC-467, certified for the amount of substance fraction of HBA0/(HBA0 + HBA1c) with >976 mmol mol−1, was used for proof of principle. The IRMM/IFCC-467 reference material is prepared from whole blood by isolating HGB using a multi-step liquid chromatographic approach.10 As the matrix, the reference material contains a buffer solution (50 mmol L−1 2-(N-morpholino)ethanesulfonic acid, 10 mmol L−1 KCN, 2 mmol L−1 ethylenedinitrilotetraacetic acid, pH 6.2) and it is shipped frozen.10 The amount-of-substance fraction was measured applying the IFCC reference measurement procedure.36 The total HGB mass fraction was determined by the ICSH reference method and is given as additional material information to be (119.7 ± 3.7) mg g−1.8,10,37The key compound for IDMS is the reference material. In this case, lyophilized FSH-HGB was used, which was characterised regarding its HGB content and HBA0 was identified by means of HPLC-ESI-MS. The traceability of the reference material was achieved by determining the purity with IDMS applying a certified primary iron reference material from the BAM (BAM-Y002, BAM-A-primary-Fe-1). Further characterisation steps did not detect additional iron sources in the material besides HGB. The purity of HGB in the reference is determined by microwave digestion of the sample and measuring the total iron content to yield wHGB = (0.4794 ± 0.0034) kg kg−1.
In the following, the determined purity of the reference is taken into account for the calculation of the results of the SS-IDMS experiments. The measurement results of the SS-IDMS were evaluated using two different IDMS approaches: double IDMS and triple IDMS.14 For both approaches the concentration of the spike is not required in the equation. In the case of the triple IDMS, which requires an additional reference blend (bz1 and bz2), the ratio of 57Fe/56Fe of the spike also cancels from the equation. Both reference samples should have a ratio of 57Fe/56Fe close to one: bz1 has a ratio slightly higher than one whereas bz2 has a ratio slightly lower. For comparison both reference samples were also used for the evaluation of double IDMS obtaining two independent results. The combined measurement uncertainties were estimated according to the Guide to the expression of uncertainty in measurement (GUM) applying the GUM Workbench Pro version 2.4 (GUM Workbench, Metrodata GmbH, Germany, http://www.metrodata.de).38 The expanded uncertainties (U) were calculated with a coverage factor of k = 2. The results of the SS-IDMS are summarised in Table 3.
| SEC-column | ||||
|---|---|---|---|---|
| Determined directly by SS-IDMS | Calculated with the ratio HBA0/total HGB from the IRMM/IFCC-467 reference material, (0.956 ± 0.005) mol mol−1 | |||
| Total HGB | U | HBA0 | U | |
| Double IDMS (n = 12) | (122.1 ± 1.8) mg g−1 | 1.5% | (116.7 ± 1.8) mg g−1 | 1.6% |
| Triple IDMS (n = 12) | (122.1 ± 1.6) mg g−1 | 1.3% | (116.7 ± 1.6) mg g−1 | 1.4% |
| MonoQ®-column | ||||
|---|---|---|---|---|
| Calculated with the ratio HBA0/total HGB from the IRMM/IFCC-467 reference material, (0.956 ± 0.005)mol mol−1 |
Determined directly by SS-IDMS | |||
| Total HGB | U | HBA0 | U | |
| Double IDMS (n = 9) | (122.4 ± 1.8) mg g−1 | 1.5% | (117.0 ± 1.6) mg g−1 | 1.4% |
| Triple IDMS (n = 9) | (122.4 ± 1.9) mg g−1 | 1.5% | (117.0 ± 1.7) mg g−1 | 1.4% |
| Certified value | ||||
|---|---|---|---|---|
| Additional material information | Calculated with the ratio HBA0/total HGB from the IRMM/IFCC-467 reference material with the certified value of 0.976 mol mol−1 | |||
| Total HGB | U | HBA0 | U | |
| IRMM/IFCC-467 | (119.7 ± 3.7) mg g−1 | 3.1% | 116.8 mg g−1 | — |
The results for total HGB obtained with the SEC-ICP-MS and MonoQ®-ICP-MS approach are within the expanded uncertainty of the value (119.7 ± 3.7) mg g−1 given in the additional material information for IRMM/IFCC-467. The main uncertainty contributions for triple IDMS are the measured isotope ratios Rbx, Rbz1 and Rbz2 with about 75% contribution, while 20% is due to the purity of the reference FSH-HGB. The remaining 5% uncertainty contribution is evenly distributed between the sample weights. In the case of the uncertainty contribution for double IDMS, about 70% is caused by the measured isotope ratios Rbx, and Rbz and 25% is generated by the purity of the reference FSH-HGB. The contributions do not change significantly whether the SEC or the MonoQ® column is used. One example for the uncertainty estimation for each approach (double and triple) can be found in the ESI.†
In the case of the approach using the MonoQ® column, the different HGB isoforms can be distinguished. Therefore, at first the ratio of HBA0 to total HGB was calculated using the peak areas of total HGB and HBA0. The ratio HBA0/total HGB was evaluated for the IRMM/IFCC-467 reference material to be (0.956 ± 0.005) mol mol−1 which is in good accordance with the certified value, 0.976 mol mol−1. Furthermore, the ratio HBA0/total HGB for FSH-HGB from Sigma-Aldrich was determined to be (0.895 ± 0.007) mol mol−1. This value for FSH-HGB was considered for the purity of HBA0 when using it as reference material in the case of the MonoQ® approach, due to the fact that not total HGB but the isoform HBA0 is quantified. In the IRMM/IFCC-467 reference material, the expected value for HBA0 is wHBA0 = 116.8 mg g−1 (wHBA0 = 119.7 mg g−1 × 0.976 mol mol−1 applying the certified amount of substance fraction of HBA0/(HBA0 + HBA1c)). The results for the HBA0 mass fraction of the MonoQ® approach and the calculated values of the SEC approach are in good agreement with the expected HBA0 values in the IFCC reference material.
Additionally, the reference material JCCRM 912-2 from ReCCS, certified for total HGB and prepared from human blood, was determined using triple IDMS. As described in the certificate, the ERY were separated; haemolysed and the cell membrane fragments were removed by centrifugation. Unstable substances were eliminated by dialysis. Except for a carbonate buffer no further buffer or preservatives were added which provides a sample whose constitution is close to real samples in routine analysis. Three different total HGB concentrations are certified.39The results for the quantification of the JCCRM 912-2 reference material are summarised in Table 4. All mass fractions obtained with the different approaches agree well with the certified values within the expanded uncertainty.
| SEC-column | ||||
|---|---|---|---|---|
| Determined directly by SS-IDMS | Calculated with the ratio HBA0/total HGB from the JCCRM 912-2 reference material, (0.870 ± 0.008) mol mol−1 | |||
| Total HGB | U | HBA0 | U | |
| JCCRM912-2L (n = 4) | (77.2 ± 1.7) mg g−1 | 2.2% | (67.2 ± 1.6) mg g−1 | 2.4% |
| JCCRM912-2M (n = 4) | (133.2 ± 2.8) mg g−1 | 2.2% | (115.9 ± 2.7) mg g−1 | 2.3% |
| JCCRM912-2H (n = 4) | (176.3 ± 3.7) mg g−1 | 2.1% | (153.5 ± 3.5) mg g−1 | 2.3% |
| MonoQ®-column | ||||
|---|---|---|---|---|
| Calculated with the ratio HBA0/total HGB from the JCCRM912-2 reference material, (0.870 ± 0.008)mol mol−1 |
Determined directly by SS-IDMS | |||
| Total HGB | U | HBA0 | U | |
| JCCRM912-2L (n = 3) | (74.7 ± 1.2) mg g−1 | 1.7% | (65.0 ± 0.9) mg g−1 | 1.4% |
| JCCRM912-2M (n = 3) | (131.0 ± 2.2) mg g−1 | 1.7% | (114.0 ± 1.6) mg g−1 | 1.4% |
| JCCRM912-2H (n = 3) | (173.4 ± 2.6) mg g−1 | 1.5% | (150.9 ± 1.8) mg g−1 | 1.2% |
| Certified value | ||||
|---|---|---|---|---|
| Certified values of the JCCRM 912-2 reference material | ||||
| Total HGB | U | HBA0 | U | |
| JCCRM912-2L | (75.1 ± 0.9) mg g−1 | 1.2% | — | — |
| JCCRM912-2M | (132.1 ± 1.4) mg g−1 | 1.1% | — | — |
| JCCRM912-2H | (172.8 ± 2.0) mg g−1 | 1.2% | — | — |
Conclusion
An innovative SS-IDMS procedure using HPLC-ICP-MS was applied successfully without prior digestion of the protein to peptides, which has the advantage that the intact protein can be analysed. The production and detailed characterisation of the HGB spike material enabled the use of species-specific IDMS guaranteeing the identical behaviour of the spike and analyte during sample preparation and separation. Complementary detection techniques (HPLC-ESI-MS, Raman spectroscopy, MonoQ®-UV-Vis-ICP-MS and SEC-UV-Vis-ICP-MS) were performed successfully for monitoring the production of the 57Fe HGB spike and for the characterisation of the spike and reference material for SS-IDMS.Two HPLC-ICP-MS methods with different potentials for quantification of HGB were successfully developed. When the SEC column is used the total HGB mass fraction can be investigated, while the application of the MonoQ® allows us to distinguish different HGB isoforms. Both methods have the potential to quantify HGB in human blood with results traceable to the SI and complete uncertainty budgets can be estimated. Furthermore, the HPLC-ICP-MS approach enables us to quantify the intact protein, not only the protohaem group as it is true for the ICSH method. As met-HGB and HGB show more or less the same retention time in the case of the SEC-ICP-MS experiment, the conversions of the sample with KCN can be forgone. Therefore, the method will be optimised in future to serve as a potential reference measurement procedure for total HGB. A total HGB quantification method that is traceable to the SI and does not require a toxic derivatising agent would be a great advantage compared to currently applied methods.
Acknowledgements
The work of this study is a part of the project HLT05, which was funded within the framework of the EMRP. The EMRP is jointly funded by the EMRP participating countries within EURAMET and the European Union.References
- W. T. Kimberly, O. Wu, E. M. Arsava, P. Garg, R. J. Ji, M. Vangel, A. Singhal, H. Ay and A. G. Sorensen, Stroke, 2011, 42, E309 Search PubMed.
- A. J. S. Coats, Heart, 2004, 90, 977–979 CrossRef CAS PubMed.
- World Health Organization, WHO/NMH/NHD/MNM, 2011, 11.1.
- W. Pschyrembel, in Klinisches Wörterbuch, ed. H. Hildebrandt, Walter de Gruyter, Berlin, 2007, vol. 261, p. 257 Search PubMed.
- C. Weykamp, W. G. John and A. Mosca, J. Diabetes Sci. Technol., 2009, 3, 439–445 CrossRef PubMed.
- J. O. Jeppsson, U. Kobold, J. Barr, A. Finke, W. Hoelzel, T. Hoshino, K. Miedema, A. Mosca, P. Mauri, R. Paroni, L. Thienpont, M. Umemoto, C. Weykamp and Int. Federation Clin. Chem. Lab. Med, Clin. Chem. Lab. Med., 2002, 40, 78–89 CrossRef CAS PubMed.
- P. Kaiser, T. Akerboom, R. Ohlendorf and H. Reinauer, Clin. Chem., 2010, 56, 750–754 CAS.
- ICSH, J. Clin. Pathol., 1978, 31, 139–143 CrossRef.
- ICSH, J. Clin. Pathol., 1978, 31, 275–279 CrossRef.
- A. Muñoz-Piñeiro, H. Schimmel and C. Klein, JRC Scientific and Technical Reports, 2007, EUR 22788 EN - 2007.
- H. U. Wolf, W. Lang and R. Zander, Clin. Chim. Acta, 1984, 136, 95–104 CrossRef CAS.
- R. Zander, W. Lang and H. U. Wolf, Clin. Chim. Acta, 1984, 136, 83–93 CrossRef CAS.
- M. E. Del Castillo Busto, M. Montes-Bayón, E. Añón and A. Sanz-Medel, J. Anal. At. Spectrom., 2008, 23, 758–764 RSC.
- C. Frank, O. Rienitz, C. Swart and D. Schiel, Anal. Bioanal. Chem., 2013, 405, 1913–1919 CrossRef CAS PubMed.
- J. Vogl and W. Pritzkow, MAPAN-Journal of Metrology Society of India, 2010, 25, 135–164 Search PubMed.
- M. Sargent, C. Harrington and R. Harte, Guidelines for achieving high accuracy in isotope dilution mass spectrometry (IDMS), Royal Sociaety of Chemistry, Cambridge, 2002 Search PubMed.
- ASTM E1840-96, Standard Guide for Raman Shift Standards for Spectrometer Calibration, West Conshohocken, 2007 Search PubMed.
- G. D. Dorough, J. R. Miller and F. M. Huennekens, Contribution from the departments of chemistry of Washington University and the University of California, 1951, vol. 73, pp. 4315–4320 Search PubMed.
- A. G. Marshall, K. M. Lee and P. W. Martin, J. Chem. Phys., 1983, 78, 1528–1532 CrossRef CAS.
- K. H. Winterhalter and E. R. Huehns, J. Biol. Chem., 1964, 239, 3699–3705 CAS.
- A. R. Fanelli, E. Antonini and A. Caputo, Biochim. Biophys. Acta, 1958, 30, 608–615 CrossRef CAS.
- Y. K. Yip, M. Waks and S. Beychok, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 64–68 CrossRef CAS.
- J. Peng, R. Mandal, M. Sawyer and X. F. Li, Clin. Chem., 2005, 51, 2274–2281 CAS.
- B. R. Wood, P. Caspers, G. J. Puppels, S. Pandiancherri and D. McNaughton, Anal. Bioanal. Chem., 2007, 387, 1691–1703 CrossRef CAS PubMed.
- H. Brunner and H. Sussner, Biochim. Biophys. Acta, 1973, 310, 20–31 CrossRef CAS.
- A. Pilinkienë, G. Biziulevièienë, A. Ulinskaitë, E. Liutkevièius, Z. Kuodis, R. Maþeikienë, G. Niaura, I. Bachmatova, L. Marcinkevièiene, V. MiliukienV and R. Meškys, Acta Medica Lituanica, 2005, 12, 47–53 Search PubMed.
- R. Janzen, M. Schwarzer, M. Sperling, M. Vogel, T. Schwerdtle and U. Karst, Metallomics, 2011, 3, 847–852 RSC.
- C. Brauckmann, C. A. Wehe, M. Kieshauer, C. Lanvers-Kaminsky, M. Sperling and U. Karst, Anal. Bioanal. Chem., 2013, 405, 1855–1864 CrossRef CAS PubMed.
- S. F. Ofori-Acquah, B. N. Green, S. C. Davies, K. H. Nicolaides, G. R. Serjeant and D. M. Layton, Anal. Biochem., 2001, 298, 76–82 CrossRef CAS PubMed.
- B. J. Wild, B. N. Green, E. K. Cooper, M. R. A. Lalloz, S. Erten, A. D. Stephens and D. M. Layton, Blood Cells, Mol., Dis., 2001, 27, 691–704 CrossRef CAS PubMed.
- A. Schmidt and M. Karas, J. Am. Soc. Mass Spectrom., 2001, 12, 1092–1098 CrossRef CAS PubMed.
- M. S. Hargrove, T. Whitaker, J. S. Olson, R. J. Vali and A. J. Mathews, J. Biol. Chem., 1997, 272, 17385–17389 CrossRef CAS PubMed.
- M. Gattoni, A. Boffi, P. Sarti and E. Chiancone, J. Biol. Chem., 1996, 271, 10130–10136 CrossRef CAS PubMed.
- H. F. Bunn, Ann. N. Y. Acad. Sci., 1973, 209, 345–353 CrossRef CAS PubMed.
- B. M. Watts and B. T. Lehmann, J. Food Sci., 1951, 17, 100–108 CrossRef.
- J. Büttner, Eur. J. Clin. Chem. Clin. Biochem., 1995, 33, 981–988 Search PubMed.
- IRMM, Certified reference material IRMM/IFCC-467 haemoglobin isolated from whole blood, 2007 Search PubMed.
- JCGM 100, Evaluation of measurement data—guide to the expression of uncertainty in measurement, 2008, BIPM Search PubMed.
- ReCCS, Certified reference material for total haemoglobin measurements JCRRM 912-2, 2015, MRF1–141 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ja00028b |
| This journal is © The Royal Society of Chemistry 2016 |