
Mass spectra of diverse organic species utilizing the liquid sampling-atmospheric pressure glow discharge (LS-APGD) microplasma ionization source
Lynn X.
Zhang
and
R. Kenneth
Marcus
*
Department of Chemistry, Clemson University, Clemson, South Carolina 29634, USA. E-mail: marcusr@clemson.edu
First published on 19th October 2015
Abstract
This laboratory has demonstrated the analytical versatility of the liquid sampling-atmospheric pressure glow discharge (LS-APGD) ionization source. Demonstrations have included elemental analysis of solutions and laser ablation (LA)-produced aerosols, ambient desorption of small organic molecules on substrates, and direct speciation of organometallic complexes in solution. Here we present initial efforts in mapping the operation space of the device as it applies to obtaining molecular mass spectra from diverse organic molecules. An initial parametric evaluation is performed using caffeine as the test molecule, with a preliminary limit of detection of 5.9 × 10−9 M (43 pg) for 20 μL injections. Conditions optimum for that molecule are applied in assessing the qualitative nature of spectra from caffeine, sinapinic acid, daidzin (a flavonoid), a FITC-labeled lipid tethered ligand, the mass marker Ultramark 1621, and the heme-protein myoglobin. The product spectra for the small molecules are very much like what would be obtained from electrospray and atmospheric pressure chemical ionization sources, predominately the production of the protonated pseudomolecular ion of the form (M + H)+. The mid-range molecules show ion signatures of the thermally stable fragments. Finally, the macromolecules yield mass spectra that are very much like those from electrospray, producing a range of protonated charged states. While much optimization and understanding are needed to best apply the source to such solutes, the fact that the same ion source can be used across the spectrum from elemental/isotopic analysis to perhaps proteomics is totally unique and is worthy of continued development.
Introduction
The use of electrical discharges (plasmas) as ionization sources is as old as the field of mass spectrometry (MS). Two of the most common ionization sources are the inductively coupled plasma (ICP) and the atmospheric pressure chemical ionization (APCI) devices. The ICP has been the benchmark method for performing elemental/isotopic analysis across solid, liquid, and gaseous samples for over 30 years.1,2 The 1–2 kW rf argon plasmas generate an intense ionization environment that efficient vaporizes and dissociates aerosol-introduced species to the atomic ion form. APCI is often the method of choice for MS analysis of small, polar, organic molecules, most often used as a detector for liquid chromatography separations.3,4 In this case, a low current, high voltage dc potential creates a corona discharge that efficiently converts mobile phase species into CI agents in the gas phase. These discharge/plasma sources operate on very different physical mechanisms and surely have two very different fields of application.More recently, low power plasma sources have been introduced for the probing of surfaces to affect ambient desorption ionization (ADI) MS analysis. Originating with the low current, atmospheric pressure helium glow discharge described as the DART (direct analysis in real time),5 there are more than a dozen plasma sources and geometries which have been applied in ADI-MS.6–12 While the mechanisms for creating these discharges are varied, thermal energy within the plasma or through heating of the gas is often used as the means of vaporizing molecular species from the sample surface. Beyond the vaporization step, ionization of volatilized species can occur via a variety of mechanisms, though predominately attributed to a CI event with agents created in the plasma.12,13
In an effort toward delivering a miniaturized plasma source alternative for elemental analysis, Marcus and co-workers developed the liquid sampling atmospheric pressure glow discharge (LS-APGD) source, first for optical emission (OES)14 and then MS elemental analysis.15,16 The LS-APGD operates at a low power (<50 W dc) with very low sample flow rates (<100 μL min−1) and small sample volumes (10 μL), wherein the microplasma is sustained between the surface of the electrolyte carrier solution and a metallic counter electrode. The gap between the electrodes (0.5–2 mm) effectively controls the power density of the discharge. In concept, this device is related to the electrolyte cathode discharge (ELCAD), designed by Cserfalvi and co-workers,17 and further characterized and improved by Hieftje and co-workers under the name of solution cathode glow discharge (SCGD).18,19 The distinction is that the size of LS-APGD yields much higher power densities (>10 W mm−3) providing sufficient energy for operation in a total-consumption mode as well as serve as a source for the elemental analysis of particles generated via laser ablation (LA).20–22
Implementation of the device as an elemental ionization source was first undertaken on a high resolution Thermo Scientific LTQ Orbitrap mass spectrometer.15,23 Further characterization took place on a Thermo Scientific LCQ, ion trap spectrometer.16 In both instances, the microplasma was simply mounted in the place of the commercial ESI source. A detailed parametric evaluation was performed to better understand factors affecting overall spectral composition, target analyte (metals) signal intensities and signal-to-background ratios (S/B), and the limits of detection (LODs).16 Of relevance to the work described here, the LS-APGD has been demonstrated to operate effectively in an ADI-MS mode, yielding high quality “molecular” mass spectra.11 More recently, the use of the microplasma to assess the metal–ligand speciation of uranyl acetate has been presented.24 In that work, use of a mixed solvent electrolyte solution was found to “cool” the plasma such that complete dissociation of solutes (required in elemental MS) did not occur. Operation in a 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 MeOH:H2O electrolyte flow allowed direct injection of the metal acetates, with the product mass spectra readily revealing the changes in ligation as a function of pH. Based on that work, it is obvious to ask the question as to whether this microplasma ionization source could generate high quality mass spectra from organic compounds injected in the solution phase. In short, can the same ionization source be employed for both “atomic” and “molecular” mass spectrometry, in fact on the same mass analyzer system?
:30 MeOH:H2O electrolyte flow allowed direct injection of the metal acetates, with the product mass spectra readily revealing the changes in ligation as a function of pH. Based on that work, it is obvious to ask the question as to whether this microplasma ionization source could generate high quality mass spectra from organic compounds injected in the solution phase. In short, can the same ionization source be employed for both “atomic” and “molecular” mass spectrometry, in fact on the same mass analyzer system?
We present here preliminary efforts in the use of the LS-APGD microplasma as an ionization source for a variety of organic molecules. As reflected in the previous ADI-MS and metallo-organic speciation reports,11,24 the predominately-water vapor microplasma environment is rich in active species such as H+ and protonated solvent clusters which act as Brønsted acid CI agents. Use of a 70:30 MeOH:H2O electrolyte yields a “softer” environment versus the 1 M HNO3 used in elemental MS, and therefore can be expected to generate molecular ions from organic compounds. The LS-APGD mass spectra for a diverse set of organic compounds are presented here, demonstrating the basic characteristics that might be achieved. Caffeine is used as a test compound to initially assess the role of microplasma operation conditions on the total ion response as well as levels of fragmentation. The ability of the LS-APGD to produce molecular ions directly from organic compounds in solution, places the device in a very unique position among the myriad of MS ionization sources. These multiple functions (elemental/isotopic/molecular) on a single platform, that can be easily interfaced with the existing commercial mass spectrometry systems, suggest a good deal of potential.
Experimental section
Sample preparation
Test samples were made as 10−5 M solutions of the selected compounds in 70:30 MeOH:H2O solution (matching the carrier electrolyte composition). The compounds were selected to represent a range in molecular weights, chemical stability, and chemical function. Caffeine (99.7%) and the common MS standard Ultramark® 1621 (95%) were purchased from Alfa Aesar (Ward Hill, MA); sinapinic acid (99%) and myoglobin (>95%) were obtained from Sigma-Aldrich, Co. LLC (St. Louis, MO, USA), daidzin (>99%) was purchased from BIOTANG Inc. (Lexington, MA), and the fluorescein isothiocyanate-labelled lipid tethered ligand (FITC-LTL) was synthesized in this laboratory.25 Methanol (HPLC grade) was purchased from Burdick & Jackson, Honeywell International (Morristown, NJ), and mixed with deionized water (DI-H2O) that prepared by a NANOpure Diamond Barnstead/Thermolyne Water System (18.2 MΩ cm−1) (Dubuque, IA). 1 M HNO3 was diluted from concentrated nitric acid (trace grade) purchased from BDH Chemicals (Poole Dorset, UK).
LS-APGD ionization source
The basic LS-APGD ionization source design and operation principles as they apply to elemental MS have been described in detail previously.16 In fact, no changes have been made in moving to molecular MS except the change in electrolyte composition from 1 M HNO3 to 70:30 MeOH:H2O. Power was provided to the counter electrode by a direct current (d.c.) Bertan Model 915 series positive polarity power supply (Hickville, NY, USA); 0–100 mA, 0–1 kV, through a 10 kΩ, 50 W resistor (HS50, Arcol UK Ltd, Truro Cornwall, UK). Helium (99.99%) was introduced between the surrounding stainless steel capillary and the glass capillary to serve as sheath/cooling gas (0.1–1.2 L min−1), while the electrolyte flow (5–50 μL min−1) was pumped through the glass capillary using a syringe pump (NE-1000, New Era Pump Systems, Farmingdale NY, USA). Four operation parameters were studied in terms of the spectral response of the caffeine test compound: liquid/sampling flow rate (5–30 μL min−1), He gas flow rate (0.1–1.1 L min−1), discharge current (5–30 mA), and plasma sampling distance (0.1–1.0 cm). In all cases, triplicate 20 μL samples were introduced using a manual six-port injector. The repeatability of these measurements generally displayed precision of better than 8% RSD.
Mass spectrometer system
A LCQ Advantage Max™ ion trap mass analyzer from Thermo Scientific (Waltham, MA, USA) was used to detect the ions produced in the glow discharge, and the accompanying Xcalibur™ data acquisition software were used for further data processing (i.e. background subtraction). As in the previous report,16 the LS-APGD stage is mounted directly in the place of the equipped ESI source; with no changes to the ion sampling interface. The system auto-tune function was employed based on the signal intensity of the molecular ion for each compound, with no form of collisional activation (e.g., CID) employed.Results and discussion
Parametric evaluation using caffeine as the test compound
Investigations into the behavior of the metallo-organic compound uranyl acetate pointed to the need to operate the plasma in less kinetically energetic conditions to preserve molecular information in the product mass spectra.24 Key in that work was the realization that use of a mixed-solvent electrolyte provided the desired spectral features. This concept is demonstrated for the test compound caffeine, with the mass spectra (Fig. 1) derived from the normal (a) 1 M HNO3 solvent and the (b) 70:30 MeOH:H2O found optimal in the uranyl acetate studies.24 Clearly seen is the shift from a spectrum that reflects electron ionization (EI)-type fragmentation where the 138 Da fragment dominates, to one which provides an (M + H)+ base peak at 195 Da and far higher analyte ion counts. This response is interpreted as the mixed-solvent plasma having a lower kinetic temperature (though this must be proven spectroscopically). The fused-ring caffeine molecule is structurally robust in comparison to uranyl acetate, which yielded virtually no molecular ion peak in the pure nitric acid solvent. It should be stated, while the 70:30 MeOH:H2O solvent had indeed been chosen through a variation of compositions in the case of the uranyl acetate studies, the same will need to be validated in the future across other “molecular” analyte systems.
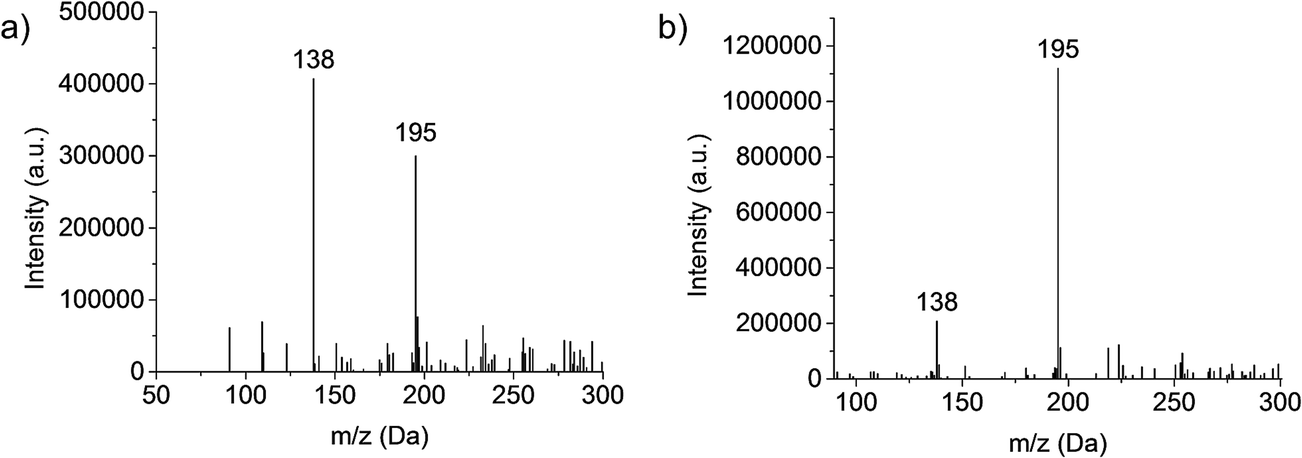 | ||
| Fig. 1 Caffeine spectral composition based on use of “elemental” and “molecular” electrolyte solutions, respectively: (a) 1 M HNO3 and (b) 70:30 MeOH:H2O. Discharge conditions: discharge current = 20 mA, solution flow rate = 10 μL min−1, sheath gas flow rate = 0.9 L min−1, and plasma sampling distance = 0.75 cm. | ||
Previous LS-APGD-MS studies demonstrated that changes in the individual parameters' effect, as well as inter-parametric co-effects, on the observed metal ion intensities, metal oxide fractions, and signal-to-background (S/B) ratios for elemental analysis.16 Likewise, detailed parametric evaluations showed that the product mass spectra could be affected by the operation and ion sampling conditions. Fig. 2 presents the responses of the caffeine pseudomolecular ion ((M + H)+) at m/z = 195 Da and the ratio of the 138 Da/195 Da signals (reflective of the fragmentation of the molecule) as a function of the microplasma operation parameters. The responses reflect the peak height of the signal transient of the two species for triplicate 20 μL injections of 10−5 M caffeine in the 70:30 MeOH:H2O carrier. The investigated parameter space was based on the previous uranyl acetate work.
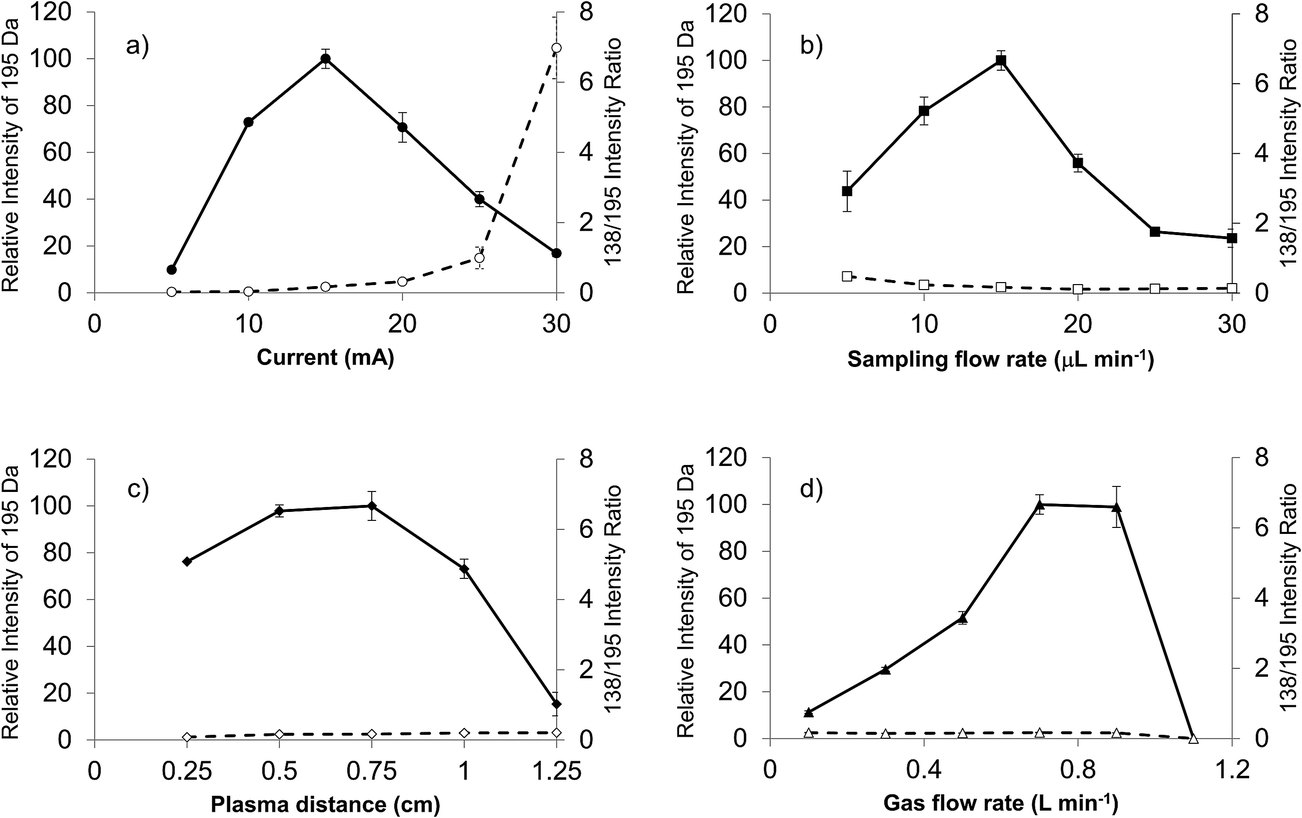 | ||
| Fig. 2 Evaluation of LS-APGD operating parameters on the response of the 195 Da (M + H) pseudomolecular ion and the ratio of the 138 Da/195 Da responses utilizing triplicate 20 μL injections of 10−5 M caffeine in 70:30 MeOH:H2O. (a) Role of discharge current (solution flow rate = 15 μL min−1, plasma sampling distance = 0.75 cm, sheath gas flow rate = 0.7 L min−1), (b) role of solution flow rate (discharge current = 15 mA, plasma sampling distance = 0.75 cm, sheath gas flow rate = 0.7 L min−1), role of plasma sampling distance (discharge current = 15 mA, solution flow rate = 15 μL min−1, sheath gas flow rate = 0.7 L min−1), and (c) role of sheath gas flow rate (discharge current = 15 mA, solution flow rate = 15 μL min−1, plasma sampling distance = 0.75 cm). | ||
The current at which the microplasma operates directly controls the energy available for solution vaporization and the subsequent gas phase processes including desolvation and ionization. As in seen in Fig. 2a, the initial increase in current yields greater M + H response, as the added energy is used to advantage. Previous optical emission studies suggest that the kinetic temperature of the microplasma does increase as a function of discharge,22 and so one would expect greater levels of gas phase desolvation will occur, likely yielding greater ion responses. Beyond a discharge current of 15 mA, there is a dramatic decrease in the pseudomolecular ion response, that is complemented by an increase in the 138/195 ratio. This pair of effects suggests increased collisional dissociation (fragmentation) as the discharge current increases. As the total response for the caffeine analyte remains fairly constant across this range, it seems clear that the added energy (via current) is channel into dissociation of previously protonated molecular species. It is important to note throughout this set of studies that the microplasma operates quite stably for the triplicate, 20 μL (∼40 ng mass) caffeine injections. While not the focus of the present work, preliminary evaluation of the limits of detection based on triplicate injections at the 10−5 M caffeine level yield a value of 5.9 × 10−9 M (43 pg) using the SNR-RSDB method.16 This value, as a single point measurement, is biased high. A more thorough analytical characterization will be the focus of future reports.
The solution flow rate into the microplasma will affect the total solvent load on the discharge, and thus the amount of work that must be done to vaporize and desolvate the solute molecules. Energy consumed to affect these processes is lost towards affecting the remainder of necessary ionization events, etc. The higher flow rate should be reflected in higher analyte delivery rates per unit time to the glow discharge, which explains the intensity increases seen in Fig. 2b for the M + H species as the electrolyte (carrier) flow rate increases from 5 μL min−1 to 15 μL min−1. Depending upon the available energy in the microplasma (set principally by the discharge current), the solvent vaporization and subsequent gas-phase desolvation consume greater proportions of the total, and so the net result is fewer observed analyte ions. This response is generally classified under the umbrella of “solvent loading” with respect to other flame and plasma sources. These same effects are seen in both the OES and MS elemental analysis modes for the LS-APGD source.23,24,26 In fact, in those works it is clear that higher flow rates, even with higher discharge currents, simply generate more solvent-related signals. For the 15 mA discharge current here, the onset of this set of effects is quite clear. Indeed, the lower energy for the downstream gas-phase processes (i.e., ionization and fragmentation) is reflected in the decreasing 138/195 ratio as a function of increasing solvent flow rate.
The roles played by the plasma sampling distance and gas flow rate are manifest in the ion delivery to the mass spectrometer ion sampling orifice, with their net effects being inter-related as seen in previous elemental MS studies.16 Longer sampling distances at a given gas flow rate result in longer residence times and lower collection efficiency due to increased solid angles of acceptance. Likewise, higher gas flow rates should reduce residence times and provide a greater exclusion of ambient species (at this point it is not clear what, if any, role the helium atoms play in the plasma ionization processes). Assuming that the M + H ions are formed in the gas phase following solute desolvation, there is some required residence time to effect the most efficient ionization (interaction with the BrØnsted acid CI agent and proton transfer). Beyond this time (distance), the propensity for ion–neutral interactions increases. These reactions could be chemical in nature causing the loss of the desired analyte ions (either through further proton transfer or adduct formation) or result in greater amounts of collisional fragmentation.
As presented in Fig. 2c, there are indeed trade-offs in the M + H response as a function of the sampling distance. The trends here are in agreement from the simple picture presented above. For example, the amounts of solvent-adduct species are most pronounced (albeit at low levels (<10%)) at sampling distances ≤ 0.5 cm, decreasing substantially with increasing separation. The previous studies showed that longer sampling distances greatly increased the proportion of metal oxide formation versus atomic ions.16 Different here, spectral interrogation for the formation of caffeine adduct ions provided no evidence that secondary gas-phase reactions was a loss mechanism. Finally, it is clear through the invariance in the 138/195 ratio that the increased distance/residence time does not add appreciably to the amount of collision dissociation observed. This makes sense as these further distances are removed from the energetic regions of the plasma, in fact they are likely at temperatures approaching ambient conditions.27 Thus, in lieu of more detailed optical and mass spectrometric mapping, it seems reasonable that the loss in signal as a function of sampling distance is dominated by dispersion effects.
For a fixed sampling distance, the role of the sheath gas flow is to improve ion transport and exclude ambient gases. As shown in Fig. 2d for a 0.75 cm sampling distance, the gas flow rate operates as projected above. At low flow rates, increases result in much improved transport as evidenced in the m/z = 195 Da response. At the highest flow rates, though, there is a dramatic drop in that signal and the onset of plasma instability. In this case, analogous to short sampling distances, the residence time in the microplasma is reduced, perhaps to levels wherein solute vaporization and ionization are not kinetically favorable prior to ion acceptance. As in the case of the sampling distance, there is little effect of gas flow rate on the observed degree of fragmentation nor is there any indication that the extent of adduct formation is influenced by the sheath gas flow.
The responses for both the pseudomolecular ion of caffeine and the relative extent of fragmentation (i.e., 138 Da/195 Da) presented in Fig. 2 yield no particular surprises. The trends here build upon insights gained in the corresponding parametric evaluation towards elemental MS use of the LS-APGD. The monitoring of the degree of fragmentation as a function of discharge conditions clearly pointed to the critical aspects that affect the energy (kinetic temperature) within the discharge region, discharge current and solvent loading. While no single molecular species can be used as a comprehensive model for the processes relevant to ionization of every potential analyte, the lessons learned here are carried over to the initial spectral characterization of diverse organic molecules. As such, a generalized set of glow discharge conditions is identified: discharge current of 15 mA, liquid sampling flow rate of 15 μL min−1, the sampling distance of 0.75 cm and the helium gas flow rate of 0.7 L min−1.
Spectral characteristics of diverse organic compounds
As described in the experimental section, six different organic compounds were selected as test samples. As there is a range of chemistries and molecular weights/structures, to be sure, the range of solutes investigated here is immense when one projects what might be expected from an energetic atomic ion source. All sample solutions were injected into the plasma at the uniform operating conditions, and the obtained spectra presented in Fig. 3. In each case, we make qualitative spectral comparisons with the most common organic, molecular ionization sources; ESI and APCI (where they are available). It must be emphasized that even for these two established methods, there will be differences in spectral features among laboratories that are attributable to specific source operation conditions, solvent composition, and MS operation parameters. As such, the comparisons are only provided for general perspective.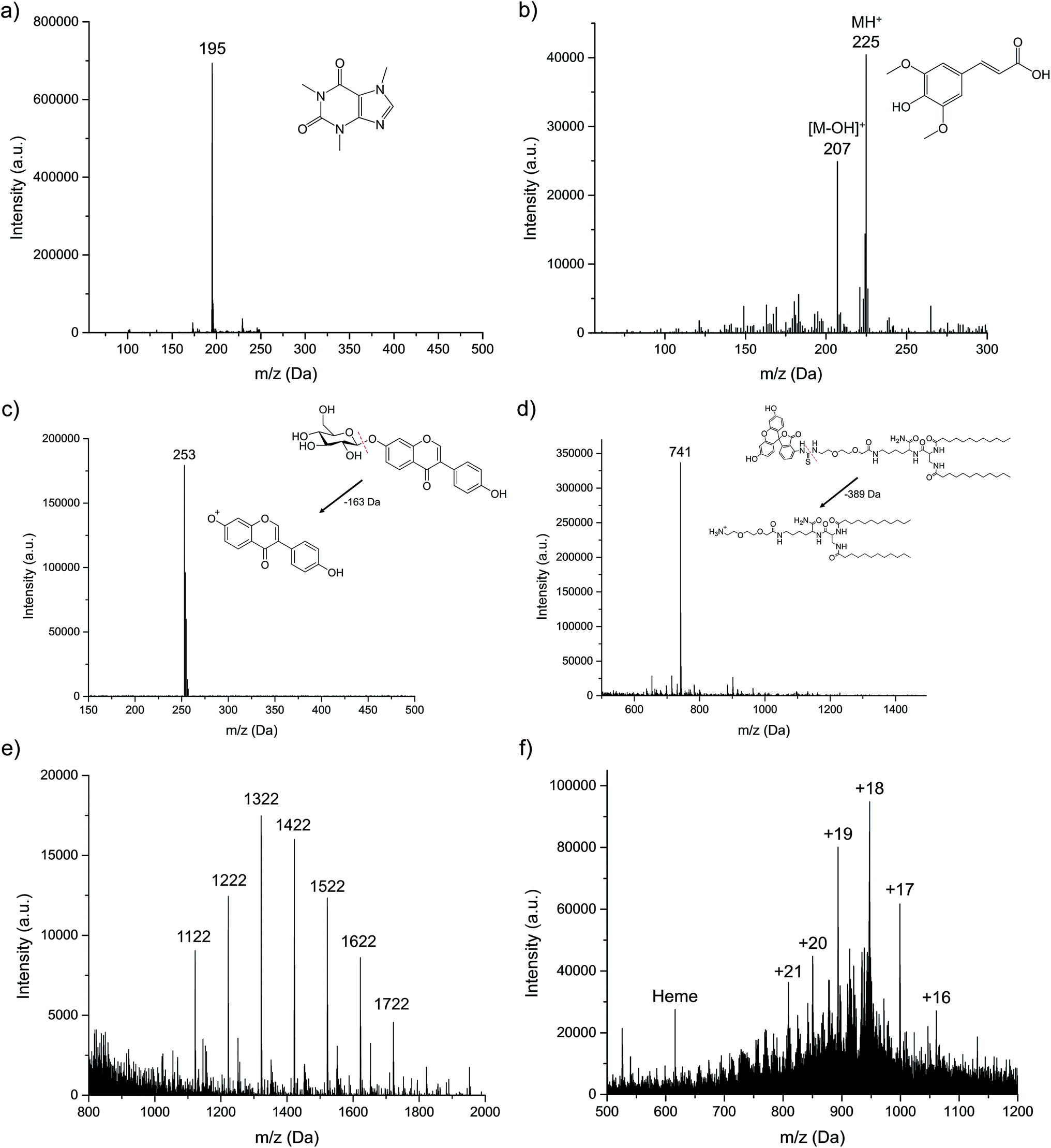 | ||
| Fig. 3 Spectral patterns for the representative organic molecule classes: (a) caffeine, (b) sinapinic acid, (c) daidzin, (d) FITC-LTL, (e) Ultramark 1621, and (f) myoglobin. Solutes introduced as 20 μL aliquots of 10−5 M dilutions in 70:30 MeOH:H2O solution flow. Discharge conditions: current = 15 mA, solution flow rate = 15 μL min−1, plasma sampling distance = 0.75 cm, and sheath gas flow rate = 0.7 L min−1. In each case, no signals above 5% relative intensity to the base peak are seen outside of the displayed mass range. | ||
For small polar molecules such as caffeine (Fig. 3a) and sinapinic acid (Fig. 3b), the base peak in the LS-APGD mass spectrum is the protonated, pseudomolecular ion ((M + H)+). Different from the conditions employed in Fig. 1, the microplasma here generates this ion almost exclusively, with almost no fragmentation observed. Given the fused ring structure of caffeine, the lack of appreciable fragmentation is not surprising. This spectrum is virtually identical to what is seen in APCI-MS,28 while for ESI-MS there is a small amount of fragmentation,29 with the 79 Da fragment being the most prominent.
In the case of sinapinic acid (Fig. 3b), a common matrix employed in MALDI-MS, the BrØnsted acid CI product is the primary ion, but there is appreciable response related to the loss of the hydroxyl group, most likely the one related to the carboxylic acid moiety. Given the structure of that molecule, and its added degrees of freedom, collisional/thermal activation is not unexpected, and thus more low-intensity fragments are seen versus caffeine. The corresponding ESI mass spectrum is composed of the same two ions (M + H and M − OH), though with the fragment ion being the base peak of the spectrum.30
Certain compounds are thermally unstable, thus no molecular ions are detected in the LS-APGD spectra. As an example of daidzin (Fig. 3c), a common flavonoid compound, the glycoside (sugar) moiety is lost (as depicted) in post-ionization degradation, resulting in highest abundance from the base aglycone fragments. Here again, such fragmentation is not unexpected, though the non-existence of any sort of molecule ion is surprising. In the case of both APCI and ESI-MS, the protonated pseudomolecular ion is the base peak, with the sole fragment being the aglycone unit.31,32 Interestingly, in both cases that unit exists as a protonated ion.
The microplasma-produced mass spectrum for the laboratory-synthesized, FITC-labeled ligand tethered ligand (FITC-LTL) (Fig. 3d) also reflects the more energetic (higher kinetic temperature) environment versus ESI-MS. In this case, the otherwise stable FITC unit is lost, as illustrated in the diagram. What is surprising in this case is the minimal amount of overall fragmentation, as certainly its structure would suggest that would be the case. The ESI mass spectrum obtained in this laboratory,25 on the other hand, is composed solely of the protonated molecular ion at m/z = 1130 Da, with a added solvent adduct ion. In both the cases of the natural project and the lipid, the observed fragmentation is far less than might be inferred based on optical emission-based temperature measurements on the order of 800–1200 K.22
The final two compounds, Ultramark 1621 (a commercially available mixture of fluorinated phosphazines) and the protein myoglobin, present a totally different set of challenges and potential ionization pathways to the other test compounds. The Ultramark spectrum in Fig. 3e depicts the expected distribution of different molecular weight species, with the specific singly-protonated mass marker appearing at 1621.9 Da. There is appreciable spectral background below 1000 Da, which could be from fragmentation of these molecules, composed of fluorocarbon chains emanating from a cyclic phosphazine core. However, the spectrum range from 1000–2000 Da is clean. ESI-MS yields a similarly-composed spectrum,33 reflective of the protonation of basic sites of the molecules in the course of the electrospray process. The mass spectrum for myoglobin (Fig. 3f) displays a range of peaks representing different degrees of protonation. As in the case of ESI-MS, it is clear that the ionization takes the form of multiple proton additions.34 As noted previously, the precise charge state distributions for ESI mass spectra are a function of many parameters, and so there is little to be gained at this stage in direct comparisons. What is decidedly different in the LS-APGD spectrum, is the clear presence of a signal corresponding to the heme group in the protein, which is not present in most ESI spectra.
Based on the ionization behavior/spectral pattern of the molecules tested, and in comparison to the literature, it is reasonable to say the collisional energetics of the LS-APGD ionization source is somewhere between an APCI source and an ESI source. With thermally stable and smaller molecules, the spectra are much cleaner and the signal intensity is strong, the spectra pattern is very similar with ones obtained from APCI. On the other hand, the LS-APGD produces mass spectra for macromolecules for which APCI is not applicable, such as the Ultramark compound. In the case of the protein, the ionization source yields spectra that composed similar to those of ESI-MS, but it would seem unlikely that in-solution/droplet ionization processes are occurring in the LS-APGD system. Indeed, based on a first-principles analysis, any sort of aerosol generation mechanism based on the formation of a Taylor cone and subsequent coulombic fission35 are not going to occur at the fields strengths used to create the LS-APGD microplasma.
Conclusions
The liquid sampling-atmospheric pressure glow discharge (LS-APGD) ionization source had previously been demonstrated to provide sensitive elemental analysis in aqueous electrolytic solution. Modification of the electrolytic solution composition provides the capacity for organic compounds analysis, yielding molecular mass spectra of high information content. The roles of the microplasma operating conditions on the basic spectral responses were evaluated for the test compound, caffeine. It was observed, that the relative extent of fragment ions corresponded to the energy input; with greater extents seen at higher discharge currents and lower liquid flow rates. The spatial profiling of the plasma suggests that a succession of processes occurs. The resultant mass spectra for the various test compounds are very much in line with a process where solution-phase solutes are vaporized and subsequently cationized via a proton transfer step, with spectral features very much like seen for ESI- and APCI-MS.Future work will concentrate on the improvement and fine-tuning of the LS-APGD source and developing a deeper level of understanding of the fundamental processes occurring the microplasma with regards to organic molecule analysis. There is a wealth of quantitative characterization that must also occur. During such studies, a wide range of compounds having different chemical/physical properties should be used to allow well-controlled, multi-parameter studies. It is believed that the LS-APGD ionization source, having multiple functions in one device, holds a unique position among atmospheric pressure discharges and has great potential to be utilized in many fields of analysis; from elemental/isotopic analysis to perhaps proteomics.
Acknowledgements
This work was supported by the Defense Threat Reduction Agency, Basic Research Award # HDTRA1-14-1-0010, to Clemson University. LXZ wishes to thank Liuwei (Jerry) Jiang for supplying the lipid test compound and many helpful discussions.References
- R. S. Houk, V. A. Fassel, G. D. Flesch, H. J. Svec, A. L. Gray and C. E. Taylor, Anal. Chem., 1980, 52, 2283–2289 CrossRef CAS
.
-
Inductively Coupled Plasma Mass Spectrometry, ed. A. Montaser, Wiley-VCH, Weinheim, 1998 Search PubMed
- A. P. Bruins, T. R. Covey and J. D. Henion, Anal. Chem., 1987, 59, 2642–2646 CrossRef CAS
- E. Rosenberg, J. Chromatogr. A, 2003, 1000, 841–849 CrossRef CAS PubMed
- R. B. Cody, J. A. Laramee and H. D. Durst, Anal. Chem., 2005, 77, 2297–2302 CrossRef CAS PubMed
- G. A. Harris, A. S. Galhena and F. M. Fernandez, Anal. Chem., 2011, 83, 4508–4538 CrossRef CAS PubMed
- J. D. Harper, N. A. Charipar, C. C. Mulligan, X. R. Zhang, R. G. Cooks and Z. Ouyang, Anal. Chem., 2008, 80, 9097–9104 CrossRef CAS PubMed
- T. L. Salter, I. S. Gilmore, A. Bowfield, O. T. Olanbanji and J. W. Bradley, Anal. Chem., 2013, 85, 1675–1682 CrossRef CAS PubMed
- L. Nyadong, A. S. Galhena and F. M. Fernandez, Anal. Chem., 2009, 81, 7788–7794 CrossRef CAS PubMed
- J. T. Shelley, J. S. Wiley, G. C. Y. Chan, G. D. Schilling, S. J. Ray and G. M. Hieftje, J. Am. Soc. Mass Spectrom., 2009, 20, 837–844 CrossRef CAS PubMed
- R. K. Marcus, C. Q. Burdette, B. T. Manard and L. X. Zhang, Anal. Bioanal. Chem., 2013, 405, 8171–8184 CrossRef CAS PubMed
- A. Albert, J. T. Shelley and C. Engelhardt, Anal. Bioanal. Chem., 2014, 406, 6111–6127 CrossRef CAS PubMed
- F. J. Andrade, J. T. Shelley, W. C. Wetzel, M. R. Webb, G. Gamez, S. J. Ray and G. M. Hieftje, Anal. Chem., 2008, 80, 2646–2653 CrossRef CAS PubMed
- R. K. Marcus and W. C. Davis, Anal. Chem., 2001, 73, 2903–2910 CrossRef CAS PubMed
- R. K. Marcus, C. D. Quarles Jr, C. J. Barinaga, A. J. Carado and D. W. Koppenaal, Anal. Chem., 2011, 83, 2425–2429 CrossRef PubMed
- L. X. Zhang, B. T. Manard, S. Konegger-Kappel and R. K. Marcus, Anal. Bioanal. Chem., 2014, 406, 7497–7509 CrossRef CAS PubMed
- T. Cserfalvi and P. Mezei, J. Anal. At. Spectrom., 1994, 9, 345–349 RSC
- M. R. Webb, F. J. Andrade, G. Gamez, R. McCrindle and G. M. Hieftje, J. Anal. At. Spectrom., 2005, 20, 1218–1225 RSC
- M. R. Webb, G. C. Y. Chan, F. J. Andrade, G. Gamez and G. M. Hieftje, J. Anal. At. Spectrom., 2006, 21, 525–530 RSC
- A. J. Carado, C. D. Quarles Jr, A. M. Duffin, C. J. Barinaga, R. E. Russo, R. K. Marcus and D. W. Koppenaal, J. Anal. At. Spectrom., 2012, 27, 385–389 RSC
- C. D. Quarles, J. Gonzalez, I. Choi, J. Ruiz, X. Mao, R. K. Marcus and R. E. Russo, Spectrochim. Acta, Part B, 2012, 76, 190–196 CrossRef
- B. T. Manard, J. J. Gonzalez, A. Sarkar, M. Dong, J. Chirinos, X. Mao, R. E. Russo and R. K. Marcus, Spectrochim. Acta, Part B, 2014, 94, 39–47 CrossRef
- C. D. Quarles Jr, A. J. Carado, C. J. Barinaga, D. W. Koppenaal and R. K. Marcus, Anal. Bioanal. Chem., 2012, 402, 261–268 CrossRef PubMed
- L. X. Zhang, B. T. Manard, B. A. Powell and R. K. Marcus, Anal. Chem., 2015, 87, 7218–7225 CrossRef CAS PubMed
- L. Jiang, A. J. Schadock-Hewitt, L. X. Zhang and R. K. Marcus, Analyst, 2015, 140, 1523–1534 RSC
- C. D. Quarles, B. T. Manard, C. Q. Burdette and R. K. Marcus, Microchem. J., 2012, 105, 48–55 CrossRef CAS
- W. C. Davis and R. K. Marcus, J. Anal. At. Spectrom., 2001, 16, 931–937 RSC
- T. Arinobu, H. Hattori, T. Kumazawa, X. P. Lee, Y. Mizutani, T. Katase, S. Kojima, T. Omori, R. Kaneko, A. Ishii and H. Seno, Forensic Toxicol., 2009, 27, 1–6 CrossRef CAS
- I. Dalmazio, L. S. Santos, R. P. Lopes, M. N. Eberlin and R. Augusti, Environ. Sci. Technol., 2005, 39, 5982–5988 CrossRef CAS PubMed
- Y. Wada, Sinapic acid; MALDI-TOF; MS; Pos, MassBank, Mass Spectrometry Society of Japan , 2011 Search PubMed.
- H. J. Rong, J. F. Stevens, M. L. Deinzer, L. de Cooman and D. de Keukeleire, Planta Med., 1998, 64, 620–627 CrossRef CAS PubMed
- T. Tohge, Daidzin; LC-ESI-QTOF; MS, MassBank, Mass Spectrometry Society of Japan , 2011 Search PubMed.
- M. Moini, Rapid Commun. Mass Spectrom., 1994, 8, 711–714 CrossRef CAS
- M. J. Powell, T. T. Razunguzwa, A. D. Biddle and G. R. Asbury, BioTechniques, 2009, 46, 373–374 CrossRef CAS
-
Electrospray and MALDI Mass Spectrometry: Fundamentals, Instrumentation, Practicalities, and Biological Applications, ed. R. B. Cole, John Wiley & Sons, Hoboken. 2010 Search PubMed
| This journal is © The Royal Society of Chemistry 2016 |