
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controlling uranyl oxo group interactions to group 14 elements using polypyrrolic Schiff-base macrocyclic ligands†
Nicola L.
Bell
,
Polly L.
Arnold
* and
Jason B.
Love
*
EaStCHEM School of chemistry, Joseph Black Building, The King's Buildings, The University of Edinburgh, West Mains Road, Edinburgh EH9 3FJ, UK. E-mail: jason.love@ed.ac.uk; polly.arnold@ed.ac.uk
First published on 29th June 2016
Abstract
Heterodinuclear uranyl/group 14 complexes of the aryl- and anthracenyl-linked Schiff-base macrocyclic ligands LMe and LA were synthesised by reaction of UO2(H2L) with M{N(SiMe3)2}2 (M = Ge, Sn, Pb). For complexes of the anthracenyl-linked ligand (LA) the group 14 metal sits out of the N4-donor plane by up to 0.7 Å resulting in relatively short M⋯OUO distances which decrease down the group; however, the solid state structures and IR spectroscopic analyses suggest little interaction occurs between the oxo and group 14 metal. In contrast, the smaller aryl-linked ligand (LMe) enforces greater interaction between the metals; only the PbII complex was cleanly accessible although this complex was relatively unstable in the presence of HN(SiMe3)2 and some organic oxidants. In this case, the equatorial coordination of pyridine-N-oxide causes a 0.08 Å elongation of the endo UO bond and a clear interaction of the uranyl ion with the Pb(II) cation in the second donor compartment.
Introduction
Uranyl [UVIO2]2+ is the most stable and prevalent form of uranium in the environment.1,2 The redox properties of uranyl are of interest in order to establish chemical routes to separate and immobilise actinide radioactive wastes.3 Reduced [UVO2]+ species are inherently unstable to disproportionation under aqueous conditions forming [UVI] and [UIV] products, with the latter being insoluble in aqueous waste streams and therefore immobilised by this process.4 Oxidised UVIO3 is also poorly soluble in water and T-shaped U-trioxo species have recently been proposed, alongside cisoid-UO2 complexes, to be important in oxo-transfer processes in aqueous media.5 As such, the synthesis of these motifs in molecular species bound within defined ligand environments can improve our understanding of actinide bonding and help us to predict uranyl speciation.We have studied extensively the reduction of uranyl incorporated within a Schiff-base macrocyclic ligand with two aryl-linked coordination pockets (Scheme 1(a)).2,6–8 Incorporation of two uranyl units into this ligand environment results in reduction to UV and oxo group rearrangement to form the homodinuclear ‘butterfly’ complex (Me3SiOUV)2{μ-(O)2}LMe which contains bridging oxo groups and acute OUO angles.2
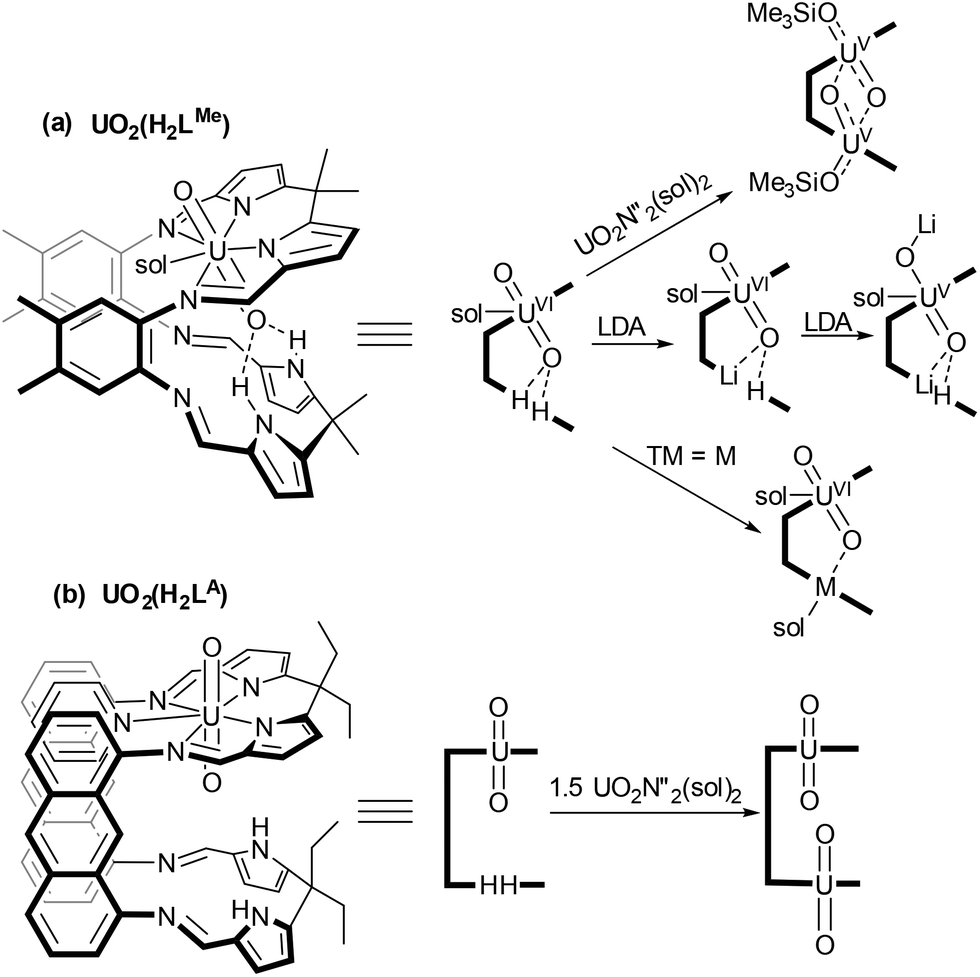 | ||
| Scheme 1 Metalation chemistry of the uranyl complexes of aryl (LMe) and anthracenyl (LA) linked ligands. (N′′ = N(SiMe3)2, Sol = THF, pyridine). | ||
We have seen similar reduction during the formation of mixed-metal complexes from UVIO2(H2LMe). Coordination of an electropositive metal within the vacant compartment of the macrocycle was shown to activate the uranyl towards reduction so forming a range of stable mixed-metal, [UVO2]+ complexes.7,8 In contrast, incorporation of a less reducing metal (e.g. Fe(II), Mn(II)) did not promote reduction and the corresponding uranyl(VI) UVIO2M(LMe) complexes were isolated.9 In this ligand, the proximity of the uranyl endo-oxygen to the lower macrocyclic pocket facilitates interaction with the second metal. More recently we have developed the chemistry of an anthracenyl-linked analogue of this ligand and synthesised mono- and dinuclear uranyl complexes (Scheme 1(b)).10,11 The anthracenyl linker enforces both a greater distance between the N4-coordination compartments and a greater degree of coplanarity.
We have now explored and compared the reactivity of the uranyl complexes of these two ligands, UO2(H2L) (L = LMe, LA) towards group 14 metal silylamides (M{N(SiMe3)2}2, M = Ge, Sn, Pb). These latter metals have been used in order to target macrocyclic complexes of UVIO3, with the chalcophilic group 14 metal potentially charge balancing through oxidation from MII to MIV. The interaction between the two metal ions in the resulting complexes and any resulting activation of the UO2 bonds has been assessed spectroscopically, as has the reactivity of these complexes towards oxo-transfer reagents.
Results and discussion
Complexes of the anthracenyl-hinged macrocycle LA
The reaction of a dark green solution of UO2(H2LA) in THF or pyridine with M{N(SiMe3)2}2 (M = Sn, Pb) results in the immediate formation of deep red solutions and the appearance of new sets of resonances in the 1H NMR spectra which are consistent with the formation of 1(Pb) and 1(Sn) (Scheme 2). In contrast, reaction of UO2(H2LA) with Ge{N(SiMe3)2}2 in pyridine takes 6 h at room temperature to reach full conversion, forming 1(Ge)-py, and in THF elevated temperatures for ca. 8 h are required to form 1(Ge)-THF cleanly. Removal of solvent and workup yielded 1(M)-sol for M = Sn, Ge, and Pb.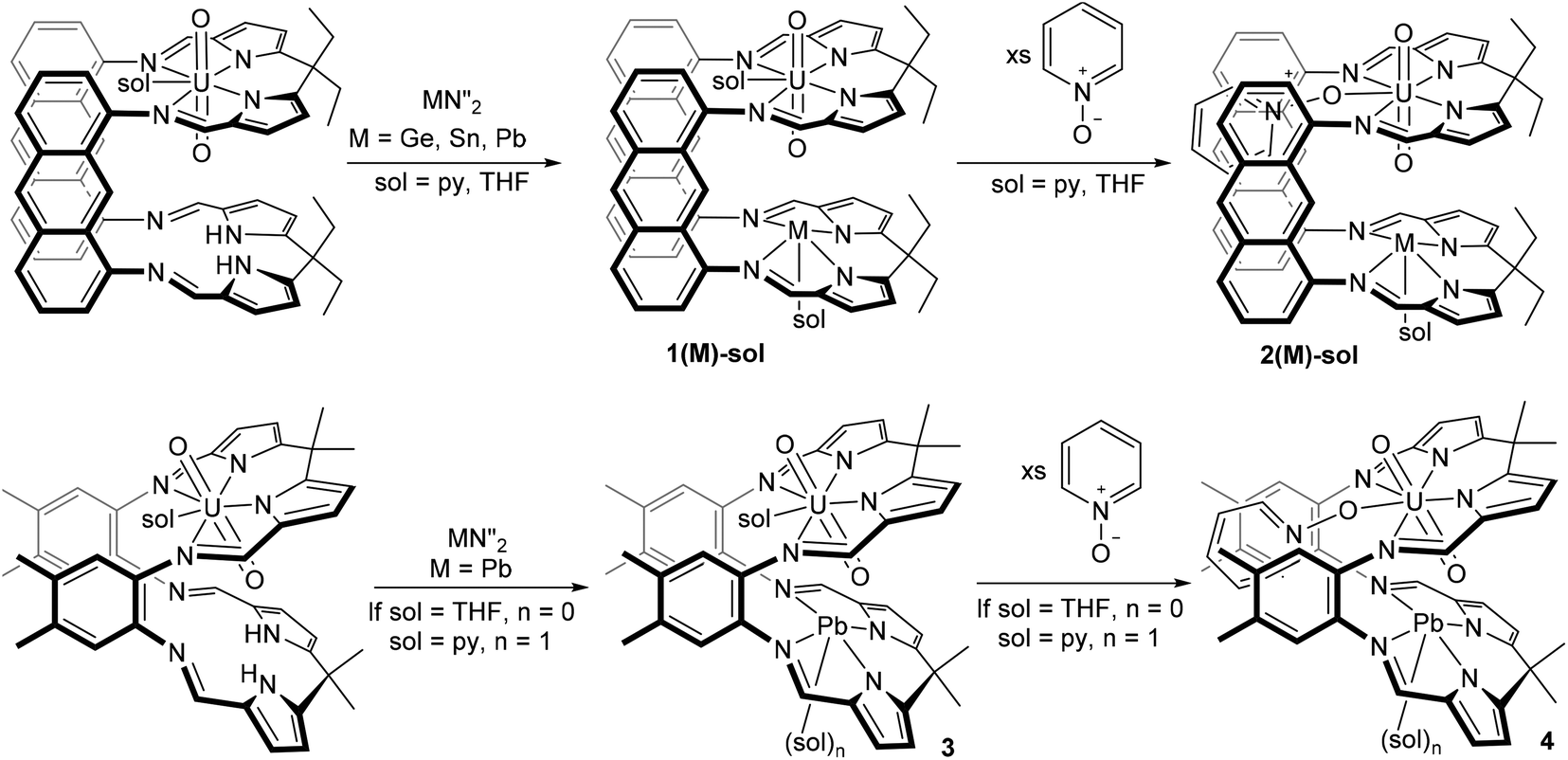 | ||
| Scheme 2 Synthesis of complexes 1–4 using ligands LA (top) and LMe (bottom). N′′ = N(SiMe3)2. | ||
X-ray quality crystals of the THF solvate of each complex were grown by either slow cooling concentrated THF/C6D6 solutions (1(Pb)-THF) or diffusion of hexanes into THF (1(Ge)-THF and 1(Sn)-THF) (Fig. 1(a): 1(Pb)-THF; Fig. S1:†1(Ge)-THF and 1(Sn)-THF). While the three structures are very similar there are some subtle differences (Table 1). Intriguingly all of the group 14 metal ions sit above the N4-donor plane within the molecular cleft, so moving closer to the uranyl endo-oxygen. We have previously demonstrated that the flexibility of the N4-donor pocket in LMe allows the coordinated metal to sit either above or below the plane (by up to ca. 0.6 Å) to accommodate the steric requirements of various co-ligands.12 The structures show that descending group 14 causes the metal to move further out of the plane and closer to the endo-oxygen. This promotes a Pb⋯OUO distance of as little as 3.06 Å, well within the range for bridging M–O–Pb bonds of 2.22–3.33 Å, although slightly longer than the M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯Pb interactions that have been characterised (all ca. 2.6 Å).13 However, the question of whether the metal interacts with, and induces any weakening of the UO2 bonding is less clear. The X-ray crystal structures show only a very slight lengthening of the UOendo bond on descending the group (ca. 0.01 Å) and no significant change in the UOexo bonds. Similarly, the X-ray crystal structure of the uranyl(VI) complex UO2(LiHLMe) (1.794(3) Å)7 also does not show significant lengthening of the UO bonds relative to its precursor UO2(H2LMe) (1.790(4) Å)14 despite there being a significant interaction between the lithium and the endo-oxygen as evidenced in the solid state IR spectra of these compounds (899 vs. 908 cm−1 respectively). The solid state IR spectra (Charts S1 and S3†) of the three THF adducts of 1(Sn), 1(Ge) and 1(Pb) are consistent with a very slight lengthening of the UO2 bond down the group; in contrast, the solution state IR spectra (Chart S6†) are inconclusive with multiple bands appearing in the [UVIO2] region (ca. 890–930 cm−1). These data may suggest the proximity of the group 14 metal to the uranyl oxo group is simply a product of crystal packing effects with the metal moving out of the N4-donor plane due to the increasing ionic radius of the metal down the group.
O⋯Pb interactions that have been characterised (all ca. 2.6 Å).13 However, the question of whether the metal interacts with, and induces any weakening of the UO2 bonding is less clear. The X-ray crystal structures show only a very slight lengthening of the UOendo bond on descending the group (ca. 0.01 Å) and no significant change in the UOexo bonds. Similarly, the X-ray crystal structure of the uranyl(VI) complex UO2(LiHLMe) (1.794(3) Å)7 also does not show significant lengthening of the UO bonds relative to its precursor UO2(H2LMe) (1.790(4) Å)14 despite there being a significant interaction between the lithium and the endo-oxygen as evidenced in the solid state IR spectra of these compounds (899 vs. 908 cm−1 respectively). The solid state IR spectra (Charts S1 and S3†) of the three THF adducts of 1(Sn), 1(Ge) and 1(Pb) are consistent with a very slight lengthening of the UO2 bond down the group; in contrast, the solution state IR spectra (Chart S6†) are inconclusive with multiple bands appearing in the [UVIO2] region (ca. 890–930 cm−1). These data may suggest the proximity of the group 14 metal to the uranyl oxo group is simply a product of crystal packing effects with the metal moving out of the N4-donor plane due to the increasing ionic radius of the metal down the group.
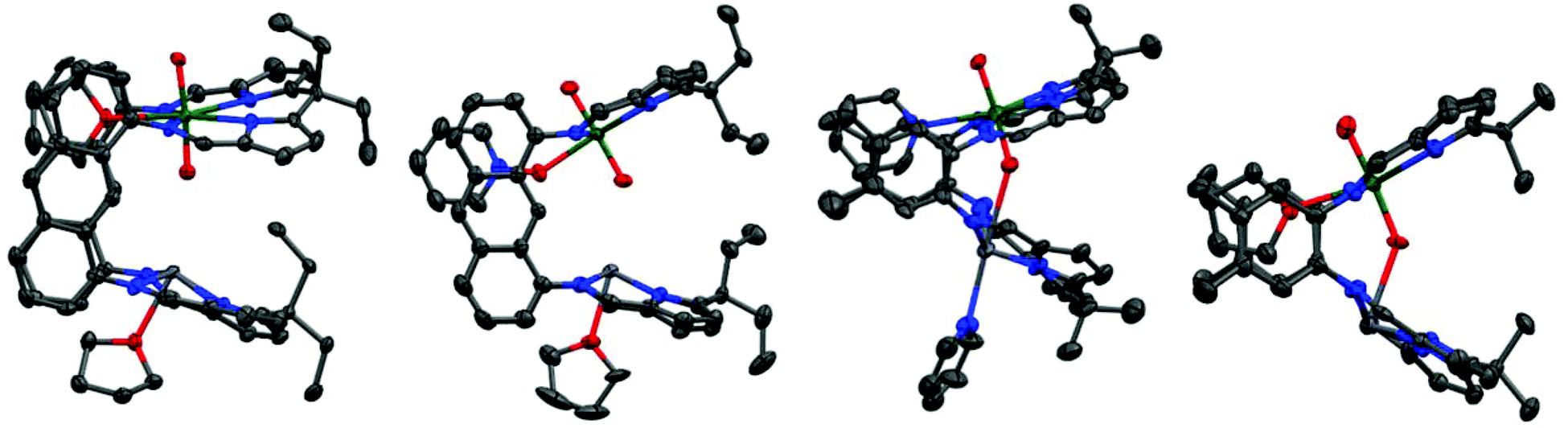 | ||
| Fig. 1 Solid-state structures of (a) 1(Pb)-THF; (b) 2(Pb)-THF; (c) 3(Pb)-py; (d) 3(Pb)-THF. For clarity, all hydrogen atoms and solvent of crystallisation are omitted (displacement ellipsoids are drawn at 50% probability). Atom colours: green = uranium; blue = nitrogen; red = oxygen; light grey = Group 14 element; dark grey = carbon. | ||
| Crystal | M⋯O (Å) | M⋯N4-plane | UO endo (Å) |
UO exo (Å) |
IR (Nujol, cm−1)a | IR (C6H6, cm−1)a | M-sol (Å) | U–O–py (Å) |
|---|---|---|---|---|---|---|---|---|
| a Resolution = 2 cm−1. b Two molecules in the asymmetric unit. | ||||||||
| UO2(H2LA)(THF) | — | — | 1.774(5) | 1.776(4) | 916 | 919/910 | — | — |
| 1(Ge)-THF | 3.36 | 0.41 | 1.757(2) | 1.762(2) | 925 | 928/910 | 2.140(2) | — |
| 1(Sn)-THF | 3.23 | 0.52 | 1.781(3) | 1.782(3) | 921 | 927/911 | 2.325(3) | — |
| 1(Pb)-THF | 3.13/3.06 | 0.55 | 1.767(4)/1.780(4) | 1.764(4)/1.761(4) | 916 | 905 | 2.487(4)/2.469(4) | — |
| 2(Pb)-THF | 3.0 | 0.59 | 1.787(7) | 1.779(7) | — | — | 2.455(7) | — |
| 2(Pb)-py | 3.0 | 0.70 | 1.779(4) | 1.776(4) | 902 | — | 2.480(5) | 2.313(3) |
| 3(Pb)-THF | 2.5 | −0.62 | 1.817(9) | 1.78(1) | 896 | 898 | — | — |
| 3(Pb)-py | 2.91(1) | −0.70 | 1.77(1) | 1.77(1) | 908 | 895 | 2.58(1) | — |
| 4(Pb)-py | 2.612(8) | −0.40 | 1.853(8) | 1.759(7) | 893 | 2.73(1) | 2.354(6) | |
The 119Sn NMR spectrum of 1(Sn)-THF shows a resonance at −459 ppm which is deshielded from that of Sn2(LA) at −527 ppm; however, with the large anthracenyl-linked molecular cleft we have previously seen endo-solvent coordination in homodinuclear complexes which may shield the metal.15Endo-coordination of solvent would not be possible in 1(Sn)-THF due to the presence of the linear UO2 group meaning the tin cation would appear relatively deshielded in the mixed metal complex.
Reaction of 1(Pb) with one equivalent of pyridine N-oxide yields a mixture of compounds in both THF and pyridine solvent which does not change upon heating. However, addition of an excess of pyridine N-oxide (>8 eq.) allowed conversion to one new product by 1H NMR spectroscopy which was assigned as 2(Pb). No free pyridine was observed in the 1H NMR spectrum suggesting no oxidation of the PbII centre has occurred and instead a broadened doublet of doublets at 6.26 ppm indicates coordination of one pyridine N-oxide molecule.
Single crystals of 2(Pb) were grown by slow diffusion of hexane into a pyridine or THF solution containing excess pyridine N-oxide. The refined structures (Fig. 1(b) for 2(Pb)-THF and Fig. S2† for 2(Pb)-py) show coordination of the pyridine N-oxide to the fifth equatorial position of uranium while THF or pyridine solvent coordinates to the Pb centre in the exo-position. While the UO2 bonds in both solvates are not significantly elongated relative to 1(Pb)-THF, the IR spectrum demonstrates a significant effect of the strong equatorial donor with the UO2 stretching frequency decreasing (902 cm−1 for 2(Pb)-pycf. 916 cm−1 for 1(Pb)-THF; Chart S7†).
Despite the lower oxidation potential of SnII (−0.13 V cf. PbII −1.8 V)16 the reaction of 1(Sn) with pyridine N-oxide similarly results in formation of the uranyl(VI) adduct 2(Sn) which was found to be in equilibrium with solvent coordinated 1(Sn) in the 1H NMR spectrum. The 119Sn NMR spectrum of 2(Sn) shows a single resonance at −480 ppm which is very slightly shielded relative to 1(Sn).
GeII is a reducing oxidation state compared with SnII and PbII but treatment of 1(Ge) with pyridine N-oxide yields a mixture of compounds in the 1H NMR spectrum. This may suggest that the relatively electropositive Ge centre may coordinate pyridine-N-oxide in a similar manner to uranium giving a competitive equilibrium between the two metals or even one that displaces a GeII cation from the macrocycle.
Complexes of the aryl-hinged macrocycle LMe
Reaction between the uranyl complex of the smaller aryl-hinged ligand UO2(H2LMe) and Pb with Pb{N(SiMe3)2}2 in pyridine yields a red solution of 3(Pb) immediately whereas in THF solvent the reaction takes several hours. The 1H NMR spectrum shows the formation of a new set of resonances consistent with 3(Pb) and the loss of the NH signal whilst the 29Si NMR spectrum shows the formation of HN(SiMe3)2. However, degradation of this complex occurs upon removal of the solvent from the reaction mixture under reduced pressure and the residue was identified as consisting of UO2(H2LMe), 3(Pb) (from pyridine solution only) and minor impurities. Pure 3(Pb) is accessed by adding an anti-solvent (hexane) to the reaction mixture and once isolated as a solid 3(Pb) is stable under reduced pressure. Treatment of crystalline 3(Pb)-py with D{N(SiMe3)2} followed by removal of the solvent under reduced pressure yielded a deep red residue which was redissolved in d5-pyridine. The 1H NMR spectrum of this material showed ligand resonances corresponding to those of UO2(H2LMe) but the NH resonance was missing suggesting that degradation was due to the presence of DN(SiMe3)2 in the reaction mixture, i.e. the silazide is sufficiently acidic to substitute the Pb from 3(Pb) by protonolysis. This behaviour contrasts to 1(Pb) which is stable in the presence of HN(SiMe3)2.Crystals suitable for X-ray diffraction of both THF and pyridine solvates of 3(Pb) were grown by diffusion of hexane into pyridine or THF solutions (Fig. 1(c) and (d) respectively). The solid state structures of these adducts are different to each other with the Pb cation coordinated by a solvent molecule in 3(Pb)-py but unsolvated in 3(Pb)-THF. This results in a significantly longer Pb–Npy bond (2.58(1) Å) compared to 2(Pb)-py (2.480(5) Å). It is interesting to note that the Pb⋯OUO distance in 3-py of 2.9 Å is very close to those in 1(Pb) and 2(Pb) demonstrating the ability of the macrocyclic ligand pockets to distort to optimise metal geometry and separation.
In 3(Pb)-THF, a short Pb⋯O bond is present and instead of THF coordinating to the Pb cation, an η5-interaction with the pyrrole π-system of an adjacent molecule exists (Fig. 1 and S3†). Both structures show bonding with the uranyl endo oxygen although the THF adduct contains a slightly lengthened UOendo bond (1.817(9) Å) relative to the pyridine adduct (1.77(1) Å). The solid state IR spectra (Charts S8 and S9†) show a UO2 asymmetric stretch for 3(Pb)-py at 907 cm−1 while that for 3(Pb)-THF is seen at 896 cm−1. The former is comparable with that for (py)UO2(H2LMe) (908 cm−1) while the latter is closer to that for (THF)UO2Li(HLMe) (899 cm−1) which contains a strong UO⋯Li interaction, known to affect the reduction chemistry of the resulting complex.
The treatment of 3(Pb) with excess pyridine N-oxide resulted in the formation of the py–O adduct 4(Pb)-py. Single crystals were grown by vapour diffusion of hexane into a concentrated pyridine solution containing excess pyridine N-oxide. The molecular structure (Fig. 2) shows the expected coordination of pyridine N-oxide to the uranium centre while the Pb centre is solvated by pyridine.
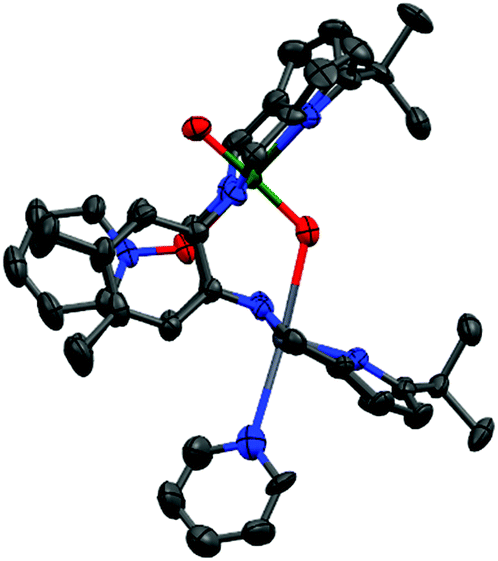 | ||
| Fig. 2 Solid state structure of 4(Pb)-py. For clarity, all hydrogen atoms are omitted (displacement ellipsoids are drawn at 50% probability). Atom colours: green = uranium; blue = nitrogen; red = oxygen; light grey = lead; dark grey = carbon. | ||
Similarly to 3(Pb)-py a short bonding interaction (2.612(8) Å) is evident between the Pb and Oendo in 4(Pb)-py which in this case causes significant elongation of the UOendo bond (1.853(8) Å) with little effect on the UOexo bond. The former UO bond distance is similar to that of reduced uranyl(V) complexes such as LiOUVOLi(HLMe) (UOendo 1.834(4) Å)7 and is slightly longer than the similar bond in (THF)2K[(HO)UVIO2(H2LMe)]17 (1.821(6) Å). This demonstrates that activation of the linear UO2 bonds is facilitated by the presence of strong oxo-donors in the equatorial plane as well as by Lewis acidic metal cations coordinating to the axial uranyl oxygen atoms. The elongation of this bond by 0.08 Å is less than seen for other uranyl UVI complexes in which a strong Lewis acid binds to an oxo group and may reflect the relatively weak Lewis acidity of PbII; the activated UO bond (1.890(4) Å) in OU(OB{C6F5}3)(Aracnac)2 (Ar = 3,5-tBu2C6H3) is significantly longer (0.14 Å) than for UO2(Aracnac)2 (1.755(5) Å),18 and in OU(OB{C6F5}3)(NCN)2 (NCN = {Me3SiN}CPh{NSiMe3}) at 1.898(3) Å the U–OB bond is elongated (0.15 Å) relative to 1.750(4) Å in UO2(NCN)2.19 The IR spectrum of 4(Pb)-py (Chart S10†) shows a UO2 asymmetric stretch at 893 cm−1, significantly shorter than for 3(Pb)-py (908 cm−1) and comparable to LiOUVOLi(HLMe) (893 cm−1) and some other reduced UVO2 complexes such as ({(py)R2AlOUVOH2LMe} and {(py)3MOUVOH2LMe}; R = Me, i-Bu, M = Li, Na, K)20 which all appear within the region 891–894 cm−1.
Attempts to oxidise the PbII centre with alternative oxidising agents, e.g. o-iodylanisole, p-iodosotoluene, bis-trimethylsilyperoxide, and trimethylamine-N-oxide resulted in the formation of UO2(H2LMe) and minor degradation products including a highly symmetrical set of resonances which were assigned as the dinuclear Pb complex 5 (eqn (1)).
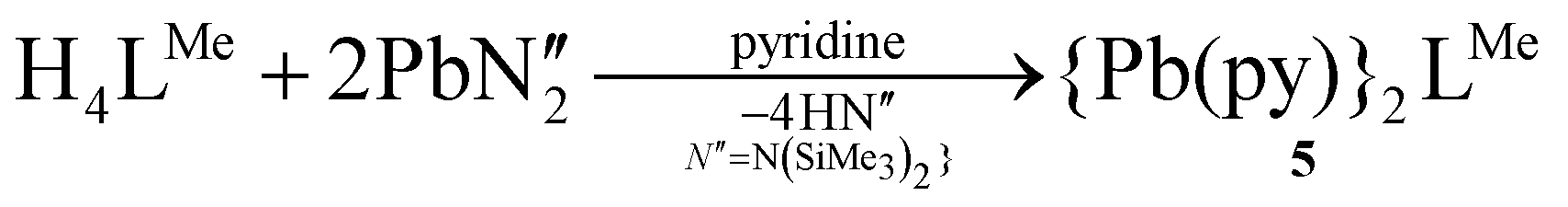 | (1) |
The reasons behind the degradation of 3(Pb) upon reaction with strong oxygen-atom oxidants are unclear; however, PbII has a relatively large ionic radius (1.2 Å)16 and therefore may be unstable within the cleft of this smaller ligand. We have previously shown that complexes of larger metal cations such as CaII adopt an alternative, bowl-shaped coordination mode with LR (R = Me, Et).15 As such, 5 was synthesised directly by treatment of H4LMe with two equivalents of Pb{N(SiMe3)2}2 in pyridine (eqn (1)).
The solid state structure of 5 (Fig. 3) shows the expected ‘bowl’ coordination geometry in which the macrocycle folds at the meso-carbon instead of the aryl linker. This provides a larger cleft which can accommodate the two Pb centres without the significant distortion out of the N4-plane seen in the solid state structure of the other, Pacman-shaped Pb complexes in this work. The Npyrrole⋯Npyrrole distance of 5.25 Å is only slightly larger than the equivalent Nimine⋯Nimine distance for 3(Pb)-py of 5.15 Å and is smaller than that in 1(Pb)-THF (5.27 Å); however, the larger Npyrrole–Pb–Npyrrole angle for 5 (152° cf. Nimine–Pb–Nimine angle of 130° for 3(Pb)-py and 138° for 1(Pb)-THF) allows more room for the metal to sit within the N4-donor plane and as a result the Pb atoms in 5 sit only 0.3 Å out of their respective N4-pockets. In addition, π-interactions between the lead centre and the aryl ring of the adjacent ligand or the pyridine solvent (see ESI Fig. S4†) add stability to this structure.
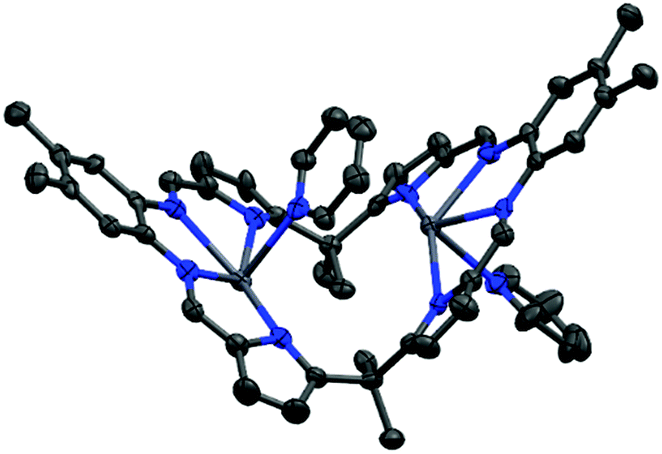 | ||
| Fig. 3 Solid state structure of 5. For clarity, all hydrogen atoms are omitted (displacement ellipsoids are drawn at 50% probability). Atom colours: blue = nitrogen; light grey = lead; dark grey = carbon. | ||
The reactions of UO2(H2LMe) in THF or pyridine with various equivalents of Sn{N(SiMe3)2}2 at different temperatures yield a complex mixture of diamagnetic and paramagnetic species as evidenced by 1H NMR spectroscopy. While slight activation of the UO22+ unit is suggested by the IR spectrum of 3(Pb)-THF, it is likely that significantly more interaction occurs with Sn due to its increased Lewis acidity compared to Pb, resulting in the activation of UO22+ towards reduction by a second molecule of Sn{N(SiMe3)2}2. We have studied extensively the reactions of UO2(H2LMe) with electropositive metal amides (e.g. LDA, Ln{N(SiMe3)2}3, and Mg{N(SiMe3)2}2) and have shown that coordination of these metals within the vacant compartment of the macrocycle results in strong interactions with the endo-oxo of the uranyl and activates this group towards reduction to UV. In contrast, ‘softer’ polarisable metal amides (e.g. Fe{N(SiMe3)2}2, Mn{N(SiMe3)2}2) yield diamagnetic transamination products. In contrast to the above reactions with SnII, no reaction between UO2(H2LMe) and Ge{N(SiMe3)2}2 occurs at ambient temperatures in either THF or pyridine. Upon heating to 80 °C a number of minor paramagnetic resonances appear suggesting that reduction of the uranyl centre occurs, whilst heating to 120 °C in pyridine results in the formation of the previously reported oxo-silylated complex [(Me3Si)OUO(H2LMe)] after 24 h as the major product along with multiple minor paramagnetic species. This may indicate that coordination of Ge{N(SiMe3)2}2 to the exogenous uranyl oxo occurs, resulting in reductive silylation as seen with ZnCl{N(SiMe3)2}.8
Conclusions
Mixed-metal uranyl/group 14 complexes of two different Schiff base macrocyclic ligands have been synthesised by transamination from UO2(H2L) (L = LMe and LA). With the larger anthracenyl-linked macrocycle GeII, SnII and PbII all coordinate in the vacant macrocyclic pocket without reduction of the uranyl. In these complexes the metal ion sits above the N4-donor plane, closer to the uranyl endo-oxygen suggesting a possible bonding interaction. However, it is clear from IR spectroscopy and solid-state structural analysis that only minor activation of the uranyl oxo groups occurs, and so this U–O–M interaction is likely a result of crystal packing forces. In contrast, similar reactions between group 14 silylamides and the uranyl complex of a smaller aryl-linked macrocycle results in reduction to multiple paramagnetic species for Sn and Ge, likely due to the closer interaction between the metal and UOendo enforced by the ligand framework. Only using Pb were we able to isolate the expected mixed-metal uranyl–Pb complex which showed similarly minor activation of the uranyl bonding in the solid state. These data suggest that the proximity of the second metal to uranyl is not an overriding factor in the reduction of uranyl(VI) to uranyl(V) and in the formation of uranyl(VI) oxo–metal bonds. However, the exchange of the equatorial ligand from THF or pyridine in 3(Pb) with pyridine N-oxide to form 4(Pb)-py results in significant elongation of the endo-oxo group and the formation of a clear OUO–Pb bonding interaction, similar to those seen in simple uranyl–perfluoroborane Lewis acid–base adducts. This is only the case when using the more constrained macrocycle environment provided by LMe, and supports the premise that the formation of uranyl(VI)-oxo Lewis acid–base interactions requires a ligand environment (i.e. the Pacman macrocycle) that not only defines the approach of the Lewis acid but also the equatorial coordination sphere of the uranyl, in this case the weak-field macrocycle N4-donor set plus the strongly donating pyridine oxide.
Experimental
General details
All manipulations were carried out under a dry, oxygen-free dinitrogen atmosphere using standard Schlenk techniques or in a glove box unless otherwise stated. Solvents (toluene, n-hexane, diethyl ether and tetrahydrofuran (THF)) were dried by passage through activated 4 Å molecular sieves or activated alumina towers and stored over activated 4 Å molecular sieves. Pyridine was distilled from potassium under dinitrogen in a solvent still prior to use. Deuterated solvents were refluxed over potassium, freeze–pump–thaw degassed three times and vacuum transferred prior to use. 1H NMR spectra were recorded at 298 K unless otherwise stated on either a Bruker AVA400 spectrometer at 399.90 MHz, or AVA500 spectrometer at 500.12 MHz. 13C NMR spectra were recorded at 298 K on a Bruker AVA500 at 125.77 MHz. 119Sn and 29Si-INEPT NMR spectra were run on a Bruker PRO500 spectrometer with a Prodigy cryoprobe at 186 MHz and a Bruker AVA400 spectrometer at 99 MHz respectively. 1H and 13C NMR spectra were referenced internally to residual protio solvent (1H) or solvent (13C) and are reported relative to tetramethylsilane (δ = 0 ppm). Chemical shifts are quoted in δ (ppm). IR spectra were acquired on a Jasco 410 FT-IR spectrophotometer as C6H6 solutions, between KBr plates, or as a Nujol mull (w = weak, m = medium, s = strong intensity). Elemental analyses were performed by Mr Stephen Boyer at London Metropolitan University or Pascher Labor, Germany.The compounds H4LMe,12 UO2(H2LMe),14 UO2(H2LA),11 Ge{N(SiMe3)2}2, Sn{N(SiMe3)2}2 and Pb{N(SiMe3)2}2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 21 were synthesised by published methods; all other chemicals were purchased from Sigma Aldrich and used as received.
21 were synthesised by published methods; all other chemicals were purchased from Sigma Aldrich and used as received.
1H NMR (500 MHz, THF-d8): δ 9.35 (s, 2H, 2 × CHim), 9.12 (s, 2H, 2 × CHim), 8.43 (s, 2H, 2 × CHAr), 7.99 (s, 2H, 2 × CHAr), 7.84 (d, J = 8.5 Hz, 2H, 2 × CH), 7.66 (d, J = 8.5 Hz, 2H, 2 × CH), 7.46 (dd, J = 8.5, 7.0 Hz, 2H, 2 × CH), 7.28 (dd, J = 8.5, 7.0 Hz, 2H, 2 × CH), 7.20 (d, J = 6.9 Hz, 2H, 2 × CH), 7.17 (d, J = 3.6 Hz, 2H, 2 × CH), 6.62 (d, J = 6.9 Hz, 2H, 2 × CH), 6.60 (d, J = 3.6 Hz, 2H, 2 × CH), 6.55 (d, J = 3.6 Hz, 2H, 2 × CH), 6.02 (d, J = 3.6 Hz, 2H, 2 × CH), 2.27 (qt, J = 7.3, 3.6 Hz, 4H, 2 × CH2), 2.09 (q, J = 7.3 Hz, 2H, CH2), 0.85 (t, J = 7.3 Hz, 3H, CH3), 0.81 (t, J = 7.3 Hz, 3H, CH3), 0.80 (t, J = 7.3 Hz, 3H, CH3), 0.22 (t, J = 7.3 Hz, 3H, CH3) ppm (one CH2 resonance hidden by THF solvent); 1H NMR (601 MHz, Pyridine-d5): δ 9.58 (s, 2H, 2 × CHim), 9.17 (s, 2H, 2 × CHim), 8.13 (s, 2H, 2 × CHAr), 7.72 (d, J = 8.5 Hz, 2H, 2 × CH), 7.67 (s, 2H, 2 × CHAr), 7.50 (d, J = 8.4 Hz, 2H, 2 × CH), 7.34 (d, J = 3.7 Hz, 2H, 2 × CH), 7.25 (m, 4H, 4 × CH), 6.94 (d, J = 6.8 Hz, 2H, 2 × CH), 6.84 (d, J = 3.9 Hz, 2H, 2 × CH), 6.61 (d, J = 3.5 Hz, 2H, 2 × CH), 6.28 (d, J = 3.6 Hz, 2H, 2 × CH), 5.68 (d, J = 6.8 Hz, 2H, 2 × CH), 2.62–2.47 (m, 4H, 2 × CH2), 2.28 (q, J = 7.2 Hz, 2H, CH2), 1.74 (q, J = 7.3 Hz, 2H, CH2), 1.25–1.04 (m, 12H, 3 × CH3), 0.67 (t, J = 7.3 Hz, 3H, CH3) ppm; 13C{1H} NMR (126 MHz, THF-d8): δ 162.7(Cq), 161.8 (CH), 154.1 (CH), 153.7 (Cq), 153.5 (Cq), 148.2 (Cq), 138.7 (Cq), 137.5 (Cq), 134.0 (Cq), 133.8 (Cq), 128.9 (Cq), 128.8 (Cq), 127.4 (CH), 127.1 (CH), 126.7 (CH), 126.3 (CH), 125.4 (CH), 124.6 (CH), 123.2 (CH), 121.6 (CH), 120.8 (CH), 113.3 (CH), 112.6 (CH), 108.4 (CH), 52.2 (Cq), 46.7 (Cq), 44.6 (CH2), 40.2 (CH2), 38.5 (CH2), 30.4 (CH2), 11.0 (CH3), 10.7 (CH3), 10.6 (CH3), 10.0 (CH3) ppm; Analysis Calcd C66H64N8O4GeU (1343.95) requires C % 58.98, H % 4.80, N % 8.34; found C % 58.40, H % 4.37, N % 8.68. FTIR (Nujol mull, cm−1): ν 1616 (m), 1591 (s), 1553 (m), 1313 (m), 1277 (s), 1266 (s), 1170 (w), 1089 (w), 1054 (m), 1027 (m), 1009 (m), 925 (s, UO2 asymmetric stretch), 875 (w), 855 (m), 742 (s), 722 (s); FTIR (C6H6, cm−1): ν 2969 (m), 2934 (m), 2875 (m), 1595 (vs), 1552 (vs), 1311 (m), 1278 (s), 1255 (m), 1090 (m), 1071 (s), 1058 (s), 1013 (m), 928 (s), 911 (m), 875 (m), 863 (m), 763 (m), 743 (m).
1H NMR (Pyridine-d5, 500 MHz): δ 9.62 (s, 2H, 2 × CHim), 9.38 (s, 2H, 2 × CHim), 8.15 (s, 2H, 2 × CHAr), 8.12 (s, 2H, 2 × CHAr), 7.74 (d, J = 8.5 Hz, 2H, 2 × CHAr), 7.58 (d, J = 8.5 Hz, 2H, 2 × CHAr), 7.51 (d, J = 3.6 Hz, 2H, 2 × CHpyrrole), 7.33 (dd, J = 7.5 Hz, 2H, 2 × CHAr), 7.26 (dd, J = 7.5 Hz, 2H, 2 × CHAr), 7.04 (d, J = 6.7 Hz, 2H, 2 × CHAr), 6.96 (d, J = 3.6 Hz, 2H, 2 × CHAr), 6.61 (d, J = 7.0 Hz, 2H, CHAr), 6.56 (d, J = 3.8 Hz, 2H, 2 × CHpyrrole), 6.16 (d, J = 3.8 Hz, 2H, 2 × CHpyrrole), 2.57 (m, 4H, 2 × CH2), 2.39 (q, J = 7.2 Hz, 2H, CH2), 1.87 (q, J = 7.2 Hz, 2H, CH2), 1.28 (t, J = 7.2 Hz, CH3), 1.15 (t, J = 7.2 Hz, CH3), 1.12 (t, J = 7.2 Hz, CH3), 0.79 (t, J = 7.2 Hz, CH3) ppm; 13C{1H} NMR (126 MHz, Pyridine-d5): δ 162.8 (Cq), 161.2 (CH), 154.4 (CH), 153.0 (Cq), 150.9 (Cq), 139.0 (Cq), 138.8 (Cq), 133.2 (Cq), 133.0 (Cq), 128.6 (Cq), 128.1 (Cq), 127.5 (CH), 126.7 (CH), 126.6 (CH), 125.9 (CH), 125.8 (CH), 125.0 (CH), 121.9 (CH), 121.2 (CH), 121.1 (CH), 114.9 (CH), 113.1 (CH), 110.0 (CH), 52.6 (Cq), 46.2 (Cq), 42.1 (CH2), 40.0 (CH2), 38.7 (CH2), 27.2 (CH2), 11.3 (CH3), 11.2 (CH3), 10.7 (CH3), 10.0 (CH3) ppm (one Cq under pyridine solvent resonances); 119Sn NMR (186 MHz, pyridine-d5): δ −459 ppm; Analysis Calcd C46H64N8O4SnU (1390.03) requires C % 57.03, H % 4.64, N % 8.06; found C % 57.20, H % 4.52, N % 8.14; FTIR (Nujol mull, cm−1): ν 1594 (s), 1554 (m), 1311 (m), 1277 (m), 1262 (m), 1171 (w), 1154 (w), 1088 (m), 1055 (s), 1016 (m), 963 (w), 921 (m, UO2 asymmetric stretch), 875 (w), 801 (m), 722 (s); FTIR (C6H6, cm−1): ν 2968 (m), 2932 (m), 2875 (m), 1599 (vs), 1553 (vs), 1311 (m), 1278 (s), 1091 (m), 1059 (s), 928 (m), 910 (m), 875 (m), 863 (m), 761 (m), 744 (m).
1H NMR (Pyridine-d5, 500 MHz): δ 9.55 (s, 2H, 2 × CHim), 9.39 (s, 2H, 2 × CHim), 8.20 (s, 2H, 2 × CHAr), 7.84 (s, 2H, 2 × CHAr), 7.75 (d, J = 8.5 Hz, 2H, 2 × CHAr), 7.50 (d, J = 8.5 Hz, 2H, 2 × CHAr), 7.45 (d, J = 3.6 Hz, 2H, 2 × CHpyrrole), 7.29 (dd, J = 7.5 Hz, 2H, 2 × CHAr), 7.26 (dd, J = 7.5 Hz, 2H, 2 × CHAr), 7.04 (d, J = 6.7 Hz, 2H, 2 × CHAr), 6.88 (d, J = 3.6 Hz, 2H, 2 × CHAr), 6.76 (d, J = 3.6 Hz, 2H, CHAr), 6.53 (d, J = 3.6 Hz, 2H, 2 × CHpyrrole), 5.79 (d, J = 6.7 Hz, 2H, 2 × CHpyrrole), 2.61 (q, J = 7.3 Hz, 2H), 2.55 (q, J = 7.3 Hz, 2H), 2.44 (q, J = 7.3 Hz, 2H), 1.97 (q, J = 7.3 Hz, 2H), 1.31 (t, J = 7.3 Hz, 3H), 1.19 (t, J = 7.3 Hz, 3H), 1.10 (t, J = 7.3 Hz, 3H), 0.83 (t, J = 7.3 Hz, 3H) ppm; 13C{1H} NMR (126 MHz, Pyridine-d5): δ 163.0 (Cq), 161.3 (CH), 158.6 (Cq), 156.1 (CH), 152.7 (Cq), 151.2 (Cq), 141.6 (Cq), 138.7 (Cq), 133.2 (Cq), 133.0 (Cq), 128.8 (Cq), 128.1 (Cq), 127.6 (CH), 126.9 (CH), 126.7 (CH), 126.0 (CH), 125.7 (CH), 122.7 (CH), 121.0 (CH), 120.2 (CH), 114.6 (CH), 113.4 (CH), 110.7 (CH), 52.6 (Cq), 47.3 (Cq), 41.0 (CH2), 39.4 (CH2), 38.7 (CH2), 26.2 (CH2), 11.3 (CH3), 11.2 (CH3), 10.4 (CH3), 9.7 (CH3) ppm; Analysis Calcd C62H56N8O3PbU (1406.41) requires C % 52.95, H % 4.01, N % 7.97; found C % 52.85, H % 4.17, N % 7.73; FTIR (Nujol mull, cm−1): ν 1597 (s), 1552 (m), 1261 (s), 1090 (m), 1055 (m), 1018 (m), 916 (m, UO2 asymmetric stretch), 874 (w), 799 (m), 722 (m); FTIR (C6H6, cm−1): ν 2971 (m), 2934 (m), 2876 (m), 1599 (vs), 1402 (w), 1315 (m), 1290 (s), 1277 (s), 1266 (s), 1090 (m), 1058 (s), 916 (m), 905 (s), 874 (s), 862 (s), 761 (m), 744 (m).
1H NMR (500 MHz, Pyridine-d5): δ 10.03 (s, 2H, 2 × CHim), 9.25 (s, 2H, 2 × CHim), 8.03 (s, 2H, 2 × CHAr), 7.86 (s, 2H, 2 × CHAr), 7.68 (d, J = 8.6 Hz, 2H, 2 × CH), 7.50 (d, J = 3.7 Hz, 2H, 2 × CHpyrrole), 7.32–7.26 (m, 4H, 4 × CH), 7.07–7.02 (m, 2H, 2 × CH), 6.88 (d, J = 3.7 Hz, 2H, 2 × CHpyrrole), 6.79 (d, J = 7.0 Hz, 2H, 2 × CH), 6.71 (d, J = 3.5 Hz, 2H, 2 × CHpyrrole), 6.46 (d, J = 3.5 Hz, 2H, 2 × CHpyrrole), 6.26 (br. dd, 1H, CH), 5.85 (d, J = 7.0 Hz, 2H, 2 × CH), 2.66–2.54 (m, 4H, 2 × CH2), 2.37 (q, J = 7.3 Hz, 2H, CH2), 1.81 (q, J = 7.3 Hz, 2H, CH2), 1.32–1.12 (m, 6H, 2 × CH3), 1.07 (t, J = 7.3 Hz, 3H, CH3), 0.69 (t, J = 7.3 Hz, 3H, CH3) ppm; 13C{1H} NMR (126 MHz, Pyridine-d5): δ 162.9 (Cq), 160.0 (CH), 159.5 (CH), 156.9 (CH), 155.6 (CH), 153.0 (Cq), 142.2 (Cq), 133.6 (CH), 133.1 (Cq), 132.7 (Cq), 129.2 (Cq), 128.5 (Cq), 127.1 (CH), 126.7 (CH), 126.3 (CH), 125.8 (CH), 125.5 (CH), 125.1 (CH), 122.3 (CH), 120.9 (CH), 120.8 (CH), 114.0 (CH), 113.6 (CH), 110.6 (CH), 52.1 (Cq), 46.9 (Cq), 41.0 (CH2), 33.1 (CH2), 25.6 (CH2),11.7 (CH3), 10.8 (CH3), 10.2 (CH3), 9.5 (CH3) ppm; Analysis Calcd C68H58N10O3PbU (1508.51) requires C % 54.14, H % 3.88, N % 9.29; found C % 54.23, H % 3.98, N % 9.40; FTIR (Nujol mull, cm−1): ν 1588 (s), 1548 (m), 1300 (m), 1276 (s), 1265 (s), 1244 (m), 1170 (m), 1158 (m), 1111 (m), 1090 (m), 1056 (m), 1040 (m), 1017 (m), 953 (m), 902 (m, UO2 asymmetric stretch), 892 (m), 871 (m), 849 (w), 756 (m), 739 (m), 722 (m).
1H NMR (400 MHz, THF-d8): δ 9.07 (s, 2H, 2 × CHim), 8.01 (s, 2H, 2 × CHim), 7.35 (s, 2H, 2 × CHAr), 6.91 (d, J = 3.6 Hz, 2H, 2 × CHpyrrole), 6.89 (s, 2H, 2 × CHAr), 6.31 (d, J = 3.4 Hz, 2H, 2 × CHpyrrole), 6.29 (d, J = 3.6 Hz, 2H, 2 × CHpyrrole), 5.93 (d, J = 3.4 Hz, 2H, 2 × CHpyrrole), 2.40 (s, 6H, 2 × CH3), 2.38 (s, 6H, 2 × CH3), 1.81 (s, 3H, CH3), 1.40 (s, 3H, CH3), 1.34 (s, 3H, CH3), 1.31 (s, 3H, CH3) ppm; 13C{1H} NMR (126 MHz, THF-d8): δ 164.6 (Cq), 161.9 (CH), 157.9 (CH), 157.4 (Cq), 146.6 (Cq), 144.8 (Cq), 139.1 (Cq), 138.3 (Cq), 134.7 (Cq), 132.4 (Cq), 124.9 (CH), 122.9 (CH), 122.2 (CH), 120.6 (CH), 109.3 (CH), 107.1 (CH), 40.7 (Cq), 38.7 (Cq), 34.3 (CH3), 33.7 (CH3), 30.1 (CH3), 27.5 (CH3), 18.7 (CH3), 18.6 (CH3)ppm; Analysis Calcd C46H50N8O3PbU (1208.19) requires C % 45.73, H % 4.17, N % 9.27; found C % 45.72, H % 4.04, N % 9.16; FTIR (Nujol mull, cm−1): ν 2725 (m), 2671 (m), 1596 (s), 1573 (m), 1299 (m), 1281 (s), 1272 (s), 1183 (m), 1050 (m), 1019 (m), 896 (m, UO2 asymmetric stretch), 722 (m); FTIR (C6H6, cm−1): ν 3375 (w), 2958 (m), 2922 (m), 2872 (w), 2862 (w), 2235 (w), 2082 (w), 1596 (vs), 1574 (s), 1357 (w), 1275 (s), 1218 (w), 1181 (w), 898 (s, UO2 asymmetric stretch) 839 (w), 821 (w), 800 (w), 764 (w), 725 (w).
1H NMR (500 MHz, Pyridine-d5): δ 9.23 (s, 2H, 2 × CHim), 8.47 (s, 2H, 2 × CHim), 7.30 (d, J = 3.3 Hz, 2H), 6.81 (d, J = 3.3 Hz, 2H), 6.66 (s, 2H, CHAr), 6.56 (s, 2H, CHAr), 6.52 (d, J = 3.3 Hz, 2H), 6.22 (d, J = 3.3 Hz, 2H), 2.17 (s, 6H, 2 × CH3), 1.93 (s, 3H, CH3), 1.84 (s, 6H, 2 × CH3), 1.81 (s, 3H, CH3), 1.34 (s, 3H, CH3), 1.27 (s, 3H, CH3) ppm; 13C{1H} NMR (101 MHz, Pyridine-d5): δ 164.7 (Cq), 163.1 (CH), 157.8 (CH), 157.6 (Cq), 145.1 (Cq), 144.3 (Cq), 139.6 (Cq), 139.1 (Cq), 134.4 (Cq), 131.9 (Cq), 126.9 (CH), 123.0 (CH), 121.6 (CH), 110.7 (CH), 108.9 (CH), 41.6 (Cq), 39.5 (Cq), 36.0 (CH3), 33.5 (CH3), 30.4 (CH3), 29.9 (CH3), 19.6 (CH3), 19.2 (CH3) ppm; IR (Nujol Mull, cm−1): ν 1600 (s), 1577 (s), 1301 (m), 1271 (s), 1219 (w), 1175 (w), 1113 (w), 1051 (m), 1018 (m), 908 (m, UO2 asymmetric stretch), 888 (m), 837 (w), 800 (w), 722 (w), 700 (w); FTIR (C6H6, cm−1): ν 2973 (w), 1594 (vs), 1573 (s), 1438 (w), 1278 (s), 1260 (m), 1052 (m), 923 (w), 895 (m, UO2 asymmetric stretch).
4(Pb)-py
To a solution of 3(Pb)-THF (10 mg, 8.3 μmol) in C6D6 (1 mL) was added pyridine N-oxide (1 mg, 10 μmol). Immediate precipitation of a brown precipitate was observed. This was centrifuged and the mother liquor removed before washing the solids with Et2O. The isolated precipitate was dried under reduced pressure to yield a light brown powder (8 mg, 6.5 μmol, 78%). Single crystals were grown from vapour diffusion of hexane into a pyridine solution of 4(Pb).
1H NMR (500 MHz, Pyridine-d5): δ 9.63 (s, 2H, 2 × CHim), 9.14 (s, 2H, 2 × CHim), 8.86 (s, 2H, 2 × CHAr), 8.12 (dd, J = 7.6 Hz, 1H, CHpy), 7.99 (s, 2H, 2 × CHAr), 7.74 (s, 2H, 2 × CHpy), 7.71 (dd, J = 7.6 Hz, 2H, 2 × CHpy), 7.62 (s, 2H, 2 × CHpyrrole), 7.36 (s, 2H, 2 × CHpyrrole), 7.08 (s, 2H, 2 × CHpyrrole), 6.65 (s, 2H, 2 × CHpyrrole), 2.55 (s, 6H, 2 × CH3), 2.49 (s, 3H, CH3), 2.32 (s, 3H, CH3), 2.28 (s, 6H, 2 × CH3), 1.87 (s, 3H, CH3), 1.58 (s, 3H, CH3) ppm; 13C{1H} NMR (126 MHz, Pyridine-d5): δ 165.0 (Cq), 161.4 (Cq), 161.4 (CH), 154.9 (CH), 147.4 (Cq), 145.9 (Cq), 139.7 (Cq), 139.6 (CH), 135.0 (Cq), 132.7 (Cq), 126.4 (CH), 125.1 (CH), 122.7 (CH), 122.3 (CH), 121.1 (CH), 110.6 (CH), 109.1 (CH), 41.8 (Cq), 39.6 (Cq), 38.5 (CH3), 37.0 (CH3), 28.3 (CH3), 27.4 (CH3), 19.43 (CH3), 19.1 (CH3) ppm; Analysis Calcd C47H45N9O3PbU (1229.17) requires C % 45.93, H % 3.69, N % 10.26; found C % 45.64, H % 3.55, N % 9.99.
1H NMR (500 MHz, C6D6): δ 8.24 (s, 2H, 2 × CHim), 8.15 (s, 2H, 2 × CHim), 6.71 (d, J = 3.5 Hz, 2H, 2 × CHpyrrole), 6.62 (d, J = 3.5 Hz, 2H, 2 × CHpyrrole), 6.48 (s, 2H, 2 × CHAr), 6.37 (d, J = 3.5 Hz, 2H, 2 × CHpyrrole), 6.34 (d, J = 3.5 Hz, 2H, 2 × CHpyrrole), 6.22 (s, 2H, 2 × CHAr), 2.03 (s, 6H, 2 × CH3), 2.02 (s, 6H, 2 × CH3), 1.76 (s, 3H, CH3), 1.73 (s, 3H, CH3), 1.71 (s, 3H, CH3), 1.47 (s, 3H, CH3) ppm; 13C{1H} NMR (126 MHz, C6D6): δ 162.8 (CH), 159.2 (CH), 158.9 (Cq), 158.4 (Cq), 142.4 (Cq), 140.9 (Cq), 140.7 (Cq), 140.0 (Cq), 133.1 (Cq), 132.5 (Cq), 123.8 (CH), 123.3 (CH), 122.5 (CH), 120.5 (CH), 110.5 (CH), 110.0 (CH), 39.6 (Cq), 39.4 (Cq), 34.9 (CH3), 32.3 (CH3), 24.1 (CH3), 24.0 (CH3), 19.5 (CH3), 19.3 (CH3) ppm; Analysis Calcd C46H48N8Pb2 (1143.35) requires C % 48.32, H % 4.23, N % 9.80; found C % 48.50, H % 4.17, N % 9.58.
Acknowledgements
The authors thank the University of Edinburgh and the EPSRC (Grant numbers EP/M010554/1 and EP/H004823/1) for funding.Notes and references
- G. R. Choppin, J. Radioanal. Nucl. Chem., 2007, 273, 695–703 CrossRef CAS.
- P. L. Arnold, G. M. Jones, S. O. Odoh, G. Schreckenbach, N. Magnani and J. B. Love, Nat. Chem., 2012, 4, 221–227 CrossRef CAS PubMed.
- J. N. Mathur, M. S. Murali and K. L. Nash, Solvent Extr. Ion Exch., 2001, 19, 357–390 CrossRef CAS; E. P. Horwitz, R. Chiarizia, M. L. Dietz and H. Diamond, Anal. Chim. Acta, 1993, 281, 361–372 CrossRef; Y. Sakamura, T. Hijikata, K. Kinoshita, T. Inoue, T. S. Storvick, C. L. Krueger, J. J. Roy, D. L. Grimmett, S. P. Fusselman and R. L. Gay, J. Alloys Compd., 1998, 271, 592–596 CrossRef.
- P. L. Arnold, J. B. Love and D. Patel, Coord. Chem. Rev., 2009, 253, 1973–1978 CrossRef CAS; J. C. Renshaw, L. J. C. Butchins, F. R. Livens, I. May, J. M. Charnock and J. R. Lloyd, Environ. Sci. Technol., 2005, 39, 5657–5660 CrossRef PubMed.
- M. Bühl and G. Schreckenbach, Inorg. Chem., 2010, 49, 3821–3827 CrossRef PubMed.
- P. L. Arnold, A.-F. Pecharman and J. B. Love, Angew. Chem., Int. Ed., 2011, 50, 9456–9458 CrossRef CAS PubMed; P. L. Arnold, E. Hollis, F. J. White, N. Magnani, R. Caciuffo and J. B. Love, Angew. Chem., Int. Ed., 2011, 50, 887–890 CrossRef PubMed; G. M. Jones, P. L. Arnold and J. B. Love, Chem. – Eur. J., 2013, 19, 10287–10294 CrossRef PubMed.
- P. L. Arnold, A.-F. Pecharman, E. Hollis, A. Yahia, L. Maron, S. Parsons and J. B. Love, Nat. Chem., 2010, 2, 1056–1061 CrossRef CAS PubMed.
- P. L. Arnold, A.-F. Pécharman, R. M. Lord, G. M. Jones, E. Hollis, G. S. Nichol, L. Maron, J. Fang, T. Davin and J. B. Love, Inorg. Chem., 2015, 54, 3702–3710 CrossRef CAS PubMed.
- P. L. Arnold, D. Patel, A. J. Blake, C. Wilson and J. B. Love, J. Am. Chem. Soc., 2006, 128, 9610–9611 CrossRef CAS PubMed.
- P. L. Arnold, G. M. Jones, Q.-J. Pan, G. Schreckenbach and J. B. Love, Dalton Trans., 2012, 41, 6595–6597 RSC.
- X. J. Zheng, N. L. Bell, C. J. Stevens, Y. X. Zhong, G. Schreckenbach, P. L. Arnold, J. B. Love and Q. J. Pan, 2016, submitted.
- G. Givaja, M. Volpe, J. W. Leeland, M. A. Edwards, T. K. Young, S. B. Darby, S. D. Reid, A. J. Blake, C. Wilson, J. Wolowska, E. J. L. McInnes, M. Schröder and J. B. Love, Chem. – Eur. J., 2007, 13, 3707–3723 CrossRef CAS PubMed.
- F. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef.
- P. L. Arnold, A. J. Blake, C. Wilson and J. B. Love, Inorg. Chem., 2004, 43, 8206–8208 CrossRef CAS PubMed.
- E. A. Connolly, J. W. Leeland and J. B. Love, Inorg. Chem., 2016, 55, 840–847 CrossRef CAS PubMed.
- Handbook of Chemistry and Physics, 61st edn, 1981 Search PubMed.
- P. L. Arnold, D. Patel, A.-F. Pecharman, C. Wilson and J. B. Love, Dalton Trans., 2010, 39, 3501–3508 RSC.
- T. W. Hayton and G. Wu, Inorg. Chem., 2009, 48, 3065–3072 CrossRef CAS PubMed.
- M. J. Sarsfield and M. Helliwell, J. Am. Chem. Soc., 2004, 126, 1036–1037 CrossRef CAS PubMed.
- M. Zegke, G. S. Nichol, P. L. Arnold and J. B. Love, Chem. Commun, 2015, 51, 5876–5879 RSC.
- T. Heidemann and S. Mathur, Eur. J. Inorg. Chem., 2014, 2014, 506–510 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1480061–1480066, 1480068, 1480069 and 1480093. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt01948j |
| This journal is © The Royal Society of Chemistry 2016 |