DOI:
10.1039/C6DT00299D
(Paper)
Dalton Trans., 2016,
45, 9860-9870
Structurally versatile phosphine and amine donors constructed from N-heterocyclic olefin units†
Received
21st January 2016
, Accepted 17th February 2016
First published on 18th February 2016
Introduction
Sterically encumbered phosphines and N-heterocyclic carbenes (NHCs) are effective ligands for supporting a variety of catalytic bond-forming processes,1 and can stabilize highly reactive molecular entities via strong coordinative interactions.2 Common traits between these two ligand classes are the presence of a strongly σ-donating atom, ease of synthesis, and a high level of structural tunability. A related ligand group that is attracting increasing attention of late are N-heterocyclic olefins (NHOs),3 which contain considerable nucleophilic character due to the highly polarized nature of the exocyclic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond, allowing these species to be strong neutral 2-electron donors (Chart 1; left). Accordingly, NHOs are now being used to intercept reactive inorganic species,4,5 as organocatalysts for various polymerization strategies,6 and as a component of pincer-type ligands.7
C double bond, allowing these species to be strong neutral 2-electron donors (Chart 1; left). Accordingly, NHOs are now being used to intercept reactive inorganic species,4,5 as organocatalysts for various polymerization strategies,6 and as a component of pincer-type ligands.7
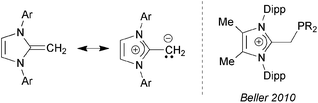 |
| | Chart 1 (Left) Canonical forms for a generic N-heterocyclic olefin (NHO); (Right) Beller's imidazolium alkylphosphines; Ar = aryl; Dipp = 2,6-iPr2C6H3. | |
In this paper, we present efficient routes to phosphine and amine donors that contain an NHO moiety [IPrCH]− directly linked to P- and N-donor sites. As shown in Chart 2, there is a possibility of coordination through either the NHO (via carbon-ligation) or the terminal P/N atoms. Our current study was motivated in part by the prior work of Beller who demonstrated that imidazolium-alkylphosphines (Chart 1; right) when combined with Pd(II) sources and base, afforded active catalysts (in situ) for the hydroxylation of arylhalides, and for both C–N (Buchwald–Hartwig) and C–C (Sonogashira) coupling reactions.8 Despite the possible formation of neutral NHO-linked phosphines (NHOPs; Chart 2; E = P) during Beller's catalytic processes, such ligands were not isolated, nor were any well-defined metal complexes with these ligands reported. As a result, we decided to explore this ligand class in more detail and consequently uncovered divergent coordination behavior towards AuCl, depending if hard amine- or soft-phosphine groups are appended to an NHO unit.
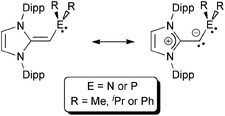 |
| | Chart 2 N-Heterocyclic olefin-phosphines (NHOP) or -amines (NHON) discussed in this paper. | |
Results and discussion
Synthesis of N-heterocyclic olefin phosphines (NHOPs)
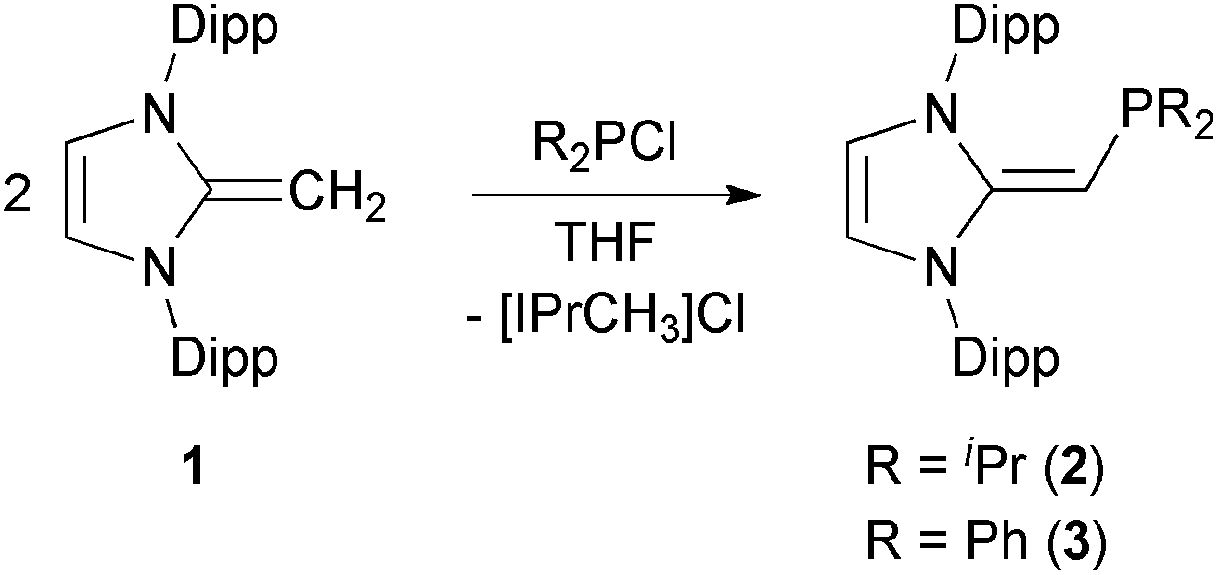
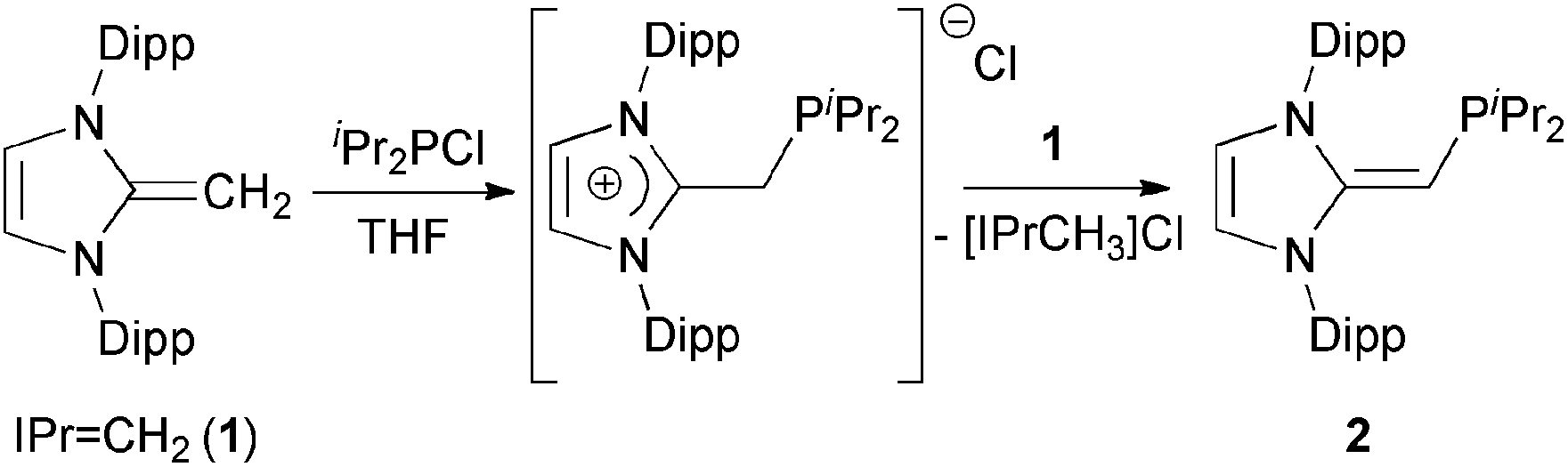
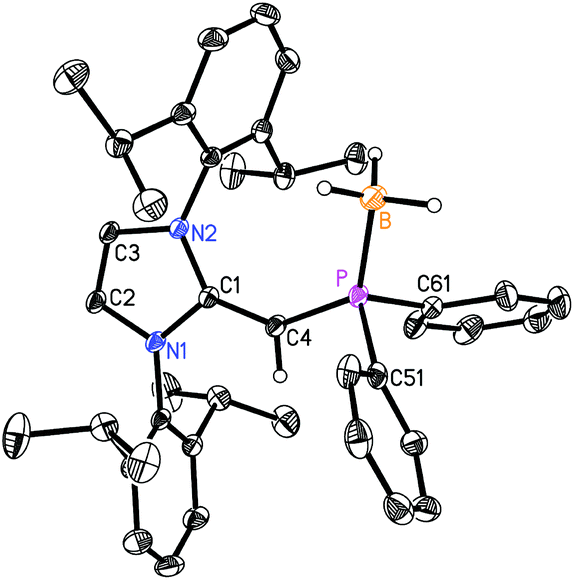
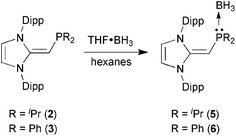
We began our studies by exploring the synthesis of the diisopropylphosphine-capped N-heterocyclic olefin (IPrCH)PiPr22 (IPr = [(HCNDipp)2C]; Dipp = 2,6-iPr2C6H3). In line with prior work from our group,9 the readily available NHO, IPrCH21,3d was combined with ClPiPr2 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio in THF (Scheme 1) with the intention of first isolating the imidazolium salt [IPr-CH2-PiPr2]Cl, which would be isostructural to Beller's pre-ligands shown in Chart 1. While there was spectroscopic evidence for the formation of the desired imidazolium salt, the starting material IPrCH21 was sufficiently basic to deprotonate [IPr-CH2-PiPr2]Cl to give 2 and the known by-product [IPrCH3]Cl.9 Fortunately 2 and [IPrCH3]Cl have quite different solubilities, allowing for their easy separation. By altering the ratio between IPrCH21 and ClPiPr2 to 2:1 (eqn (1)) and conducting the reaction in THF at room temperature for 20 h, we were able to isolate pure (IPrCH)PiPr22 in 81% yield after extracting 2 from the product mixture (containing [IPrCH3]Cl) with hexanes. Following a similar procedure, the phenyl-substituted NHOP (IPrCH)PPh23 was prepared in an isolated yield of 83% (eqn (1)). The new NHOPs 2 and 3 were each characterized by NMR spectroscopy, elemental analysis and X-ray crystallography (colorless crystals grown from hexanes at −30 °C; Fig. 1 and 2).
:1 ratio in THF (Scheme 1) with the intention of first isolating the imidazolium salt [IPr-CH2-PiPr2]Cl, which would be isostructural to Beller's pre-ligands shown in Chart 1. While there was spectroscopic evidence for the formation of the desired imidazolium salt, the starting material IPrCH21 was sufficiently basic to deprotonate [IPr-CH2-PiPr2]Cl to give 2 and the known by-product [IPrCH3]Cl.9 Fortunately 2 and [IPrCH3]Cl have quite different solubilities, allowing for their easy separation. By altering the ratio between IPrCH21 and ClPiPr2 to 2:1 (eqn (1)) and conducting the reaction in THF at room temperature for 20 h, we were able to isolate pure (IPrCH)PiPr22 in 81% yield after extracting 2 from the product mixture (containing [IPrCH3]Cl) with hexanes. Following a similar procedure, the phenyl-substituted NHOP (IPrCH)PPh23 was prepared in an isolated yield of 83% (eqn (1)). The new NHOPs 2 and 3 were each characterized by NMR spectroscopy, elemental analysis and X-ray crystallography (colorless crystals grown from hexanes at −30 °C; Fig. 1 and 2).| | 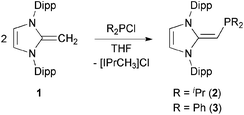 | (1) |
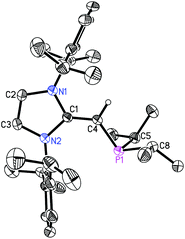 |
| | Fig. 1 . Molecular structure of 2 with thermal ellipsoids at the 30% probability level. Only one of the four crystallographically-independent molecules in the unit cell is presented. The hydrogen atom attached to C(4) is shown with an arbitrarily small thermal parameter; all other hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°): P–C(4) 1.780(3)–1.788(3), P–C(5) 1.835(4)–1.883(3), P–C(8) 1.859(4)–1.896(4), C(1)–C(4) 1.364(4)–1.366(4); P–C(4)–C(1) 126.9(2)–129.8(2), N(1)–C(1)–N(2) 104.0(2), C(4)–P–C(5) 101.03(14)–105.72(18), C(4)–P–C(8) 99.71(15)–104.29(17), C(5)–P–C(8) 99.12(14)–101.57(17). | |
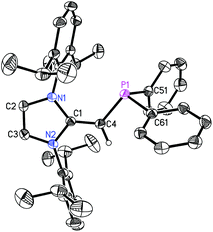 |
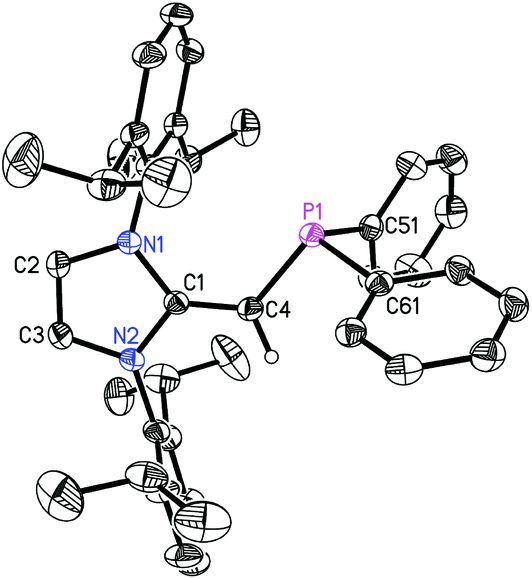
| | Fig. 2 Molecular structure of 3 with thermal ellipsoids at the 30% probability level. The hydrogen atom attached to C(4) is shown with an arbitrarily small thermal parameter; all other hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°) with values corresponding to a second molecule in the asymmetric unit in square brackets: P(1)–C(4) 1.7762(15) [1.782(2)], P(1)–C(51) 1.8411(16) [1.839(2)], P(1)–C(61) 1.8406(15) [1.8448(18)], C(1)–C(4) 1.365(2) [1.377(2)]; C(4)–P(1)–C(51) 104.37(7) [104.50(9)], C(4)–P(1)–C(61) 99.75(6) [99.70(8)], P(1)–C(4)–C(1) 126.25(11) [126.16(16)], N(1)–C(2)–N(2) 104.08(12) [104.29(12)]. | |
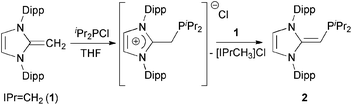 |
| | Scheme 1 In the reaction of IPrCH2 (1) and iPr2PCl in a 1:1 ratio, we observe the formation of the imidazolium-alkylphosphine salt [IPr-CH2-PiPr2]Cl as well as the desired neutral NHO-appended phosphine 2. | |
The refined structure of (IPrCH)PiPr22 is presented in Fig. 1 with only one of the four crystallographically-independent molecules in the unit cell shown; thus, selected bond lengths and angles are provided as a range. The exocyclic CC bonds in 2 [C(1)–C(4) = 1.364(4) to 1.366(4) Å] are considerably shorter than the typical C–C single bond length of ca. 1.530 Å,10 and is only slightly elongated compared to the exocyclic CC bond length of 1.332(4) Å in free IPrCH2.3c As expected, the crystallographically determined C(sp2)–P linkages in 2 [C(4)–P(1) = 1.780(3) to 1.788(2) Å] are contracted with respect to the C(sp3)–P bonds involving the iPr substituents [1.835(4) to 1.896(4) Å]. For comparison the C(olefin)–P distances in cis-Ph2P–CHCH–PPh2 are 1.817(3) and 1.825(3) Å,11 suggesting a possible increase in C(π) → P–C(σ*) hyperconjugation in 2, leading to shorter C(sp2)–P bonds. The overall metrical parameters in (IPrCH)PPh2 (3) (Fig. 2) are quite similar to that of 2 with an average C(4)–P(1) distance of 1.780(2) Å in 3 and average C(1)–C(4)–P(1) angles of 126.0(2)°, compared to a range of 126.9(2) to 129.8(2)° in 2.
Synthesis of the N-heterocyclic olefin amine (IPr![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) CH)NMe24
CH)NMe24
In addition to preparing NHOPs, we wanted to see if a harder amine donor could be incorporated onto an NHO scaffold. The dimethylamino-substituted NHO, (IPrCH)NMe24, was prepared by combining two equivalents of the commercially available carbene IPr with one equivalent of Eschenmoser's salt [H2CNMe2]I12 in toluene (eqn (2)). In this process, the first equivalent of IPr is believed to undergo a nucleophilic attack on the iminium moiety to form [IPr-CH2–NMe2]I which is then subsequently deprotonated by a second equivalent of IPr to yield (IPrCH)NMe24 and the imidazolium by-product [IPrH]I (which can be recycled for the preparation of IPr) (eqn (2)). In a similar fashion as the syntheses of 2 and 3, the salt by-product [IPrH]I is much less soluble than the target ligand 4, thus separation could be achieved by filtering the reaction mixture. One drawback with this synthesis is that the crude samples of 4 occasionally contains ca. 5–10% of unreacted IPr (as determined by 1H NMR), which is difficult to separate from (IPrCH)NMe24 due to their similar solubilities in common organic solvents. However, a successful way to remove the IPr contaminant involves adding a small amount of BPh3 to form the known adduct IPr·BPh3,13 which is much less soluble in hexanes than 4.| |  | (2) |
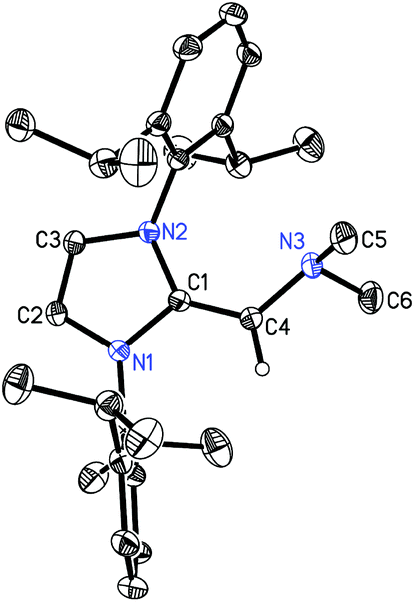
The structure of (IPrCH)NMe24 was authenticated by X-ray crystallography (Fig. 3) and this study revealed an exocyclic C(1)–C(4) bond length of 1.3463(14) Å which is slightly shorter than the corresponding distances in the NHOPs 2 and 3, suggesting the retention of substantial C–C π-bonding in this unit. The C(1)–C(4)–N(1) angle was also consistent with sp2-hybridization at C(4) [122.98(9)°], while the nitrogen atom of the –NMe2 group is significantly pyramidalized [Σ°(N) = 333.35(17)°] consistent with a lack of substantial N(3)–C(4) π-bonding.
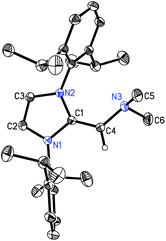 |
| | Fig. 3 Molecular structure of 4 with thermal ellipsoids at the 30% probability level. The hydrogen atom attached to C(4) is shown with an arbitrarily small thermal parameter; all other hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°): N(1)–C(1) 1.4024(12), N(2)–C(1) 1.4009(12), C(1)–C(4) 1.3463(14), C(4)–N(3) 1.4299(13), N(3)–C(5) 1.4563(16), N(3)–C(6) 1.4570(16); N(1)–C(1)–N(2) 104.44(8), N(1)–C(1)–C(4) 125.71(9), C(1)–C(4)–N(3) 122.98(9), C(4)–N(3)–C(6) 110.19(10). | |
Coordination of the NHOPs 2 and 3 to BH3 and AuCl
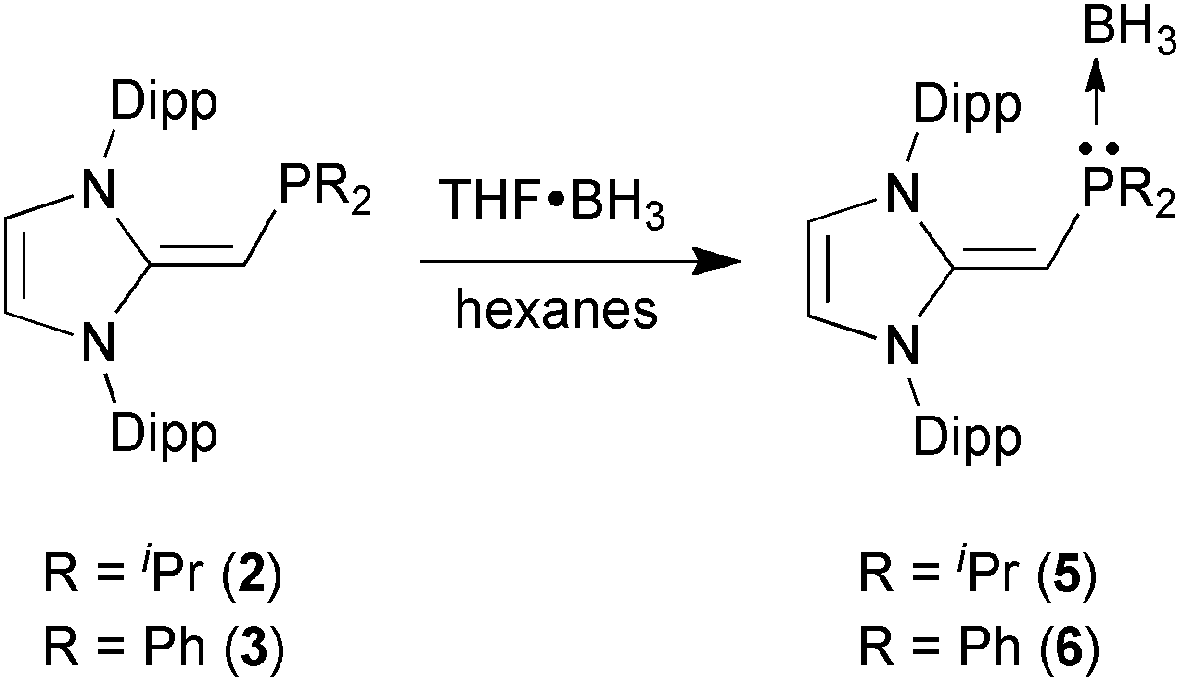
With the new NHOPs in hand, we first tested their reactivity with the Lewis acid source THF·BH3. When either (IPrCH)PiPr22 or (IPrCH)PPh23 was combined with THF·BH3 in hexanes (eqn (3)), the reaction mixture went from yellow to colorless after 90 min at room temperature. After the volatiles were removed, the respective phosphine–borane adducts (IPrCH)iPr2P·BH35 and (IPrCH)Ph2P·BH36 were isolated as colorless crystals in 52 and 56% yields after recrystallization from cold (−30 °C) hexanes or toluene (slow evaporation), respectively. As expected, coordination of a BH3 unit was evident by NMR spectroscopy, which showed broad 11B NMR resonances at −42.0 and −35.8 ppm for 5 and 6, respectively, consistent with the presence of four-coordinate boron environments. In addition, considerable downfield shifts in the 31P resonances were noted within the NHOPs upon BH3 coordination: from −17.4 ppm in 2 to 21.9 ppm in (IPrCH)iPr2P·BH35; from −31.4 ppm in 3 to 7.3 ppm in (IPrCH)Ph2P·BH36. Such a substantial change in 31P NMR chemical shift indicated the likely presence of BH3 units bound to the phosphorus centers; this postulate was confirmed by performing single-crystal X-ray crystallography (5: Fig. 4; 6: Fig. 5).
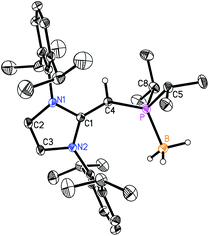 |
| | Fig. 4 Molecular structure of 5 with thermal ellipsoids at the 30% probability level. The hydrogen atom attached to C(4) is shown with an arbitrarily small thermal parameter; all other hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°): P–B 1.9166(18), P–C(4) 1.7504(14), C(1)–C(4) 1.3749(18), P–C(5) 1.8486(14), P–C(8) 1.8495(16); C(4)–P–B 124.56(7), P–C(4)–C(1) 138.02(11), C(4)–P–C(5) 102.07(7), C(4)–P–C(8) 106.76(7), B–P–C(5) 110.56(8), B–P–C(8) 107.50(8). | |
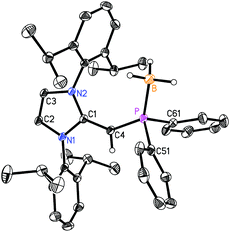 |
| | Fig. 5 Molecular structure of 6 with thermal ellipsoids at the 30% probability level. The hydrogen atom attached to C(4) is shown with an arbitrarily small thermal parameter; all other hydrogens have been omitted for clarity. Selected bond lengths (Å) and angles (°): P–B 1.9242(18), P–C(4) 1.7479(11), C(1)–C(4) 1.3811(15), P–C(51) 1.8292(12), P–C(61) 1.8300(13); C(4)–P–B 125.76(6), P–C(4)–C(1) 138.31(9), C(4)–P–C(51) 101.10(5), C(4)–P–C(61) 107.67(6), B–P–C(51) 109.21(7), B–P–C(61) 108.15(7). | |
As shown in Fig. 4, (IPrCH)iPr2P·BH35 contains a P-bound borane residue with a P–B bond length of 1.9166(18) Å; for comparison, the dialkylphosphine–borane adduct tBu2PH·BH3 has a P–B bond length of 1.936(2) Å.14 In the case of (IPrCH)iPr2P·BH35, the P(1)–C(4) length [1.7504(14) Å] is contracted in comparison to the corresponding distance in the free phosphine (IPrCH)PiPr22 [1.780(3) to 1.788(2) Å]. The exocyclic C(1)–C(4) double bond within the NHO unit in 5 [1.3749(18) Å] is essentially the same length within experimental error as the exocyclic CC bond distances in the phosphine 2 [1.364(4) to 1.366(4) Å]. The main structural change noted upon coordination of BH3 is a widening of the P–C(4)–C(1) from 126.9(2)° in the free ligand 2 to 138.02(11)° in adduct 5. Similarly, the P–C(4)–C(1) angle in the phenyl analogue (IPrCH)Ph2P·BH3 (6) [138.31(9)°] (Fig. 5) is wider than in the free phosphine (IPrCH)PPh2 (3) [126.0(2)° avg.]. In both compounds 5 and 6, the BH3 unit is oriented in an anti-fashion with respect to the exocyclic olefinic C–H group, placing the BH3 group in close proximity to one of the flanking Dipp aryl groups of the NHO ligand; such a coordination mode could enhance aryl⋯metal interactions within NHOP–metal complexes.1e
| |  | (3) |
After demonstrating the successful coordination of the small Lewis acid BH3 to the NHOPs 2 and 3, we decided to expand our studies to include transition metals. Our initial explorations focused on the noble metals Pd and Pt since complexes bearing these elements in conjunction with bulky phosphines15 and NHCs16 are often used in metal-mediated cross-coupling reactions. Despite the presence of a potentially strongly coordinating terminal –PiPr2 unit in (IPrCH)PiPr22, no discernable reaction was noted when excess 2 (2–3 equiv.) was combined with either Pd(PPh3)4 or Pt(PPh3)4 in hot C6D6 (50 °C) for 4 days (monitored by 31P NMR spectroscopy). A similar lack of reactivity was found with the two coordinate Pt(0) complex Pt(PtBu3)2. Attempts to form a bis NHOP–PdCl2 pre-catalyst17 by treating PdCl2(NCPh)2 with two equiv. of 2 in toluene, led to an immediate color change of the reaction mixture from yellow to dark red, however 31P NMR analysis revealed the formation of six spectroscopically distinct products from which a single clean product could not be isolated.
Reports of using [PdCl(cinnamyl)]2 (cinnamyl = η3-H2CCHCH(Ph)) as a palladium source to generate active L·Pd(cinnamyl) pre-catalysts (L = ligand)18 in cross-coupling reactions led us to combine [PdCl(cinnamyl)]2 with (IPrCH)PiPr22. When 2 was mixed with [PdCl(cinnamyl)]2 in toluene several new species were found by 31P NMR spectroscopy. In one case, layering of the crude reaction mixture with hexanes, followed by cooling to −30 °C gave a small batch of yellow crystals (2–3 mg) that were identified by X-ray crystallography as the target Pd(II) complex (IPrCH)PiPr2·PdCl(cinnamyl) 7 (Fig. 6).
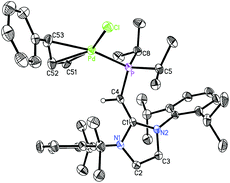 |
| | Fig. 6 Molecular structure of (IPrCH)PiPr2·PdCl(cinnamyl) 7 with thermal ellipsoids at the 30% probability level. The hydrogen atom attached to C(4) is shown with an arbitrarily small thermal parameter; all other hydrogens have been omitted for clarity. Selected bond lengths (Å) and angles (°): Pd–P 2.3086(12), Pd–Cl 2.3582(12), Pd–C(51) 2.113(6), Pd–C(52) 2.143(5), Pd–C(53) 2.261(5), P–C(4) 1.765(4), C(1)–C(4) 1.386(6), P–C(5) 1.847(5), P–C(8) 1.863(5); P–Pd–Cl 102.23(4), C(4)–P–Pd 103.85(15), P–C(4)–C(1) 136.1(3), C(4)–P–C(5) 105.8(2), C(4)–P–C(8) 110.7(2), Pd–P–C(5) 117.51(16), Pd–P–C(8) 113.69(18), C(51)–C(52)–C(53) 120.4(6). | |
Upon closer inspection of the structure of 7 (Fig. 6) it is clear that the –PiPr2 unit is free to rotate with respect to the bulky IPrCH– group. In the BH3 adduct 5, the isopropropyl groups are rotated away from the IPr unit, while in (IPrCH)PiPr2·PdCl(cinnamyl) 7 the phosphorus bound iPr substituents are positioned toward one Dipp group, enabling the more hindered PdCl(cinnamyl) array to occupy a more open side of the NHOP ligand coordination sphere. Therefore despite the bulk of (IPrCH)PiPr2, there exists sufficient torsional flexibility to allow different coordination pockets to be formed (a useful property for catalysis when various intermediates need to be stabilized). The Pd–cinnamyl bonding interactions in 7 range from 2.113(6) Å to 2.261(5) Å with the longest Pd–C bond to C(53) positioned trans to the phosphine donor. In the NHC complex IPr·PdCl(cinnamyl), the related trans-positioned Pd–C bond length (with respect to the IPr donor) is 2.201(17) Å,19 indicating that the ligand (IPrCH)PiPr2 exerts a similar degree of trans-influence as IPr.
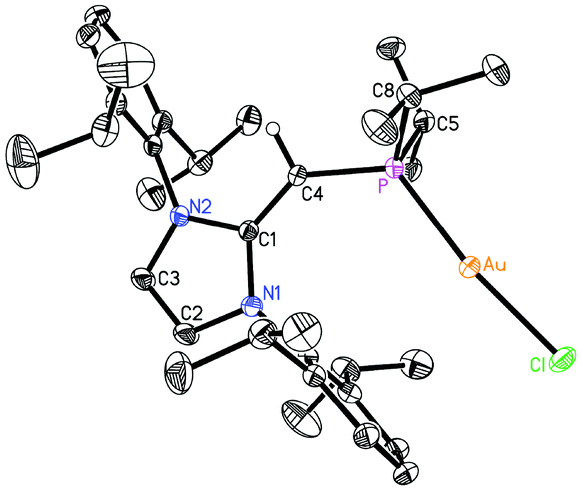
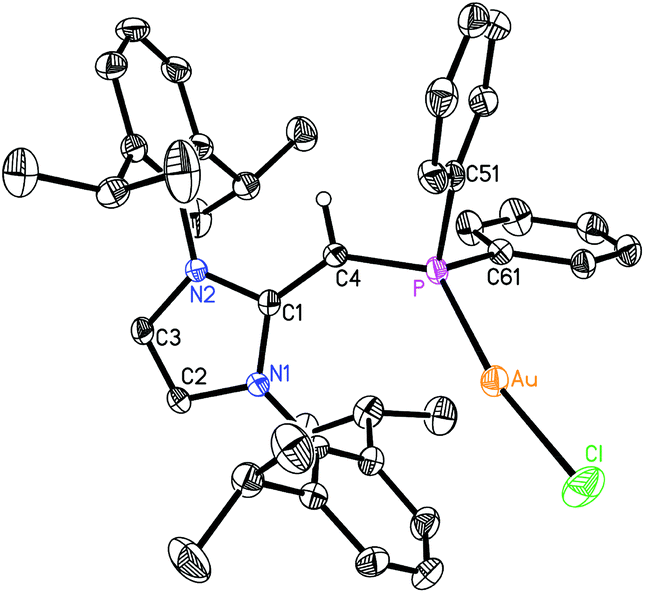
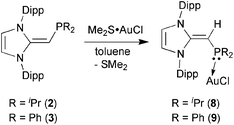
Given the difficulties faced in introducing an NHOP as a ligand to Pd and Pt centers, we decided the explore the coordination of this ligand class to gold(I) centers. Added motivation for this work stems from the rapidly growing use of Au(I) complexes in catalysis (e.g. in the hydroamination of alkynes).20 A toluene solution of (IPrCH)PiPr22 was added to a molar equivalent of Me2S·AuCl, and after stirring at room temperature for 2 h, (IPrCH)iPr2P·AuCl 8 was obtained as a pale yellow solid in an 85% yield after filtration of the reaction mixture and removal of the volatiles (eqn (4)); the resulting product was analytically pure as judged by satisfactory C, H and N analyses. Compound 8 was characterized by X-ray crystallography and the refined molecular structure is shown in Fig. 7. The metrical parameters within the IPrCH– unit in 8 are similar to those in the BH3 adduct (IPrCH)iPr2P·BH35, with comparable P–C(4) and exocyclic C(1)–C(4) bond lengths [1.7742(19) and 1.376(3) Å, respectively]. Interestingly, the –PiPr2 unit in 8 is rotated in such as fashion as to place the hindered isopropyl groups away from the Dipp groups within the IPrCH– unit; as a result the Au(I) center lies over the π-face of a Dipp substituent (Au⋯C(ipso) distance = 3.507 Å), and accordingly the P–Au–Cl angle [171.40(2)°] is distorted from the expected linear geometry. For comparison, shorter arene⋯Au(I) interactions have been noted within a series of Buchwald biarylphosphine–Au(I) complexes L·Au(NCMe)+ (3.04–3.19 Å).21 The corresponding diphenylphosphine-capped NHO complex (IPrCH)PPh2·AuCl 9 was prepared (98% yield) in a similar straightforward manner as 8, and exhibited the same overall geometry as in 8 (Fig. 8) with a slightly narrower P–Au–Cl angle of 168.72(4)°.
| |  | (4) |
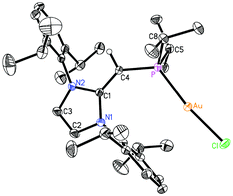 |
| | Fig. 7 Molecular structure of 8 with thermal ellipsoids at the 30% probability level. The hydrogen atom attached to C(4) is shown with an arbitrarily small thermal parameter; all other hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°): Au–P 2.2348(5), Au–Cl 2.2991(5), P–C(4) 1.7442(19), C(1)–C(4) 1.376(3), P–C(5) 1.848(2), P–C(8) 1.845(2); P–Au–Cl 171.40(2), C(4)–P–Au 122.82(7), P–C(4)–C(1) 136.20(15), C(4)–P–C(5) 105.69(10), C(4)–P–C(8) 104.31(10), Au–P–C(5) 106.89(7), Au–P–C(8) 110.83(7). | |
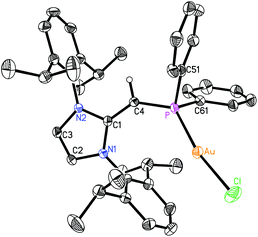 |
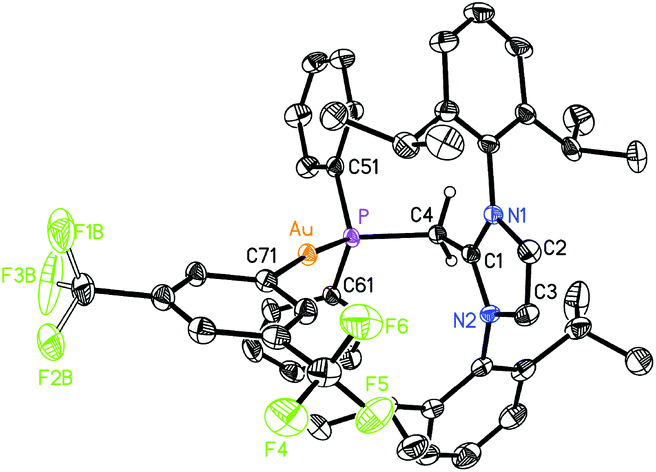
| | Fig. 8 Molecular structure of 9 with thermal ellipsoids at the 30% probability level. The hydrogen atom attached to C(4) is shown with an arbitrarily small thermal parameter; all other hydrogens have been omitted for clarity. Selected bond lengths (Å) and angles (°): Au–P 2.2334(8), Au–Cl 2.2914(10), P–C(4) 1.741(3), C(1)–C(4) 1.381(4), P–C(51) 1.831(3), P–C(61) 1.826(3); P–Au–Cl 168.72(4), C(4)–P–Au 128.48(11), P–C(4)–C(1) 136.9(3), C(4)–P–C(51) 99.84(15), C(4)–P–C(61) 109.43(15), Au–P–C(51) 106.45(11), Au–P–C(61) 106.46(11). | |
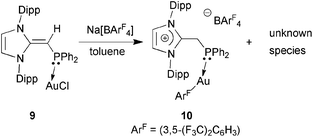
In an attempt to prepare a more reactive Au(I) complex for future catalytic trials,20d the NHO–Au complex (IPrCH)Ph2P·AuCl 9 was treated with Na[BArF4] ([BArF4]− = (3,5-(F3C)2C6H3)4B) in toluene. This reaction afforded a gummy orange precipitate from which a product of [BArF4]− anion activation [IPr-CH2-PPh2·Au(3,5-(F3C)2C6H3)]BArF410 could be isolated and structurally characterized (eqn (5); Fig. 9). While the mechanism of this process is under investigation, protonation of the exocyclic olefin within the NHO unit occurred to yield an imidazolium-alkylphosphine ligand,8 along with the removal of one ArF unit from the generally unreactive weakly coordinating [BArF4]− anion. One possible source of the proton would be C–H activation of the backbone olefin within the IPr unit.22 The generation of a highly electron deficient Au(I) center during the reaction process could facilitate the abstraction of ArF from the [BArF4]− anion; although rare, related processes have been noted with both phosphine and NHC-bound Au(I) centers.23 The structure of 10 is shown in Fig. 9 and, as expected, a nearly linear coordination geometry exists at the Au(I) center [P–Au–C(71) angle = 174.82(11)°]. The coordinative Au–P interaction in 10 [2.2798(8) Å] is only marginally elongated in relation to the Au–P distance in (IPrCH)iPr2P·AuCl 8 [2.2348(5) Å], while the adjacent P–C(4) bond length in 10 is longer by ca. 0.12 Å when compared to the P–C(4) distance in 8 as a result of a hybridization change at carbon from sp2 in 8 to sp3 in 10. No reaction was observed when (IPrCH)Ph2P·AuCl 9 was treated with Na[SbF6].
| |  | (5) |
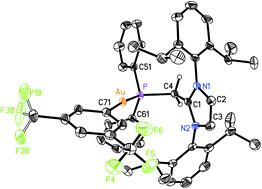 |
| | Fig. 9 Molecular structure of 10 with thermal ellipsoids at the 30% probability level. The hydrogen atoms attached to C(4) is shown with an arbitrarily small thermal parameter; all other hydrogen atoms and the B(3,5-(F3C)2C6H3)4− anion have been omitted for clarity. Selected bond lengths (Å) and angles (°): Au–P 2.2798(8), Au–C(71) 2.070(23), P–C(4) 1.864(4), C(1)–C(4) 1.487(4); P–Au–C(71) 174.82(11), C(4)–P–Au 112.43(11), C(1)–C(4)–P 116.5(2). | |
To further evaluate the donation abilities of the new phosphines, we targeted the preparation of NHOP·Rh(CO)2Cl complexes with the hope of obtaining informative ν(CO) IR data.3d When the NHOPs 2 and 3 were each combined with 0.5 equiv. of [RhCl(CO)2]2, three different Rh–P containing products were found, as evidenced by 31P{1H} NMR spectroscopy in the form of doublet resonances due to coupling to Rh (I = 1/2). Despite multiple attempts, we could not separate the products due to their similar solubilities in common organic solvents, and as such further investigations were not pursued.
Divergent coordination chemistry of (IPrCH)NMe24
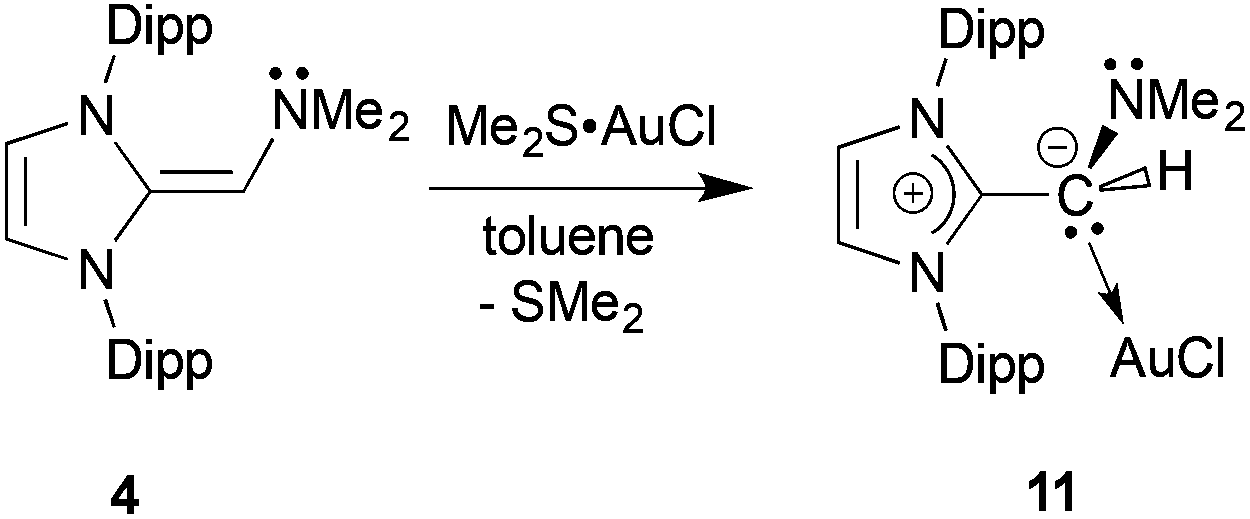
As presented above, the NHOPs 2 and 3 exclusively bind to Lewis acidic units through the terminal phosphine residues. However in the corresponding amine-capped NHOs (such as 4) featuring hard N-donor sites, there exists a chance that olefin coordination could transpire with soft Lewis acids (Chart 1). Somewhat to our surprise, (IPrCH)NMe24 did not yield clean reactivity with THF·BH3, with multiple products identified by 11B NMR spectroscopy. In contrast, an isolable 1:1 complex (IPrCH)NMe2·AuCl 11 formed as a yellow solid in 89% yield when 4 was combined with Me2S·AuCl in toluene (eqn (6)). The most drastic change in the 13C{1H} NMR spectra of the (IPrCH)NMe2 units was the upfield shift of the olefinic CHNMe2 carbon from 89.0 ppm in free (IPrCH)NMe24 to a position of 58.4 ppm in 11; this latter spectroscopic signature suggested possible olefin coordination to gold in 11. Crystals of 11 were obtained for X-ray crystallographic analysis and despite the lower quality of the data, (IPrCH)NMe2 coordination through a C–Au linkage was confirmed with a distance of 2.044(15) Å; moreover a nearly linear geometry was present at gold [C(3)–Au–Cl angle = 177.6(4)°; see Fig. S32 in the ESI†]. Therefore one can see direct evidence for the two possible binding modes of NHO-supported amines and phosphines in this study (Chart 2).| | 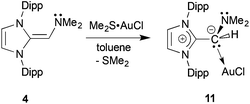 | (6) |
Conclusion
We have reported efficient syntheses of neutral N-heterocyclic olefin-appended phosphines and amine donors, and present preliminary coordination behavior with the Lewis acids BH3 and AuCl. Interestingly, modulation of the donor properties enables either NHO-based coordination (via an olefinic carbon atom) or standard phosphine binding modes to be adopted. As a result, we are exploring these coordinatively versatile ligands within the context of late metal-mediated catalysis.
Experimental
General
All reactions were performed in either an inert atmosphere glove box (Innovative Technology, Inc.) or using Schlenk techniques. Solvents were dried using a Grubbs-type solvent purification system24 manufactured by Innovative Technologies, Inc. and stored under an atmosphere of nitrogen prior to use. Chlorodiisopropylphosphine, chlorodiphenylphosphine, N,N-dimethyliminium iodide ([Me2NCH2]I), borane tetrahydrofuran complex, dimethylsulfide gold(I) chloride, Na[SbF6], and [PdCl(cinnamyl)]2 were used as received from Sigma Aldrich. Na[(3,5-(F3C)2C6H3)4B] was obtained from Sigma Aldrich and dried under vacuum at 100 °C for 12 h prior to use. IPrCH213d and IPr25 were prepared according to literature procedures. 1H, 1H{31P}, 13C{1H}, 31P{1H}, 11B, and 11B{1H} NMR spectra were recorded on a Varian VNMRS-400 or Varian VNMRS-500 spectrometer and referenced externally to SiMe4, 85% H3PO4, or F3B·OEt2. Elemental analyses were performed by the Analytical and Instrumentation Laboratory at the University of Alberta. Melting points were measured in sealed glass capillaries under nitrogen using a MelTemp melting point apparatus and are uncorrected.
X-ray crystallography
Crystals for X-ray diffraction studies were removed from a vial and immediately coated with thin a layer of hydrocarbon oil (Paratone-N). A suitable crystal was then mounted on a glass fiber, and quickly placed in a low temperature stream of nitrogen on the X-ray diffractometer. All data (Tables 1 and 2) were collected using a Bruker APEX II CCD detector/D8 diffractometer using Mo Kα or Cu Kα radiation with the crystals cooled to −80 °C or −100 °C. The data was corrected for absorption through Gaussian integration from the indexing of the crystal faces. Crystal structures were solved using intrinsic phasing SHELXT26 (2, 4, 5, 6, 9 and 11), direct methods (3), or Patterson/structure expansion (7 and 8)27 and refined using full-matrix least-squares on F2. The assignment of hydrogen atoms positions were based on the sp2 or sp3 hybridization of their attached carbon atoms, and were given thermal parameters 20% greater than those of their parent atoms.
Table 1 Crystallographic data for compounds 2–6
| |
(IPrCH)PiPr2 (2) |
(IPrCH)PPh2 (3) |
(IPrCH)NMe2 (4) |
(IPrCH)iPr2P·BH3 (5·0.5C6H14) |
(IPrCH)Ph2P·BH3 (6) |
|
R
1 = ∑||Fo| − |Fc||/∑|Fo|; wR2 = [∑w(Fo2 − Fc2)2/∑w(Fo4)]1/2.
|
| CCDC number |
1448851
|
1448848
|
1448850
|
1448846
|
1448845
|
| Formula |
C34H51N2P |
C40H47N2P |
C30H43N3 |
C35.50H57.50BN2P |
C40H50BN2P |
| Formula weight |
518.73 |
586.76 |
445.67 |
554.11 |
600.60 |
| Cryst. dimens. (mm) |
0.34 × 0.17 × 0.17 |
0.21 × 0.18 × 0.07 |
0.20 × 0.15 × 0.08 |
0.49 × 0.08 × 0.06 |
0.24 × 0.20 × 0.12 |
| Crystal system |
Monoclinic |
Monoclinic |
Monoclinic |
Trigonal |
Monoclinic |
| Space group |
P21/c |
P21/c |
P21/n |
R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) |
P21/n |
| Unit cell dimensions |
|
a (Å) |
21.3382(6) |
10.7803(2) |
9.3774(2) |
42.1750(6) |
10.7033(2) |
|
b (Å) |
18.3587(5) |
16.7651(3) |
20.2169(4) |
|
18.7813(3) |
|
c (Å) |
17.2832(4) |
39.0845(6) |
20.2169(4) |
10.4933(2) |
17.9853(3) |
|
β (°) |
33.3817(9) |
95.7606(9) |
100.0916(11) |
|
92.3243(8) |
|
V (Å3) |
13053.4(6) |
7028.2(2) |
2799.90(10) |
16164.1(6) |
3612.46(11) |
|
Z
|
16 |
8 |
4 |
18 |
4 |
|
ρ calcd (g cm−3) |
1.056 |
1.109 |
1. 057 |
1.025 |
1.104 |
|
μ (mm−1) |
0.897 |
0.894 |
0.463 |
0.835 |
0. 874 |
| Temperature (°C) |
−100 |
−100 |
−100 |
−100 |
−100 |
| 2θmax (°) |
146.35 |
146.98 |
148.31 |
148.11 |
145.02 |
| Total data |
75117 |
48771 |
108108 |
38050 |
24968 |
| Unique data (Rint) |
25575 (0.0449) |
13861 (0.0370) |
5659 (0.0433) |
7292 (0.0561) |
7118 (0.0257) |
| Obs [I > 2σ(I)] |
17632 |
11433 |
5104 |
6290 |
6558 |
|
R
1 [Fo2 ≥ 2σ (Fo2)]a |
0.0698 |
0.0461 |
0.0417 |
0.0497 |
0.0371 |
| wR2 [all data] |
0.2012 |
0.1292 |
0.1151 |
0.1428 |
0.1037 |
| Max/min Δr (e Å−3) |
0.954/−0.511 |
0. 444/−0.299 |
0.238/−0.256 |
0.552/−0.453 |
0.311/−0.366 |
Table 2 Crystallographic data for compounds 7–10
| |
(IPrCH)iPr2P·PdCl(cinnamyl) (7) |
(IPrCH)iPr2P·AuCl (8) |
(IPrCH)Ph2P·AuCl (9·0.5C7H8) |
[IPr-CH2–PPh2·AuArF]BArF4 (10) |
|
R
1 = ∑||Fo| − |Fc||/∑|Fo|; wR2 = [∑w(Fo2 − Fc2)2/∑w(Fo4)]1/2.
|
| CCDC number |
1448843
|
1448847
|
1448849
|
1448844
|
| Formula |
C50H68ClN2PPd |
C34H51AuClN2P |
C43.50H51AuClN2P |
C80H63AuBF30N2P |
| Formula weight |
869.88 |
751.15 |
865.25 |
1861.07 |
| Cryst. dimens. (mm) |
0.14 × 0.13 × 0.10 |
0.32 × 0.18 × 0.16 |
0.19 × 0.18 × 0.06 |
0.48 × 0.13 × 0.01 |
| Crystal system |
Triclinic |
Monoclinic |
Monoclinic |
Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/n |
P21/c |
P21/n |
| Unit cell |
|
a (Å) |
10.3535(3) |
10.4781(4) |
9.7488(6) |
14.0570(4) |
|
b (Å) |
12.5649(4) |
16.3684(6) |
20.0872(13) |
25.1328(6) |
|
c (Å) |
18.9321(6) |
20.9256(7) |
20.8717(14) |
22.7564(7) |
|
α (°) |
72.828(2) |
|
|
|
|
β (°) |
100.0916(11) |
101.6635(4) |
98.4585(10) |
102.1003(18) |
|
γ (°) |
89.231(2) |
|
|
|
|
V (Å3) |
2304.58(13) |
3514.8(2) |
4042.8(5) |
7861.0(4) |
|
Z
|
2 |
4 |
4 |
4 |
|
ρ calcd (g cm−3) |
1.254 |
1.419 |
1.422 |
1.573 |
|
μ (mm−1) |
4.357 |
4.330 |
3.776 |
4.750 |
| Temperature (°C) |
−100 |
−80 |
−80 |
−100 |
| 2θmax (°) |
147.88 |
56.66 |
55.22 |
148.42 |
| Total data |
87008 |
32818 |
36511 |
55754 |
| Unique data (Rint) |
8901 (0.1174) |
8717 (0.0238) |
9373 (0.0430) |
12 419(0.0808) |
| Obs [I > 2σ(I)] |
7672 |
7584 |
7578 |
7672 |
|
R
1 [Fo2 ≥ 2σ (Fo2)]a |
0.0566 |
0.0200 |
0.0316 |
0.0420 |
| wR2 [all data] |
0.1547 |
0.0532 |
0.0855 |
0.1156 |
| Max/min Δr (e Å−3) |
1.436/−1.103 |
1.076/−0.505 |
1.671/−1.292 |
1.078/−2.636 |
Special refinement conditions
(IPr![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) CH)PiPr2·BH35.
Attempts to refine peaks of residual electron density as disordered or partial-occupancy solvent hexane carbon atoms were unsuccessful. The data were corrected for disordered electron density through use of the SQUEEZE procedure as implemented in PLATON.28 A total solvent-accessible void volume of 1145 Å3 with a total electron count of 212 (consistent with 4.24 molecules of solvent hexane, or ∼0.25 molecules per formula unit of 5) was found in the unit cell.
CH)PiPr2·BH35.
Attempts to refine peaks of residual electron density as disordered or partial-occupancy solvent hexane carbon atoms were unsuccessful. The data were corrected for disordered electron density through use of the SQUEEZE procedure as implemented in PLATON.28 A total solvent-accessible void volume of 1145 Å3 with a total electron count of 212 (consistent with 4.24 molecules of solvent hexane, or ∼0.25 molecules per formula unit of 5) was found in the unit cell.
(IPrCH)PiPr2·PdCl(cinnamyl) 7.
The crystal used for data collection was found to display non-merohedral twinning. Both components of the twin were indexed with the program CELL_NOW (Bruker AXS, Inc., Madison, WI, 2004). The second twin component can be related to the first component by a 7.4° rotation about the [0.2 1 −0.35] axis in real space and about the [0.1 1 −0.4] axis in reciprocal space. Integrated intensities for the reflections from the two components were written into a SHELXL-201426 HKLF 5 reflection file with the data integration program SAINT (version 8.34A), using all reflection data (exactly overlapped, partially overlapped and non-overlapped). The refined value of the twin fraction (SHELXL-2014 BASF parameter) was 0.3198(17).
(IPrCH)PPh2·AuCl 9.
Attempts to refine peaks of residual electron density as disordered or partial-occupancy solvent toluene or hexane carbon atoms were unsuccessful. The data were corrected for disordered electron density through use of the SQUEEZE procedure as implemented in PLATON.28 A total solvent-accessible void volume of 517 Å3 with a total electron count of 110 (consistent with 2 molecules of solvent toluene, or 0.5 molecules per formula unit of the Au complex) was found in the unit cell.
Synthetic details
Synthesis of (IPrCH)PiPr22.
iPr2PCl (100 μL, 0.77 mmol) was added dropwise to IPrCH21 (0.508 g, 1.26 mmol) in 8 mL of THF. The resulting mixture was stirred for 20 h to give an orange suspension. The mixture was then filtered and the volatiles were removed from the filtrate to afford an orange solid that was extracted with 4 mL of hexanes and filtered again. Removal of the volatiles from the filtrate gave 2 as a light brown solid (0.267 g, 81%). Crystals suitable for X-ray crystallography were obtained by cooling (−30 °C) a saturated solution of 2 in hexanes. 1H NMR (400 MHz, C6D6): δ 7.26–7.11 (m, 6H, ArH), 5.88 (dd, 3JHH = 2.4 Hz, 5JHH = 0.8 Hz, 1H, NCHCHN), 5.85 (dd, 3JHH = 2.4 Hz, 5JHH = 0.8 Hz, 1H, NCHCHN), 3.28 (overlapping septets, 4H, ArCH(CH3)2), 2.66 (d, 2JHP = 5.6 Hz, 1H, CHPiPr2), 1.45 (d, 3JHH = 7.2 Hz, 6H, ArCH(CH3)2), 1.33 (d, 3JHH = 7.2 Hz, 6H, ArCH(CH3)2), 1.25 (broad septet, 3JHH = 7.2 Hz, 2H, PCH(CH3)2), 1.18 (d, 3JHH = 6.8 Hz, 6H, ArCH(CH3)2), 1.15 (d, 3JHH = 6.8 Hz, 6H, ArCH(CH3)2), 0.96 (dd, 3JHH = 7.2 Hz, 3JHP = 11.6 Hz, 6H, PCH(CH3)CH3), 0.90 (dd, 3JHH = 6.8 Hz, 3JHP = 12.8 Hz, 6H, PCH(CH3)CH3). 13C{1H} (126 MHz, C6D6): δ 154.5 (Ar–C), 154.3 (Ar–C), 148.7 (Ar–C), 148.1 (Ar–C), 134.6 (NCN), 129.7 (Ar–C), 129.4 (Ar–C), 124.6 (Ar–C), 123.9 (Ar–C), 117.8 (HCCH), 115.0 (HCCH), 51.4 (d, 1JCP = 114.7 Hz, HCPiPr2), 29.1 (ArCH(CH3)2), 28.7 (ArCH(CH3)2), 26.5 (d, 2JCP = 11.1 Hz, PCH(CH3)2), 25.9 (ArCH(CH3)2), 24.9 (ArCH(CH3)2), 23.4 (ArCH(CH3)2), 22.6 (ArCH(CH3)2). 31P{1H} NMR (160 MHz, C6D6): δ −17.4. Mp (°C): 132–135. Anal. Calcd for C34H51N2P: C, 78.72; H, 9.91; N, 5.40. Found: C, 77.76; H 9.85; N, 5.21.
Synthesis of (IPrCH)PPh23.
Ph2PCl (41.2 μL, 0.16 mmol) was added dropwise to a solution of IPrCH21 (0.150 g, 0.37 mmol) in 3 mL of THF. The resulting mixture was stirred overnight to give an orange suspension. The precipitate was allowed to settle and the mother liquor was isolated after filtration. The volatiles were removed from the mother liquor to afford (IPrCH)PPh23 as a brown solid (0.078 g, 83%). Crystals suitable for X-ray crystallography were obtained by cooling (−30 °C) a saturated solution in hexanes. 1H NMR (400 MHz, C6D6): δ 7.35–6.92 (m, 16H, ArH and PhH), 5.92 (s, 2H, N(CH)2N), 3.34 (d, 3JHP = 5.6 Hz, 1H, CHPPh2), 3.26 (overlapping septets, 4H, ArCH(CH3)2), 1.33 (d, 3JHH = 7.2 Hz, 6H, ArCH(CH3)2), 1.19 (d, 3JHH = 7.0 Hz, 6H, ArCH(CH3)2), 1.18 (d, 3JHH = 7.0 Hz, 6H, ArCH(CH3)2), 1.12 (d, 3JHH = 7.2 Hz, 6H, ArCH(CH3)2). 13C{1H} NMR (126 MHz, C6D6): δ 154.0 (d, 1JCP = 35 Hz, Ph–C), 148.5 (Ar–C), 147.9 (Ar–C), 146.2 (d, 2JCP = 13 Hz, Ph–C), 136.6 (Ar–C), 134.2 (Ar–C), 132.3 (d, 2JCP = 20 Hz, Ph–C), 130.0 (Ar–C), 129.7 (Ar–C), 127.7 (Ar–C), 126.8 (Ar–C), 124.5 (d, 1JCP = 41 Hz, Ph–C), 117.1 (HCCH), 115.5 (HCCH), 52.7 (HCPPh2), 29.1 (CH(CH3)2), 28.8 (CH(CH3)2), 25.1 (CH(CH3)2), 24.7 (CH(CH3)2), 23.4 (CH(CH3)2), 22.5 (CH(CH3)2). 31P{1H} NMR (162 MHz, C6D6): δ −31.4. Mp (°C): 172–176. Anal. Calcd for C40H47N2P: C, 81.87; H, 8.07; N, 4.77. Found: C, 81.34; H 8.33; N, 5.05.
Synthesis of IPrCHNMe24.
A solution of IPr (0.481 g, 1.24 mmol) in 3 mL of toluene was added to finely ground [H2CN(CH3)2]I (0.115 g, 0.62 mmol). The resulting mixture was stirred overnight to give a cloudy yellow reaction mixture. The mother liquor was isolated after filtration. The volatiles were then removed from the mother liquor to afford a yellow solid that was extracted with 2 mL of hexanes and filtered. Removal of the volatiles from the filtrate afforded 4 as a yellow solid (227 mg, 82%, product also contained 7% of unreacted IPr). Further purification can be performed by adding BPh3 (ca. 2 mg) to 4 (0.050 g) in minimal amount of benzene (ca. 0.5 mL). The solution was stirred for 15 min and 2 mL of hexanes was added to yield a white precipitate. The mother liquor was isolated after filtration and the volatiles were removed from the filtrate to afford 4 (0.040 g) containing <1% of unreacted IPr. Crystals suitable for X-ray crystallography were obtained by cooling (−30 °C) a saturated solution in hexanes. 1H NMR (500 MHz, C6D6): δ 7.23 (t, 3JHH = 7.5 Hz, 2H, ArH), 7.14 (d, 3JHH = 8.0 Hz, 4H, ArH), 5.86 (dd, 3JHH = 2.5 Hz, 5JHH = 1.0 Hz, 1H, HCCH), 5.77 (d, 3JHH = 2.0 Hz, 1H, HCCH), 3.51 (septet, 3JHH = 7.0 Hz, 2H, CH(CH3)2), 3.47 (s, 1H, CHN(CH3)2), 3.36 (septet, 3JHH = 7.0 Hz, 2H, CH(CH3)2), 1.97 (s, 6H, N(CH3)2), 1.41 (d, 3JHH = 7.0 Hz, 6H, CH(CH3)2), 1.38 (d, 3JHH = 7.0 Hz, 6H, CH(CH3)2), 1.27 (d, 3JHH = 7.0 Hz, 6H, CH(CH3)2), 1.22 (d, 3JHH = 7.0 Hz, 6H, CH(CH3)2). 13C{1H} NMR (126 MHz, C6D6): δ 149.0 (Ar–C), 148.1 (Ar–C), 145.0 (Ar–C), 138.1 (NCN), 129.1 (Ar–C), 128.3 (Ar–C), 124.6 (Ar–C), 123.1 (Ar–C), 117.4 (HCCH), 114.4 (HCCH), 89.0 (HCN(CH3)2), 49.8 (CH(CH3)2), 28.7 (N(CH3)2), 28.6 (N(CH3)2), 25.6 (CH(CH3)2), 24.4 (CH(CH3)2), 23.8 (CH(CH3)2), 22.9 (CH(CH3)2). Mp (°C): 89–94. Anal. Calcd for C30H43N3: C, 80.85; H, 9.72; N, 9.43. Found: C, 79.04; H 9.43; N, 8.52. Despite repeated attempts, analyses were consistently low in the carbon content. See Fig. 7 and 8 in the ESI† for a copy of the NMR spectra of 4.
Preparation of (IPrCH)PiPr2·BH35.
106 μL of THF·BH3 (1.0 M solution in THF, 0.11 mmol) was added dropwise to a solution of IPrCHPiPr22 (50 mg, 0.096 mmol) in 2 mL of hexanes. The reaction mixture was stirred for 1.5 h and then filtered. The volatiles were removed from the filtrate and the resulting solid was dissolved in approximately 0.5 mL of hexanes and cooled (−30 °C) to afford (IPrCH)PiPr2·BH3 as a white microcrystalline solid (27 mg, 52%). Crystals suitable for X-ray crystallography were obtained by cooling (−30 °C) a saturated solution in hexanes. 1H NMR (500 MHz, C6D6): δ 7.24 (t, 3JHH = 7.5 Hz, 2H, ArH), 7.17 (d, 3JHH = 8.0 Hz, 4H, ArH), 5.87 (s, 2H, N(CH)2N), 3.16 (septet, 3JHH = 7.0 Hz, 4H, ArCH(CH3)2), 2.09 (d, 2JHP = 10.0 Hz, 1H, CHPiPr2), 1.44 (d, 3JHH = 6.5 Hz, 12H, CH(CH3)2), 1.44 (broad septet, 2H, P(CH(CH3)2)2), 1.14 (d, 3JHH = 6.5 Hz, 12H, ArCH(CH3)2), 1.06 (dd, 3JHH = 7.0 Hz, 3JHP = 14.5 Hz, 6H, PCH(CH3)2), 0.93 (dd, 3JHH = 7.0 Hz, 3JHP = 13.5 Hz, 6H, PCH(CH3)2), 0.25 (broad d, 2JHP = 15.0 Hz, 3H, BH3). 13C{1H} NMR (126 MHz, C6D6): δ 154.3 (d, 2JCP = 11.6 Hz, NCN), 147.9 (Ar–C), 130.2 (Ar–C), 128.4 (Ar–C), 128.2 (Ar–C), 128.0 (Ar–C), 124.8 (N(CH)2N), 40.2 (d, 1JCP = 73.8 Hz, HCPiPr2), 28.8 (ArCH(CH3)2), 26.7 (d, 2JCP = 39.5 Hz, P(CH(CH3)2)2), 25.3 (ArCH(CH3)2), 22.9 (ArCH(CH3)2), 17.2 (d, 1JCP = 58.9 Hz, P(CH(CH3)2)2). 11B{1H} NMR (160 MHz, C6D6): δ −42.0. 31P{1H} NMR (201 MHz, C6D6): δ 21.9. Mp (°C): 154–156. Anal. Calcd for C34H54BN2P: C, 76.67; H, 10.22; N, 5.26. Found: C, 75.94; H 10.10; N, 5.42.
Preparation of (IPrCH)PPh2·BH36.
93.8 μL of THF·BH3 (1.0 M solution in THF, 0.094 mmol) was added dropwise to a solution of IPrCHPPh23 (50.0 mg, 0.085 mmol) in 2 mL of hexanes. The reaction mixture was stirred for 90 min. The solvent volume was reduced in vacuo until the mixture just turned cloudy and then cooled (−30 °C) to afford 6 as an off-white microcrystalline solid (29 mg, 56%). Crystals suitable for X-ray crystallography were obtained by slow evaporation of a saturated solution of (IPrCH)PPh2·BH36 in toluene at room temperature. 1H NMR (500 MHz, C6D6): δ 7.69–7.71 (m, 4H, PhH), 7.72 (t, 3JHH = 7.5 Hz, 2H, ArH), 7.12 (d, 3JHH = 8.0 Hz, 4H, ArH), 6.98–6.95 (m, 6H, PhH), 5.93 (s, 2H, N(CH)2N), 3.15 (septet, 3JHH = 7.0 Hz, 4H, CH(CH3)2), 2.83 (d, 2JHP = 9.5 Hz, 1H, CHPPh2), 1.24 (d, 3JHH = 7.0 Hz, 12H, CH(CH3)2), 1.12 (d, 3JHH = 7.0 Hz, 12H, CH(CH3)2), 0.98 (broad d, 2JHP = 16.0 Hz, 3H, BH3). 13C{1H} NMR (126 MHz, C6D6): δ 153.2 (d, 2JCP = 15.6 Hz, NCN), 147.7 (Ar–C), 137.7 (d, 1JCP = 58.7 Hz, Ph–C), 132.0 (d, JCP = 9.3 Hz, Ph–C), 130.4 (ArC), 129.1 (d, JCP = 2.0 Hz, Ph–C), 128.3 (Ar–C), 128.0 (d, JCP = 9.5 Hz, Ph–C), 125.0 (N(CH)2N), 117.6 (Ar–C), 44.2 (d, 1JCP = 84.8 Hz, HCPPh2), 29.0 (CH(CH3)2), 25.2 (CH(CH3)2), 22.8 (CH(CH3)2). 11B{1H} NMR (128 MHz, C6D6): δ −35.8. 31P{1H} NMR (162 MHz, C6D6): δ 7.3. Mp (°C): 164–170. Anal. Calcd for C40H50BN2P: C, 79.99; H, 8.39; N, 4.66. Found: C, 79.36; H 8.38; N, 4.68.
Reaction of IPrCHNMe2 with THF·BH3.
63.8 μL of THF·BH3 (1.0 M solution in THF, 0.058 mmol) was added dropwise to a solution of IPrCHNMe24 (26 mg, 0.058 mmol) in 1 mL of hexanes. Once THF·BH3 was added the yellow solution became colorless. The reaction was stirred for approximately 2 hours and then the volatiles were removed. 11B NMR analysis showed that there was no THF·BH3 remaining, however 5 new unidentifiable products were formed; attempts to obtain pure products were not successful.
Reaction of (IPrCH)PiPr2 with [PdCl(cinnamyl)]2.
[PdCl(cinnamyl)]2 (0.024 g, 0.046 mmol) was combined with (IPrCH)PiPr22 (0.048 g, 0.093 mmol) in 2 mL of toluene. The reaction mixture rapidly became yellow in color. The solution was left to stir overnight to yield a red solution and the volatiles were removed. 1H and 31P NMR spectroscopy showed a mixture of several products. On one occasion, yellow crystals (2–3 mg) suitable for X-ray crystallography were obtained by cooling (−30 °C) a saturated solution of the reaction mixture in toluene/hexanes. Data for (IPrCH)PiPr2·PdCl(cinnamyl) 7. 1H NMR (400 MHz, C6D6): δ 7.55 (d, 3JHH = 8.0 Hz, 2H, ArH), 7.22–7.00 (m, 8H, PhH and ArH), 5.90 (s, 2H, N(CH)2N), 5.41 (ddd, JHH = 13.2 Hz, JHH = 9.2 Hz, JHH = 9.2 Hz, 1H, CH2CHCHPh), 4.98 (m, 3H, CH2CHCHPh), 4.01 (broad d, 3JHP = 11.2 Hz, 3H, PCH(CH3)2), 3.49 (broad d, 3JHP = 6.8 Hz, 3H, PCH(CH3)2), 3.23 (broad septet, 4H, ArCH(CH3)2), 2.79 (broad s, (IPrCH)PiPr2), 2.44 (broad d, 3JHH = 12.0 Hz, 3H, PCH(CH3)2), 1.88 (broad s, 3H, PCH(CH3)2), 1.38 (broad m, 12H, ArCH(CH3)2), 1.14 (broad m, 1H, PCH(CH3)2), 1.06 (d, 3JHH = 6.8 Hz, 12H, ArCH(CH3)2). 31P{1H} NMR (162 MHz, C6D6): δ 34.0. We did not have enough sample to record a meaningful 13C{1H} NMR spectrum.
Synthesis of (IPrCH)PiPr2·AuCl 8.
A solution of (IPrCH)PiPr22 (99 mg, 0.19 mmol) in 5 mL of toluene was added dropwise to solid Me2S·AuCl (56 mg, 0.19 mmol) to give a yellow solution. This reaction mixture was stirred at room temperature for 2 hours and a small amount of metallic precipitate was observed. The mixture was then filtered and the volatiles were then removed from the filtrate to afford (IPrCH)PiPr2·AuCl 8 as a pale yellow solid (121 mg, 85%). Crystals suitable for X-ray crystallography were obtained by cooling (−30 °C) a saturated solution in a 1:1 mixture of toluene/hexanes. 1H NMR (500 MHz, C6D6): δ 7.49 (t, 3JHH = 7.0 Hz, 2H, ArH), 7.23 (d, 3JHH = 8.0 Hz, 4H, ArH), 5.78 (s, 2H, N(CH)2N), 3.01 (septet, 3JHH = 7.0 Hz, 4H, ArCH(CH3)2), 2.22 (d, 2JHP = 6.0 Hz, 1H, CHPiPr2), 1.38 (d, 3JHH = 7.0 Hz, 12H, ArCH(CH3)2), 1.28 (septet, 3JHH = 8.0 Hz, 2H, P(CH(CH3)2)2), 1.07 (d, 3JHH = 7.0 Hz, 12H, ArCH(CH3)2), 0.87 (dd, 3JHH = 7.0 Hz, 3JHP = 18.0 Hz, 6H, PCH(CH3)2), 0.80 (dd, 3JHH = 7.0 Hz, 3JHP = 16.0 Hz, 6H, PCH(CH3)2). 13C{1H} NMR (126 MHz, C6D6): δ 153.7 (d, 2JCP = 10.3 Hz, NCN), 147.0 (Ar–C), 134.3 (Ar–C), 131.2 (Ar–C), 129.3 (Ar–C), 125.4 (Ar–C), 117.5 (N(CH)2N), 40.7 (d, 1JCP = 81.3 Hz, HCPiPr2), 29.3 (d, 2JCP = 41.6 Hz, P(CH(CH3)2)2), 28.8 (ArCH(CH3)2), 25.0 (ArCH(CH3)2), 23.2 (ArCH(CH3)2), 19.1 (d, 1JCP = 3.8 Hz, P(CH(CH3)2)2), 18.3 (ArCH(CH3)2). 31P{1H} NMR (201 MHz, C6D6): δ 28.7. Mp (°C): 90 (decomp., turned black). Anal. Calcd for C34H51AuClN2P: C, 54.36; H, 6.84; N, 3.73. Found: C, 54.82; H 6.86; N, 3.61.
Synthesis of (IPrCH)PPh2·AuCl 9.
A solution of (IPrCH)PPh23 (78 mg, 0.13 mmol) in 5 mL of toluene was slowly added to solid Me2S·AuCl (40 mg, 0.14 mmol) to give a yellow solution. This reaction mixture was stirred at room temperature for 90 minutes and a small amount of metallic precipitate was observed. The mixture was filtered and the volatiles were then removed from the filtrate to afford (IPrCH)PPh2·AuCl 9 as a pale yellow solid (108 mg, 98%). Crystals suitable for X-ray crystallography were obtained by cooling (−30 °C) a saturated solution in a 2:1 mixture of toluene/hexanes. 1H NMR (400 MHz, C6D6): δ 7.47–7.49 (m, 4H, PhH), 7.41 (t, 3JHH = 8.0 Hz, 2H, ArH), 7.18 (d, 3JHH = 8.0 Hz, 4H, ArH), 6.86–6.87 (m, 6H, PhH), 5.83 (s, 2H, N(CH)2N), 3.00 (septet, 3JHH = 6.8 Hz, 4H, CH(CH3)2), 2.80 (d, 2JHP = 6.4 Hz, 1H, CHPPh2), 1.20 (d, 3JHH = 6.8 Hz, 12H, CH(CH3)2), 1.08 (d, 3JHH = 6.8 Hz, 12H, CH(CH3)2). 13C{1H} NMR (126 MHz, C6D6): δ 152.3 (d, 2JCP = 13.6 Hz, NCN), 146.9 (Ar–C), 139.1 (d, 1JCP = 63.9 Hz, Ph–C), 137.8 (Ar–C), 133.7 (Ar–C), 132.6 (d, 2JCP = 14.1 Hz, Ph–C), 131.4 (N(CH)2N), 129.7 (d, JCP = 2.3 Hz, Ph–C), 117.5 (Ar–C), 44.6 (d, 1JCP = 92.6 Hz, HCPPh2), 29.0 (CH(CH3)2), 24.6 (CH(CH3)2), 23.1 (CH(CH3)2). 31P{1H} NMR (162 MHz, C6D6): δ 8.1. Mp (°C): 122 (decomp., turned black). Anal. Calcd for C40H47AuN2P: C, 58.65; H, 5.78; N, 3.42. Found: C, 58.83; H 5.89; N, 3.13.
Reaction of IPrCHPPh2·AuCl and Na[BArF4]: isolation of [IPr–CH2–PPh2·Au(3,5-(F3C)2C6H3)]BArF410.
(IPrCH)PPh2·AuCl 9 (17 mg, 0.020 mmol) and Na[BArF4] (18 mg, 0.020 mmol) were combined in 2 mL of toluene and stirred at room temperature overnight. A pale orange solution formed along with a gummy orange precipitate. The mother liquor was decanted away and the precipitate was exposed to prolonged vacuum to yield an orange solid. This solid was then extracted with CH2Cl2 (2 × 1 mL) and the combined extracts were filtered. The filtrate was then layered with 2 mL of hexanes before cooling to −30 °C, leading to colorless crystals of 10 (19 mg). 1H NMR (500 MHz, CDCl3): δ 7.42 (broad d, 3JHH = 5.5 Hz, 2H, ArF–H), 7.69 (broad s, 8H, ArH in BArF4), 7.66 (s, 2H, N(CH)2N), 7.64 (broad s, 1H, ArF–H), 7.52 (m, 6H, Ph–H), 7.40 (t, 3JHH = 7.5 Hz, 2H, ArH), 7.29 (d, 3JHH = 8.0 Hz, 4H, ArH), 7.23–7.25 (m, 4H, Ph–H), 6.94–6.98 (m, 4H, ArH in BArF4), 3.82 (d, 2JHP = 10.4 Hz, 2H, CH2PPh2), 2.82 (br, 4H, CH(CH3)2), 1.18 (d, 3JHH = 6.8 Hz, 12H, CH(CH3)2), 1.13 (d, 3JHH = 6.8 Hz, 12H, (CH(CH3)2). 11B{1H} NMR (128 MHz, CDCl3): δ −6.6. 19F NMR (376 MHz, CDCl3): δ −62.3 [BArF4−], −62.6 [Au–ArF]. 31P{1H} NMR (162 MHz, CDCl3): δ 34.4. 13C{1H} NMR (126 MHz, CDCl3): δ 161.7 (q, 1JCB = 49.6 Hz, Ar–C in BArF4−), 146.7 (Ar–C), 144.9 (Ar–C), 137.9 (Ar–C in Au–ArF), 134.8 (Ar–C in BArF4−), 133.7 (Ar–C), 132.8 (Ar–C), 131.9 (d, JCP = 14.0 Hz, Ar–C, 129.9 (d, JCP = 19.5 Hz, Ar–C), 128.8 (q, 1JCB = 49.6 Hz in BArF4−), 126.2 (Ar–C), 125.6 (d, 2JCP = 12.7 Hz, Ar–C), 123.5 (Ar–C), 121.3 (N(CH)2N), 119.9 (Ar–C), 117.5 (Ar–C in BArF4−), 30.0 (CH(CH3)2), 26.9 (HCPPh2), 26.1 (CH(CH3)2), 22.7 (CH(CH3)2). The CF3 groups in the Au–ArF unit could not be located in the 13C{1H} NMR spectrum. Mp (°C): 97 (decomp.; turned brown). Anal. Calcd for C80H63AuBF30N2P: C, 51.63; H, 3.41; N, 1.51. Found: C, 51.31; H, 3.62; N, 1.52.
Synthesis of (IPrCH)NMe2·AuCl 11.
A solution of IPrCHNMe24 (98 mg, 0.22 mmol) in 5 mL of toluene was added dropwise to solid Me2S·AuCl (65 mg, 0.22 mmol) to give a dark yellow reaction mixture. This reaction mixture was stirred at room temperature for 90 minutes and a metallic precipitate was observed. The reaction mixture was then filtered and the volatiles were then removed from the filtrate to afford (IPrCH)NMe2·AuCl as a pale yellow solid (133 mg, 89%). Crystals of 11 were obtained by cooling a 2:1 toluene/hexanes solution overnight to −30 °C, however the low quality of the data prevents a detailed discussion of the metrical parameters (see Fig. S32 in the ESI†). 1H NMR (400 MHz, C6D6): δ 7.13 (d, 3JHH = 9.0 Hz, 2H, ArH), 7.01–7.04 (m, 4H, ArH), 6.10 (s, 2H, N(CH)2N), 4.02 (s, 1H, CHNMe2), 3.04 (septet, 3JHH = 6.4 Hz, 4H, CH(CH3)2), 1.79 (broad s, 6 H, N(CH3)2), 1.47 (d, 3JHH = 6.4 Hz, 6H, CH(CH3)2), 1.47 (d, 3JHH = 6.4 Hz, 6H, CH(CH3)2), 0.95 (d, 3JHH = 7.2 Hz, 6H, CH(CH3)2), 0.95 (d, 3JHH = 7.2 Hz, 6H, CH(CH3)2). 13C{1H} NMR (126 MHz, C6D6): δ 161.0 (NCN), 147.5 (Ar–C), 145.3 (Ar–C), 132.2 (Ar–C), 131.2 (Ar–C), 124.7 (Ar–C), 124.5 (Ar–C), 120.8 (HCCH), 58.4 (HCN(CH3)2) 29.8 (CH(CH3)2), 29.4 (CH(CH3)2), 26.0 (CH(CH3)2), 25.9 (CH(CH3)2), 23.2 (N(CH3)2), 23.1 (N(CH3)2). Mp (°C): 123 (decomp., turned dark brown). Anal. Calcd C30H43AuN3: C, 53.14; H, 6.39; N, 6.20. Found: C, 52.68; H, 6.33; N, 6.00.
Acknowledgements
This work was supported by the Natural Sciences and Engineering Research Council of Canada (Discovery Grant to E. R.; postgraduate scholarship to M. W. L.), Alberta Innovates-Technology Futures (New Faculty Award to E. R.), the Donors of the American Chemical Society Petroleum Research Fund, and The Killam Trust (postgraduate fellowship to M. W. L.).
Notes and references
-
(a) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485 CrossRef CAS PubMed;
(b) M. C. Jahnke and F. E. Hahn, Coord. Chem. Rev., 2015, 293, 95 CrossRef;
(c) D. J. Nelson and S. P. Nolan, Chem. Soc. Rev., 2013, 42, 6723 RSC;
(d) A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722 CrossRef CAS PubMed;
(e) R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461 CrossRef CAS PubMed;
(f) C. M. Crudden and D. P. Allen, Coord. Chem. Rev., 2004, 248, 2247 CrossRef CAS;
(g) D. Bourissou, O. Guerret, F. P. Gabbaï and G. Bertrand, Chem. Rev., 2000, 100, 39 CrossRef CAS PubMed;
(h) For a review on cyclic(alkyl)aminocarbenes, see: M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2015, 48, 256 CrossRef CAS PubMed.
- For selected review articles in this area, see:
(a)
L. J. Murphy, K. N. Robertson, J. D. Masuda and J. A. C. Clyburne, in N-Heterocyclic Carbenes, ed. S. P. Nolan, Wiley-VCH, Weinheim, 2014, pp. 427–497 Search PubMed;
(b) Y. Wang and G. H. Robinson, Inorg. Chem., 2014, 53, 11815 CrossRef CAS PubMed;
(c) E. Rivard, Dalton Trans., 2014, 43, 8577 RSC;
(d) G. Prabusankar, A. Sathyanarayana, P. Suresh, C. N. Babu, K. Srinivas and B. P. R. Metla, Coord. Chem. Rev., 2014, 269, 96 CrossRef CAS;
(e) N. Kuhn and A. Al-Sheikh, Coord. Chem. Rev., 2005, 249, 829 CrossRef CAS;
(f) N. Burford and P. J. Ragogna, J. Chem. Soc., Dalton Trans., 2002, 4307 RSC;
(g) C. Jones, Chem. Commun., 2001, 2293 RSC.
-
(a) N. Kuhn, H. Bohnen, J. Kreutzberg, D. Bläser and R. Boese, J. Chem. Soc., Chem. Commun., 1993, 1136 RSC;
(b) C. E. I. Knappke, J. M. Neudörfl and A. J. von Wangelin, Org. Biomol. Chem., 2010, 8, 1695 RSC;
(c) S. M. I. Al-Rafia, A. C. Malcolm, S. K. Liew, M. J. Ferguson, R. McDonald and E. Rivard, Chem. Commun., 2011, 47, 6987 RSC;
(d) For an improved synthetic route to NHOs, see: K. Powers, C. Hering-Junghans, R. McDonald, M. J. Ferguson and E. Rivard, Polyhedron, 2016 DOI:10.1016/j.poly.2015.07.070.
-
(a) A. C. Malcolm, K. J. Sabourin, R. McDonald, M. J. Ferguson and E. Rivard, Inorg. Chem., 2012, 51, 12905 CrossRef CAS PubMed;
(b) S. M. I. Al-Rafia, M. R. Momeni, M. J. Ferguson, R. McDonald, A. Brown and E. Rivard, Angew. Chem., Int. Ed., 2013, 52, 6390 CrossRef CAS PubMed;
(c) Y. Wang, M. Y. Abraham, R. J. Gilliard Jr., D. R. Sexton, P. Wei and G. H. Robinson, Organometallics, 2013, 32, 6639 CrossRef CAS;
(d) R. S. Ghadwal, S. O. Reichmann, F. Engelhardt, D. M. Andrada and G. Frenking, Chem. Commun., 2013, 49, 9440 RSC;
(e) S. M. I. Al-Rafia, M. R. Momeni, M. J. Ferguson, R. McDonald, A. Brown and E. Rivard, Organometallics, 2013, 32, 6201 CrossRef;
(f) S. Kronig, P. G. Jones and M. Tamm, Eur. J. Inorg. Chem., 2013, 2301 CrossRef CAS;
(g) R. S. Ghadwal, C. J. Schürmann, F. Engelhardt and C. Steinmetzger, Eur. J. Inorg. Chem., 2014, 4921 CrossRef CAS;
(h) R. S. Ghadwal, C. J. Schürmann, D. M. Andrada and G. Frenking, Dalton Trans., 2015, 44, 14359 RSC;
(i) W.-H. Lee, Y.-F. Lin, G.-H. Lee, S.-M. Peng and C.-W. Chiu, Dalton Trans., 2016 10.1039/c5dt03847b.
- For an important early study on the coordination of an NHO with Au(I) centers, see: A. Fürstner, M. Alcarazo, R. Goddard and C. W. Lehmann, Angew. Chem., Int. Ed., 2008, 47, 3210 CrossRef PubMed.
-
(a) R. D. Crocker and T. V. Nguyen, Chem. – Eur. J., 2016, 22, 2208 CrossRef CAS PubMed;
(b) S. Naumann, A. W. Thomas and A. P. Dove, ACS Macro Lett., 2016, 5, 134 CrossRef CAS;
(c) Y.-B. Wang, D.-S. Sun, H. Zhou, W.-Z. Zhang and X.-B. Lu, Green Chem., 2015, 17, 4009 RSC;
(d) Y.-B. Jia, Y.-B. Wang, W.-M. Ren, T. Xu, J. Wang and X.-B. Lu, Macromolecules, 2014, 47, 1966 CrossRef CAS.
- M. Iglesias, A. Iturmendi, P. J. Sanz Miguel, V. Polo, J. Pérez-Torrente and L. A. Oro, Chem. Commun., 2015, 51, 12431 RSC.
-
(a) A. Dumrath, X.-F. Wu, H. Neumann, A. Spannenberg, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 8988 CrossRef CAS PubMed;
(b) A. Dumrath, C. Lübbe, H. Neumann, R. Jackstell and M. Beller, Chem. – Eur. J., 2011, 17, 9599 CrossRef CAS PubMed.
- S. M. I. Al-Rafia, M. J. Ferguson and E. Rivard, Inorg. Chem., 2011, 50, 10543 CrossRef CAS PubMed.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1 RSC.
- S. J. Berners-Price, L. A. Colquhoun, P. C. Healy, K. A. Byriel and J. V. Hanna, J. Chem. Soc., Dalton Trans., 1992, 3357 RSC.
- J. Schreiber, H. Maag, N. Hashimoto and A. Eschenmoser, Angew. Chem., Int. Ed. Engl., 1971, 10, 330 CrossRef CAS.
- J. Monot, M. Makhlouf Brahmi, S.-H. Ueng, C. Robert, M. Desage-El Murr, D. P. Curran, M. Malacria, L. Fensterbank and E. Lacôte, Org. Lett., 2009, 11, 4914 CrossRef CAS PubMed.
- H. Dorn, E. Vejzovic, A. J. Lough and I. Manners, Inorg. Chem., 2001, 40, 4327 CrossRef CAS PubMed.
-
(a) C. W. Cheung, D. S. Surry and S. L. Buchwald, Org. Lett., 2013, 15, 3734 CrossRef CAS PubMed;
(b) J. D. Hicks, A. M. Hyde, A. M. Cuezva and S. L. Buchwald, J. Am. Chem. Soc., 2009, 131, 16720 CrossRef CAS PubMed;
(c) R. J. Lundgren, B. D. Peters, P. G. Alsabeh and M. Stradiotto, Angew. Chem., Int. Ed., 2010, 49, 4071 CrossRef CAS PubMed;
(d) S. M. Crawford, C. B. Lavery and M. Stradiotto, Chem. – Eur. J., 2013, 19, 16760 CrossRef CAS PubMed.
-
(a) C. Valente, S. Çalimsiz, K. H. Hoi, D. Mallik, M. Sayah and M. G. Organ, Angew. Chem., Int. Ed., 2012, 51, 3314 CrossRef CAS PubMed;
(b) S. Meiries, G. Le Duc, A. Chartoire, A. Collado, K. Speck, K. S. A. Arachchige, A. M. Z. Slawin and S. P. Nolan, Chem. – Eur. J., 2013, 19, 17358 CrossRef CAS PubMed.
- W. A. Herrmann, M. Elison, J. Fischer, C. Köcher and G. R. J. Artus, Angew. Chem., Int. Ed. Engl., 1995, 34, 2371 CrossRef CAS.
-
(a) A. R. Martin, A. Chartoire, A. M. Z. Slawin and S. P. Nolan, Beilstein J. Org. Chem., 2012, 8, 1637 CrossRef CAS PubMed;
(b) P. R. Melvin, D. Balcells, N. Hazari and A. Nova, ACS Catal., 2015, 5, 5596 CrossRef CAS.
- M. S. Viciu, O. Navarro, R. F. Germaneau, R. A. Kelly III, W. Sommer, N. Marion, E. D. Stevens, L. Cavallo and S. P. Nolan, Organometallics, 2004, 23, 1629 CrossRef CAS.
-
(a) A. S. K. Hashmi, Angew. Chem., Int. Ed., 2005, 44, 6990 CrossRef CAS PubMed;
(b) D. J. Gorin, B. D. Sherry and F. D. Toste, Chem. Rev., 2008, 108, 3351 CrossRef CAS PubMed;
(c) C. Obradors and A. M. Echavarren, Acc. Chem. Res., 2014, 47, 902 CrossRef CAS PubMed;
(d) H. Yang and F. P. Gabbaï, J. Am. Chem. Soc., 2015, 137, 13425 CrossRef CAS PubMed.
- P. Pérez-Galán, N. Delpont, E. Herrero-Gómez, F. Maseras and A. E. Echavarren, Chem. – Eur. J., 2010, 16, 5324 CrossRef PubMed.
-
(a) A. Sidiropoulos, C. Jones, A. Stasch, S. Klein and G. Frenking, Angew. Chem., Int. Ed., 2009, 48, 9701 CrossRef CAS PubMed;
(b) S. M. I. Al-Rafia, P. A. Lummis, A. K. Swarnakar, K. C. Deutsch, M. J. Ferguson, R. McDonald and E. Rivard, Aust. J. Chem., 2013, 66, 1235 Search PubMed.
-
(a) H. K. Lenker, T. G. Gray and R. A. Stockland, Dalton Trans., 2012, 41, 13274 RSC;
(b) S. G. Weber, D. Zahner, F. Rominger and B. F. Straub, Chem. Commun., 2012, 48, 11325 RSC;
(c) N. Phillips, T. Dodson, R. Tirfoin, J. I. Bates and S. Aldridge, Chem. – Eur. J., 2014, 20, 16721 CrossRef CAS PubMed.
- A. B. Pangborn, M. A. Giardello, R. H. Grubbs, R. K. Rosen and F. J. Timmers, Organometallics, 1996, 15, 1518 CrossRef CAS.
- L. S. Jafarpour, E. D. Stevens and S. P. Nolan, J. Organomet. Chem., 2000, 606, 49 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
-
P. T. Beurskens, G. Beurskens, R. de Gelder, J. M. M. Smits, S. Garcia-Granda and R. O. Gould, DIRDIF-2008 Program. Crystallographic Laboratory, Radboud University, Nijmegen, The Netherlands Search PubMed.
-
(a) A. L. Spek, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 9 CrossRef CAS PubMed;
(b)
A. L. Spek, PLATON - a multipurpose crystallographic tool, Utrecht University, Utrecht, The Netherlands Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR spectra for all compounds and refined structure of 11. CCDC 1448842–1448851. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt00299d |
| ‡ These authors contributed equally to this study. |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a