
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The design of second generation MOP-phosphonites: efficient chiral hydrosilylation of functionalised styrenes†
James T.
Fleming
,
Arne
Ficks
,
Paul G.
Waddell
,
Ross W.
Harrington
and
Lee J.
Higham
*
School of Chemistry, Bedson Building, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK. E-mail: lee.higham@ncl.ac.uk
First published on 25th November 2015
Abstract
A series of enantiopure MOP-phosphonite ligands, with tailored steric profiles, have been synthesised and are proven to be very successful in high-yielding, regio- and enantioselective catalytic hydrosilylation reactions of substituted styrenes, affording important chiral secondary alcohols.
Chiral mono- and bidentate phosphorus ligands have achieved considerable acclaim in a variety of transition metal-catalysed asymmetric transformations.1 Atropisomeric monophosphines with a binaphthyl backbone2 have proven to be particularly successful in chiral hydrosilylations of alkenes.3
We recently reported a new class of MOP-phosphonite ligands (Ia and Ib), based on a MOP/XuPhos-type hybrid (Chart 1).4 Chiral phosphonites are ligands with good π-acceptor properties and are effective in a range of asymmetric hydrocyanations,5 hydroformylations6 and hydrogenations.7 Ligands Ia and Ib possess a phosphorus donor bound directly to a binaphthyl backbone – the a series has a hydrogen in the 2′ position of the lower naphthyl ring whilst a methoxy group occupies the site in the b series (Chart 1). This difference has profound implications for the catalytic performance of the parent (S)-H-MOP and (R)-MeO–MOP ligands, and we wished to examine this effect for phosphonite derivatives. Importantly, we also wanted to examine the effect of incorporating a second chiral moiety, which we achieved by appending either (R)- or (S)-binol to isolate the diastereomeric pairs (S,Rb)-Ia/(S,Sb)-Ia and (R,Rb)-Ib/(R,Sb)-Ib. By investigating the coordination chemistry and catalytic performance of the four ligand pairs, we were able to demonstrate that subtle differences in the position of the palladium atom relative to the lower naphthyl ring in these complexes appears to have important consequences for chiral induction in the catalytic hydrosilylation of styrene.4 These results led us to question whether the large chiral binol group was in fact necessary for effective asymmetric catalysis.
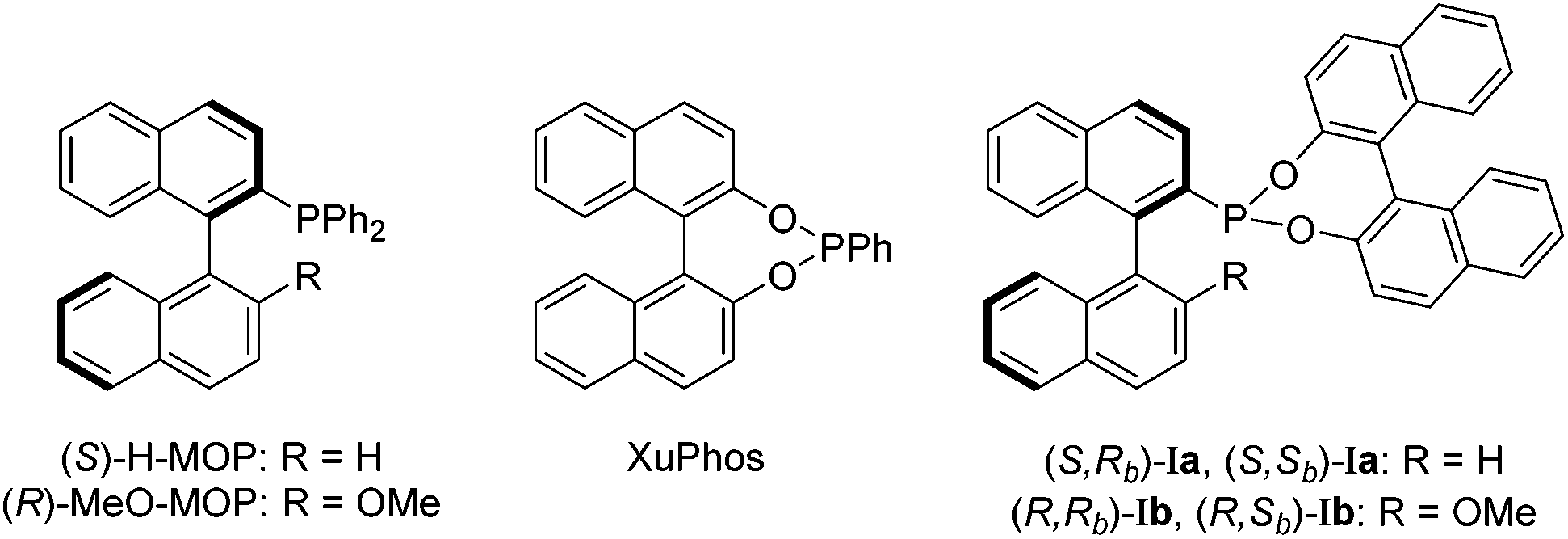 | ||
| Chart 1 A selection of binaphthyl-containing monophosphorus-donor ligands. | ||
In this study we sought to investigate the effect of replacing the binol moiety of Ia and Ib with achiral ancillary aryloxides, in order to establish if the enantioselectivities remain high by virtue of the phosphorus-bonded binaphthyl backbone, and thus eliminate the requirement for a second chiral centre. To the best of our knowledge this has not been demonstrated for MOP phosphonites, which are likely to effectively catalyse alternate substrates than their phosphine cousins, by virtue of their differing electronic properties. Herein we describe the synthesis of the phenoxy derivatives (S)-3a and (R)-3b, which allowed us to probe the effect of removing the chelating element of the added aryloxide, before describing how the sensitivity of these ligands to hydrolysis demanded the design of next generation biphenoxy-based MOP-phosphonites (S)-4a and (R)-4b, which were further improved upon to yield the user-friendly, chiral phosphonites (S)-5a and (R)-5b, that perform better than the corresponding MOP phosphines in the hydrosilylation of functionalised styrenes (Chart 2 and Table 2).
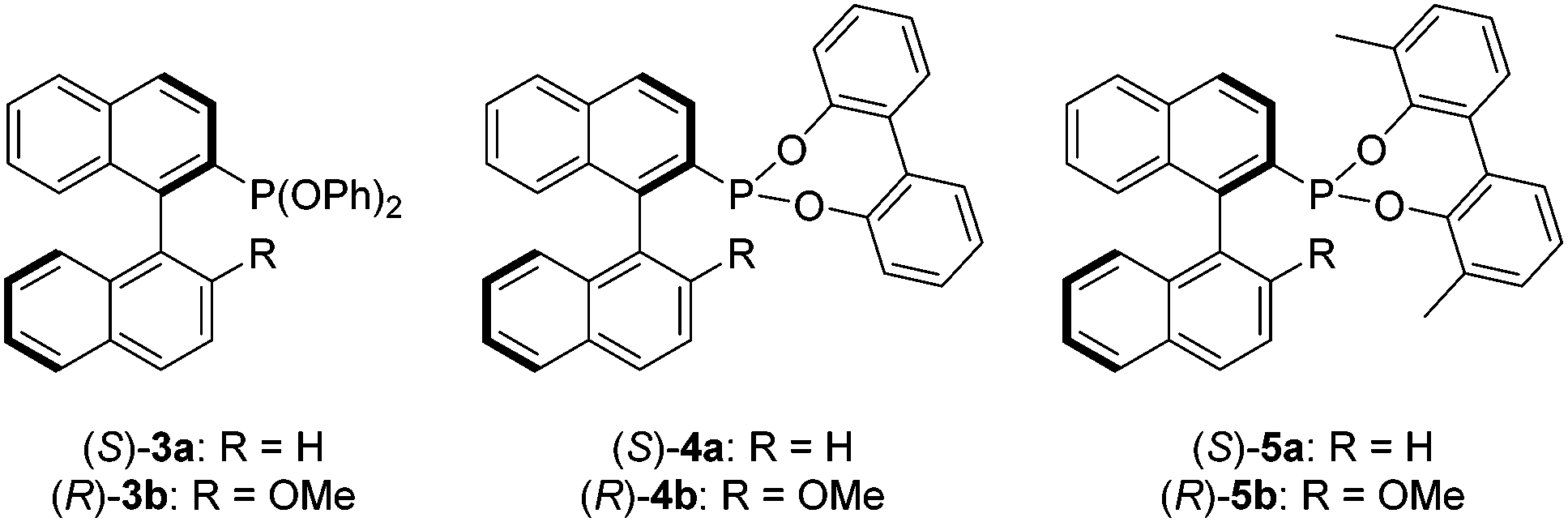 | ||
| Chart 2 Phosphonite ligands (S)-3a, (R)-3b, (S)-4a, (R)-4b, (S)-5a and (R)-5b. | ||
The six new MOP-phosphonite ligands were synthesised following a procedure we have developed, starting with the air-stable, chiral primary phosphine precursors (S)-1a and (R)-1b.8 In a two-step, one-pot reaction, the primary phosphine was treated with phosphorus pentachloride, quantitatively yielding the dichlorophosphines (S)-2a and (R)-2b (Scheme 1).4 Our initial targets (S)-3a and (R)-3b were synthesised by employing two equivalents of phenol, however both syntheses proved to be temperamental as a result of their sensitivity to hydrolysis, with (S)-3a particularly low-yielding. Therefore we sought to replace the two phenol groups with the achiral, chelating 2,2′-biphenol, in order to ascertain whether we could inhibit this decomposition. This approach led to the successful preparation of the enantiopure compounds (S)-4a and (R)-4b; the ligands can be stored under nitrogen for a period of weeks with only minimal decomposition. However, during our initial investigations into the coordination chemistry of these ligands, we noticed slow decomposition in solution and their performance in catalytic hydrosilylation required improvement (vide infra). Thus, in order to push the design concept still further and optimise both ligand stability and catalytic capability, we synthesised the bulky ortho-methyl substituted starting material 2,2′-dihydroxyl-3,3′-dimethyl-1,1′-biphenyl (ESI†), and used it to prepare the ligands (S)-5a and (R)-5b which possess a more imposing steric profile. Both phosphonites were isolated in high yield, and are purified by passing through a plug of alumina in air. They can be stored under nitrogen without decomposition over several months, and their coordination complexes also exhibit better stability.
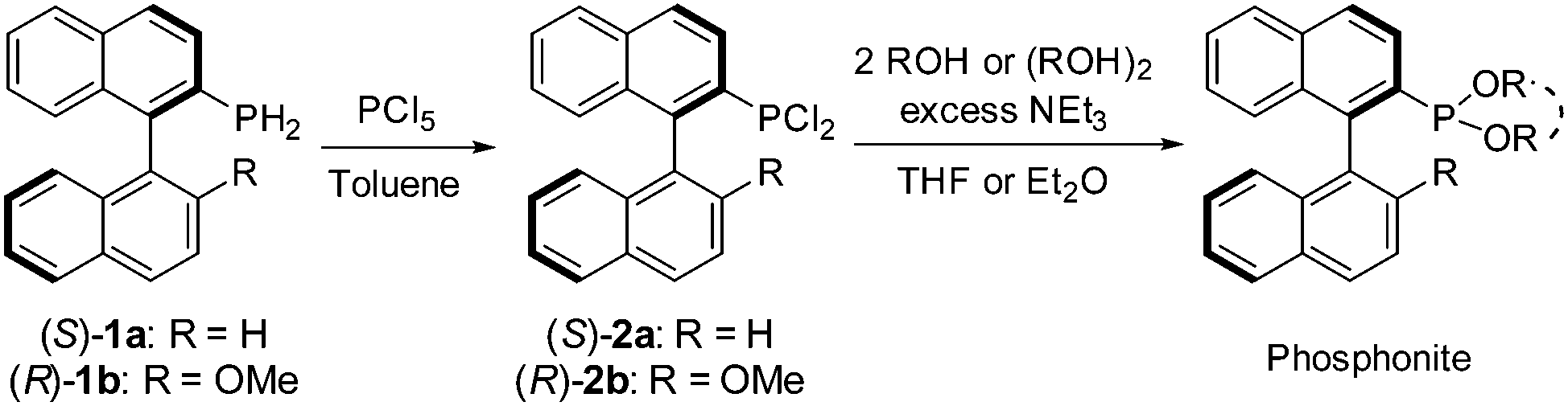 | ||
| Scheme 1 The synthesis of the phosphonite ligands from (S)-1a and (R)-1b. | ||
The phosphonites were fully characterised by multinuclear NMR spectroscopy and High Resolution Mass Spectrometry. Their 31P{1H} spectra show characteristic singlet peaks; δ (ppm) = (S)-3a (154.8), (R)-3b (155.6), (S)-4a (177.7), (R)-4b (180.0), (S)-5a (172.4) and (R)-5b (174.2). The resonances for the phenoxyl-derived phosphonites have a notable upfield shift compared to the biphenoxyl analogues. Single crystals of (R)-3b and (R)-4b suitable for X-ray crystallographic analysis were obtained (Fig. 1 and 2); there are two crystallographically independent molecules in the asymmetric unit of (R)-3b. Density Functional Theory calculations at the B3LYP/6-31G* level of theory (ESI†) revealed that the phosphonites have lower HOMO (Highest Occupied Molecular Orbital) and LUMO (Lowest Unoccupied Molecular Orbital) energies than their aryl counterparts, and have a lower phosphorus contribution to the HOMO, implying the phosphonites are weaker P-ligand σ-donors. To study the coordination chemistry of our ligands with Pd(II), we reacted them with [Pd(η3-C4H7)Cl]2 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, P:Pd ratio, to quantitatively synthesise the complexes [(η3-C4H7)PdCl(LP)].
:1, P:Pd ratio, to quantitatively synthesise the complexes [(η3-C4H7)PdCl(LP)].
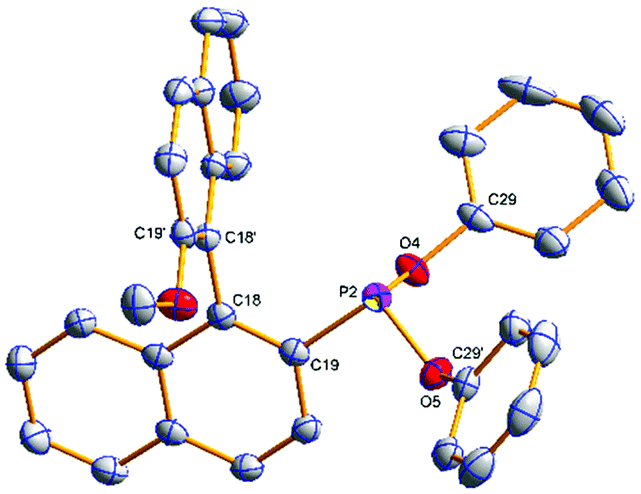 | ||
| Fig. 1 Molecular structure of (R)-3b. Hydrogen atoms have been omitted for clarity. Selected average bond distances (Å) and angles (°): P2–C19 1.830(2), P2–O4 1.6461(17), P2–O5 1.6619(18), O4–C29 1.402(3), O5–C29′ 1.391(3); O4–P2–O5 99.13(9), O4–P2–C19 93.63(9), O5–P2–C19 97.85(10), C19–C18–C18′–C19′ –86.5(3). | ||
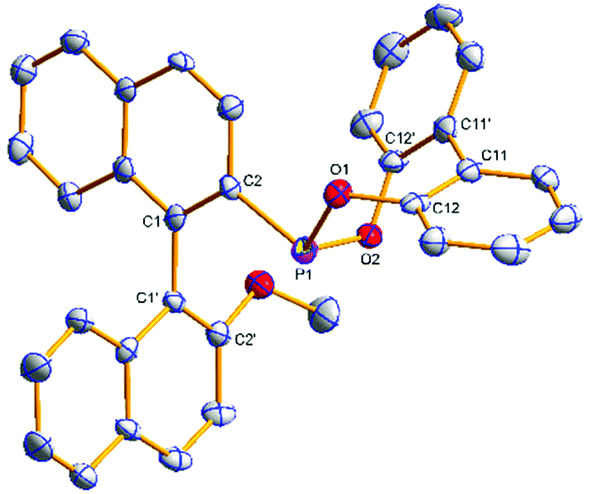 | ||
| Fig. 2 Molecular structure of (R)-4b. Hydrogen atoms have been omitted for clarity. Selected average bond distances (Å) and angles (°): P1–C2 1.812(7), P1–O1 1.658(4), P1–O2 1.657(4), O1–C12 1.411(7), O2–C12′ 1.396(7), C11–C11′ 1.482(9); O1–P1–O2 99.5(2), O1–P1–C2 96.6(2), O2–P1–C2 100.3(3), C2–C1–C1′–C2′ –86.5(7), C12–C11–C11′–C12′ –48.6(8). | ||
Single crystals of [(η3-C4H7)PdCl(S)-(4a)] ((S)-6a) and [(η3-C4H7)PdCl((S)-(5a))] ((S)-7a) suitable for X-ray analysis were grown (Fig. 3 and 4); the asymmetric unit of (S)-7a comprises two molecules in different conformations (the second structure and an overlay is provided in the ESI†) and there is twofold disorder of the methallyl ligand and the palladium atom in one of these independents. The phosphonites show monodentate coordination through the phosphorus, and there is a pseudo-square-planar geometry around the palladium.
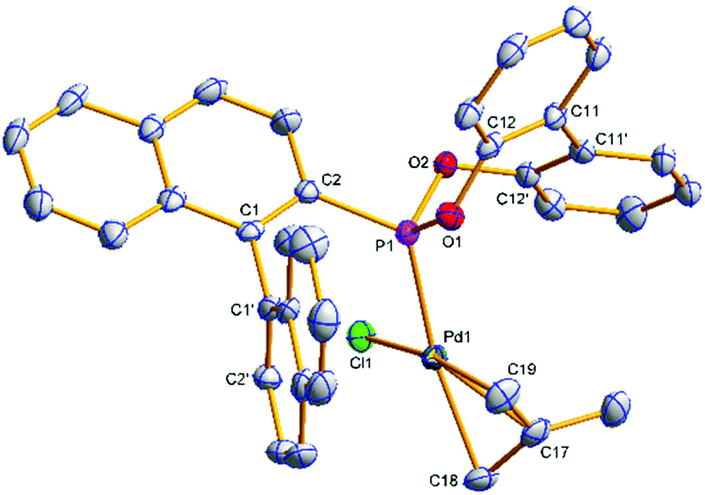 | ||
| Fig. 3 Molecular structure of (S)-6a. Hydrogen atoms have been omitted for clarity. Selected average bond distances (Å) and angles (°): Pd1–P1 2.2478(10), Pd1–Cl1 2.3583(9), Pd1–C17 2.162(4), Pd1–C18 2.195(4), Pd1–C19 2.096(4), P1–C2 1.815(4), P1–O1 1.619(3), P1–O2 1.610(3), C11–C11′ 1.477(5); P1–Pd1–Cl1 91.55(4), P1–Pd1–C18 165.51(11), P1–Pd1–C19 98.27(13), C18–Pd1–C19 67.46(16), C18–C17–C19 114.6(4), C2–C1–C1′–C2′ –80.8(5), C12–C11–C11′–C12′ 42.3(6). | ||
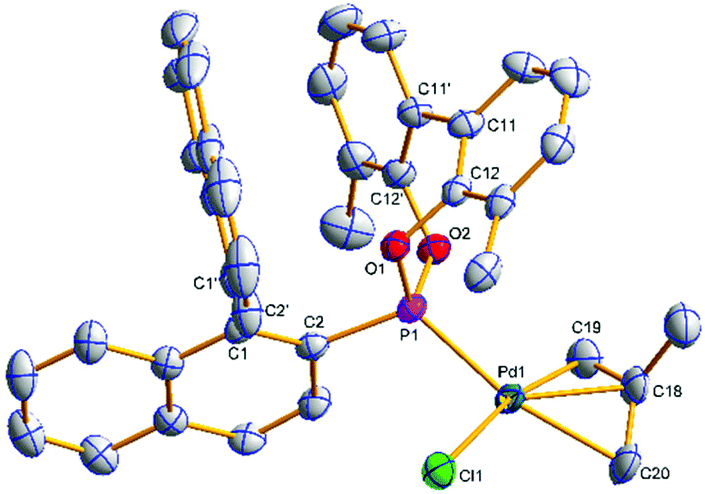 | ||
| Fig. 4 Molecular structure of (S)-7a. Hydrogen atoms have been omitted for clarity. Selected average bond distances (Å) and angles (°): Pd1–P1 2.2368(11), Pd1–Cl1 2.3739(11), Pd1–C18 2.152(4), Pd1–C19 2.095(4), Pd1–C20 2.206(4), P1–C2 1.804(4), P1–O1 1.610(3), P1–O2 1.622(3), C11–C11′ 1.485(6); P1–Pd1–Cl1 94.59(4), P1–Pd1–C19 96.50(15), P1–Pd1–C20 162.95(15), C19–Pd1–C20 66.8(2), C19–C18–C20 117.4(5), C2–C1–C1′–C2′ –81.9(6), C12–C11–C11′–C12′ –46.0(7). | ||
The allyl carbons trans to phosphorus exhibit a longer Pd–C bond length (2.195, 2.206 Å) than the allyl carbons cis to the phosphorus (2.096, 2.095 Å), which is consistent with the stronger trans influence of the phosphonite compared to the chloride ligand. In (S)-6a the lower naphthyl fragment of the binaphthyl backbone is face-to-face with the palladium centre (Fig. 3). Neither of the two independents of (S)-7a display this feature, with the lower naphthyl group being orientated away from the palladium centre and facing the biphenyl moiety (Fig. 4 and ESI†). In the two independents of (S)-7a the torsion angle of the dimethyl substituted biphenyl moiety is of opposite sign, and also when comparing the biphenyl moiety in (R)-4b and (S)-6a (Fig. 2 and 3), implying no restriction to rotation about the C11–C11′ bond.
The 31P{1H} NMR peaks of (S)-6a and (S)-7a experience slight shifts compared to the free ligands, and show the presence of two independent resonances due to the two isomers formed; δ (ppm) = (S)-6a (172.0 and 173.6) and (S)-7a (172.9 and 177.6). The isomers are a result of the rotation of the allyl moiety, via a selective syn/anti exchange of the allyl protons cis to the phosphorus, by a η3–η1–η3 mechanism, due to the stronger trans effect of the phosphonite donor ligand.4,9 The rapid exchange process resulted in broadened peaks in the NMR spectra at room temperature; cooling of CD2Cl2 solutions of the complexes to −20 °C sharpened the resonances and allowed for full characterisation of the methallyl ligands in both isomers (ESI†). Reaction of (S)-7a with NaBArF resulted in loss of NaCl and the formation of [(η3-C4H7)Pd(S)-(5a)]BArF ((S)-8a), where (S)-5a acts as a chelating P,C-π-donor. The upfield 13C{1H} NMR coordination chemical shifts for both C1′ and C2′ suggest a η2-binding mode (Fig. 5), which is in agreement with the results of an NMR study reported by Pregosin and co-workers.9
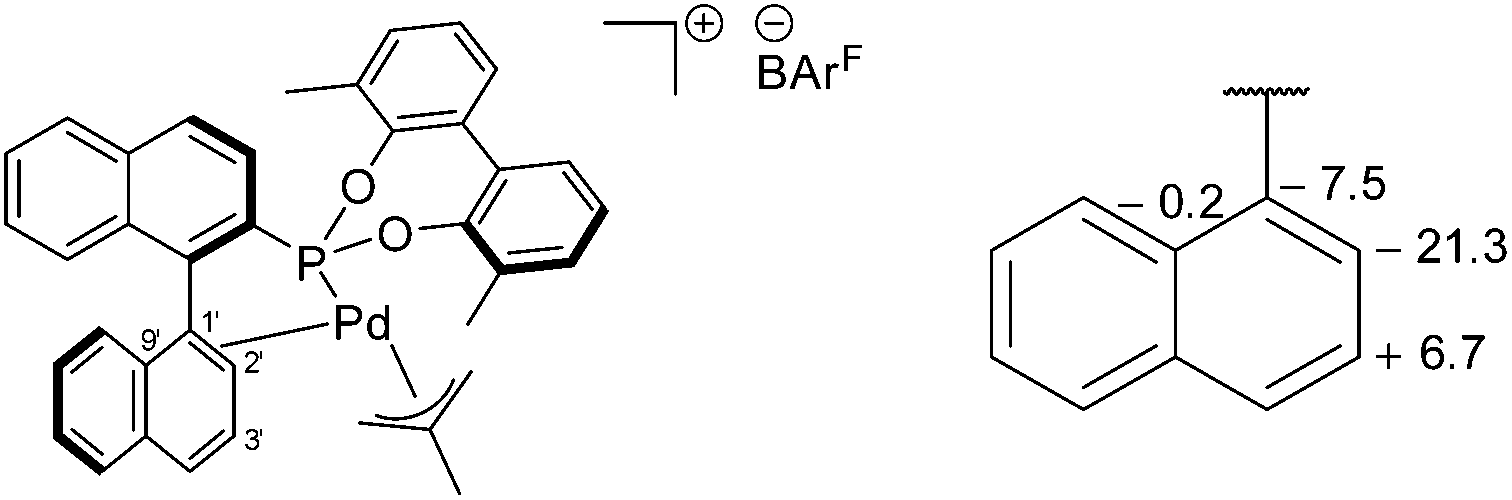 | ||
| Fig. 5 Proposed structure of (S)-8a (left) and a fragment showing the 13C{1H} NMR coordination chemical shift (ppm) between the major isomers of (S)-7a and (S)-8a (right). | ||
As discussed, MOP phosphine ligands are known to give high enantioselectivity in the palladium-catalysed asymmetric hydrosilylation of alkenes, particularly styrene, to give chiral secondary alcohols (Scheme 2).3 To gain an insight into how the different stereoelectronic profiles of our phosphonites impacts upon their catalytic activity in the same transformation, we prepared catalysts by reacting each phosphonite with [Pd(η3-C3H5)Cl]2 (Table 1). We chose to test our phosphonites against the well-known H-MOP and MeO–MOP phosphines (the latter is a commercial compound), employing P:Pd ratios of 1:1 and 2:1 at room temperature – full conversion was obtained in all cases. We noted a general increase in enantioselectivity and reaction rate in the order (S)-5a/(R)-5b>(S)-4a/(R)-4b>(S)-3a/(R)-3b, with the introduction of the biphenyl moiety, and subsequently the methyl groups, markedly improving the ligand performance. Phosphonite (S)-5a gave excellent enantioselectivity for (R)-1-phenylethanol (Table 1, 95%, entry 18), which is slightly higher than that for the (S)-H-MOP phosphine (Table 1, 94%, entry 4), although the latter reaction reached conversion more quickly. We also tested the H-MOP phosphonite ligands (S,Rb)-Ia and (S,Sb)-Ia at 0 °C, these ligands are far less selective than (S)-5a.4
 | ||
| Scheme 2 The synthesis of chiral secondary alcohols via the palladium catalysed asymmetric hydrosilylation and subsequent oxidation of styrene derivatives. | ||
| L | P:Pd |
T | Timeb | Conv.c | eed | |
|---|---|---|---|---|---|---|
| a The catalyst was generated in situ from the ligand (0.25 or 0.50 mol%) and [Pd(η3-C3H5)Cl]2 (0.125 mol%), and reacted with styrene (10 mmol) and trichlorosilane (12 mmol). b Time taken for reaction to reach completion. c Determined by 1H NMR spectroscopy. d % ee determined by chiral GC; absolute configuration assigned by comparison of the sign of optical rotation to literature (ESI). e Reaction stopped. | ||||||
| 1 | (S)-H-MOP | 1:1 |
rt | 1 min | >99% | 74 (R) |
| 2 | (S)-H-MOP | 1:1 |
0 °C | 4 h | >99% | 93 (R) |
| 3 | (S)-H-MOP | 2:1 |
rt | 1 h | >99% | 75 (R) |
| 4 | (S)-H-MOP | 2:1 |
0 °C | 12 h | >99% | 94 (R) |
| 5 | (R)-MeO-MOP | 1:1 |
rt | 10 min | >99% | 22 (R) |
| 6 | (R)-MeO-MOP | 2:1 |
rt | 1 h | >99% | 20 (R) |
| 7 | (S)-3a | 1:1 |
rt | 24 h | >99% | 63 (R) |
| 8 | (S)-3a | 2:1 |
rt | 24 h | >99% | 73 (R) |
| 9 | (R)-3b | 1:1 |
rt | 24 h | >99% | 7 (S) |
| 10 | (R)-3b | 2:1 |
rt | 24 h | >99% | 1 (S) |
| 11 | (S)-4a | 1:1 |
rt | 6 min | >99% | 73 (R) |
| 12 | (S)-4a | 2:1 |
rt | 1 h | >99% | 79 (R) |
| 13 | (R)-4b | 1:1 |
rt | 10 min | >99% | 23 (R) |
| 14 | (R)-4b | 2:1 |
rt | 1 h | >99% | 27 (R) |
| 15 | (S)-5a | 1:1 |
rt | 2 min | >99% | 79 (R) |
| 16 | (S)-5a | 1:1 |
0 °C | 5 h | >99% | 92 (R) |
| 17 | (S)-5a | 2:1 |
rt | 2 h | >99% | 80 (R) |
| 18 | (S)-5a | 2:1 |
0 °C | 72 h | >99% | 95 (R) |
| 19 | (R)-5b | 1:1 |
rt | 1 h | >99% | 45 (R) |
| 20 | (R)-5b | 2:1 |
rt | 70 min | >99% | 51 (R) |
| 21 | (S,Rb)-Ia | 1:1 |
0 °C | 168 h | 88%e | 4 (S) |
| 22 | (S,Rb)-Ia | 2:1 |
0 °C | 168 h | >99% | 60 (R) |
| 23 | (S,Sb)-Ia | 1:1 |
0 °C | 72 h | >99% | 59 (R) |
| 24 | (S,Sb)-Ia | 2:1 |
0 °C | 168 h | 22%e | 77 (R) |
Interestingly, although the methoxy-substituted ligands (R)-MeO–MOP phosphine and (R)-5b performed poorly compared to their H-substituted counterparts – an established trend for this ligand family10 – the phosphonite ligand gave much higher enantioselectivities than the corresponding phosphine (51% versus 20%, Table 1, entries 20 and 6), demonstrating further the potential these derivatives have to improve existing toolkits.
It is notable that at room temperature, the hydrosilylation occurs as a highly exothermic, quite violent reaction which is accompanied by a spontaneous colour change to deep black; similar observations with arylmonophosphinoferrocenes have also been made.11 When a 2:1, P:Pd ratio was used there was often a significant delay in the time taken to observe this phenomenon (from 1–8 h, Table 1). However these exothermic colour changes were not observed when the external temperature of the flask was kept at 0 °C. Consequently, the catalysis performed at 0 °C using both (S)-H-MOP and the sterically bulky (S)-5a resulted in longer reaction times, however this was offset by an increased ee of the product – phosphonite (S)-5a gave the best ee with a value of 95%.
The final objective in this study was to extend our styrene screening regimen to substrates that MOP phosphines struggle to catalyse, or confer only low or moderate enantioselectivity in the product, with the aim that the unique stereoelectronic properties of the phosphonites would allow for improvements to be made. As such we extended our screening with (S)-5a to include the para-substituted styrenes listed in Table 2. Introducing an electron withdrawing chloride group resulted in much longer reaction times, however full conversion and a high ee, better than (S)-H-MOP, were again obtained (Table 2, entries 1 and 3; note entry 2 for improving the ee generated by the latter). The chloro-substituted chiral secondary alcohol product was used to synthesise the antihistamine Clemastine enantioselectively.12 More significantly, when an electron donating 4-methoxy substituent was incorporated onto the styrene precursor, (S)-5a was found to be a remarkably superior ligand, generating a much higher ee than (S)-H-MOP, albeit more slowly (85% versus 58%, Table 2, entries 8 and 6). It is of note that the product alcohol from this reaction was used to prepare antagonists of lysophosphatidic receptors, which may have implications in the treatment of lung fibrosis, liver disease and cancer.13 Our future work will seek to extend the substrate scope to styrenes bearing other important functional groups.
| L | Ar | T | Timeb | Conv.c | eed | |
|---|---|---|---|---|---|---|
| a The catalyst was generated in situ from the ligand (0.50 mol%) and [Pd(η3-C3H5)Cl]2 (0.125 mol%) and reacted with styrene (10 mmol) and trichlorosilane (12 mmol). b Time taken for reaction to reach completion. c Determined by 1H NMR spectroscopy. d % ee determined by chiral GC; absolute configuration assigned by comparison of the sign of optical rotation to literature (ESI). e Reaction stopped. | ||||||
| 1 | (S)-H-MOP | 4-ClC6H4 | rt | 1 h | >99% | 78 (R) |
| 2 | (S)-H-MOP | 4-ClC6H4 | 0 °C | 24 h | >99% | 94 (R) |
| 3 | (S)-5a | 4-ClC6H4 | rt | 18 h | >99% | 86 (R) |
| 4 | (S)-5a | 4-ClC6H4 | 0 °C | 336 h | <5%e | — |
| 5 | (S)-H-MOP | 4-MeOC6H4 | rt | 10 min | >99% | 35 (R) |
| 6 | (S)-H-MOP | 4-MeOC6H4 | 0 °C | 12 h | >99% | 58 (R) |
| 7 | (S)-5a | 4-MeOC6H4 | rt | 70 min | >99% | 78 (R) |
| 8 | (S)-5a | 4-MeOC6H4 | 0 °C | 240 h | >99% | 85 (R) |
In summary, we have synthesised a series of novel MOP-phosphonite ligands from air-stable, chiral primary phosphine precursors, and have demonstrated the ease with which both the sterics and the hydrolytic nature of the ligands can be tuned, by modifying the aryl groups of the aryloxy moiety. In comparison to our previous report on Ia/Ib, a major finding here is that a second chiral element is not necessary for the chiral induction, crucially both the ee and reaction rates can be increased without it.4 In the palladium-catalysed asymmetric hydrosilylation of functionalised styrenes, our ligands proved to be high yielding, and both regio- and enantioselective; phosphonite (S)-5a achieved ees of up to 95%. When the substrate used was 4-methoxystyrene, (S)-5a is significantly more enantioselective than Hayashi's benchmark phosphine (S)-H-MOP, demonstrating how MOP-phosphonite ligands have the potential to fill voids left by their phosphine counterparts in the field of asymmetric catalysis.
We wish to acknowledge the financial support of the EPSRC for a Fellowship award (LJH: EP/G005206/1) and its National Mass Spectrometry Facility at Swansea, and the School of Chemistry, Newcastle University for a Studentship (JTF).
Notes and references
- Phosphorus Ligands in Asymmetric Catalysis, ed. A. Börner, Wiley-VCH, Weinheim, 2008 Search PubMed; Phosphorus(III) Ligands in Homogeneous Catalysis: Design and Synthesis, ed. P. C. J. Kamer and P. W. N. M. van Leeuwen, John Wiley & Sons, Chichester, 2012 Search PubMed.
- T. Hayashi, Acc. Chem. Res., 2000, 33, 354 CrossRef CAS PubMed; F. Lagasse and H. B. Kagan, Chem. Pharm. Bull., 2000, 48, 315 CrossRef PubMed; R. M. Hiney, A. Ficks, H. Müller-Bunz, D. G. Gilheany and L. J. Higham, in Organometallic Chemistry, ed. I. J. S. Fairlamb and J. M. Lynam, The Royal Society of Chemistry, Cambridge, 2011, vol. 37, pp. 27–45 Search PubMed.
- T. Hayashi, Catal. Surv. Asia, 1999, 3, 127 CrossRef CAS; J. W. Han and T. Hayashi, Tetrahedron: Asymmetry, 2014, 25, 479 CrossRef.
- A. Ficks, R. M. Hiney, R. W. Harrington, D. G. Gilheany and L. J. Higham, Dalton Trans., 2012, 41, 3515 RSC.
- L. Bini, C. Muller and D. Vogt, Chem. Commun., 2010, 46, 8325 RSC.
- J. I. van der Vlugt, J. M. J. Paulusse, E. J. Zijp, J. A. Tijmensen, A. M. Mills, A. L. Spek, C. Claver and D. Vogt, Eur. J. Inorg. Chem., 2004, 4193 CrossRef CAS.
- C. Claver, E. Fernandez, A. Gillon, K. Heslop, D. J. Hyett, A. Martorell, A. G. Orpen and P. G. Pringle, Chem. Commun., 2000, 961 Search PubMed; M. T. Reetz and T. Sell, Tetrahedron Lett., 2000, 41, 6333 CrossRef CAS.
- R. M. Hiney, L. J. Higham, H. Müller-Bunz and D. G. Gilheany, Angew. Chem., Int. Ed., 2006, 45, 7248 CrossRef CAS PubMed; A. Ficks, C. Sibbald, S. Ojo, R. W. Harrington, W. Clegg and L. J. Higham, Synthesis, 2013, 265 Search PubMed.
- P. G. A. Kumar, P. Dotta, R. Hermatschweiler, P. S. Pregosin, A. Albinati and S. Rizzato, Organometallics, 2005, 24, 1306 CrossRef CAS.
- K. Kitayama, Y. Uozumi and T. Hayashi, J. Chem. Soc., Chem. Commun., 1995, 1533 RSC.
- H. L. Pedersen and M. Johannsen, J. Org. Chem., 2002, 67, 7982 CrossRef CAS PubMed.
- A. M. Fournier, R. A. Brown, W. Farnaby, H. Miyatake-Onzozabal and J. Clayden, Org. Lett., 2010, 12, 2222 CrossRef CAS PubMed.
- J. H. Hutchinson, T. J. Seiders, B. Wang, J. M. Arruda, J. R. Roppe and T. Parr, Br. Pat., GB2470833, 2010 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental and crystallographic data. CCDC 1416469 (R)-3b, 1416470 (R)-(4b), 1416471 (S)-(6a) and 1416472 (S)-(7a). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt04475h |
| This journal is © The Royal Society of Chemistry 2016 |