
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tris(pyrazolyl)phosphines with copper(I): from monomers to polymers†
Cornelis G. J.
Tazelaar
a,
Emmanuel
Nicolas
 a,
Tom
van Dijk
a,
Daniël L. J.
Broere
a,
Mitchel
Cardol
a,
Martin
Lutz
b,
Dietrich
Gudat
c,
J. Chris
Slootweg
*a and
Koop
Lammertsma
*ad
a,
Tom
van Dijk
a,
Daniël L. J.
Broere
a,
Mitchel
Cardol
a,
Martin
Lutz
b,
Dietrich
Gudat
c,
J. Chris
Slootweg
*a and
Koop
Lammertsma
*ad
aDepartment of Chemistry and Pharmaceutical Sciences, Vrije Universiteit Amsterdam, De Boelelaan 1083, 1081 HV Amsterdam, The Netherlands. E-mail: j.c.slootweg@vu.nl; k.lammertsma@vu.nl
bBijvoet Center for Biomolecular Research, Crystal and Structural Chemistry, Utrecht University, Padualaan 8, 3584 CH Utrecht, The Netherlands
cInstitute for Inorganic Chemistry, Universität Stuttgart, Pfaffenwaldring 55, 70569 Stuttgart, Germany
dDepartment of Chemistry, University of Johannesburg, Auckland Park, Johannesburg, 2006 South Africa
First published on 9th November 2015
Abstract
The parent tris(pyrazolyl)phosphine and its 3,5-Me2, 3-Ph, and 3-t-Bu derivatives have been prepared by a simple procedure and show modest Lewis basicity of the phosphorus apex as was established by the magnitude of the 1JP,Se coupling constant of the phosphine selenides. Because of the chelating properties of both the N- and P-sites, neutral phosphorus-centered scorpion ligands allow coordination modes that are unavailable to the abundantly used anionic tris(pyrazolyl)borate scorpionates as we established for Cu(I)-complexation. The substituted P-scorpion ligands only allow for N-coordination, as the P-apex is presumably less accessible. Two X-ray crystal structures were obtained for the Cu-complex of tris(3,5-dimethylpyrazolyl)-phosphine with acetonitrile and triphenylphosphine in the fourth coordination site. The parent P-scorpion ligand can chelate with both its pyrazolyl groups and its P-apex with the product depending on the ratio in which it is mixed with the Cu(I) complex. Reacting two equivalents of the ligand with [Cu(MeCN)4][PF6] resulted in a complex in which Cu is coordinated to the three pyrazolyl groups of one ligand and to the P-apex of the other ligand as confirmed by an X-ray crystal structure determination and a DFT computational analysis. Reacting the ligand and the Cu(I) complex in an equimolar ratio resulted in a remarkable one-dimensional P-scorpion coordination polymer for which a single crystal X-ray structure could be determined. A detailed analysis of the structural features is presented.
Introduction
Tris(pyrazolyl)borates (A, Fig. 1) were first introduced by Trofimenko1 in 1966 and became one of the most widely applied polydentate anionic ligands in coordination chemistry and catalysis because of their versatility and stability.2–5 Neutral ligands result from changing the boron apex to a carbon one as in tris(pyrazolyl)methane (B)6 for which ample syntheses have been reported.7 The neutral phosphorus-centered analogues C and their oxides OC were introduced in the mid-1970s, but hardly used.8–10 Tolman's group explored chiral versions of OC ligands for asymmetric catalysis,11–14 while Weigand's group applied tris(3,5-dimethylpyrazolyl)phosphine (CMe2) as a synthon for cationic phosphorus compounds.15–19 Recently, we reported a simple method to synthesize the PO-centered OC ligands and studied their coordination chemistry.20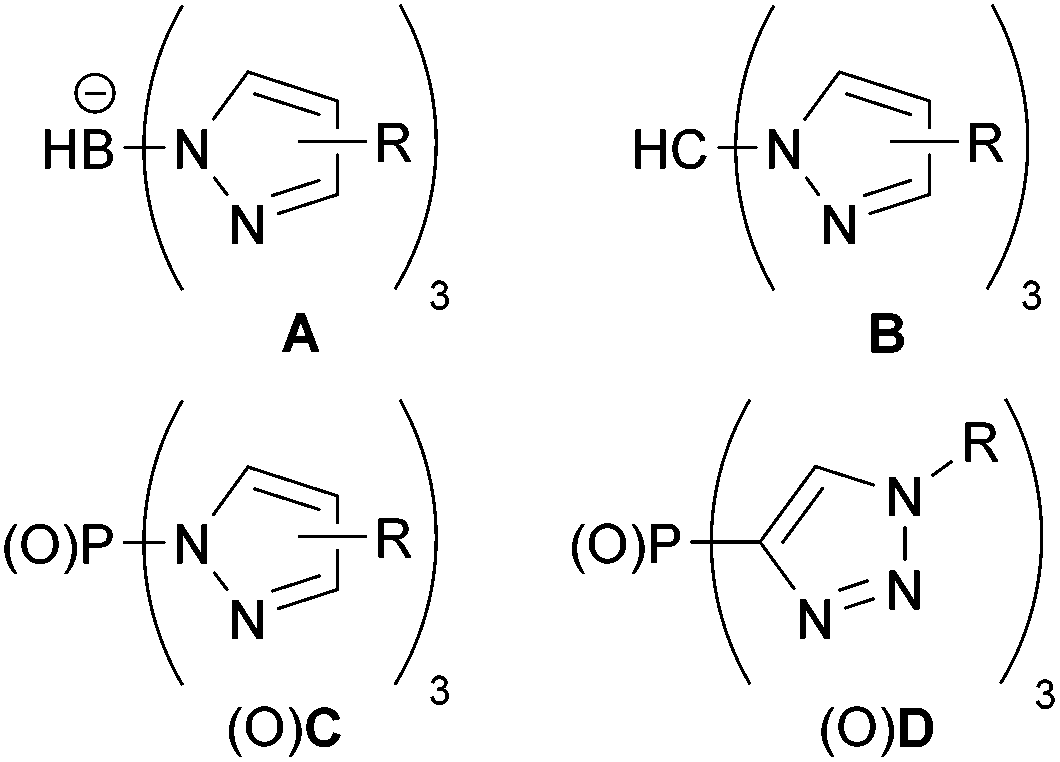 | ||
| Fig. 1 Scorpion-type ligands. | ||
Tris(pyrazolyl)phosphines have multiple coordination sites, making them well suited for ligation with more than one metallic complex. Recently, Hazari and coworkers showed C coordinating to Mg in a κ3 fashion, leaving the phosphorus apex available for additional coordination.21 We were able to use this coordinating ability in a study on the related tris(triazolyl)phosphine D to form the bimetallic complex (OC)5WDMo(CO)3 (Fig. 2).22 We also showed that the two oxidized ligands OC and OD influence the coordinated metal similarly,23 but OC has more substitution options because of the position of the carbon atoms in its heterocyclic rings.
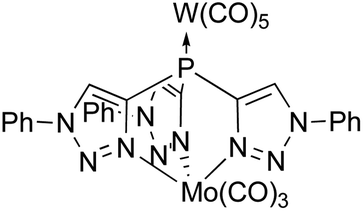 | ||
| Fig. 2 A bimetallic complex supported by ligand D. | ||
Besides multimetallic complexes, scorpion ligands B also enable access to coordination macrocycles and polymers as shown in the seminal work of Reger,24 who used tethered, multitopic tris(pyrazolyl) moieties (Fig. 3a) with silver(I) complexes to generate an abundance of well-identified supramolecular structures,25–29 including argentachains with κ2–κ1 coordination modes (Fig. 3b).26,28–33 One-dimensional coordination polymers are also accessible by the embedded use of cyano-substituted pyrazoles as has been demonstrated for the coordination of A(t-Bu/Ph,4-CN) with Cu(I).34 Similarly, S2N-coordination polymers result when Ir complexes react with heteroscorpionates akin to B, but with methimazolyl instead of pyrazolyl groups.35 A ‘metallic tape’ forms on reacting a thallium salt with tris(mercaptothiadiazolyl)borate36,37 that can be viewed as a Janus scorpionate ligand because of the combined soft and hard binding sites.38 Janus-type ligands like tris(amidomethyl)phosphines with N- and P-coordination sites (Fig. 3c) have been deployed to form bimetallic complexes39,40 and coordination polymers41 with the Co–Y, Co–Gd, and Rh–Ti transition metal pairs.
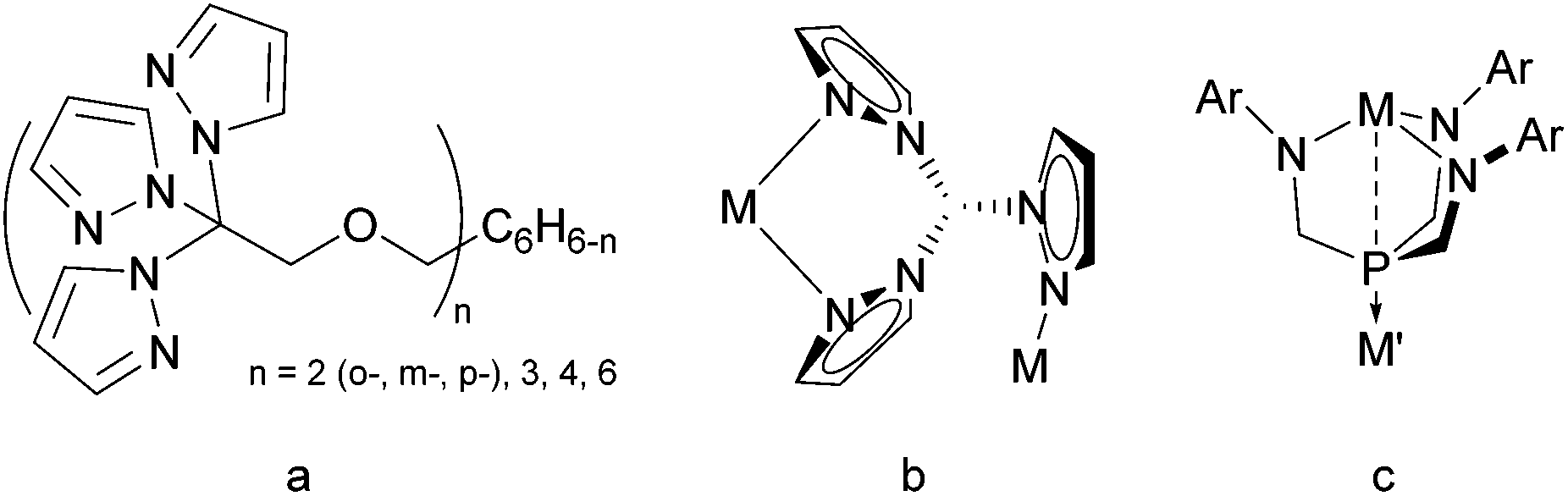 | ||
| Fig. 3 (a) Multitopic tris(pyrazolyl)methane ligands, (b) the κ2–κ1 bonding mode, and (c) a bimetallic Janus-type complex. | ||
Herein we present the synthesis of tris(pyrazolyl)phosphines C, their coordination with copper(I), and their Janus property to generate a one-dimensional coordination polymer.
Results and discussion
Ligand synthesis
The known tris(pyrazolyl)phosphine CH and tris(dimethylpyrazolyl)phosphine CMe2,8,9 and the new, more congested tris(3-phenylpyrazolyl)phosphine CPh and tris(3-tert-butylpyrazolyl)phosphine Ct-Bu were synthesized in modest to excellent yields using our recently reported protocol (Scheme 1).20 The products were obtained by slow addition of phosphorus trichloride to a mixture of three equivalents of the appropriate pyrazole and a slight excess of triethylamine in THF cooled to 0 °C, followed by prolonged stirring, either at room temperature for CH and CMe2 or under reflux for the bulkier CPh and Ct-Bu. The reaction progress was monitored by 31P NMR spectroscopy using the characteristic singlet for the phosphorus apex of the products (δ 61.1 (CH), 72.0 (CMe2), 60.2 (CPh), and 58.2 (Ct-Bu)). After filtering off by-products and removal of the volatiles, colorless to yellow solids were obtained for which additional purification was only needed for CH (recrystallization) and CPh (washings), reducing their isolated yields significantly. The 1H and 13C NMR spectra revealed simple signal patterns, reflecting the expected equivalence of the three pyrazolyl groups; the 13C{1H} NMR spectra showed the characteristic doublets (JC,P ∼ 10 Hz) for the C3 and C5 carbons. | ||
| Scheme 1 Synthesis of tris(pyrazolyl)phosphines CH–Ct-Bu. | ||
Relative Lewis basicities of P-centered systems, and thus their ligating ability, can be examined by means of their phosphine selenides since the magnitude of the 1JP,Se coupling constant is inversely related to the σ donating character of the free phosphine.42–44 To examine the P-donor capacity of compounds Cx, we have investigated selenides SeCH and SeCMe2 (Scheme 2). Both selenides were obtained as yellow powders after reaction of the parent compounds with an excess of selenium for two to three days in refluxing toluene, followed by filtration over silica, evaporation of the volatiles, and washing with pentane. Unsubstituted SeCH (∼24%) showed a resonance at δ(31P) 37.7 ppm with a 1JP,Se coupling constant of 1014 Hz with the corresponding values for SeCMe2 (∼88%) of 40.7 ppm and 872 Hz (see ESI p. S-17 and S-19† respectively). The latter 1JP,Se coupling is at the high end of those reported for selenophosphoramides, indicating CMe2 to be a weaker donor than, e.g., P(NMe2)3 (cf., 1JP,Se = 784 Hz for the corresponding selenide).45 Unsubstituted CH turns out to be a still weaker donor that compares better with weakly donating phosphonates (cf., 1JP,Se = 1025 Hz for SeP(OPh)3).46 The weak σ donating nature of the P apices of CH and CMe2 is also reflected by the harsh conditions required for the formation of their selenides.
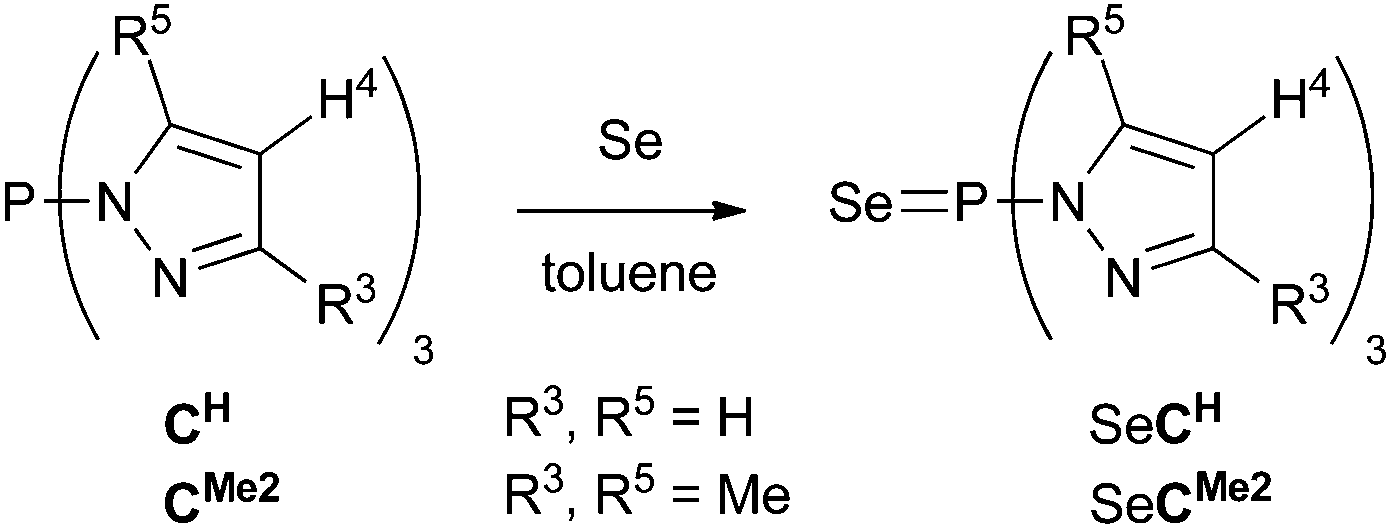 | ||
| Scheme 2 Synthesis of tris(pyrazolyl)phosphine selenides SeCH and SeCMe2. | ||
Complexation to copper(I)
For tris(pyrazolyl)phosphines (C) to be suitable Janus-type ligands for generating one-dimensional coordination polymers they must be able to ligate in a head-to-tail fashion with Cu(I). Hence, their N- and P-donor sites must have complementary affinities for the copper complex. Earlier, it has been shown that the three pyrazolyl groups of carbon-centered Bt-Bu and phosphine oxide-centered OCPh,Me and OCt-Bu coordinate with Cu(I) with acetonitrile completing the coordination sphere.20,47 For OCMe2 we have shown that the coordination can also be completed with a phosphorus ligand such as triphenylphosphine.20 However, the donor ability of PPh3 differs significantly from that of C (e.g., 1JP,Se = 735 Hz for SeP(Ph)3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 48). Ideally, a head-to-tail Cu(I)-complex with C would result from simply treating a Cu(I) complex with two equivalents of C, one providing the three coordinating pyrazolyl groups and the other the phosphorus apex. However, this approach did not give the desired result when [Cu(NCMe)4][PF6] was treated overnight in THF with the dimethyl substituted ligand CMe2. A colorless solid precipitated, yielding crystals suitable for X-ray diffraction upon recrystallization (CH2Cl2/pentane). The determined molecular structure reveals [CMe2Cu(NCMe)][PF6] (Fig. 4, left), which must have been formed by replacing three acetonitrile ligands of [Cu(NCMe)4][PF6] for one CMe2. Apparently, the phosphorus apex of the second CMe2 is unable to replace the remaining acetonitrile. Reacting CMe2 and the Cu-complex in an equimolar ratio gave the same product in 67.7% isolated yield. In the crystal, the metal complex is located at a general position and has only a very approximate non-crystallographic C3v symmetry. The molecular structure shows the CMe2 ligand to be bound in a tridentate fashion via its pyrazolyl rings with Cu–N bond lengths ranging from 2.071(3) to 2.080(3) Å and N–Cu–N angles ranging from 88.49(11) to 94.52(10)°. The acetonitrile is linearly coordinated to copper with a Cu–N14 bond length of 1.888(3) Å that is typical for tris(pyrazolyl)acetonitrile Cu(I)-complexes.49 The 1H and 13C NMR data are also similar to those of the comparable acetonitrile containing Cu-complexes of Bt-Bu, OCPh,Me and OCt-Bu, but the acetonitrile ligand could not be observed due to a rapid exchange with the CD3CN solvent; the solid material was poorly soluble in common NMR solvents. It is interesting to note that, while the 13C chemical shifts are hardly effected by coordination of Cu, the JC,P coupling constants show typical changes, suggesting an altered electron density in the Pz rings. For [CMe2Cu(NCMe)][PF6], ΔJC3,P = −8.5, ΔJC4,P = 4.3, and ΔJC5,P = 20.4 Hz. Similar changes are observed for all complexes reported here (see the Experimental section). The observed 31P NMR chemical shift at 23.3 ppm is substantially shielded compared to 72.0 ppm for the uncomplexed CMe2 ligand, reflecting the significant influence of Cu-complexation, even though the P1–Cu distance of 3.3176(12) Å is well outside the range of a direct interaction between the two atoms. To ascertain the presence of the acetonitrile ligand, a better soluble complex could be obtained by either anion or ligand exchange. For example, treating [CMe2Cu(NCMe)][PF6] with Na[BArF24] in CH2Cl2 gave in 57% isolated yield colorless [CMe2Cu(NCMe)][BArF24] that readily dissolves in solvents less polar than acetonitrile.
48). Ideally, a head-to-tail Cu(I)-complex with C would result from simply treating a Cu(I) complex with two equivalents of C, one providing the three coordinating pyrazolyl groups and the other the phosphorus apex. However, this approach did not give the desired result when [Cu(NCMe)4][PF6] was treated overnight in THF with the dimethyl substituted ligand CMe2. A colorless solid precipitated, yielding crystals suitable for X-ray diffraction upon recrystallization (CH2Cl2/pentane). The determined molecular structure reveals [CMe2Cu(NCMe)][PF6] (Fig. 4, left), which must have been formed by replacing three acetonitrile ligands of [Cu(NCMe)4][PF6] for one CMe2. Apparently, the phosphorus apex of the second CMe2 is unable to replace the remaining acetonitrile. Reacting CMe2 and the Cu-complex in an equimolar ratio gave the same product in 67.7% isolated yield. In the crystal, the metal complex is located at a general position and has only a very approximate non-crystallographic C3v symmetry. The molecular structure shows the CMe2 ligand to be bound in a tridentate fashion via its pyrazolyl rings with Cu–N bond lengths ranging from 2.071(3) to 2.080(3) Å and N–Cu–N angles ranging from 88.49(11) to 94.52(10)°. The acetonitrile is linearly coordinated to copper with a Cu–N14 bond length of 1.888(3) Å that is typical for tris(pyrazolyl)acetonitrile Cu(I)-complexes.49 The 1H and 13C NMR data are also similar to those of the comparable acetonitrile containing Cu-complexes of Bt-Bu, OCPh,Me and OCt-Bu, but the acetonitrile ligand could not be observed due to a rapid exchange with the CD3CN solvent; the solid material was poorly soluble in common NMR solvents. It is interesting to note that, while the 13C chemical shifts are hardly effected by coordination of Cu, the JC,P coupling constants show typical changes, suggesting an altered electron density in the Pz rings. For [CMe2Cu(NCMe)][PF6], ΔJC3,P = −8.5, ΔJC4,P = 4.3, and ΔJC5,P = 20.4 Hz. Similar changes are observed for all complexes reported here (see the Experimental section). The observed 31P NMR chemical shift at 23.3 ppm is substantially shielded compared to 72.0 ppm for the uncomplexed CMe2 ligand, reflecting the significant influence of Cu-complexation, even though the P1–Cu distance of 3.3176(12) Å is well outside the range of a direct interaction between the two atoms. To ascertain the presence of the acetonitrile ligand, a better soluble complex could be obtained by either anion or ligand exchange. For example, treating [CMe2Cu(NCMe)][PF6] with Na[BArF24] in CH2Cl2 gave in 57% isolated yield colorless [CMe2Cu(NCMe)][BArF24] that readily dissolves in solvents less polar than acetonitrile.
 | ||
| Fig. 4 Displacement ellipsoid plot of [CMe2Cu(NCMe)][PF6] (left) and [CMe2Cu(PPh3)][PF6] (right) drawn at the 50% probability level. The PF6 anions and hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°] for [CMe2Cu(NCMe)][PF6]: Cu1–N21 2.077(3), Cu1–N14 1.888(3), P1–N11 1.713(3), P1⋯Cu1 3.3176(12), N21–Cu1–N22 94.52(10), N14–Cu1–N21 120.02(12), N11–P1–N12 101.63(13), Cu1–N14–C14 176.4(3). Only one of the two independent cations is displayed for [CMe2Cu(PPh3)][PF6], without PF6 anions and CH2Cl2 solvent molecules: Cu1–N12 2.110(2), Cu1–P12 2.1819(8), P11–N11 1.713(3), P11⋯Cu1 3.3479(9), N12–Cu1–N22 89.54(9), P12–Cu1–N12 123.57(7), N11–P11–N21 100.04(12). | ||
Its 31P NMR chemical shift at 20.0 ppm (CDCl3) compares with the complex having PF6− as the counterion (23.3 ppm; CD3CN), while the 1H NMR chemical shift at 2.23 ppm confirms the presence of the acetonitrile ligand.
The loosely bound acetonitrile of [CMe2Cu(NCMe)][PF6] could be exchanged for PPh3 upon stirring an equimolar mixture in CH2Cl2 for one hour. Following workup, the product (81%) showed NMR spectra with features similar to its precursor, but with additional signals in the aromatic region of both 1H and 13C NMR spectra. The 31P NMR spectrum showed in addition to the resonance for the CMe2 apex at δ 21.4 ppm one at δ 6.5 ppm for the new PPh3 ligand. Crystals obtained from CH2Cl2/pentane were suitable for a crystal structure determination by X-ray diffraction. The asymmetric unit showed two independent complexes with two CH2Cl2 molecules. Fig. 4 (right) displays one [CMe2Cu(PPh3)][PF6] complex. Its molecular structure confirms CMe2 as a tridentate ligand with PPh3 completing the distorted tetrahedral coordination around copper. The Cu–N bond lengths range from 2.079(2) to 2.110(2) Å (2.056(2) to 2.103(2) Å in the second independent molecule) with the Cu(1)–P(12) bond being 2.1819(8) Å (2.1692(8) Å in the second independent molecule). A comparison with the molecular structure of [OCMe2Cu(PPh3)][PF6]20 reveals the structural effect of the phosphorus apex. While the two geometries are very similar, the differences are more pronounced around the apex with [CMe2Cu(PPh3)][PF6] having longer P–N bonds (1.710(3)–1.714(3) Å) than its oxidized form (1.673(2)–1.681(2) Å) and a longer P1⋯Cu separation of 3.3479(9) Å (versus 3.2301(5) Å) due to the circa 4° more acute P–N angles.
As for CMe2, treating the more congested ligands CPh and Ct-Bu with [Cu(NCMe)4][PF6] in either a uni- or a bimolecular ratio gives only the acetonitrile adducts [CPhCu(NCMe)][PF6] and [Ct-BuCu(NCMe)][PF6] in 56 and 43% isolated yields, respectively. Their 1H and 13C NMR spectra confirmed a 1:1 ratio of acetonitrile and the scorpion ligand. For both complexes the 31P NMR resonance of the P-apex is shielded by about 15 ppm compared to the free ligands [CPhCu(NCMe)][PF6] δ 46.4, CPhδ 60.2 ppm; [Ct-BuCu(NCMe)][PF6] δ 43.3, Ct-Buδ 58.2 ppm) which is much less than 49 ppm observed for the dimethyl derivative CMe2. This difference is likely due to the methyl groups at the 5-position of CMe2, which shield the P-apex upon κ3-N3 complexation (Fig. 4).
Next, we turned to tris(pyrazolyl)phosphine CH, because its substituted derivatives are apparently too congested around the phosphorus apex for Cu(I) complexation. Reacting CH and [Cu(NCMe)4][PF6] in a 2:1 ratio in CH2Cl2 also gave a colorless solid, isolated in 88% yield, but its spectroscopic properties showed it to differ substantially from the Cu-complexes of the other C ligands. Whereas the 1H and 31P NMR spectra recorded in CD2Cl2 at ambient temperature gave only very broad signals that could not be interpreted, 1H and 13C NMR spectra at 201 K both showed two sets of signals for the pyrazolyl groups in a 1:1 ratio (see ESI p. S-30 and 31†). The 31P NMR spectrum at the same temperature demonstrated the different effects Cu coordination has on both sides of the CH ligand. It displayed two singlets for the complex, and a septet at δ −144.7 ppm (1JP,F = 712 Hz) for the PF6− anion. The phosphorus at δ 39.0 is shielded compared to the apex of the free ligand (δ 61.1 ppm) due to adoption of the paddlewheel conformation required for κ3-N3 coordination. The other phosphorus, at δ 62.0 ppm, is deshielded, as is common for coordinating phosphines. The small change in the chemical shift reflects the modest interaction of this weakly donating phosphorus with the Cu(I) center. The connectivity for this dimeric Cu-complex [(CH)2Cu][PF6] could be confirmed by a single crystal X-ray structure determination at 110 K. The crystal structure contains two independent metal complex molecules. One metal complex is located at an exact, crystallographic threefold axis, the other molecule is located at a general position with an approximate, non-crystallographic C3 symmetry (r.m.s. deviation 0.149 Å, see Fig. 5). Its molecular structure confirms κ3-coordination of the three pyrazolyl groups of one CH ligand to Cu(I), forming Cu–N bonds and N–Cu–N angles that range from 2.035(3) to 2.051(3) Å and 91.25(14) to 92.96(13)°, respectively, and κ1-coordination of the phosphorus apex of the other CH ligand with a Cu–P bond length of 2.0938(11) Å. Interestingly, the pyrazolyl rings of this second ligand are all oriented with their nitrogens toward the metal, with non-bonding Cu⋯N distances in the range 3.403(6) to 3.499(4) Å. All distances around copper are shorter than those for [CMe2Cu(PPh3)][PF6], reflecting reduced steric crowding, while the non-bonding Cu1⋯P1 distance of 3.3201(12) Å is only marginally shorter. Surprisingly, while the solution NMR data are in harmony with the determined crystal structure of [(CH)2Cu][PF6], repetitively recorded solid state 13C and 31P NMR data at 297 K are not. Only one singlet is observed at δ 40.3 ppm for the two phosphorus apices and the 13C NMR spectrum suggests the presence of two sets of signals for the pyrazolyl rings in a 2:1 ratio. This may suggest that both CH ligands are bound to Cu(I) with two pyrazolyl groups, leaving the third one uncoordinated. Density functional calculations at B3PW91/6-31G(d) (LANL2DZ for Cu) showed the C2 symmetric form of the [(CH)2Cu] cation to be only 2.8 kcal mol−1 less stable than the C3 isomer (Fig. 6).50 This energy difference may even be less as the C2 form can be stabilized by π–π stacking in the solid state as has been demonstrated for Ag(I) complexes of tris(pyrazolyl)methane ligands B.25,26 Since both isomers are so close in energy, it may well be that isomerisation has occurred due to the large difference in the measurement temperature (187 K) or due to loss of the co-crystallized solvent during solid state NMR sample preparation. The geometry of the calculated κ3,κ1-N3P isomer corresponds reasonably well with the structures found in the X-ray crystal structure, but the gas phase calculations overestimate all bond lengths around the Cu center; the calculated Cu–N and Cu–P bonds are 2.115 Å and 2.204 Å, respectively, whereas in the crystal structure Cu–N bonds of 2.035(3)–2.051(3) Å and Cu–P bonds of 2.074(2)–2.0938(11) Å are found. It seems fair to conclude that the Cu–P interaction is readily disturbed, which concurs with the low P-donor ability of CH and its inability to form the substituted analogues [(Cx)2Cu][PF6] (x = Me2, Ph, t-Bu).
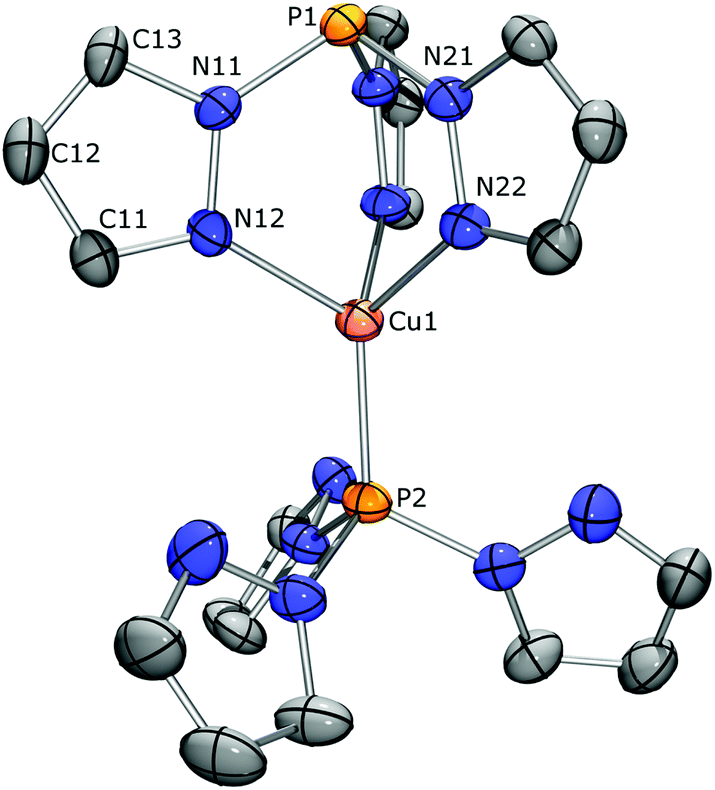 | ||
| Fig. 5 Displacement ellipsoid plot of [(CH)2Cu][PF6] drawn at the 50% probability level. One of the two independent cations is displayed, PF6 anions, disordered solvent molecules, and all hydrogen atoms have been omitted for clarity. Selected bond lengths [Å] and angles [°]: Cu1–N12 2.038(3), Cu1–P2 2.0938(11), P1–N11 1.715(4), P1⋯Cu1 3.3201(12), N12–Cu1–N22 91.25(14), P2–Cu1–N12 123.55(10), N11–P1–N21 99.20(17). | ||
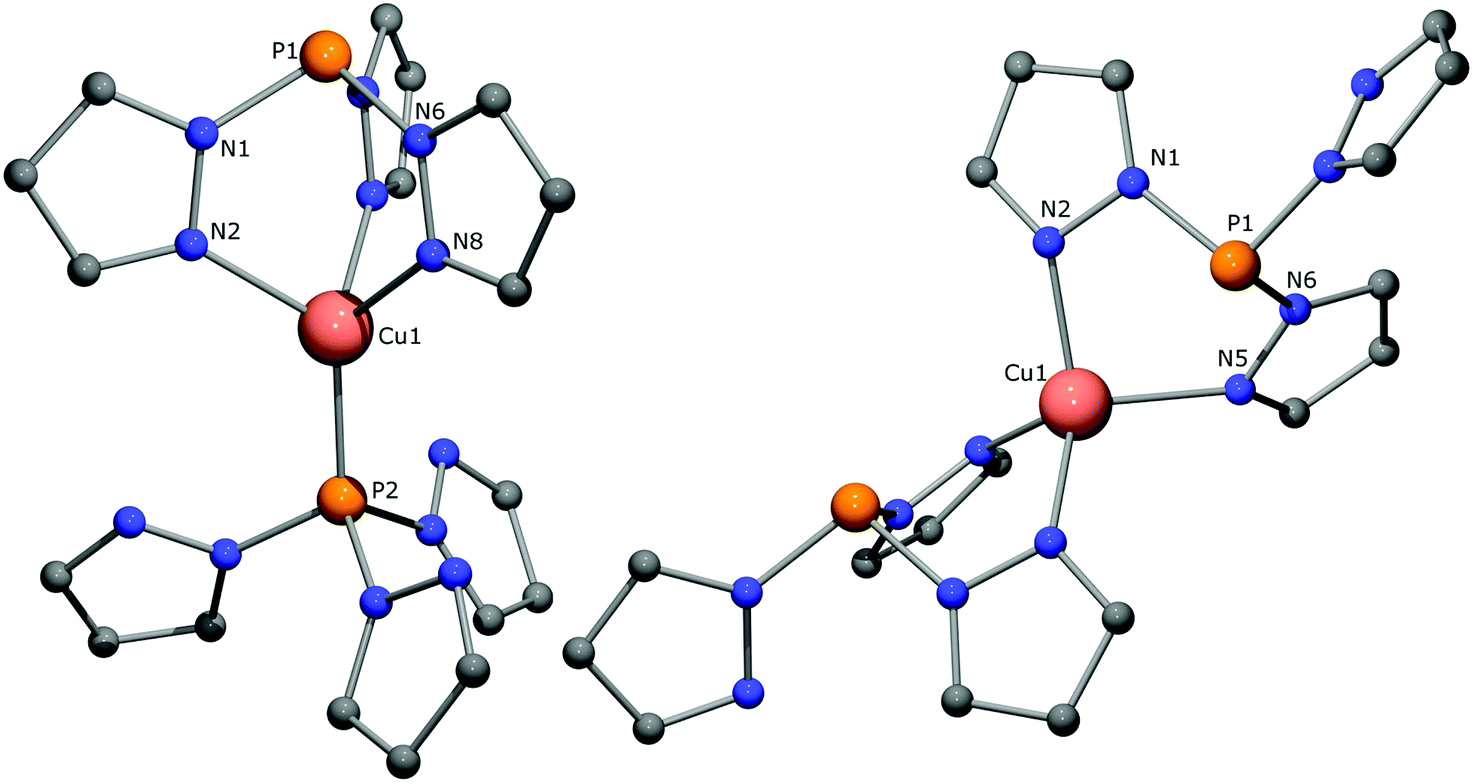 | ||
| Fig. 6 Optimized structures for the κ3,κ1-N3P (left) and κ2,κ2-N4 (right) bound isomers of [(CH)2Cu]+ as calculated at the B3PW91/6-31G(d) (LANL2DZ for Cu) level of theory. All hydrogen atoms have been omitted for clarity. Selected bond lengths [Å] and angles [°] for κ3,κ1-N3P: Cu1–N2 2.115, Cu1–P2 2.204, P1–N1 1.735, P1–Cu1 3.365, N2–Cu1–N8 90.09, P2–Cu1–N2 125.20, N1–P1–N6 100.67. Selected bond lengths [Å] and angles [°] for κ2,κ2-N4: Cu1–N2 2.091, Cu1–N5 2.141, P1–N1 1.756, P1–Cu1 3.053, N2–Cu1–N5 96.80, N1–P1–N6 99.00. | ||
Coordination polymer
With the promising [(CH)2Cu][PF6] result in hand, we were eager to learn whether a one-dimensional coordination polymer ([CHCu][PF6])n could be formed. The logical step was to attempt equimolar mixing of CH and [Cu(NCMe)4][PF6]. In CH2Cl2, this gave a beige suspension from which a solid material was isolated that was insoluble in common NMR solvents like CD2Cl2 and CDCl3. Solid state NMR was employed to identify its nature. 31P MAS NMR spectroscopy revealed a slightly asymmetric (due to residual dipolar interactions) quartet at δ 42.0 ppm (1JP,Cu = 2250 Hz) and a septet for the PF6− anion at δ −145.4 ppm (1JP,F = 713 Hz) (see ESI p. S-33†). The quartet, due to coupling with the two Cu isotopes (I = 3/2), clearly indicates P–Cu coordination. The chemical shift at δ 42.0 ppm suggests a tridentate N-coordinated CH ligand, as found for [(CH)2Cu][PF6] in solution (δ 39.0 ppm) and for [CHCu(PPh3)][PF6] in both solution and the solid state (δ 42.4 and 40.6 ppm, respectively; vide infra). These NMR data are supportive of the formation of a polymeric complex ([CHCu][PF6])n with alternating ligands and Cu(I) centers in which each CH acts on one side as a tridentate N-ligand and on the other side as a monodentate P-ligand. Elemental analysis of the solid matches the proposed stoichiometry (see the Experimental section).The crystal structure determination of ([CHCu][PF6])n turned out to be rather difficult. A needle-shaped crystal obtained from dichloromethane was cracked into two fragments. There is severe disorder in the PF6 anions and co-crystallized solvent molecules. More importantly, we detected “whole molecule” disorder in the one-dimensional Cu coordination polymer. It appears that a chain running in the uvw = [1, −1, 1] direction has very similar packing properties as a chain running in the opposite direction. Least-squares refinement of this crystal structure in the triclinic space group P1 (no. 1) could only be handled by introducing a large number of geometry restraints. Details are given in the ESI.†
A first crystal obtained from 1,2-dichloroethane had a trigonal symmetry and was merohedrally twinned (see the ESI†). Fortunately we were able to obtain a non-twinned crystal with the same unit cell parameters. The crystal structure contains two independent polymeric Cu coordination chains which are located on threefold axes, respectively. Both polymers show the “whole molecule” disorder described above. In one of the chains this disorder could be ignored because of rather low residual electron densities (Fig. 7). In the other chain, a disorder model could be found and refined (Fig. 8). Because of the disorder, it was not possible to obtain reliable bond distances and angles, but the coordination mode could be proofed unambiguously. The two independent one-dimensional coordination polymers are oriented in the direction of the crystallographic c-axis. Both chains have two monomeric units in the crystallographic unit cell, respectively. Consequently, the axis length of c = 10.6155(4) Å leads to an average Cu⋯Cu distance of c/2 = 5.3078(2) Å. The polymeric nature of the structure is also reflected in the shape of the crystal. The c-direction corresponds to the long dimension of the needle shaped crystal (Fig. S4 in the ESI†).
 | ||
| Fig. 7 One-dimensional Cu coordination chain in the crystallographic c-direction of ([CHCu][PF6])n. Only one of the two independent polymers is shown. Location in the unit cell at (0, 0, z), Wyckoff position a. A slight disorder in this chain has been ignored in the structure refinement. In the drawing, hydrogen atoms, PF6 anions and C2H4Cl2 solvent molecules have been omitted for clarity. Symmetry codes: (i) y, x, z − 1/2; (ii) y, x, z + 1/2; (iii) x, y, z + 1. Selected bond lengths [Å] and angles [°]: Cu3–N13 2.034(5), Cu3–P3′ 2.081(2), P3–N23 1.700(4), P3⋯Cu3 3.227(2), N13–Cu3–N13 93.22(17), P3′–Cu3–N13 122.95(12), N23–P3–N23′ 101.8(2). | ||
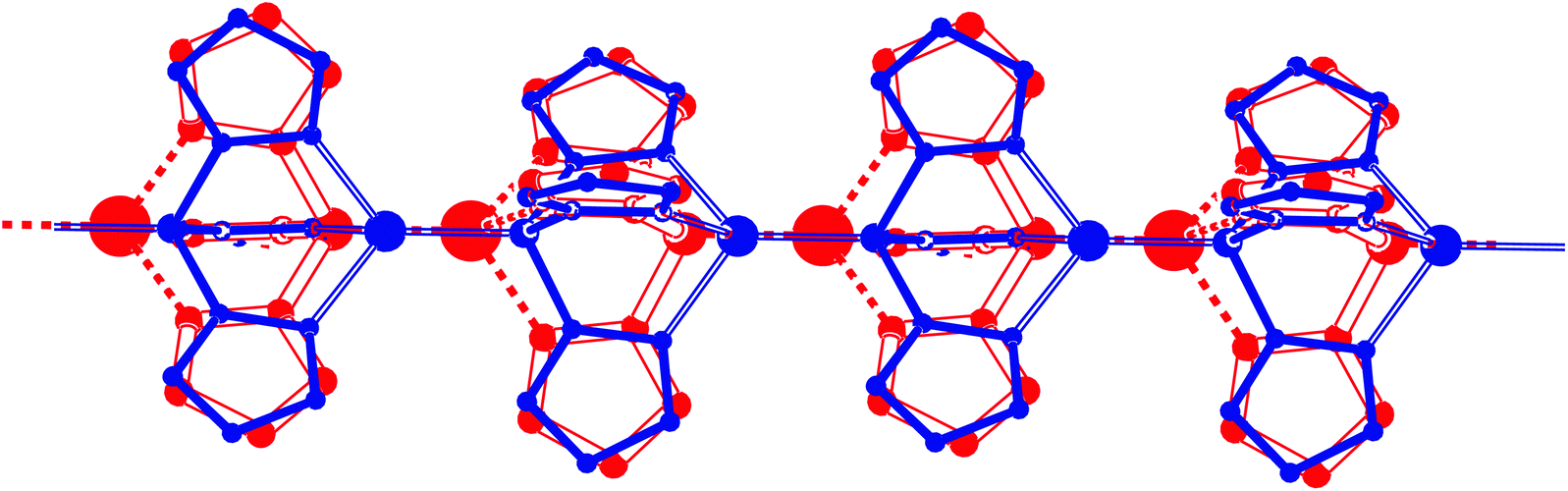 | ||
| Fig. 8 Disorder in the one-dimensional Cu coordination chain of ([CHCu][PF6])n. Location in the unit cell at (2/3, 1/3, z), Wyckoff position b. The major disorder component is drawn in red, the minor component in blue. The estimated ratio between the two components is 65:35%. | ||
The fact that tris(pyrazolyl)phosphine CH but not CMe2 gives access to a Cu(I)-coordination polymer highlights that even modest steric factors can be inhibiting. A similar observation was made by Dougherty et al. who were unable to obtain a polymetallic species from tris-[2-(1-methylimidazolyl)phosphine] and Cu(ClO4)2·6H2O.51 Apparently, methyl groups at 3- and/or 5-positions of the imidazolyl and pyrazolyl groups hamper sufficiently the weak ligation of the copper center to the P-donor site.
Formation of the coordination polymer ([CHCu][PF6])n was further supported by ligand exchange experiments to trap monomeric units (Scheme 3). For example, reaction of the polymer with one equivalent of CH in CH2Cl2 gave a clear solution from which a yellow powder was isolated. This was identified by NMR spectroscopy to be identical to the separately synthesized [(CH)2Cu][PF6] (vide supra), now with an isolated yield of 81%. Likewise, cleaving the polymeric chain with one equivalent of PPh3 in CH2Cl2 gave a 97% isolated yield of [CHCu(PPh3)][PF6]. The 31P NMR spectrum resembles that of [CMe2Cu(PPh3)][PF6] (vide supra) and showed a broad singlet at δ 7.8 ppm for PPh3 bound to Cu(I) and a sharp singlet at δ 42.4 ppm for the P-apex of CH. The 1H NMR spectrum showed a 1:1 stoichiometric ratio for the set of signals for CH and a multiplet for the phenyl hydrogens of PPh3. The 31P MAS NMR spectrum resembled that recorded in solution, but the Cu(I)-coordinated PPh3 appeared as a quartet at δ 14.2 ppm (1JP,63Cu = 1862 Hz), while the P-apex of the CH ligand gives a singlet at δ 40.6 ppm that resembles its chemical shift of δ 42.0 ppm found for the polymer.
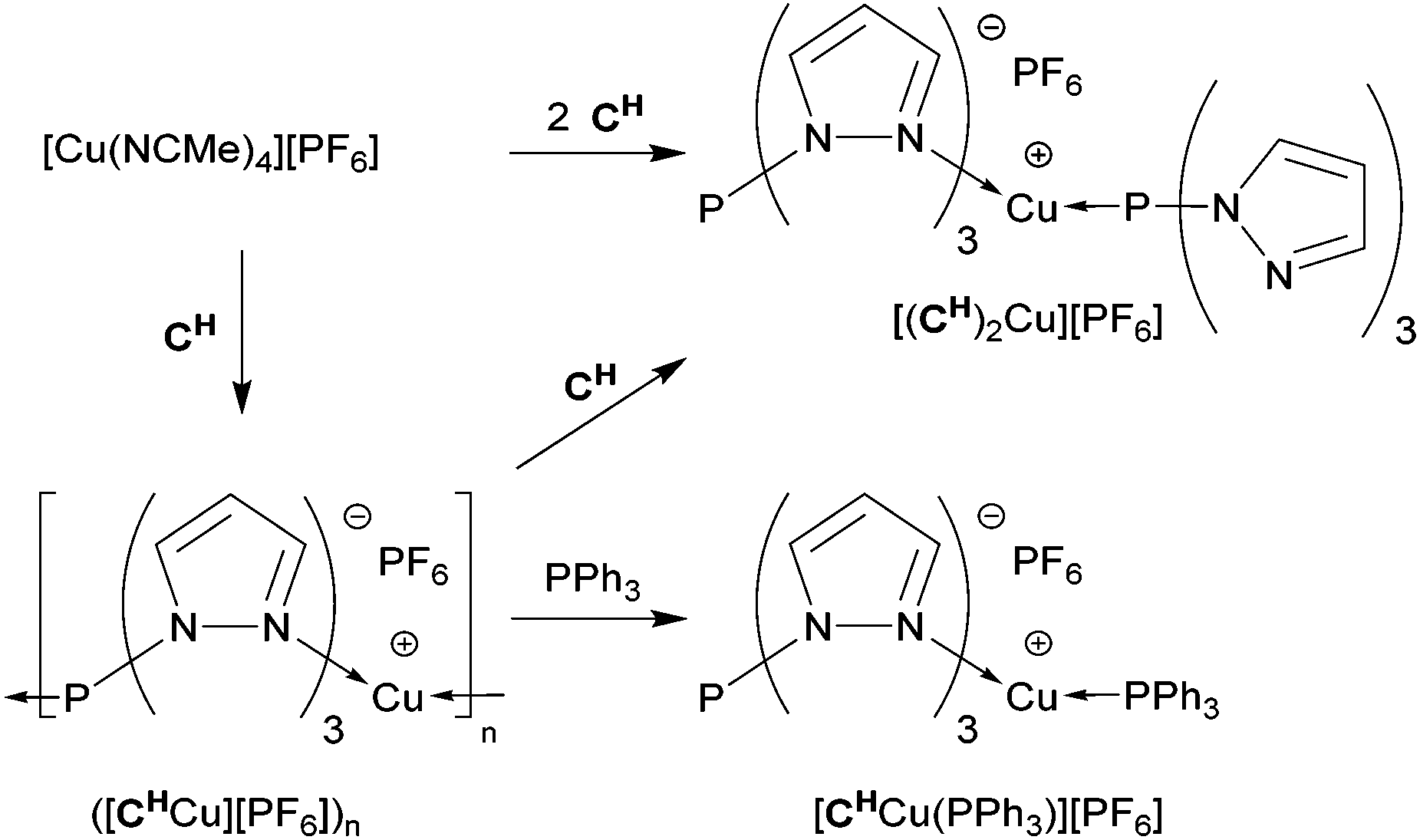 | ||
| Scheme 3 Formation and reactivity of Cu complexes of tris(pyrazolyl)phosphine CH. | ||
Conclusions
We have demonstrated the different binding modes of tris(pyrazolyl)phosphine C. It can bind metals as a tridentate nitrogen donor and as a monodentate phosphine donor. Both binding modes of this Janus-type ligand can be combined when choosing the appropriate steric requirements, making it a suitable building block for coordination polymers. The fact that ligands C are readily available and that a great variety of substituted pyrazoles is already known3,4 make tris(pyrazolyl)phosphines very promising candidates for further studies.Experimental section
General procedures
All experiments were performed under an atmosphere of dry nitrogen. Solvents were purified, dried, and degassed by standard techniques. 3-Phenyl-1H-pyrazole,52 3-tert-butyl-1H-pyrazole,52 [Cu(NCMe)4][PF6],53 and [Na][BArF24]54 were prepared according to literature procedures. Triethyl amine was dried over sodium, and phosphorus trichloride was distilled under nitrogen before use. Other reagents were obtained commercially and used as received.NMR spectra were recorded at 298 K with a Bruker Avance 250, a Bruker Avance 400, or a Bruker Ultrashield 500 spectrometer. 1H and 13C NMR spectra were referenced internally to residual solvent resonances (CDCl3: 1H: 7.26 ppm (CHCl3), 13C{1H}: 77.16 ppm; CD2Cl2: 1H: 5.32 ppm (CDHCl2), 13C{1H}: 53.84 ppm; CD3CN: 1H: 1.94 ppm (CD2HCN), 13C{1H}: 1.32 ppm). Other nuclei were referenced to external standards: 19F: BF3·Et2O (0.0 ppm) and 31P: 85% H3PO4 (0.0 ppm). Most coupling constants in the 1H NMR spectra were determined after applying line narrowing. Solid-state CP-MAS NMR spectra were recorded on a Bruker Avance 400 spectrometer (31P: 161.9 MHz) equipped with a 4 mm MAS probe. Solid samples were prepared in standard ZrO2 rotors. MAS experiments were carried out using spinning speeds between 6 and 15 kHz at 297 K. Cross polarization was applied using a ramp-shaped contact pulse and a mixing time between 3 and 5 ms. 31P chemical shifts were referenced to 85% H3PO4 as the external standard (Ξ = 40.480747 MHz). The given values for 1J(P,Cu) refer to the averaged value of couplings to the two isotopes 63Cu and 65Cu if not specified otherwise. High-resolution electrospray ionization mass spectrometry (HR ESI-MS) was performed using a Bruker MicroTOFQ, with ESI in positive mode (capillary voltage 4.5 kV). Melting points were determined on a Stuart Scientific SMP3 melting point apparatus using sealed capillaries. Elemental analyses were performed at the Microanalytical Laboratory of the Laboratorium für Organische Chemie, ETH Zürich, Switzerland.
:0.3) according to 1H NMR spectroscopy. Further purification of SeCH was not successful.
1H NMR (400.1 MHz, CDCl3): δ 6.51 (ddd, 4JH,P = 2.8 Hz, 3JH,H = 2.8 Hz, 3JH,H = 1.5 Hz, 3H; PzH-4), 7.96 (ddd, 4JH,P = 2.1 Hz, 3JH,H = 1.5 Hz, 4JH,H = 0.5 Hz, 3H; PzH-3), 8.04 (ddd, 3JH,H = 2.8 Hz, 3JH,P = 1.4 Hz, 4JH,H = 0.5 Hz, 3H; PzH-5). 13C{1H} NMR (125.8 MHz, CDCl3): δ 109.7 (d, 3JC,P = 6.9 Hz; PzC-4), 136.6 (d, 2JC,P = 13.9 Hz; PzC-5), 147.4 (d, 3JC,P = 14.9 Hz; PzC-3). 31P{1H} NMR (101.3 MHz, CDCl3): δ 37.7 (s, 1JP,Se = 1014 Hz).
X-ray crystal structure determination
A table containing the experimental details of the crystal structure determination is included in the ESI (ESI p. S-6†).58 against F2 of all reflections based on a HKLF-5 file.59 Non-hydrogen atoms were refined freely with anisotropic displacement parameters. All hydrogen atoms were introduced in calculated positions and refined with a riding model. 306 parameters were refined with no restraints. R1/wR2 [I > 2σ(I)]: 0.0484/0.1198. R1/wR2 [all refl.]: 0.0704/0.1328. S = 1.023. Residual electron density between −0.87 and 1.24 e Å−3. Batch scale factor for the second crystal fragment BASF = 0.518(2). Geometry calculations and checking for higher symmetry were performed with the PLATON program.60
58 against F2 of all reflections. Non-hydrogen atoms were refined freely with anisotropic displacement parameters. All hydrogen atoms were introduced in calculated positions and refined with a riding model. The PF6 anions and CH2Cl2 solvent molecules were refined with disorder models. 1014 parameters were refined with 934 restraints (distances, angles and displacement parameters of the disordered groups). R1/wR2 [I > 2σ(I)]: 0.0481/0.1134. R1/wR2 [all refl.]: 0.0729/0.1250. S = 1.026. Residual electron density between −1.31 and 1.39 e Å−3. Geometry calculations and checking for higher symmetry were performed with the PLATON program.60
529 reflections were measured on a Bruker Kappa ApexII diffractometer with a sealed tube and a Triumph monochromator (λ = 0.71073 Å) at a temperature of 110(2) K up to a resolution of (sin θ/λ)max = 0.65 Å−1. X-ray intensities were integrated with the Saint software.64 Multiscan absorption correction and scaling were performed with SADABS56 (correction range 0.67–0.75). 9095 reflections were unique (Rint = 0.027), of which 8301 were observed [I > 2σ(I)]. The structure was solved with direct methods using the program SHELXS-97.57 Least-squares refinement was performed with SHELXL-201458 against F2 of all reflections. The crystal structure contains large voids (930 Å3 per unit cell) filled with disordered solvent molecules. Their contribution to the structure factors was secured by back-Fourier transformation with the Squeeze routine65 resulting in 338 electrons per unit cell. Non-hydrogen atoms were refined freely with anisotropic displacement parameters. All hydrogen atoms were introduced in calculated positions and refined with a riding model. 481 parameters were refined with 1 restraint (floating origin). R1/wR2 [I > 2σ(I)]: 0.0317/0.0799. R1/wR2 [all refl.]: 0.0377/0.0830. S = 1.050. Flack parameter66x = 0.003(3). Residual electron density between −1.04 and 0.99 e Å−3. Geometry calculations and checking for higher symmetry were performed with the PLATON program.60
58 against F2 of all reflections. One polymeric coordination chain was refined with a disorder model [see the text]. A slight disorder in the other coordination chain has been ignored. Also, the carbon atoms of one dichloroethane solvent molecule were refined with a disorder model. Non-hydrogen atoms were refined freely with anisotropic displacement parameters with the exception of the minor disorder component of the Cu chain which was refined isotropically. All hydrogen atoms were introduced in calculated positions and refined with a riding model. 394 parameters were refined with 208 restraints (floating origin, distances and angles of the disordered groups, displacement parameters of the disordered dichloroethane). R1/wR2 [I > 2σ(I)]: 0.0462/0.1100. R1/wR2 [all refl.]: 0.0627/0.1176. S = 1.030. Refinement as a two-component inversion twin resulted in a Flack parameter66x = 0.03(2). Residual electron density between −0.57 and 2.36 e Å−3 (non-modeled disorder). Geometry calculations and checking for higher symmetry were performed with the PLATON program.60
Acknowledgements
This work was supported by the Council for Chemical Sciences of the Netherlands Organization for Scientific Research (NWO/CW), European Union (Marie Curie ITN SusPhos, Grant Agreement No. 317404), and benefitted from EU COST Action CM1302.Notes and references
- S. Trofimenko, J. Am. Chem. Soc., 1966, 88, 1842–1844 CrossRef CAS.
- S. Trofimenko, Chem. Rev., 1993, 93, 943–980 CrossRef CAS.
- S. Trofimenko, Scorpionates: The Coordination Chemistry of Polypyrazolylborate Ligands, Imperial College Press, London, 1999 Search PubMed.
- C. Pettinari, Scorpionates II: Chelating Borate Ligands, Imperial College Press, London, 2008 Search PubMed.
- V. Gandin, F. Tisato, A. Dolmella, M. Pellei, C. Santini, M. Giorgetti, C. Marzano and M. Porchia, J. Med. Chem., 2014, 57, 4745–4760 CrossRef CAS PubMed.
- H. R. Bigmore, S. C. Lawrence, P. Mountford and C. S. Tredget, Dalton Trans., 2005, 635–651 RSC.
- D. L. Reger, T. C. Grattan, K. J. Brown, C. A. Little, J. J. S. Lamba, A. L. Rheingold and R. D. Sommer, J. Organomet. Chem., 2000, 607, 120–128 CrossRef CAS.
- S. Fischer, L. K. Peterson and J. F. Nixon, Can. J. Chem., 1974, 52, 3981–3985 CrossRef CAS.
- S. Fischer, J. Hoyano and L. K. Peterson, Can. J. Chem., 1976, 54, 2710–2714 CrossRef CAS.
- H. B. Davis, J. K. Hoyano, P. Y. Leung, L. K. Peterson and B. Wolstenholme, Can. J. Chem., 1980, 58, 151–158 CrossRef CAS.
- C. J. Tokar, P. B. Kettler and W. B. Tolman, Organometallics, 1992, 11, 2737–2739 CrossRef CAS.
- D. D. Lecloux and W. B. Tolman, J. Am. Chem. Soc., 1993, 115, 1153–1154 CrossRef CAS.
- D. D. Lecloux, C. J. Tokar, M. Osawa, R. P. Houser, M. C. Keyes and W. B. Tolman, Organometallics, 1994, 13, 2855–2866 CrossRef CAS.
- M. C. Keyes, B. M. Chamberlain, S. A. Caltagirone, J. A. Halfen and W. B. Tolman, Organometallics, 1998, 17, 1984–1992 CrossRef CAS.
- J. J. Weigand, K. O. Feldmann, A. K. C. Echterhoff, A. W. Ehlers and K. Lammertsma, Angew. Chem., Int. Ed., 2010, 49, 6178–6181 CrossRef CAS PubMed.
- K. O. Feldmann, S. Schulz, F. Klotter and J. J. Weigand, ChemSusChem, 2011, 4, 1805–1812 CrossRef CAS PubMed.
- K. O. Feldmann and J. J. Weigand, Angew. Chem., Int. Ed., 2012, 51, 7545–7549 CrossRef CAS PubMed.
- K. O. Feldmann, R. Frohlich and J. J. Weigand, Chem. Commun., 2012, 48, 4296–4298 RSC.
- K. O. Feldmann and J. J. Weigand, J. Am. Chem. Soc., 2012, 134, 15443–15456 CrossRef CAS PubMed.
- C. G. J. Tazelaar, V. Lyaskovskyy, T. van Dijk, D. L. J. Broere, L. A. Kolfschoten, R. O. H. Khiar, M. Lutz, J. C. Slootweg and K. Lammertsma, Organometallics, 2012, 31, 3308–3315 CrossRef CAS.
- A. K. Bartholomew, L. M. Guard, N. Hazari and E. D. Luzik, Aust. J. Chem., 2013, 66, 1455–1458 CrossRef CAS.
- S. G. A. van Assema, C. G. J. Tazelaar, G. B. de Jong, J. H. van Maarseveen, M. Schakel, M. Lutz, A. L. Spek, J. C. Slootweg and K. Lammertsma, Organometallics, 2008, 27, 3210–3215 CrossRef CAS.
- C. G. J. Tazelaar, V. Lyaskovskyy, I. M. van Doorn, X. Schaapkens, M. Lutz, A. W. Ehlers, J. C. Slootweg and K. Lammertsma, Eur. J. Inorg. Chem., 2014, 2014, 1836–1842 CrossRef CAS.
- D. L. Reger, T. D. Wright, R. F. Semeniuc, T. C. Grattan and M. D. Smith, Inorg. Chem., 2001, 40, 6212–6219 CrossRef CAS PubMed.
- D. L. Reger, R. F. Semeniuc and M. D. Smith, Inorg. Chem., 2001, 40, 6545–6546 CrossRef CAS PubMed.
- D. L. Reger, R. F. Semeniuc, I. Silaghi-Dumitrescu and M. D. Smith, Inorg. Chem., 2003, 42, 3751–3764 CrossRef CAS PubMed.
- D. L. Reger, R. F. Semeniuc and M. D. Smith, Inorg. Chem., 2003, 42, 8137–8139 CrossRef CAS PubMed.
- D. L. Reger, R. F. Semeniuc, V. Rassolov and M. D. Smith, Inorg. Chem., 2004, 43, 537–554 CrossRef CAS PubMed.
- D. L. Reger, E. A. Foley, R. F. Semenluc and M. D. Smith, Inorg. Chem., 2007, 46, 11345–11355 CrossRef CAS PubMed.
- D. L. Reger, R. F. Semeniuc and M. D. Smith, Eur. J. Inorg. Chem., 2002, 543–546 CrossRef CAS.
- D. L. Reger, R. F. Semeniuc and M. D. Smith, Dalton Trans., 2003, 285–286 RSC.
- D. L. Reger, R. F. Semeniuc and M. D. Smith, Eur. J. Inorg. Chem., 2003, 3480–3494 CrossRef CAS.
- D. L. Reger, R. F. Semeniuc, C. A. Little and M. D. Smith, Inorg. Chem., 2006, 45, 7758–7769 CrossRef CAS PubMed.
- N. F. Zhao, J. C. Bullinger, M. J. Van Stipdonk, C. L. Stern and D. M. Eichhorn, Inorg. Chem., 2008, 47, 5945–5950 CrossRef CAS PubMed.
- M. J. Lopez-Gomez, N. G. Connelly, M. F. Haddow, A. Hamilton, M. Lusi, U. Baisch and A. G. Orpen, Dalton Trans., 2011, 40, 4647–4659 RSC.
- R. M. Silva, C. Gwengo, S. V. Lindeman, M. D. Smith and J. R. Gardinier, Inorg. Chem., 2006, 45, 10998–11007 CrossRef CAS PubMed.
- J. R. Gardinier, R. M. Silva, C. Gwengo and S. V. Lindeman, Chem. Commun., 2007, 1524–1526 RSC.
- For a review on the chemistry of tripodal Janus ligands see: I. Kuzu, I. Krummenacher, J. Meyer, F. Armbruster and F. Breher, Dalton Trans., 2008, 5836–5865 RSC.
- H. Han, M. Elsmaili and S. A. Johnson, Inorg. Chem., 2006, 45, 7435–7445 CrossRef CAS PubMed.
- R. Raturi, J. Lefebvre, D. B. Leznoff, B. R. McGarvey and S. A. Johnson, Chem. – Eur. J., 2008, 14, 721–730 CrossRef CAS PubMed.
- H. Han and S. A. Johnson, Eur. J. Inorg. Chem., 2008, 471–482 CrossRef CAS.
- D. W. Allen and B. F. Taylor, J. Chem. Soc., Dalton Trans., 1982, 51–54 RSC.
- T. S. Barnard and M. R. Mason, Organometallics, 2001, 20, 206–214 CrossRef CAS.
- P. W. Dyer, J. Fawcett, M. J. Hanton, R. D. W. Kemmitt, R. Padda and N. Singh, Dalton Trans., 2003, 104–113 RSC.
- R. D. Kroshefsky, R. Weiss and J. G. Verkade, Inorg. Chem., 1979, 18, 469–472 CrossRef CAS.
- S. M. Socol and J. G. Verkade, Inorg. Chem., 1984, 23, 3487–3493 CrossRef CAS.
- D. L. Reger, J. E. Collins, A. L. Rheingold and L. M. Liable-Sands, Organometallics, 1996, 15, 2029–2032 CrossRef CAS.
- R. P. Pinnell, C. A. Megerle, S. L. Manatt and P. A. Kroon, J. Am. Chem. Soc., 1973, 95, 977–978 CrossRef CAS.
- Search result (30-03-2015) in the Cambridge Structural Database V5.36: 22 structures identified displaying an average Cu–N distance of 1.881 Å.
- Calculations at the BP86/6-311G(dp) (SDD for Cu) and B3PW91/6-311G(dp)D3 (SDD for Cu) levels of theory result in lower relative stabilities of the κ2,κ2-N4 isomer of 5.0 and 11.0 kcal mol−1 respectively.
- W. G. Dougherty and W. S. Kassel, Inorg. Chim. Acta, 2010, 364, 120–124 CrossRef CAS.
- S. Trofimenko, J. C. Calabrese and J. S. Thompson, Inorg. Chem., 1987, 26, 1507–1514 CrossRef CAS.
- G. J. Kubas, Inorg. Synth., 1990, 28, 68–70 CrossRef CAS.
- N. A. Yakelis and R. G. Bergman, Organometallics, 2005, 24, 3579–3581 CrossRef CAS PubMed.
- A. M. M. Schreurs, X. Y. Xian and L. M. J. Kroon-Batenburg, J. Appl. Crystallogr., 2010, 43, 70–82 CrossRef CAS.
- G. M. Sheldrick, SADABS and TWINABS: Area-Detector Absorption Correction, Universität Göttingen, Göttingen, 1999 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed.
- R. Herbst-Irmer and G. M. Sheldrick, Acta Crystallogr., Sect. B: Struct. Sci., 1998, 54, 443–449 CrossRef.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- Z. Otwinowski and W. Minor, in Methods in Enzymology, ed. J. C. W. Carter and R. M. Sweet, Academic Press, New York, 1997, vol. 276, pp. 307–326 Search PubMed.
- P. T. Beurskens, G. Beurskens, R. d. Gelder, S. Garcia-Granda, R. O. Gould and J. M. M. Smits, The DIRDIF2008 program system, Crystallography Laboratory, University of Nijmegen, Nijmegen, 2008 Search PubMed.
- Derived values do not contain the contribution of the disordered solvent molecules.
- SAINT-Plus, Bruker AXS Inc., Madison, WI, 2001 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 9–18 CrossRef CAS PubMed.
- S. Parsons, H. D. Flack and T. Wagner, Acta Crystallogr., Sect. B: Struct. Sci., 2013, 69, 249–259 CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2015, 71, 3–8 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental data for the crystal structure determination of the other crystals of ([CHCu][PF6])n, NMR spectra, and computational details. CCDC 1427170–1427175. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt03994k |
| This journal is © The Royal Society of Chemistry 2016 |