
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bis-(benzothiazol-2-yl)-amines and their metal amides: a structural comparison in the solid state†‡
David-R.
Dauer
,
Melchior
Flügge
,
Regine
Herbst-Irmer
and
Dietmar
Stalke
*
Institut für Anorganische Chemie, Georg-August-Universität Göttingen, Tammannstraße 4, 37077 Göttingen, Germany. E-mail: dstalke@chemie.uni-goettingen.de
First published on 19th November 2015
Abstract
Within this work, the field of amide ligand platforms for group 13 metal complexation, especially for Al(III) is investigated in a synthetic as well as in a structural comparative approach. Starting from bis-heterocyclo methanides, which mimic the omnipresent nacnac ligand, the next enhancement in this class of ligands includes the exchange of the central methylene bridge by an amine nitrogen atom. With this modification three different sec. amines, each symmetrically substituted, could be synthesised as parent neutral benzothiazole containing ligand systems: (NCSC6H4)2NH (1), (4-MeNCSC6H3)2NH (2) and (4-OMeNCSC6H3)2NH (3). Apart from these compounds also a lithiated species and a row of group 13 metal complexes of the deprotonated ligands could be examined by applying single crystal X-ray diffraction analyses. In this course three new dimethyl aluminium containing complexes [Me2Al{(NCSC6H4)2N}] (4), [Me2Al{(4-MeNCSC6H3)2N}] (5) and [Me2Al{(4-OMeNCSC6H3)2N}·AlMe3] (6) as well as two lithiated compounds [Li{(NCSC6H4)2N}]4 (7), [Li{(4-MeNCSC6H3)2N}]4 (8) were structurally and spectroscopically characterised. A subsequent structural comparison of 1–6 and 8 in the solid state shows that the parent ligand systems prefer a planar cis-trans alignment due to hydrogen bond formation. In contrast to that, the metallated species favour a planar but trans–trans or cis–cis alignment depending on the metal cation.
Introduction
The ubiquitous β-diketiminate ligand (nacnac) is mostly known for its ability to stabilise metal ions in low oxidation states. It enjoys great popularity because the steric and electronic properties can easily be modified by using different substituents at the imine moieties.1 A significant advantage of those nacnac ligand systems is e.g. that in the case of the most wide-spread Dipp-substituted derivative (Dipp = 2,6-diisopropylphenyl) the phenyl entities are twisted nearly perpendicular with respect to the chelating plane of the nacnac backbone. This twisting of the residues causes the coordinated metal ion to be maximally shielded and thus prevented from oligomerisation or electrophilic attacks.2 In previous work a binding motif as in nacnac metal complexes was mimicked by replacing the acyclic imine moieties with fused cycles and subsequent deprotonation resulting in a delocalised six electron containing π-system. Formally the substituents in the backbone of the nacnac ligand are fixed to the imine residue, so that a cyclic heteroaromatic compound is formed. Referring to this, in the early 1990s pyridyl groups were introduced in such kind of ligand systems to generate lithium and group 13 complexes of the monoanionic bis-(pyrid-2-yl)-methanide.3 As a further consequence of tuning the coordination ability and electronic properties of this new ligand class also the bridging atom between the two heteroaromates was switched for a group 15 element, wherein the CH2 group of the parent ligand systems is isoelectronically replaced by an NH, PH or even AsH group.4 Subsequently in the case of the sec. phosphanes the pyridyl moieties were substituted by larger heteroaromates like benzothiazole. This sec. phosphane (NCSC6H4)2PH was the scaffold of several investigations, wherein the corresponding phosphanide anion acts as a Janus head ligand, containing the soft P-centred and the hard N-centred coordination site.5 Furthermore, the corresponding bis-heterocyclo methanides are part of current research.6 To continue those investigations the new ligand systems 1–3 were synthesised and characterised in detail. These amine species represent the lighter homologues of the benzothiazole containing phosphanes by switching the bridging atom to the lighter congener. Similarly they can be compared to the dipyridyl amine, because they just vary in the heterocyclic side arms while the bridging moiety (NH) stays the same (Scheme 1).7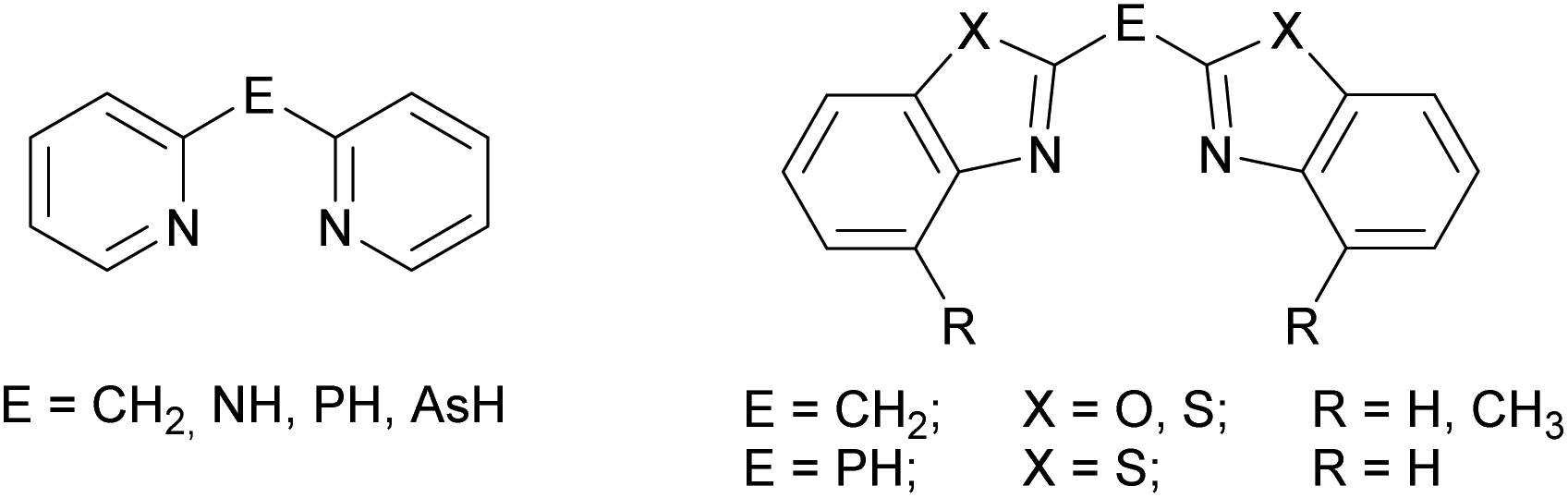 | ||
| Scheme 1 Introduced comparable ligand types. | ||
The derived amides 4–6 (Scheme 5, vide infra) are of special interest regarding their coordination ability, because they offer a magnitude of permutations of suitable donor sites.8 Furthermore, the catalytic behaviour of metal complexes containing related amines/amides as ligands plays a vital role in current research. An iridium complex encumbering a phosphane functionalised (di-) pyridyl amine offers easy N-alkylation of anilines or amino pyridines9 and another iridium complex with 1,3,5-triazine that links between two phosphane functionalised amine moieties acts as a catalyst in pyrrole synthesis.10 Moreover, in addition to the well-known bis-(oxazoline)-methane ligand, which is prone to its activity towards asymmetric catalysis,11 related species were investigated, wherein the bridging carbon atom is replaced by a nitrogen atom. For example bis-(oxazoline)-amines,12 5-aza-semicorrins13 and chiral bis-(2-pyridylimino)-isoindoles should be mentioned in this context.14
Results and discussion
Syntheses of the ligands
First the syntheses of the bis-heterocyclo amines will be discussed with respect to the equi-structural bis-heterocyclo methanes.6 The vital difference between those two ligand systems is the bridging moiety, which links the two identical benzannulated heterocycles. The methylene unit in the first is now replaced by a sec. amine function.Different to the methane derivatives, which were synthesised by cyclocondensation reactions for building up the five-membered heterocycles,15 for the generation of the symmetrically substituted amines 1–3 two equivalents of the 2-aminobenzothiazole, which could further be substituted at the 4-position of the annulated C6-perimeter, in the presence of 1.4 eq. phenol were employed (Scheme 2).16 Thus three different ligands could be synthesised and characterised. X-ray crystallographic studies of the parent ligands enable to quantify their arrangement in the solid state. The molecular structures of the benzothiazole containing compounds 1–3 could be obtained and discussed in detail. Also the synthesis of the benzoxazole containing derivative 9 was achieved by using 2-aminobenzoxazole as starting material, but due to the small yields and poor solubility no crystals suitable for single crystal X-ray diffraction could be grown.
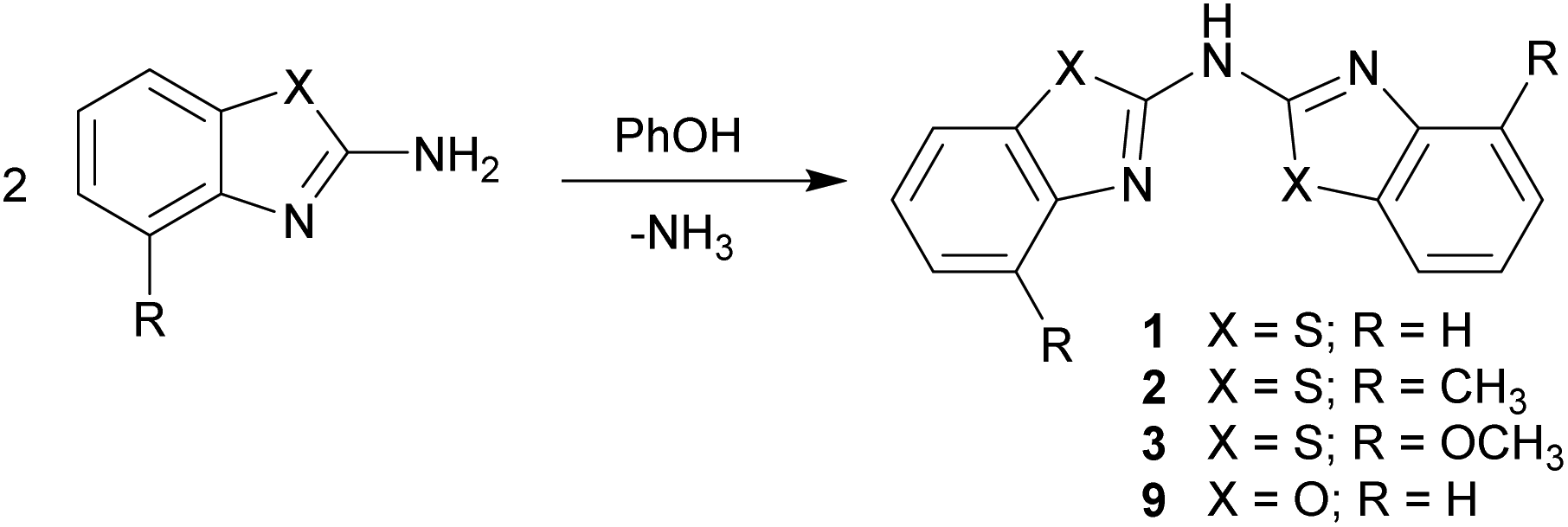 | ||
| Scheme 2 Synthetic route to the parent ligand systems 1–3 and 9. | ||
Structural comparison of 1–3
Compound 1 (Fig. 1 and 2) crystallises in the monoclinic space group P21 and the asymmetric unit contains a single molecule. The methyl substituted parent ligand 2 however (Fig. 3) crystallises in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with two molecules and half a toluene molecule located at an inversion centre in the asymmetric unit, while the methoxy derivative 3 (Fig. 4) crystallises in the monoclinic space group C2/c. The asymmetric unit here consists also of two ligands with a whole co-crystallised toluene molecule (Table 4). Each determined crystal structure of the three related ligand systems 1–3 shows the presence of structure-determining hydrogen bonds between the amine moiety as a hydrogen bond donor and an imine moiety of a neighbouring molecule as a hydrogen acceptor.17 Furthermore, both heteroaromatic residues are oriented to different directions in the solid state. Referring to the hydrogen bonding properties the position of the amine proton is not fixed and varies within the discussed ligand systems (vide infra).
with two molecules and half a toluene molecule located at an inversion centre in the asymmetric unit, while the methoxy derivative 3 (Fig. 4) crystallises in the monoclinic space group C2/c. The asymmetric unit here consists also of two ligands with a whole co-crystallised toluene molecule (Table 4). Each determined crystal structure of the three related ligand systems 1–3 shows the presence of structure-determining hydrogen bonds between the amine moiety as a hydrogen bond donor and an imine moiety of a neighbouring molecule as a hydrogen acceptor.17 Furthermore, both heteroaromatic residues are oriented to different directions in the solid state. Referring to the hydrogen bonding properties the position of the amine proton is not fixed and varies within the discussed ligand systems (vide infra).
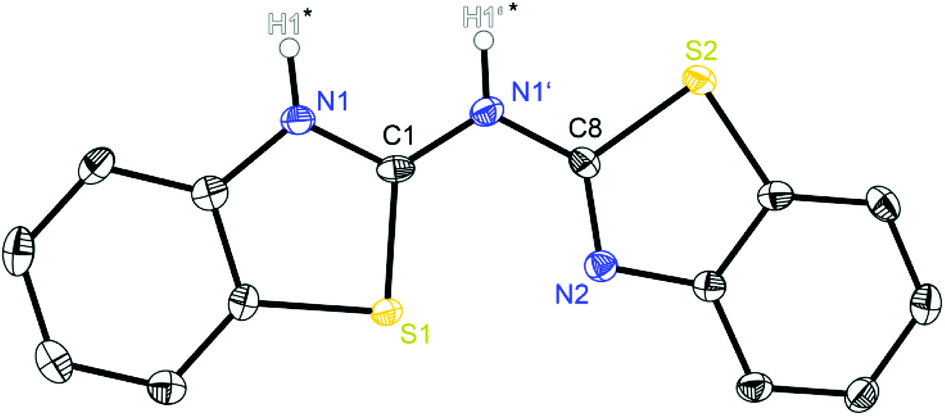 | ||
| Fig. 1 Molecular structure of (NCSC6H4)2NH (1). Anisotropic displacement parameters are depicted at the 50% probability level. C–H hydrogen atoms are omitted for clarity. Both hydrogen atoms of the amine moieties highlighted with a star are only partially occupied due to their positional disorder. Structural data are given in Tables 1–3. | ||
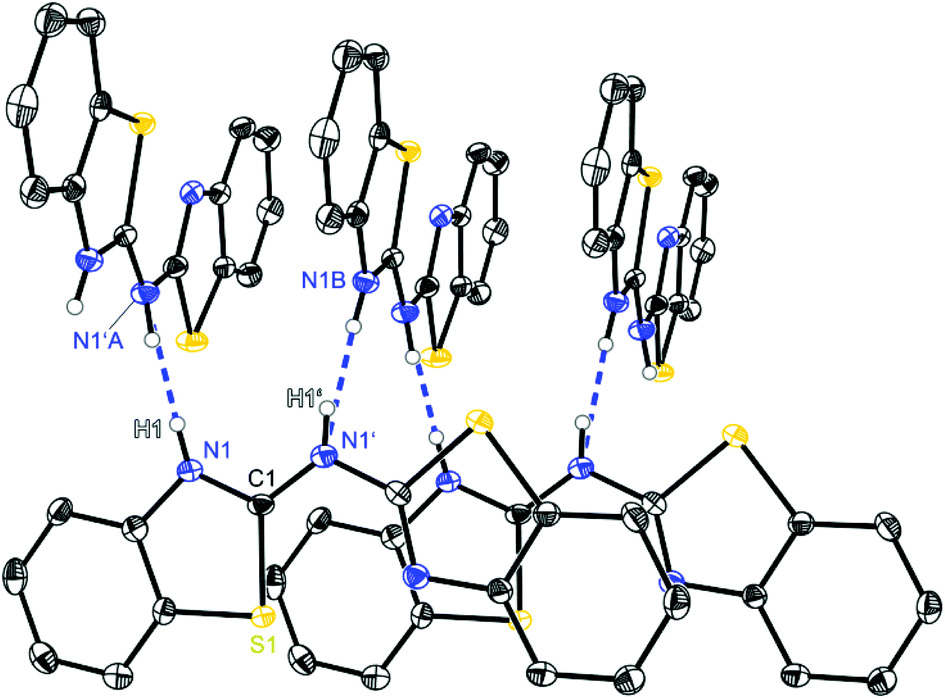 | ||
| Fig. 2 Molecular structure of a hydrogen bridged chain of (NCSC6H3)2NH (1). Anisotropic displacement parameters are depicted at the 50% probability level. C–H hydrogen atoms are omitted for clarity. Structural data are given in Tables 1–3. | ||
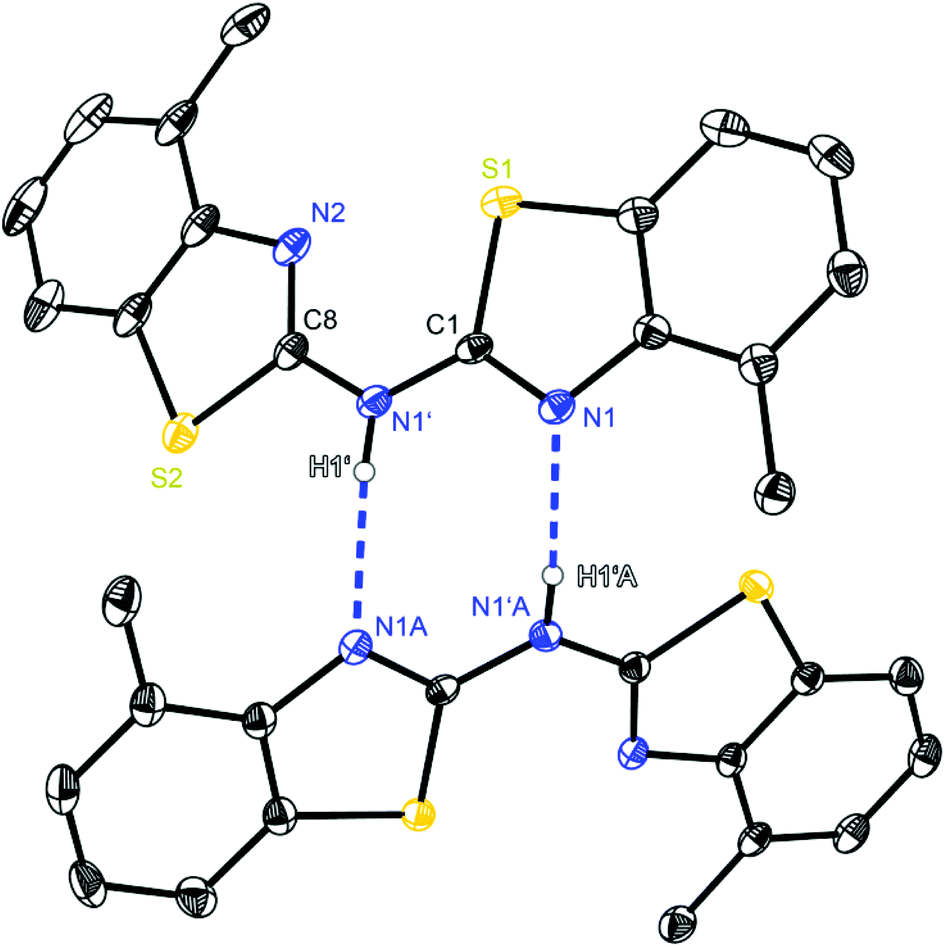 | ||
| Fig. 3 Molecular structure of a hydrogen bridged dimer of (4-MeNCSC6H3)2NH (2). Anisotropic displacement parameters are depicted at the 50% probability level. C–H hydrogen atoms are omitted for clarity. Structural data are given in Tables 1–3. | ||
 | ||
| Fig. 4 Molecular structure of a hydrogen bridged tetramer of (4-OMeNCSC6H3)2NH (3). Anisotropic displacement parameters are depicted at the 50% probability level. C–H hydrogen atoms are omitted for clarity. Structural data are given in Tables 1–3. | ||
All ligands adopt a nearly planar arrangement (Fig. 1–4) because the central trivalent nitrogen atom exhibits a sp2-hybridisation. The angular sum at the bridging nitrogen atom with respect to the ring pivot carbon atoms and the freely refined hydrogen atom position gives 360(2)° in 2 and in 3, which clearly indicates sp2-hybridisation. The angular sum in 1 at N1′ cannot be determined unambiguously due to the positional disorder of the amine hydrogen atom. The positions of the other hydrogen atoms are taken from the difference Fourier map and refined freely with their site occupation factors adding up to 1. Thus three different configurational isomers are conceivable with each benzothiazole substituent inclined to two possible orientations in such a planar setting: cis or trans. Analogue to earlier structural investigations of related ligand systems like di(pyrid-2-yl)-amides and -phosphanides a classification for those three configurational isomers is described in Scheme 3.4c,18 In contrast to this, the related methylene bridged derivatives (NCOC6H4)2CH2,6b (NCSC6H4)2CH2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 6b and (4-MeNCOC6H3)2CH26a do not show a planar arrangement of the heterocyclic residues, but rather the coordination geometry of the bridging carbon atom is distorted tetrahedral.
6b and (4-MeNCOC6H3)2CH26a do not show a planar arrangement of the heterocyclic residues, but rather the coordination geometry of the bridging carbon atom is distorted tetrahedral.
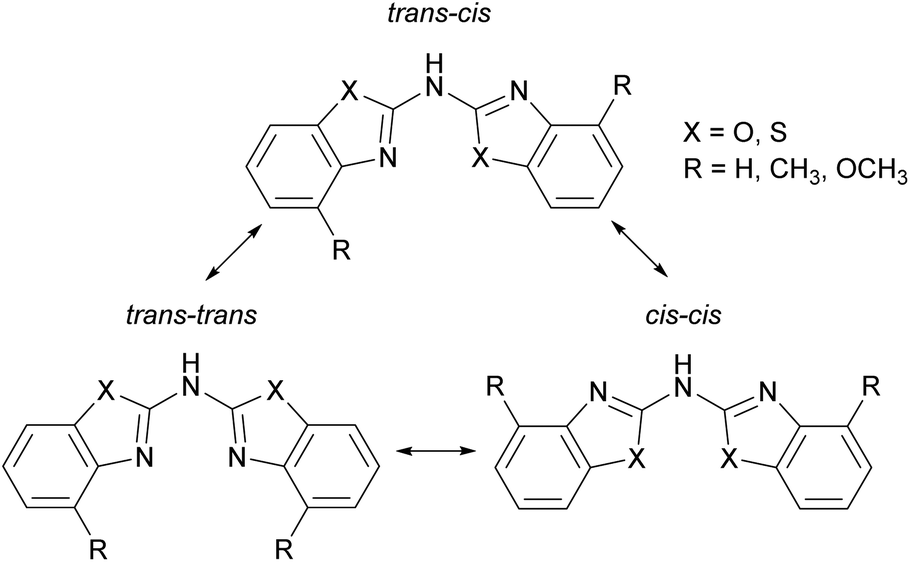 | ||
| Scheme 3 Three possible configuration isomers of the bis-heterocyclic substituted amines 1–3. | ||
The used cis–trans nomenclature distinguishes the arrangement of the highest priority atom, the chalcogen atom, with respect to the partially present Cipso![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Nbridge double bond. The most stable conformation of 1–3 is the one with one heterocycle cis- and the other trans-arranged as deduced from the crystal structures.
Nbridge double bond. The most stable conformation of 1–3 is the one with one heterocycle cis- and the other trans-arranged as deduced from the crystal structures.
Following the description of the different conformers in principal there is the additional option for an imine-enamine-tautomerism, depicted in Scheme 4 for the observed cis–trans arrangement. This implies that three different tautomeric isomers might occur, in which one of them encloses two CipsoNhet double bonds (form b) and both others consist each of one CipsoNhet and one exocyclic CipsoNbridge double bond (forms a and c). These structures determine the position of the amine hydrogen atom. In form b there is a clear separation between the two conjugated, aromatic heterocycles by the bridging NH spacer. In a and c one of the endocyclic CipsoNhet double bonds has tautomerised to an exocyclic CipsoNbridge double bond. Thus the bridging nitrogen atom is left two-coordinated and one heterocyclic N-atom gets protonated to adopt the sec. amine functionality. Indeed, such a tautomerism is already established with heterocyclic substituted sec. phosphanes.4a,19 With a diimine form b and the two imino-enamine forms a and c it was only discussed recently at the well-known ligand platform of bis-(oxazoline)-methane.20
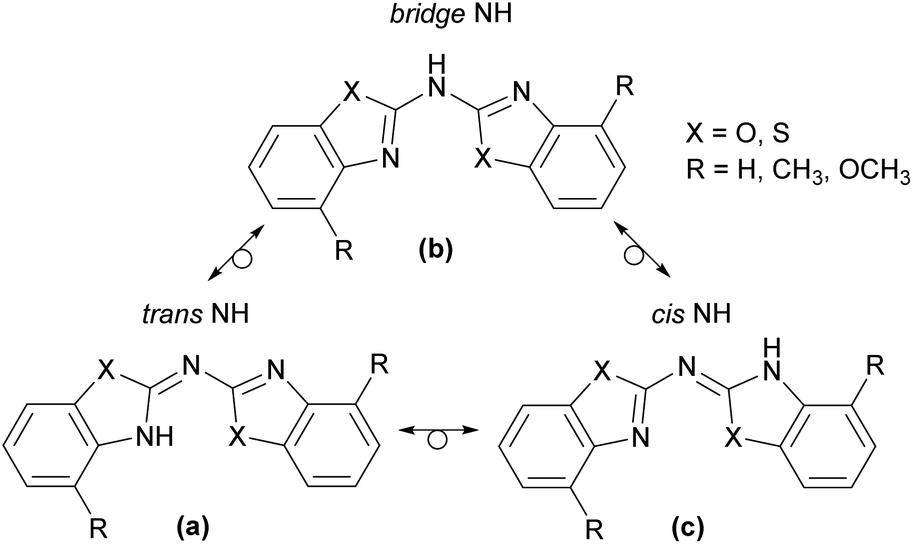 | ||
| Scheme 4 Imine-enamine tautomerism in cis–trans conformers of 1–3. | ||
With regard to the latter cases two possible positions for the NH hydrogen atom can be envisaged, depending on whether the hydrogen atom is bonded to the cis or to the trans aligned part of the molecule. So the trans NH in a is prone to intramolecular hydrogen bonding to the adjacent chalcogen acceptor within the other heterocyclic substituent and the cis NH is more exposed to intermolecular hydrogen bonding to neighbouring molecules in the unit cell.
Considering this conformational and tautomeric freedom, the following decisive observations in the solid state can be made: in any case of the parent protonated ligands 1–3 the cis–trans arrangement is favoured associated with the organic substituents at the annulated benzene perimeters pointing to opposite directions to reduce steric interaction. This cis–trans configuration is also present in most polymorphs known of the dipyridyl amine ligand.7 However, in bis-(benzothiazol-2-yl)-phosphane the trans–trans configuration is preferred in the solid state, where it is shown that the hydrogen atom is located at one nitrogen atom of the benzothiazole unit to form an intramolecular hydrogen bond between both ring nitrogen atoms. The bridging phosphorus remains two-coordinate.5c,d This difference can be explained by the higher Lewis basicity of the nitrogen atoms compared to phosphorus, so that the formed N–H bonds and the resulting N–H⋯N hydrogen bonds are energetically more advantageous.
The crystal structure of 1 in Fig. 1 reveals a significant fraction of the tautomers b and c because the remaining electron density at the nitrogen atoms N1 and N1′ is a clear sign for the presence of a NH hydrogen atom. Due to the positional disorder in the solid state the hydrogen atoms H1′ and H1 can be refined in a ratio of approximately 1:2, indicating that form c is slightly favoured in the solid state. Table 1 shows two different Cipso–Nbridge bond lengths with the C1–N1′ distance to be about 0.03 Å shorter than the C8–N1′ bond (1.336 Å vs. 1.367 Å). Furthermore, the endocyclic Cipso–Nhet bond lengths deviate significantly (C1–N1: 1.331 Å; C8–N2: 1.301 Å). These findings document alternating C–N and CN bonds, with C1N1′ and C8N2 to be double bonds and C1–N1 and C8–N1′ single bonds. This pattern would also explain the tautomeric form c and the deviations of the observed bond lengths from C(sp2)–N(sp2) single (1.40 Å) and C(sp2)N(sp2) double bonds (1.29 Å)21 are due to the presence of the other isomer b.
| Amines | 1 | 2 (av.) | 3 (av.) |
|---|---|---|---|
| C1–N1′ | 1.336(4) | 1.374(2) | 1.363(3) |
| C8–N1′ | 1.367(4) | 1.370(2) | 1.365(2) |
| C1–N1 | 1.331(4) | 1.293(2) | 1.292(2) |
| C8–N2 | 1.301(4) | 1.307(2) | 1.301(2) |
| C1–N1′–C8 | 120.3(2) | 124.89(14) | 125.59(17) |
| Amides | 4 (av.) | 5 | 6 |
|---|---|---|---|
| C1–N1′ | 1.337(3) | 1.332(2) | 1.366(3) |
| C8–N1′ | 1.342(3) | 1.331(2) | 1.365(3) |
| C1–N1 | 1.339(3) | 1.341(2) | 1.334(3) |
| C8–N2 | 1.323(3) | 1.344(2) | 1.328(3) |
| Al1–N1 | 1.929(2) | 1.956(1) | 1.995(2) |
| Al1–N2 | 1.927(2) | 1.967(1) | 1.996(2) |
| Al2–N1′ | — | — | 2.066(2) |
| C1–N1′–C8 | 119.42(19) | 122.97(12) | 118.44(18) |
| N1–Al1–N2 | 91.32(8) | 96.52(5) | 87.75(8) |
In the structures 2 and 3 (Fig. 3 and 4) the amine functionality could only be observed at the bridging position at N1′ indicating the presence of exclusively form b. Additionally the Cipso–Nbridge bond lengths compiled in Table 1 show nearly the same values: 1.374/1.370 Å for 2 and 1.363/1.365 Å for 3. These bond lengths range between a C(sp2)–N(sp2) single (1.40 Å) and C(sp2)N(sp2) double bond length (1.29 Å),21 which indicates some evidence for CN tautomerism.
Interestingly in none of the studied compounds any evidence for form a in Scheme 4 was detected in the solid state. Another remarkable aspect of the amines 1–3 is the option to form different stable N–H⋯N bonded aggregates (Table 2).22
| 1 | 2 (av.) | 3 (av.) | |
|---|---|---|---|
| H1′⋯N1 (Å) | 2.15(5) | 2.08(2) | 2.12(2) |
| H1⋯N1′ (Å) | 2.15(5) | — | — |
| N1⋯N1′ (Å) | 2.910(4) | 2.920(2) | 2.926(2) |
| N1′–H1′⋯N1 (°) | 167(5) | 171.5(2) | 161(2) |
| N1–H1⋯N1′ (°) | 177(6) | — | — |
Starting with bis-(benzothiazol-2-yl)-amine 1 (Fig. 2) infinite chains of the molecules can be observed because each molecule forms two hydrogen bonds to two neighbours. In any case the bridging N1′ of one molecule and the ring nitrogen atom N1 of another form a suitable hydrogen donor–acceptor pair. Due to the positional disorder of the NH hydrogen atom two hydrogen bridges are feasible: either N1–H1⋯N1′ or N1′–H1′⋯N1.
The amine hydrogen atom can easily be switched among N1 and N1′ because of the rather short intramolecular N1/N1′ distance (2.910 Å) and the chain-like formation of hydrogen bridged aggregates.
Focusing on the ligand bis-(4-methylbenzothiazol-2-yl)-amine 2 (Fig. 3), which differs from 1 just by the two additional methyl groups, also hydrogen bonding can be observed. The substitution prunes the aggregation to discrete dimers, only N1′ acting as a hydrogen donor, because in 2 and 3 the amine H-atom is exclusively located at the bridging position. Consequently the ring nitrogen atom N1 of the cis-aligned 4-methylbenzothiazole unit acts as the corresponding hydrogen acceptor of the adjacent molecule. Even the related CH2 linked ligand system (4-MeNCOC6H4)2 reveals the presence of C–H⋯N hydrogen bonds, in which the bridging methylene moiety is acting as hydrogen donor to form a 3D network. The strongest hydrogen bond interactions within the solid state structure of (4-MeNCOC6H4)2 result in a C–H⋯N angle of 171.1° and a H⋯N distance of 2.39 Å.6a
Complementary to the abovementioned ligand the bis-(4-methoxybenzothiazol-2-yl)-amine 3 shows methoxy residues instead of simple methyl groups in 2. Again, a different aggregation in the solid state is present since compound 3 forms a tetrameric hydrogen bonded species (Fig. 4). Within this motif also only N1′ acts as the hydrogen donor. Remarkably the methoxy oxygen atoms are not involved in any hydrogen bonding. Although 3 forms two intermolecular hydrogen bonds with two other molecules like in 1, it does not generate infinite chains but forms a discrete tetramer. This is due to the fact that both adjacent hydrogen bonded molecules are additionally coordinated by another amine for their part, so that four amine derivatives are ordered in a cyclic fashion. In contrast to the other ligand systems this arrangement of hydrogen bonds in 3 is not as favoured as in the other cases, which is confirmed by the disadvantageous D–H⋯A angle and the long H⋯A distance (Table 2).
Syntheses of the aluminium complexes
Analogue to the procedure described earlier for metallated bis-heterocyclo methanides like [Me2Al{(NCOC6H4)2CH}]6b or [Me2Al{Dipp2nacnac}],23 the amines 1–3 were treated with a slight excess of pure trimethyl aluminium at about 0 °C, dissolved in toluene (Scheme 5). After slow addition of the organometallic reagent the reaction mixture was allowed to warm to room temperature and kept stirring over night for optimised conversion.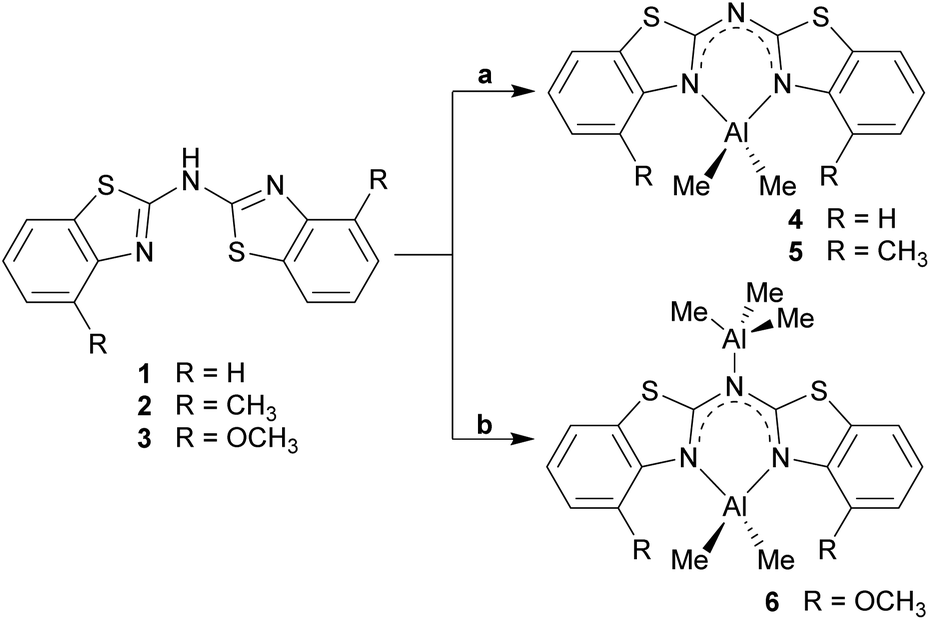 | ||
| Scheme 5 Synthesis of the dimethyl aluminium containing compounds 4–6 (toluene, 0 °C; a: 1.1 eq. AlMe3; b: exc. AlMe3). | ||
Three different bis-heterocyclo amides containing an AlMe2 unit could be synthesised in moderate yields. Scheme 5 shows the reaction of the starting materials with one equivalent AlMe3 to give the monoanionic amide chelating the dimethyl aluminium moiety and with the second equivalent an additionally AlMe3 molecule to be co-coordinated by the bridging amide nitrogen atom. Apart from NMR spectroscopic characterisation in solution also X-ray structure determination was applied to the received single crystals.
Structural comparison of 4–6
At first glance the crystal structures of all three metallated species look almost the same except for the additional AlMe3 coordination in 6. The asymmetric unit of 4 contains two molecules, in 5 just one and in 7 one target molecule and a lattice toluene molecule. In all cases the amine functionality is deprotonated and the organometallic AlMe2 residue in each case is coordinated by the two ring nitrogen donor atoms N1/N2 within the two benzothiazole rings. This chelating ability is comparable to that of the bis-heterocyclo methanides, where formally the bridging nitrogen atom is iso-valence-electronically replaced by a CH-group.6 The main difference of 4–6 (Fig. 5–7) and the corresponding methanides equipped with benzoxazole or benzothiazole moieties is, that rather than the amide atom no further involvement of the bridging CH-functionality in coordination was observed yet. Just one example of this kind of coordination motif was obtained by synthesis of [MeZn{(Py)2CH}]2, Py = pyrid-2-yl. In that dimeric structure the organozinc part is chelated by two ring nitrogen atoms of one ligand as well as coordinated by the bridging carbon atom of the second half of the head-to-tail dimer.24 With compound 6 it was also possible to generate an additional binding site for another metal. This is very advantageous in respect to the Janus head ligands or scorpionates, which offer two different coordination sites and facilitate homo- or heterobimetallic complexes as well.5a–c,e,25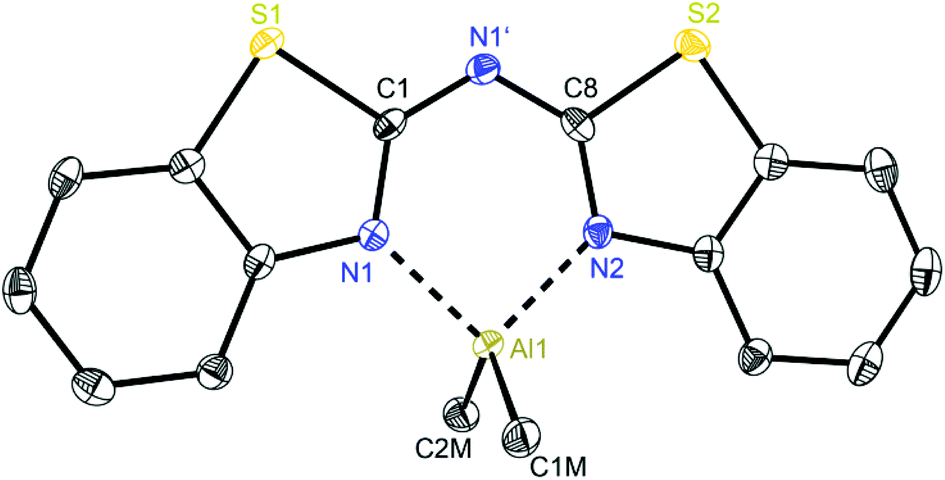 | ||
| Fig. 5 Molecular structure of one molecule of [Me2Al{(NCSC6H4)2N}] (4). Anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms and positional disorder of the whole molecule are omitted for clarity. Structural data are given in Tables 1 and 3. | ||
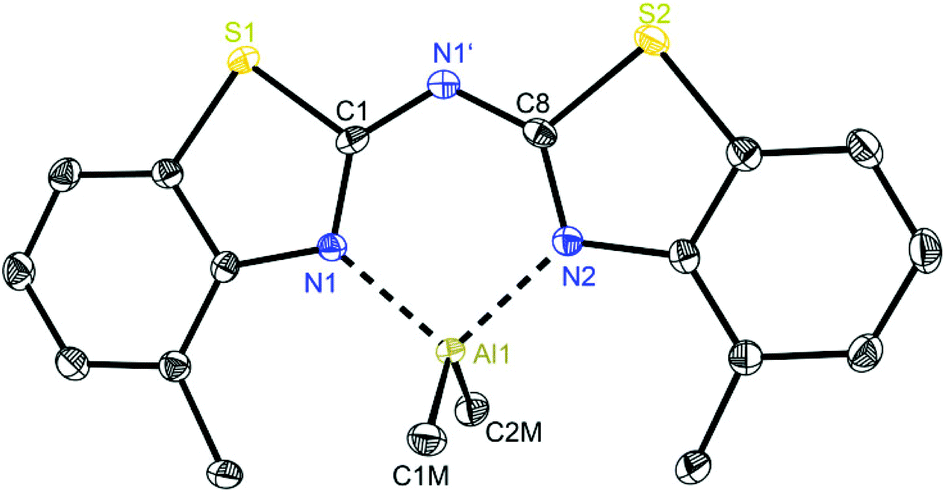 | ||
| Fig. 6 Molecular structure of [Me2Al{(4-MeNCSC6H3)2N}] (5). Anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms are omitted for clarity. Structural data are given in Tables 1 and 3. | ||
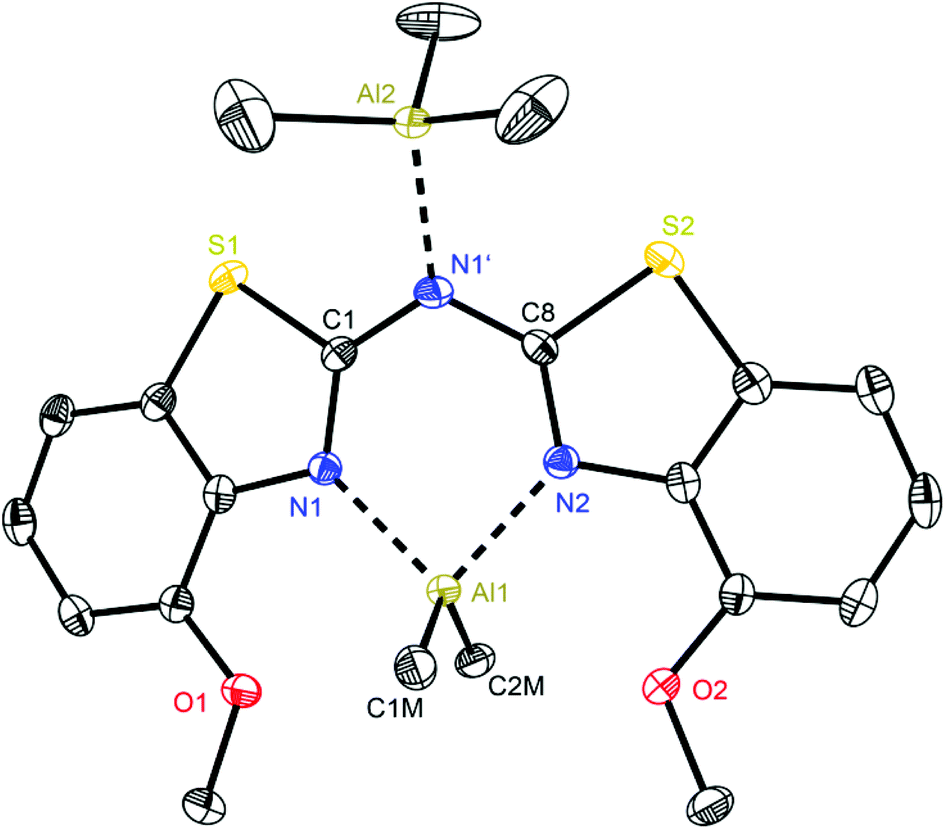 | ||
| Fig. 7 Molecular structure of [Me2Al{(4-OMeNCSC6H3)2N}·AlMe3] (6). Anisotropic displacement parameters are depicted at the 50% probability level. Co-crystallised solvent molecules, hydrogen atoms and positional disorder of the whole molecule are omitted for clarity. Structural data are given in Tables 1 and 3. | ||
In consideration of some paradigmatic bond lengths and angles within the formed six-membered metalla heterocycles, which are displayed in Table 1, some concluding statements can be made:
• From comparison 4 to 5 it is evident, that the C–N bond lengths within the metalla heterocycle cover the narrow range of 1.323 to 1.342 Å for 4 and 1.331 to 1.344 Å for 5.
• The angle in the backbone of the deprotonated ligand system as well as the bite angle of the chelating nitrogen atoms and the aluminium fragment are widened from 4 (119.42° and 91.32°) to 5 (122.97° and 96.52°).
• The deviation of the metal atom from the C2N3 main ligand plane, the folding as well as the torsion angles are decreasing from 4 to 5, indicating that the bis-(4-methylbenzothiazol-2-yl)-amide 5 is most suitable for close coordination of an Al(III) cation (Table 3). According to this also the bis-heterocyclo methanide derivative with additional methyl substituents [(4-MeNCOC6H4)2CH]− shows a distinct coordination preference for Al(III), which is displayed in a small folding angle (3.04 to 4.43°) and just slight deviations of the metal atom from the C3N2 plane (0.009 to 0.048 Å) observed for a series of corresponding aluminium complexes.6a
| Metal dist. from C2N3 plane (Å) | Folding angle (°) | Torsion angle (°) | |
|---|---|---|---|
| a Deviation from the N1–C1–C8–N2 plane. The extent of folding is calculated by measuring the angle between the planes, which are spanned by each heteroaromatic substituent including the amide bridge, and the overall torsion angle is distinguished by the averaged sum of the single torsion angles N1–C1–N1′–C8 and C1–N1′–C8–N2. | |||
| 1 | — | 1.18(10) | 175.1(8) |
| 2 | — | 3.11(6) | 172.0(4) |
| 3 | — | 5.76(7) | 175.0(5) |
| 4 | 0.100(2) | 7.58(17) | 6.4(6) |
| 5 | 0.038(2) | 6.34(4) | 0.9(4) |
| 6 | 0.158(3)a | 25.46(5) | 0.6(7) |
| 1 | 2·0.25 tol | 3·0.5 tol | 4 | 5 | 6·0.42 tol | 8·0.25 tol | |
|---|---|---|---|---|---|---|---|
a
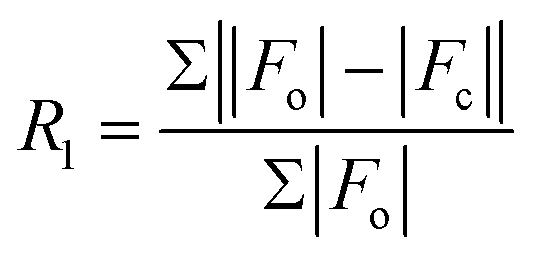 b
b
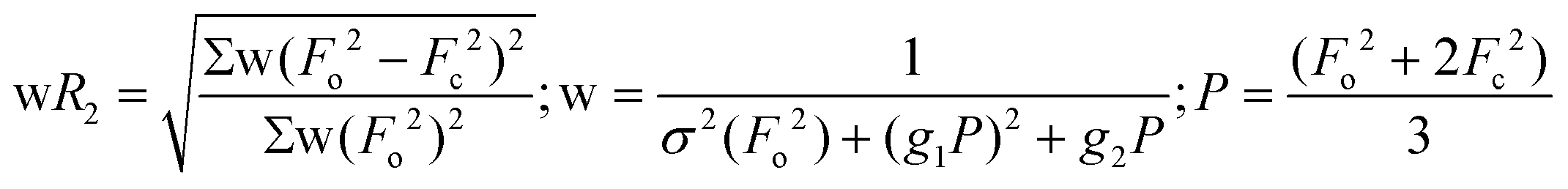
|
|||||||
| Formula | C14H9N3S2 | C16H13N3S2·0.25 C7H8 | C16H13N3O2S2·0.5 C7H8 | C16H14AlN3S2 | C18H18AlN3S2 | C21H27Al2N3O2S2·0.42 C7H8 | C16H12LiN3S2·0.25 C7H8 |
| Mol. w., g mol−1 | 283.37 | 334.45 | 389.48 | 339.40 | 367.47 | 509.92 | 340.38 |
| CCDC no. | 1042655 | 1042656 | 1042657 | 1042658 | 1042659 | 1042660 | 1042661 |
| Wavelength, Å | 0.71073 | 0.71073 | 0.71073 | 0.56086 | 0.56086 | 0.71073 | 0.71073 |
| Crystal system | Monoclinic | Triclinic | Monoclinic | Triclinic | Orthorhombic | Monoclinic | Triclinic |
| Space group | P21 |
P |
C2/c |
P |
Pccn | P21/c |
P |
| a, Å | 10.853(3) | 8.317(2) | 28.107(4) | 8.678(2) | 16.013(3) | 11.136(2) | 14.094(2) |
| b, Å | 4.587(2) | 12.009(3) | 12.581(2) | 14.569(3) | 16.930(3) | 17.335(3) | 16.125(3) |
| c, Å | 12.513(4) | 16.306(4) | 21.980(3) | 14.732(3) | 12.855(2) | 14.122(2) | 16.706(3) |
| α, ° | 90 | 85.55(2) | 90 | 63.90(2) | 90 | 90 | 71.00(3) |
| β, ° | 93.90(2) | 75.83(2) | 112.93(3) | 88.93(3) | 90 | 95.96(2) | 73.41(3) |
| γ, ° | 90 | 89.23(3) | 90 | 74.02(2) | 90 | 90 | 69.20(2) |
| V, Å3 | 621.5(4) | 1574.3(7) | 7158(2) | 1596.6(7) | 3485.0(10) | 2711.4(8) | 3293.3(13) |
| Z | 2 | 4 | 16 | 4 | 8 | 4 | 8 |
| Refl. measured | 14858 |
25733 |
58842 |
38457 |
100934 |
23794 |
38560 |
| Refl. Unique | 2785 | 7109 | 8090 | 7063 | 3588 | 5593 | 9464 |
| R 1 [I > 2σ(I)]a | 0.0309 | 0.0344 | 0.0400 | 0.0349 | 0.0267 | 0.0432 | 0.0571 |
| wR2 (all refl.)b | 0.0801 | 0.0871 | 0.1027 | 0.0800 | 0.0725 | 0.1351 | 0.1491 |
| R int | 0.0240 | 0.0358 | 0.0592 | 0.0280 | 0.0346 | 0.0351 | 0.0862 |
| Abs. stru. para.35 | 0.02(2) | — | — | — | — | — | — |
| Δρfin, e Å−3 | 0.365/−0.281 | 0.366/−0.340 | 0.428/−0.397 | 0.381/−0.234 | 0.370/−0.210 | 0.730/−0.666 | 0.323/−0.317 |
With regard to the crystal structure of 6 several different features have to be considered in addition to the already stated. On the one hand this is a unique structure within the bis-(benzothiazol-2-yl)-methanide and -amide complexes, which contains a second Lewis acidic AlMe3 fragment. However, this motif of an additional Lewis acid base adduct formation was also noticed in [Et2Al{(Py)2N}·AlEt3] from previous work.4b
This additional coordination causes special properties referring to the folding parameters and the bonding situation. The resulting six-membered metalla heterocycle deviates most from the other structures 4 and 5 and the isoelectronic methanide equivalents.6b The lengths of the bridging Cipso–Nbridge bonds stay nearly the same upon metallation (1.363(3) and 1.365(2) Å in the protonated ligand 3vs. 1.366(3) and 1.365(3) Å in 6), whereas the endocyclic Cipso–Nhet bond lengths are elongated (Table 1) due to the loss of electron density by coordination of the electrophilic AlMe2+ cation. By introducing the methoxy moieties to the ligand framework the bite angle of the chelating site N1–Al1–N2 shrinks to 87.8° and as a consequence the Al–N distances are elongated to 1.995(2) Å and among the longest observed in this series of group 13 complexes.
On the other hand it is noticeable, that the central six-membered metalla heterocycle differs most from planarity when compared to 4 and 5. The bridging nitrogen atom N1′ is no longer located within the plane made up from N1, C1, C8 and N2 like in the other structures. N1′ and Al1 are displaced from the C2N2 plane to the same side, whereby even the deviation from that plane is more pronounced for N1′ than for Al1 (0.0211(3) vs. 0.0158(3) Å) to result in a pronounced butterfly-arrangement like in the comparable crystal structure of [Et2Al{(Py)2N}·AlEt3].4b The C1–N1 and C8–N2 bonds are almost aligned parallel, resulting in a torsion angle of only 0.5°, but the folding of the whole framework, which is also indicated by the large displacement of the bridging amide functionality, is very distinct (about 25.5°, Table 3).
These findings confirm the assumption, that the lone pair generated by deprotonation of the parent ligand is not fully delocalised among the other C–N bonds but rather predominantly localised at the central nitrogen atom N1′. This amidic resonance structure a in Scheme 6 seems to suit best this binding situation, because the amide anion is similar to the carbanion sp3-hybridised26 and there is an obvious difference between the bond lengths concerning the endocyclic and exocyclic C–N bonds, whereat the latter are significantly elongated.
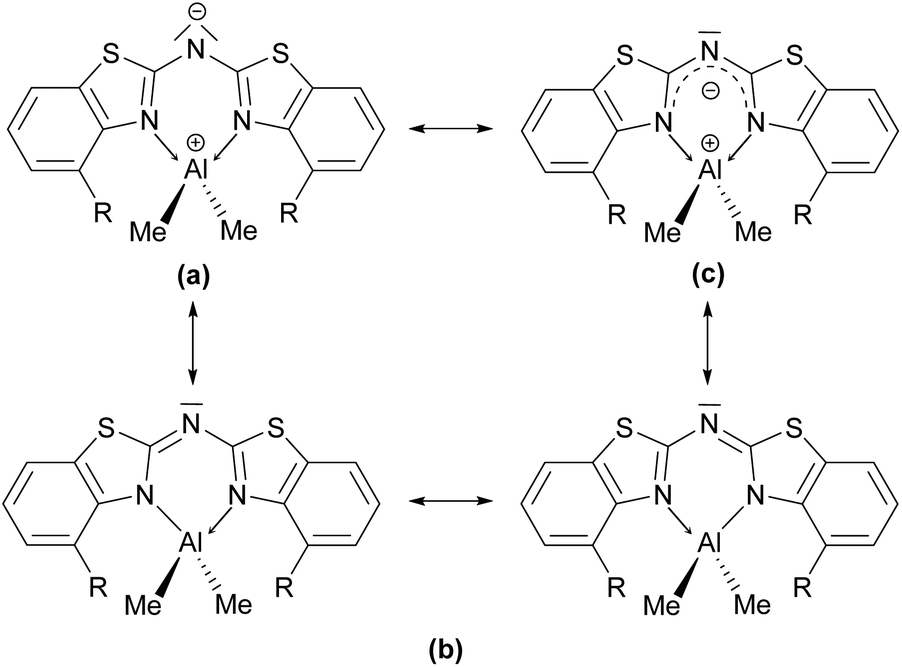 | ||
| Scheme 6 Mesomeric resonance structures of group 13 bis-(benzothiazol-2-yl)-amides 4–6. | ||
The referring 1H-NMR data of 6 show also the presence of the AlMe2 fragment and the additional AlMe3 Lewis acidic group. Furthermore, the apparent cross peak for the 3J-coupling between the protons of the AlMe2 unit and N1 from an 1H–15N-HMBC NMR experiment proves, that the AlMe2 unit is still coordinated by the chelating endocyclic nitrogen donors in solution. The same is valid for the observed 4J-coupling between the aromatic H5 and the same nitrogen donor. Those cross peaks were also detected in the case of the corresponding methanide derivatives.6b However, no interaction of the AlMe3 towards the bridging N1′ was detected suggesting that the Lewis acid base adduct 6 is not maintained in solution.
Lithiated species
As depicted in Scheme 7 the lithiation of the parent ligand system bis-(4-methylbenzothiazol-2-yl)-amine 2 in the absence of Lewis-bases leads to the formation of a tetrameric species. The lithium cations in the centre of 8 are arranged in a distorted tetrahedral fashion, where the distance between the two opposite perpendicularly aligned Li⋯Li edges is considerably elongated. Because of the limited crystal quality of 8 the geometrical features are not discussed in any detail but only presented in a qualitative manner. For 7 no suitable crystals for X-ray diffraction could be obtained, but this species could sufficiently be characterised and identified in solution by NMR spectroscopic techniques.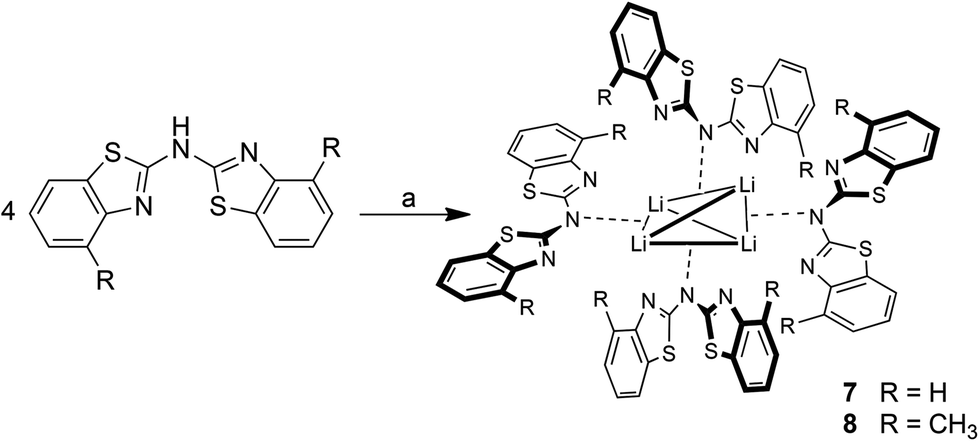 | ||
| Scheme 7 Synthesis of the lithiated compounds 7–8 (a: 4.4 eq. nBuLi, toluene, 0 °C). | ||
Due to the limited solubility of the lithiated compounds in non-donating solvents and for a better comparability of the recorded NMR data deuterated THF was used as appropriate solvent. Presumably this courses the deaggregation of the tetramer 8 in solution, which was obtained upon recrystallisation from non-donating toluene.
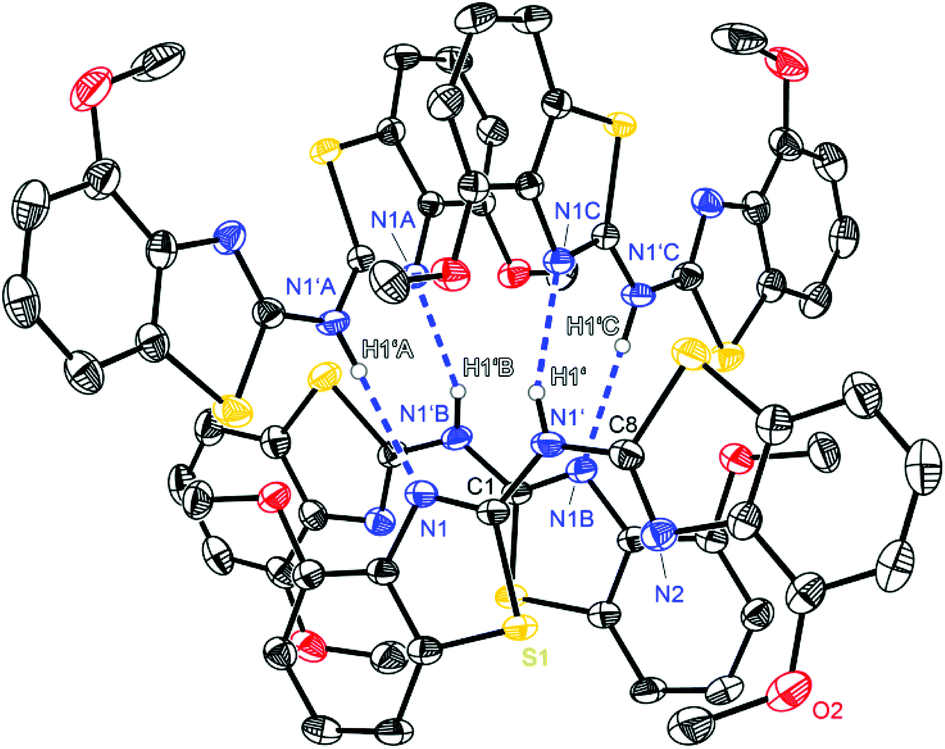
The crystal structure permits to discuss the binding motif in this lithiated species. Interestingly in contrast to all other metallated species 4–6 and the compounds in earlier investigations of the related bis-heterocyclomethanides6b the ligand in 8 has switched to the cis–cis configuration (see also Schemes 3 and 8). In all other cases the trans–trans isomer was favoured for the coordination of the AlMe2 moiety to the two nitrogen donor atoms of the heterocycles. The distorted Li4 tetrahedron (Li⋯Li edges labelled from a–f in Fig. 8) is coordinated by four bis-(4-methylbenzothiazol-2-yl)-amide ligands (labelled from L1–L4), each containing three nitrogen atoms for coordination. The central, bridging nitrogen atoms of each ligand are pointing towards the centre of a Li⋯Li edge, so that over all four (a–d) of the six tetrahedral edges are coordinated (indicated in Fig. 8 as turquoise dashed lines) by one central nitrogen donor atom. The remaining two opposite long edges e and f are addressed by the nitrogen atoms in the periphery of the ligands L3 and L4. No coordination via the sulfur atoms in the backbone is observed.
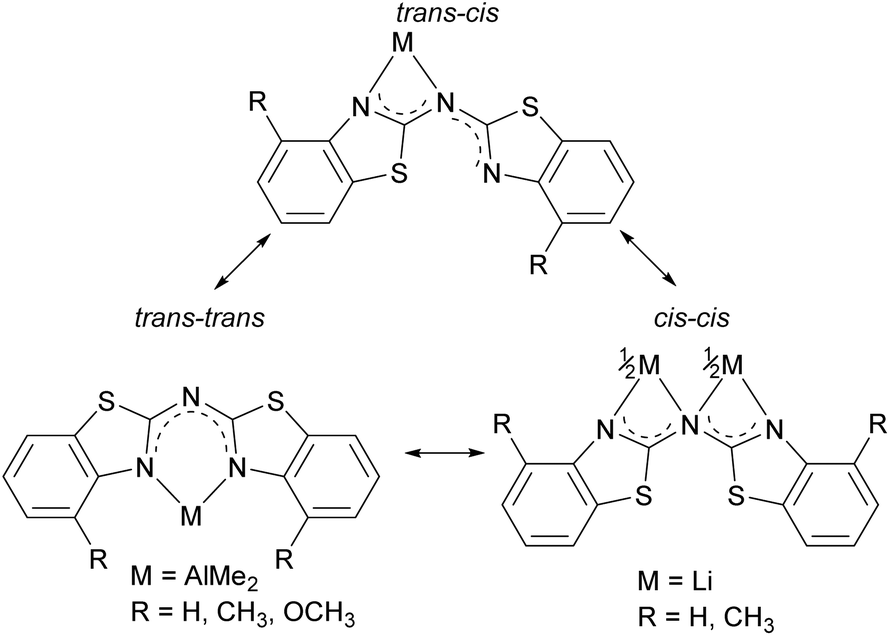 | ||
| Scheme 8 Possible binding motifs upon metallation within 4–8. | ||
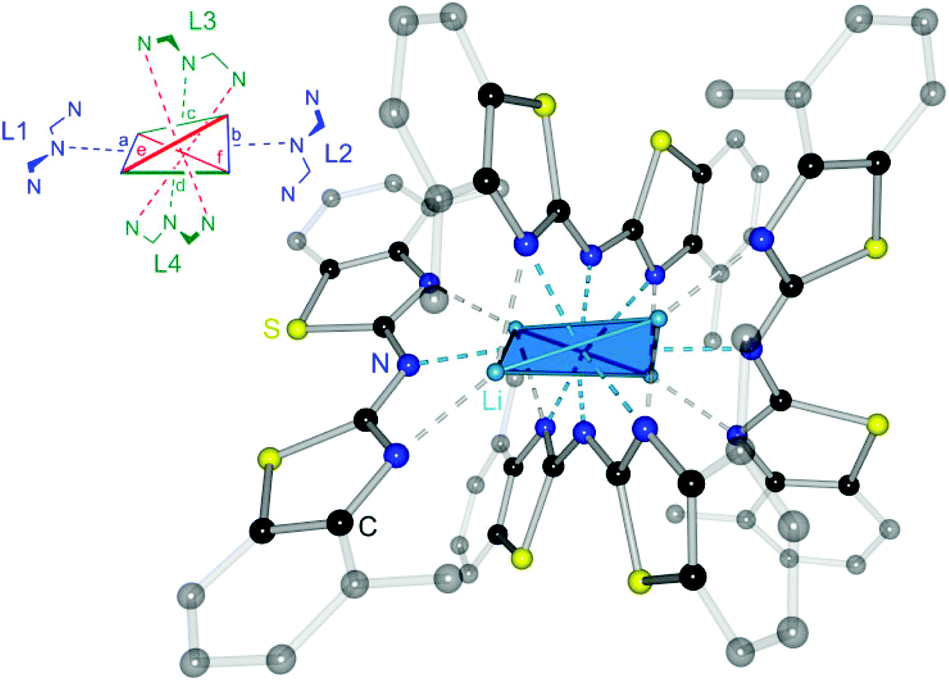 | ||
| Fig. 8 Molecular structure of [Li{(4-MeNCSC6H3)2N}]4 (8). To clarify the binding motif co-crystallised solvent molecules and hydrogen atoms are omitted as well as the annulated moieties are represented in faded colour. In the upper left corner a simplified model of the coordination environment is depicted. | ||
Apart from the coordination of the Li⋯Li edges also the apical lithium cations are addressed by the cis-arranged nitrogen donors of the peripheral benzothiazole rings to gain fourfold coordination (grey dashed lines).
The ligands L1 and L2 show the same coordination behaviour, whereas the central nitrogen atoms are pointing towards the edges a and b, respectively, the neighbouring nitrogen atoms in the heterocycle are pointing to the lithium corners of the same edge. L3 and L4 however, show a different binding motif.
Each central amide moiety also points to the edges c and d, respectively, but the geminal bonded nitrogen donors are oriented to the centres of the other edges e and f. In these cases the thiazole units bridge equally between the edges as well as one of those corners.
So each edge is coordinated by at least one bridging amide (a–d) or two thiazole nitrogen atoms (e and f) and the corners are addressed each by two thiazole nitrogen atoms (one from L1 or L2 and the other one from L3 or L4).
A short comparison to other wide-spread Li4-tetrahedra containing organolithiums27 like [EtLi]4,28 [MeLi]429 or [t-BuLi]430 or to lithium amide rings and ladders31 shows, that in 8 a highly distorted Li4-tetrahedron instead of the expected ring stack or ladder is formed. In contrast to the abovementioned tetrahedra with quite regular Li⋯Li edges (averaged values: [EtLi]4: 2.53 Å; [MeLi]4: 2.59 Å; [t-BuLi]4: 2.41 Å) 8 exhibits four shorter Li⋯Li (2.71(1) to 3.00(1) Å) and two elongated (3.48(1) to 4.13(1) Å) edges.
As depicted in Scheme 8 in total three different binding motifs can occur in the case of the metallated species 4–8. Comparing these motifs to the experimentally determined crystal structures the trans–trans conformation was mostly observed within in the row of the investigated amides as well as methanide derivatives and the cis–cis aligned ligand was just detected once by means of 8. In contrast to this the cis–trans conformation shown at the left hand side could not be observed yet in any bis-(benzothiazol-2-yl)-amide and corresponding methanide.6b Instead the trans–trans conformation facilitates the formation of a six-membered metalla heterocycle in contrast to the cis–cis arrangement, where a higher strained four-membered ring would result. However, this cis–trans arrangement was present in the molecular structure of [Me2Tl{(Py)2N}]∞ due to the larger and easier to polarise cation.4b,c
Conclusions
In summary, three neutral bis-heterocyclo amines 1–3 and the corresponding metal amides 4–8 could be synthesised and characterised by single crystal X-ray diffraction.In contrast to the corresponding methane derivatives the amines 1–3 always form a co-planar arrangement of the heteroaromatic substituents preferentially in the cis–cis conformer with a torsion angle of almost 180°. Especially the formation of different hydrogen bonded aggregates helps to maintain this planarity. An overall comparison of the hydrogen bonding abilities of the investigated systems shows, that the steric demand of the substituent at the C6-perimeter has an increasing influence on the aggregation and the strength of the formed hydrogen bonds. While 1 with the smallest steric demand forms the most linear but also longest hydrogen bonds in a 2D chain-like aggregation motif, the implementation of methyl groups augments the steric demand of the ligand 2 compared to 1. This leads to the formation of a dimeric species, causing an almost linear D–H⋯A angle and the shortest distances between the acceptor and the bridging hydrogen. Further increasing the steric congestion in 3 the methoxy groups force the molecule to divide its hydrogen bonding pattern between to two neighbouring molecules instead of just satisfying one in 2.
The metallated species 4–5 are comparable to the related methanides with the nitrogen atoms of the benzothiazole units chelating in a trans–trans fashion to the AlMe2 moiety (Scheme 8). The experimentally determined values for the C–N bond lengths suggest a delocalised six electron π-system, because they are half way between C(sp2)–N(sp2) single (1.40 Å) and C(sp2)N(sp2) double bonds (1.29 Å). The widening of the backbone and the bite angle from 4–5 can be attributed to the additional strain introduced by the methyl groups in 5 which causes the benzothiazole units to bent away from each other. This evidence is mirrored by the transannular N1⋯N2 distance of 2.764(3) Å for 4 and 2.927(2) Å for 5 and by the N1′⋯Al1 distance (4: 3.334(2) Å; 5: 3.250(2) Å), illustrating the deeper inserted organometallic moiety in 5 compared to 4.
The coordination of the bridging amide to an additional Lewis acid in terms of Al2 in 6 causes the whole ligand framework to fold and hence lowers the orbital overlap of the concerned π-system. This folding can be explained, since N1′ is slightly inclined towards Al2. This distortion leads to an environment of N1′, which is indicating a partially stereochemically active lone-pair at the nitrogen atom. The angular sum around N1′ of about 359° is marginally smaller than for an expected ideal sp2-hybridised nitrogen atom. For further applications of such metal complexes to generate low valent metal fragments the criterion of maximum orbital overlap is vital, because the tentative lone pair at Al(I) from the reduction of the Al(III) can best be stabilised by planar arranged ligand systems.
The lithiated compounds 7 and 8 are different in comparison to the group 13 complexes: by means of the crystal structure of 8 it was possible to determine the ligand arrangement also in the presence of a non-polar, aprotic solvent like toluene. A tetrameric metal complex is formed upon lithiation of 2 in the absence of any Lewis donor base, which shows a clear preference to promote a cis–cis configuration of the amide ligand, never observed in similar systems before. Due to the lack of other donors the active ligand periphery has to suite the lithium tetrahedron in the centre of the molecular structure of 8. As a consequence each available nitrogen donor site of the bis-heterocyclo amides has to be involved in coordination to provide the required electron density to lithium.
Moreover, in each molecular structure the metal coordination only involves the nitrogen atoms, no metal–sulfur interaction could be observed so far. Referring to the HSAB principle32 this fact was expected in this paper, because Al3+ and Li+ cations are regarded hard cations and therefore preferentially coordinate the harder nitrogen rather than the soft sulfur atom.
Experimental section
General procedures
All manipulations were carried out under an atmosphere of N2 or Ar by using Schlenk techniques.33 All solvents used within metallation reactions were distilled from Na or K before use. The starting materials were purchased commercially und used as received. 1H, 13C, 15N and 27Al NMR spectroscopic data were recorded on a Bruker Avance 500 MHz, Bruker Avance 400 MHz and a Bruker Avance 300 MHz spectrometer and referenced to the deuterated solvent (thf-d8).34 Elemental analyses (C, H, N and S) were carried out on a Vario EL3 at the Mikroanalytisches Labor, Institut für Anorganische Chemie, University of Göttingen. Several compounds are containing lattice solvent confirmed by X-ray diffraction, because the crystals were grown in the mother liquor. As a result of drying these samples not the whole amount of incorporated lattice solvent could be removed, so that no solvent free compounds are yielded. The remaining solvent leads to slightly enhanced values in the elemental analyses for C and H. All EI-MS spectra (70 eV) were recorded on a Finnigan MAT 95.Ligand syntheses
In general the four different ligand systems were synthesised by coupling of two equivalents of 1-amino-benzoxazole respectively the corresponding C4-substituted 1-amino-benzothiazoles in the presence of phenol.Metallation reactions
To a solution of the corresponding ligand 1, 2 or 3 (1.00 eq.) in toluene a slight excess of the pure organometallic reactant AlMe3 and nBuLi, respectively, (1.10 eq.) was slowly added at 0 °C. The reaction mixture was stirred overnight and allowed to warm to rt. Afterwards the volume of the solution was reduced to a few mL and the resulting concentrated solution stored at −32 °C in a refrigerator. Overnight crystals suitable for X-ray diffraction experiments could be obtained. The crystals thus formed were filtered, washed twice with pre-cooled toluene or hexane (0 °C) and finally dried in vacuum. The given yields below are just based on the received crystals unless stated otherwise. No further improvement of the yields was attempted because the solutions might have contained impurities upon repeated precipitation.X-ray crystallographic studies
Single crystals were selected from a Schlenk flask under argon or nitrogen atmosphere and covered with perfluorinated polyether oil on a microscope slide, which was cooled with a nitrogen gas flow using the X-TEMP2 device.36 An appropriate crystal was selected using a polarise microscope, mounted on the tip of a MiTeGen©MicroMount or glass fibre, fixed to a goniometer head and shock cooled by the crystal cooling device. The data for 1–8 were collected from shock-cooled crystals at 100(2) K. The data of 1, 2, 6 and 8 were collected on an Incoatec Mo Microsource37 and compounds 4 and 5 were collected on an Incoatec Ag Microsource,38 each equipped with mirror optics and APEX II detector with a D8 goniometer. The data of 3 were measured on a Bruker TXS-Mo rotating anode with mirror optics and APEX II detector with a D8 goniometer. All diffractometers were equipped with a low-temperature device and used either MoKα radiation of λ = 0.71073 Å or AgKα radiation of λ = 0.56086 Å. The data were integrated with Saint39 and an semi-empirical absorption correction (Sadabs)38 was applied. The structures were solved by direct methods (Shelxt)40 and refined by full-matrix least-squares methods against F2 (Shelxl2014).41 All non-hydrogen-atoms were refined with anisotropic displacement parameters. The hydrogen atoms were refined isotropically on calculated positions using a riding model with their Uiso values constrained to equal to 1.5 times the Ueq of their pivot atoms for terminal sp3 carbon atoms and 1.2 times for all other carbon atoms. Disordered moieties were refined using bond lengths restraints and isotropic displacement parameter restraints. The positions of the amine hydrogen atoms are taken from the difference Fourier map and refined freely.Crystallographic data for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre. The CCDC numbers, crystal data and experimental details for the X-ray measurements are listed in the ESI.‡
Conflict of interest
The authors declare no competing financial interest.Acknowledgements
Thanks to the Danish National Research Foundation (DNRF93) funded Center for Materials Crystallography (CMC) for partial support and the Land Niedersachsen for providing a fellowship in the GAUSS PhD program.Notes and references
- L. Bourget-Merle, M. F. Lappert and J. R. Severn, Chem. Rev., 2002, 102, 3031–3065 CrossRef CAS PubMed.
- W. W. Schoeller, Inorg. Chem., 2011, 50, 2629–2633 CrossRef CAS PubMed.
- (a) H. Gornitzka and D. Stalke, Angew. Chem., 1994, 106, 695–698 ( Angew. Chem., Int. Ed. Engl. , 1994 , 33 , 693–695 ) CrossRef CAS; (b) H. Gornitzka and D. Stalke, Organometallics, 1994, 13, 4398–4405 CrossRef CAS.
- (a) M. Pfeiffer, T. Stey, H. Jehle, B. Klüpfel, W. Malisch, V. Chandrasekhar and D. Stalke, Chem. Commun., 2001, 337–338 RSC; (b) M. Pfeiffer, F. Baier, T. Stey, D. Leusser, D. Stalke, B. Engels, D. Moigno and W. Kiefer, J. Mol. Model, 2000, 6, 299–311 CrossRef CAS; (c) H. Gornitzka and D. Stalke, Eur. J. Inorg. Chem., 1998, 1998, 311–317 CrossRef; (d) A. Steiner and D. Stalke, Angew. Chem., Int. Ed. Engl., 1995, 107, 1908–1910 CrossRef; (e) A. Steiner and D. Stalke, Organometallics, 1995, 14, 2422–2429 CrossRef CAS; (f) A. Steiner and D. Stalke, J. Chem. Soc., Chem. Commun., 1993, 444–446 RSC.
- (a) C. Kling, D. Leusser, T. Stey and D. Stalke, Organometallics, 2011, 30, 2461–2463 CrossRef CAS; (b) T. Stey, J. Henn and D. Stalke, Chem. Commun., 2007, 413–415 RSC; (c) T. Stey, M. Pfeiffer, J. Henn, S. K. Pandey and D. Stalke, Chem. – Eur. J., 2007, 13, 3636–3642 CrossRef CAS PubMed; (d) T. Stey and D. Stalke, Z. Anorg. Allg. Chem., 2005, 631, 2931–2936 CrossRef CAS; (e) L. Mahalakshmi and D. Stalke, in Group 13 Chemistry I, ed. H. W. Roesky and D. A. Atwood, Springer, Berlin, Heidelberg, 2002, ch. 3, vol. 103, pp. 85–115 Search PubMed.
- (a) D.-R. Dauer, M. Flügge, R. Herbst-Irmer and D. Stalke, Dalton Trans., 2015 10.1039/C5DT03913D; (b) D.-R. Dauer and D. Stalke, Dalton Trans., 2014, 43, 14432–14439 RSC.
- H. Schödel, C. Näther, H. Bock and F. Butenschön, Acta Crystallogr., Sect. B: Struct. Sci., 1996, 52, 842–853 CrossRef.
- R. Kempe, Angew. Chem., 2000, 112, 478–504 ( Angew. Chem., Int. Ed. , 2000 , 39 , 468–493 ) CrossRef.
- (a) S. Michlik and R. Kempe, Chem. – Eur. J., 2010, 16, 13193–13198 CrossRef CAS PubMed; (b) B. Blank, S. Michlik and R. Kempe, Chem. – Eur. J., 2009, 15, 3790–3799 CrossRef CAS PubMed; (c) B. Blank, S. Michlik and R. Kempe, Adv. Synth. Catal., 2009, 351, 2903–2911 CrossRef CAS.
- S. Michlik and R. Kempe, Nat. Chem., 2013, 5, 140–144 CrossRef CAS PubMed.
- (a) B. D. Ward and L. H. Gade, Chem. Commun., 2012, 48, 10587–10599 RSC; (b) G. Desimoni, G. Faita and K. A. Jørgensen, Chem. Rev., 2011, 111, 284–437 CrossRef PubMed.
- H. Werner, R. Vicha, A. Gissibl and O. Reiser, J. Org. Chem., 2003, 68, 10166–10168 CrossRef CAS PubMed.
- U. Leutenegger, G. Umbricht, C. Fahrni, P. von Matt and A. Pfaltz, Tetrahedron, 1992, 48, 2143–2156 CrossRef CAS.
- (a) D. C. Sauer, R. L. Melen, M. Kruck and L. H. Gade, Eur. J. Inorg. Chem., 2014, 2014, 4715–4725 CrossRef CAS; (b) D. C. Sauer, H. Wadepohl and L. H. Gade, Inorg. Chem., 2012, 51, 12948–12958 CrossRef CAS PubMed; (c) B. K. Langlotz, H. Wadepohl and L. H. Gade, Angew. Chem., 2008, 120, 4748–4752 CrossRef.
- H. Ben Ammar, J. Le Nôtre, M. Salem, M. T. Kaddachi and P. H. Dixneuf, J. Organomet. Chem., 2002, 662, 63–69 CrossRef CAS.
- T. Papenfuhs and A. G. Hoechst, Verfahren zur Herstellung von 2,2-Imino-bis-benzthiazol-Verbindungen, DE29 47 489 A1, Germany, 1979 Search PubMed.
- (a) J. Overgaard and B. B. Iversen, Charge Density Methods, in Hydrogen Bond Studiesed. ed. D. Stalke, Springer, 2012, vol. 146 Search PubMed; (b) E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Pure Appl. Chem., 2011, 83, 1619–1636 CAS; (c) G. R. Desiraju, Acc. Chem. Res., 2002, 35, 565–573 CrossRef CAS PubMed; (d) T. Steiner, Angew. Chem., 2002, 114, 50–80 ( Angew. Chem., Int. Ed. , 2002 , 41 , 48–76 ) CrossRef; (e) T. Steiner, Angew. Chem., Int. Ed., 2002, 114, 50–80 CrossRef; (f) D. Braga, F. Grepioni and G. R. Desiraju, Chem. Rev., 1998, 98, 1375–1406 CrossRef CAS PubMed; (g) G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford University Press, New York, 1997 Search PubMed; (h) G. A. Jeffrey and W. Saenger, Hydrogen Bonding in Biological Structures, Springer, Berlin, 1991 Search PubMed; (i) J. Emsley, Chem. Soc. Rev., 1980, 9, 91–124 RSC.
- M. Pfeiffer, A. Murso, L. Mahalakshmi, D. Moigno, W. Kiefer and D. Stalke, Eur. J. Inorg. Chem., 2002, 2002, 3222–3234 CrossRef.
- J. Hey, D. Leusser, D. Kratzert, H. Fliegl, J. M. Dieterich, R. A. Mata and D. Stalke, Phys. Chem. Chem. Phys., 2013, 15, 20600–20610 RSC.
- A. Walli, S. Dechert and F. Meyer, Eur. J. Org. Chem., 2013, 7044–7049 CrossRef CAS.
- (a) P. Müller, R. Herbst-Irmer, A. L. Spek, T. R. Schneider and M. R. Sawaya, Crystal Structure Refinement A Crystallographer's Guide to SHELXL, Oxford University Press, New York, 2006 Search PubMed; (b) P. Rademacher, Strukturen organischer Moleküle, VCH, Weinheim, 1987 Search PubMed.
- G. R. Desiraju, J. Am. Chem. Soc., 2013, 135, 9952–9967 CrossRef CAS PubMed.
- (a) D. Li, Y. Peng, C. Geng, K. Liu and D. Kong, Dalton Trans., 2013, 42, 11295–11303 RSC; (b) M. Stender, B. E. Eichler, N. J. Hardman, P. P. Power, J. Prust, M. Noltemeyer and H. W. Roesky, Inorg. Chem., 2001, 40, 2794–2799 CrossRef CAS PubMed; (c) B. Qian, D. L. Ward and M. R. Smith, Organometallics, 1998, 17, 3070–3076 CrossRef CAS.
- H. Gornitzka, C. Hemmert, G. Bertrand, M. Pfeiffer and D. Stalke, Organometallics, 2000, 19, 112–114 CrossRef CAS.
- (a) M. M. Meinholz, S. K. Pandey, S. M. Deuerlein and D. Stalke, Dalton Trans., 2011, 40, 1662–1671 RSC; (b) M. M. Meinholz and D. Stalke, Eur. J. Inorg. Chem., 2011, 2011, 4578–4584 CrossRef CAS; (c) A. Murso and D. Stalke, Dalton Trans., 2004, 2563–2569 RSC; (d) F. Baier, Z. Fei, H. Gornitzka, A. Murso, S. Neufeld, M. Pfeiffer, I. Rüdenauer, A. Steiner, T. Stey and D. Stalke, J. Organomet. Chem., 2002, 661, 111–127 CrossRef CAS.
- E. S. Wallis and F. H. Adams, J. Am. Chem. Soc., 1933, 55, 3838–3851 CrossRef CAS.
- (a) D. Stalke and E. Carl, in Lithium Compounds in Organic Synthesis, ed. R. Luisi and V. Capriati, Wiley-VCH, Weinheim, 2014, pp. 1–32 Search PubMed; (b) H. J. Reich, Chem. Rev., 2013, 113, 7130–7178 CrossRef CAS PubMed; (c) V. H. Gessner, C. Däschlein and C. Strohmann, Chem. – Eur. J., 2009, 15, 3320–3334 CrossRef CAS PubMed; (d) D. B. Collum, A. J. McNeil and A. Ramirez, Angew. Chem., 2007, 119, 3060–3077 ( Angew. Chem., Int. Ed. , 2007 , 46 , 3002–3017 ) CrossRef; (e) T. Stey and D. Stalke, in The Chemistry of Organolithium Compounds, Wiley, Chichester, 2004, pp. 47–120 Search PubMed; (f) M. C. Whisler, S. MacNeil, V. Snieckus and P. Beak, Angew. Chem., Int. Ed., 2004, 43, 2206–2225 ( Angew. Chem. , 2004 , 116 , 2256–2276 ) CrossRef CAS PubMed; (g) B. L. Lucht and D. B. Collum, Acc. Chem. Res., 1999, 32, 1035–1042 CrossRef CAS.
- H. Dietrich, J. Organomet. Chem., 1981, 205, 291–299 CrossRef CAS.
- E. Weiss, T. Lambertsen, B. Schubert, J. K. Cockeroft and A. Wiedenmann, Chem. Ber., 1990, 123, 79–81 CrossRef CAS.
- (a) T. Kottke and D. Stalke, Angew. Chem., 1993, 105, 619–621 ( Angew. Chem., Int. Ed. Engl. , 1993 , 32 , 580–582 ) CrossRef CAS; (b) T. Kottke and D. Stalke, Angew. Chem., Int. Ed. Engl., 1993, 105, 619–621 CrossRef CAS.
- (a) R. E. Mulvey, Chem. Soc. Rev., 1998, 27, 339–346 RSC; (b) K. Gregory, P. v. R. Schleyer and R. Snaith, Adv. Inorg. Chem., 1991, 37, 47–142 CrossRef CAS; (c) R. E. Mulvey, Chem. Soc. Rev., 1991, 20, 167–209 RSC; (d) D. R. Armstrong, D. Barr, R. Snaith, W. Clegg, R. E. Mulvey, K. Wade and D. Reed, J. Chem. Soc., Dalton Trans., 1987, 1071–1081 RSC.
- (a) R. G. Pearson, J. Am. Chem. Soc., 1985, 107, 6801–6806 CrossRef CAS; (b) R. G. Parr and R. G. Pearson, J. Am. Chem. Soc., 1983, 105, 7512–7516 CrossRef CAS.
- Georg-August-Universität Göttingen, Virtuelles Labor, http://www.stalke.chemie.uni-goettingen.de/virtuelles_labor/advanced/13_de.html.
- Georg-August-Universität Göttingen, Virtuelles Labor, http://www.stalke.chemie.uni-goettingen.de/virtuelles_labor/nmr/de.html.
- S. Parsons, H. D. Flack and T. Wagner, Acta Crystallogr., Sect. B: Struct. Sci., 2013, 69, 249–259 CAS.
- (a) D. Stalke, Chem. Soc. Rev., 1998, 27, 171–178 RSC; (b) T. Kottke, R. J. Lagow and D. Stalke, J. Appl. Crystallogr., 1996, 29, 465–468 CrossRef CAS; (c) T. Kottke and D. Stalke, J. Appl. Crystallogr., 1993, 26, 615–619 CrossRef; (d) Georg-August-Universität Göttingen, Virtuelles Labor, http://www.stalke.chemie.uni-goettingen.de/virtuelles_labor/special/22_de.html.
- T. Schulz, K. Meindl, D. Leusser, D. Stern, J. Graf, C. Michaelsen, M. Ruf, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2009, 42, 885–891 CrossRef CAS.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10 CAS.
- Bruker AXS Inc., in Bruker Apex CCD, SAINT v8.30C, ed. Bruker AXS Inst. Inc., WI, USA, Madison, 2013 Search PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2015, 71, 3–8 CrossRef PubMed.
- (a) G. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed; (b) G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2014, 70, C1437 Search PubMed; (c) G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
Footnotes |
| † In Memory of Prof. Heinrich Nöth. |
| ‡ Electronic supplementary information (ESI) available: Tables of data collection parameters, bond lengths and angles of compounds 1–6 and 8. CCDC 1042655–1042661. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt03911h |
| This journal is © The Royal Society of Chemistry 2016 |