
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structure and properties of Al-MIL-53-ADP, a breathing MOF based on the aliphatic linker molecule adipic acid†
Helge
Reinsch
*a,
Renjith S.
Pillai
b,
Renée
Siegel
c,
Jürgen
Senker
c,
Alexandra
Lieb
d,
Guillaume
Maurin
b and
Norbert
Stock
*e
aDepartment of Chemistry, University of Oslo, P.O. Box 1033 Blindern, 0315 Oslo, Norway. E-mail: helge.reinsch@smn.uio.no
bInstitut Charles Gerhard Montpellier, UMR-5253 Université Montpellier CNRS ENSCM, Place E. Bataillon 34095, Montpellier cedex 05, France. E-mail: guillaume.maurin@univ-montp2.fr
cInorganic Chemistry III, University Bayreuth, Universitätsstaße. 30, 95447 Bayreuth, Germany
dInstitute of Chemistry, Universitätsplatz 2, 39106, Magdeburg, Germany
eInstitute of Inorganic Chemistry, Christian-Albrechts-Universität Kiel, Max-Eyth-Straße 2, 24118 Kiel, Germany. E-mail: stock@ac.uni-kiel.de
First published on 16th October 2015
Abstract
The new aluminium based metal–organic framework [Al(OH)(O2C–C4H8–CO2)]·H2O denoted as Al-MIL-53-ADP-lp (lp stands for large pore) was synthesised under solvothermal conditions. This solid is an analogue of the archetypical aluminium terephthalate Al-MIL-53 based on the aliphatic single-chain linker molecule adipic acid (H2ADP, hexanedioic acid). In contrast to its aromatic counterparts, Al-MIL-53-ADP exhibits a structural breathing behaviour solely upon dehydration/rehydration. The crystal structure of the anhydrous compound denoted as Al-MIL-53-ADP-np (np stands for narrow pore) was determined by a combination of forcefield-based computations and Rietveld refinement of the powder X-ray diffraction data while the structure of the hydrated form Al-MIL-53-ADP-lp was derived computationally by a combination of force field based methods and Density Functional Theory calculations. Both structures were further supported by 1H, 13C and 27Al high-resolution NMR MAS 1D data coupled again with simulations. Al-MIL-53-ADP was further characterised by means of vibrational spectroscopy, elemental analysis, thermogravimetry and water vapour sorption.
Introduction
Metal–organic frameworks (MOFs) are hybrid porous materials that have been widely studied due to their promises in several fields of applications like gas storage,1 separation of mixtures of gases2 or liquids,3 catalysis4 or sensing.5 Among the variety of reported MOFs a sub-group of materials exhibits a rare structural phenomenon denoted as “breathing”.6 This corresponds to a fully reversible transition between different framework conformations triggered by an external stimulus which leads to large changes of the cell parameters. Such a stimulus is typically given by temperature, external pressure or the presence of certain guest molecules inside the pores of the framework.7 A particularly interesting series of compounds that shows this structural flexibility is the MIL-53-series (MIL stands for Materials of Institute Lavoisier). These solids are built up by the interconnection of infinite chains of trans corner sharing MO6 polyhedra (where M designates a trivalent metal cation) and linear dicarboxylate ions, leading to rhombic channels. The pore dimensions are changed during such a breathing phenomenon and this is mostly affected by a rotation around the O–O-axis of the carboxylate moieties, the so-called knee-cap mechanism.8 Until now several metal ions were proven to form this framework structure like Sc3+,9 V3+,10 Cr3+,11 Fe3+,12 Al3+,13 Ga3+ (ref. 14) and In3+.15 Their structures and properties have been studied under a wide variety of conditions and especially the nature of the linker molecule was shown to have a profound impact on the breathing. Employing functionalised terephthalates for the synthesis of MOFs based on, for example, Fe3+,16 Al3+ (ref. 17) or V3+ (ref. 18) strongly affects the sorption properties and micropore volumes. Moreover the different framework conformations are massively affected by the size of the linker molecules. Extending the linker molecule by employing 2,6-naphthalenedicarboxylic acid or 4,4′-biphenyldicarboxylic acid leads to frameworks which do not show any breathing behaviour, as it was demonstrated for example for Al-19,20 and V-MOFs.21 In a similar manner the use of shorter linker molecules such as fumaric acid was proven to drastically decrease the breathing in an Al-MOF with MIL-53 topology to 3–4% of the cell volume.22 Very few studies of MOFs related to MIL-53 deal with aliphatic linker molecules. Employing trans-1,4-cyclohexanedicarboxylic acid (H2CDC) and In3+ or Cr3+, such materials could be obtained.23,24 However, these compounds were not reported to show any kind of breathing. More recently it was also possible to synthesise the CDC2− based aluminium MOF with MIL-53 structure, denoted as CAU-13 (where CAU stands for Christian-Albrechts-Universität).25 Remarkably it was shown that the knee-cap mechanism is not dominant in the breathing of this framework upon adsorption. Instead, the dimension of the channels is mainly altered due to conformational changes of the cyclohexane ring.26 When water or no guest molecules are present in the pores, half of the linker molecules adopt a,a-conformation and the other half the e,e-conformation. Upon adsorption of xylene, all linker molecules adopt the e,e-conformation and thus the channel diameter is increased from 3.5 to 5 Å. The same effect was observed for Ga3+ based CAU-13.27Herein, we report the aluminium based metal–organic framework [Al(OH)(O2C–C4H8–CO2)] or Al-MIL-53-ADP (ADP stands for adipate) and its breathing behaviour, which is the first example of a MIL-53 framework based on a single-chain saturated aliphatic linker molecule.
Experimental
Materials
All chemicals are commercially available and were used without further purification.Methods
The synthesis was carried out in Pyrex glass bottles with screw cap and a volume of 100 mL. Powder X-ray diffraction (PXRD) data was collected on a STOE Stadi P diffractometer equipped with a linear position sensitive detector (PSD) using monochromated CuKα1 radiation in transmission geometry. All processing of the crystallographic data was done with TOPAS.28 PXRD data depending on humidity was collected on a Bruker PANalytical Empyrean diffractometer (CuKα-radiation). Elemental analysis was measured with an Eurovektor EuroEA Elemental Analyzer. IR-spectra were measured on a Bruker ALPHA-FT-IR A220/D-01 spectrometer equipped with an ATR-unit. The thermogravimetric analyses were recorded using a TA instruments Q500 with a heating rate of 10 K min−1 under oxygen flow. Sorption experiments were performed using a BEL JAPAN INC. Belsorpmax.All solid-state NMR experiments were acquired on Bruker Avance-III HD spectrometers operating at a B0 field of 14.1 T (1H and 27Al) and 9.4 T (13C). 1H (600.15 MHz) high-resolution spectra were acquired after a 90° pulse of 1.4 μs with a spinning speed of 62.5 kHz using a commercial 1.3 mm MAS double resonance probe (Bruker). 13C (100.6 MHz) MAS spectra were obtained with a ramped cross-polarization (CP) experiment where the nutation frequency νnut on the proton channel was varied linearly by 50%. The samples were spun at 12.5 kHz in a 4 mm MAS double resonance probe (Bruker). The corresponding νnut on the 13C channel and the contact time were adjusted to 70 kHz and 3.0 ms, respectively. Proton broadband decoupling with spinal-64 was applied during acquisition. Both the 1H and 13C spectra are referenced with respect to TMS (tetramethylsilane) using the secondary standard adamantane. Finally, the 27Al (156.4 MHz) 1D MAS experiments were acquired using a rotor-synchronized spin-echo with central transition selective 90° and 180° pulses of 12.5 and 25 μs (to remove the resonances from the satellite transitions) at a spinning speed of 20 kHz (3.2 mm MAS triple resonance probe, Bruker). The reference was set using a solution of AlCl3 adjusted to a pH of one with hydrochloric acid. The 27Al spectra were fitted using the simulation package SOLA included in Bruker's Topsin3.2 software.
The force field based-geometry optimizations were conducted using the Forcite software available in the Materials Studio package and the generic Universal Force Field Lennard–Jones parameters for all atoms of the MOF framework.29
The DFT geometry optimizations were performed with the QUICKSTEP module, available as part of the CP2K code using the PBE functional and the triple zeta basis set (TZVP-MOLOPT) for all atoms, except for the Al centres, where double zeta functions (DZVP-MOLOPT) were considered. Semi-empirical dispersion corrections as implemented in the DFT-D3 method were taken into account.30 These calculations considered simulation boxes consisting of (1 × 2 × 2) and (2 × 2 × 2) unit cells for the anhydrous and hydrated phases respectively. The first-principles calculations of the NMR parameters were performed from the structural models for both the large and the narrow pore forms with CASTEP31,32 using the PBE functional33 and ultra-soft pseudo-potentials generated ‘on the fly’.34 The wave functions were expanded on a plane wave basis set with a kinetic energy cut-off of 610 eV. The projector augmented waves (PAW)35 and gauge included projector augmented waves (GIPAW)36 algorithms for the EFGs and NMR chemical shifts, respectively.
Synthesis
Al-MIL-53-ADP was synthesised from a mixture of 10.88 g (16.33 mmol) Al2(SO4)3·18H2O, 2.4 g (16.33 mmol) adipic acid, 21 mL H2O and 9 mL dimethylformamide. The slurry was thoroughly mixed and heated to 130 °C for 12 h. After cooling down to room temperature the raw product was filtered off and thoroughly ground to obtain a fine powder. This powder was redispersed in 50 mL H2O by stirring at room temperature for 1 h and filtrated again. Despite this treatment the product still contains minor impurities of sulphur, probably originating from the sulphate ions in the starting mixture. Thus the elemental analysis detected 32.9% C, 5.1% H and 1.0% S. The values calculated for the fully hydrated form [Al(OH)(O2C–C4H8–CO2)]·H2O are slightly deviating and amount to 34.9% C and 5.3% H. Several attempts for complete removal of the impurities by boiling in water or dimethylformamide with or without stirring at various temperatures did not lower the sulphur content any further. For the PXRD measurements the samples were prepared directly after filtration from aqueous dispersion (Al-MIL-53-ADP-lp) or after heating the MOF at 180 °C in air for three hours (Al-ML-53-ADP-np).Results and discussion
Structure determination
The PXRD pattern of the anhydrous Al-MIL-53-ADP (Fig. 1) exhibits much higher crystallinity than the pattern of the hydrated compound (Fig. S1†) and therefore the structure determination started with this framework conformer. The indexed unit cell shows a monoclinic symmetry (a = 15.828(2) Å, b = 7.947(2) Å, c = 6.611(2) Å, β = 93.78(2)°) with extinction conditions suitable for space group P21. A first model for successful refinement was deduced starting from the structure of the iron pyromellitate MIL-8237 using supergroup/subgroup relationships. This MIL-53-type framework structure was converted from C2/c into the respective subgroups P21/m and P21 using Powdercell.38 The so-obtained structure with lower symmetry was adapted to the indexed cell parameters and the pyromellitate ions were replaced by adipate ions. This structural model was geometry-optimised at the force field level and subsequently refined by Rietveld methods. The final plot is shown in Fig. 1 and relevant parameters are summarised in Table 1. The crystallographic information file and the most relevant bond distances are given in the ESI.† | ||
| Fig. 1 The Rietveld plot for the anhydrous Al-MIL-53-ADP-np. The black line is the experimental data, the red line is the fit and the blue line the difference curve. Vertical bars mark the Bragg reflection positions. | ||
| Ideal formula | [Al(OH)(O2C–C4H8–CO2)] | [Al(OH)(O2C–C4H8–CO2)]·H2O |
| Crystal system | Monoclinic | Triclinic |
| Space group | P21 | P1 |
| a/Å | 15.8741(8) | 6.6289 |
| b/Å | 7.9646(3) | 8.6458 |
| c/Å | 6.6187(3) | 9.0811 |
| α/° | 90 | 62.429 |
| β/° | 93.669(5) | 87.743 |
| γ/° | 90 | 88.372 |
| V/Å3 | 835.1(3) | 461.0 |
| Method | Rietveld refinement | DFT calculation |
| R wp/% | 8.1 | — |
| R Bragg/% | 1.5 | — |
| GoF | 2.2 | — |
For the hydrated form Al-MIL-53-ADP-lp we started with the indexed triclinic cell parameters (a = 6.623(2) Å, b = 8.624(2) Å, c = 9.058(2) Å, α = 117.38(2)°, β = 92.54(1)°, γ = 88.27(2)°) which were obtained after a structure-less cell parameter refinement.
These lattice parameters are similar to the ones reported for CAU-13,25 which contains cyclohexanedicarboxylate ions and also exhibits the MIL-53-type structure. Hence, a ligand replacement strategy was applied to the structure of CAU-13 while the space group was converted from P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) to P1. This structural model without water molecules was then geometry optimized at the force field level by imposing the cell parameters to the indexed values. The resulting structural model could not be refined by Rietveld methods due to a lower crystallinity of this form compared to the anhydrous solid. Hence, this structural model was completed by including one water molecule per formula unit as determined from TGA and water sorption experiments and subsequently optimised by DFT calculations, retaining the indexed cell parameters and taking also the hydrogen atoms into account in order to get a more reliable model. Such an approach has been proven highly valuable for the structure elucidation of complex and weakly ordered materials.22,39 The final optimised model was converted to conventional cell parameter settings (Table 1) and led to a simulated PXRD pattern in reasonable agreement with that observed (Fig. S1†). However, the intensities especially at low 2θ values are much lower than expected for a highly crystalline structure. We attribute this to severe disorder of the H2O guest molecules as well as to the relatively easy conformational changes of the aliphatic linker molecules induced by small rotational movements which require small activation barriers to be overcome. Accounting for this by using the spherical harmonics corrections for an initial fit without any structural refinement gives a reasonable agreement of the calculated and simulated PXRD pattern (see ESI†). The spherical harmonics correction is usually applied for modelling of deviating intensities in case of strong textural effects, however, in this case we attribute these deviations to the pronounced disorder in Al-MIL-53-ADP-lp.
to P1. This structural model without water molecules was then geometry optimized at the force field level by imposing the cell parameters to the indexed values. The resulting structural model could not be refined by Rietveld methods due to a lower crystallinity of this form compared to the anhydrous solid. Hence, this structural model was completed by including one water molecule per formula unit as determined from TGA and water sorption experiments and subsequently optimised by DFT calculations, retaining the indexed cell parameters and taking also the hydrogen atoms into account in order to get a more reliable model. Such an approach has been proven highly valuable for the structure elucidation of complex and weakly ordered materials.22,39 The final optimised model was converted to conventional cell parameter settings (Table 1) and led to a simulated PXRD pattern in reasonable agreement with that observed (Fig. S1†). However, the intensities especially at low 2θ values are much lower than expected for a highly crystalline structure. We attribute this to severe disorder of the H2O guest molecules as well as to the relatively easy conformational changes of the aliphatic linker molecules induced by small rotational movements which require small activation barriers to be overcome. Accounting for this by using the spherical harmonics corrections for an initial fit without any structural refinement gives a reasonable agreement of the calculated and simulated PXRD pattern (see ESI†). The spherical harmonics correction is usually applied for modelling of deviating intensities in case of strong textural effects, however, in this case we attribute these deviations to the pronounced disorder in Al-MIL-53-ADP-lp.
Structure description
The framework structures of Al-MIL-53-ADP-np and Al-MIL-53-ADP-lp are based on the connection of infinite chains of trans corner sharing AlO6-polyhedra via adipate anions (Fig. 2). The flexible adipate molecules adopt conformations that lead to an uncommon “Z”-shape of the one-dimensional pores. Taking the H-atoms of the linker molecules into account, which were not localised during the Rietveld refinement, Al-MIL-53-ADP-np must be considered as a dense structure. The packing of these channels is substantially different compared to the ones observed in most other MIL-53-type structures.13,19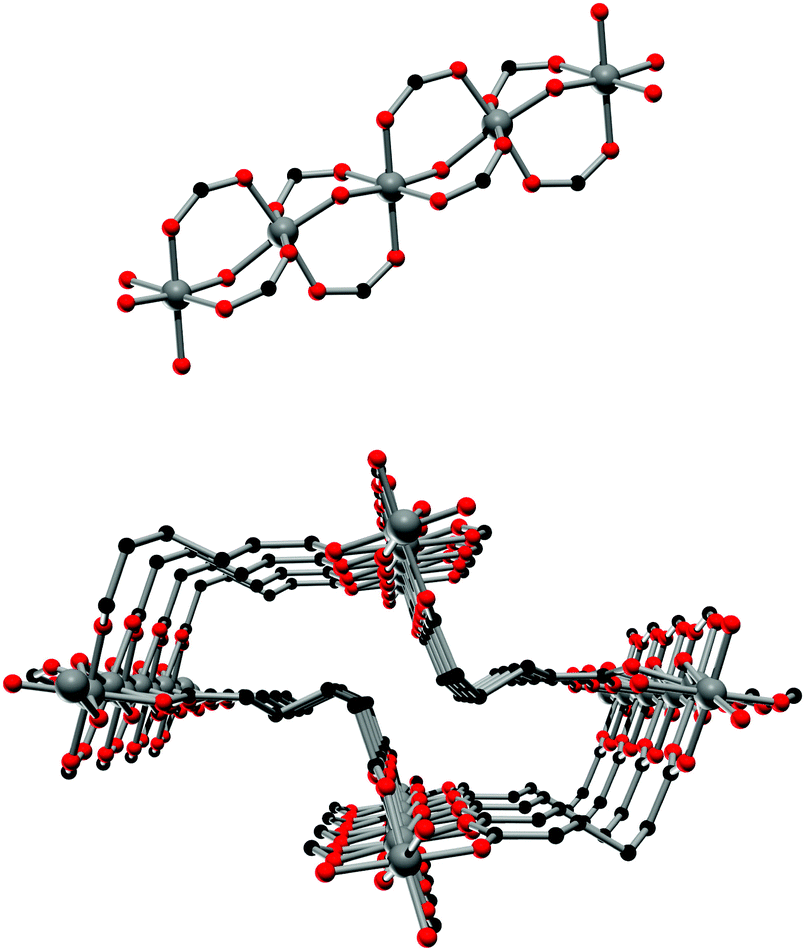 | ||
| Fig. 2 Top: The infinite chain of trans corner sharing AlO6-polyhedra. Bottom: one channel in the structure of Al-MIL-53-ADP-np as seen along the c-axis, showing also the conformation of the adipate linker molecules. Aluminium atoms in grey, oxygen atoms in red and carbon atoms in black. | ||
For compounds based on aromatic linker molecules, the channels exhibit rectangular shape and opposite channel walls that are strictly parallel to each other. The shape of the channels in Al-MIL-53-ADP-np rather resembles a “Z” and thus the observed packing does not show parallel opposite channel walls (Fig. 3).
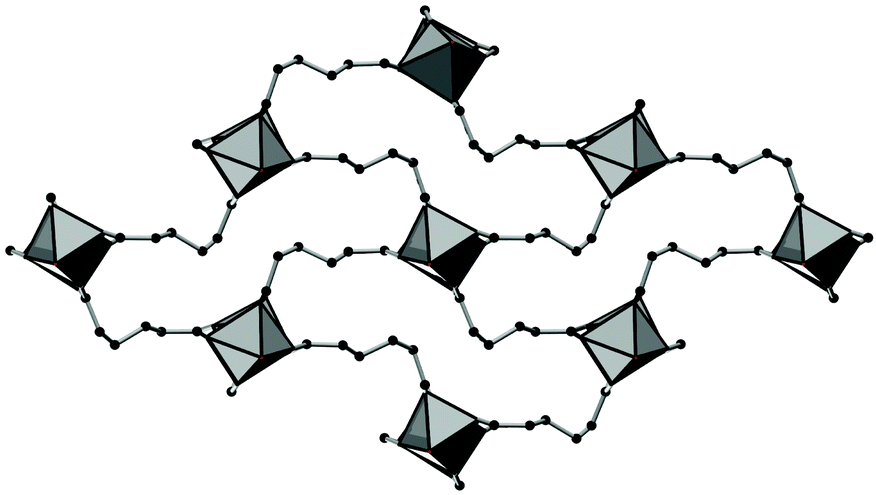 | ||
| Fig. 3 The structure of Al-MIL-53-ADP-np as seen along the c axis, showing the packing of the “Z”-shaped channels. AlO polyhedra in grey and carbon atoms in black. | ||
The crystal structure of Al-MIL-53-ADP-lp does not show any change in connectivity but represents a hydrated analogue of the narrow pore form. Due to the insertion of water molecules the channels open up to a size suitable to accommodate water molecules. Thus the channel diameter as determined by pore size distribution analysis carried out on the structural model is ca. 3.2 Å (Fig. 4 and S3†). The shape of the channels rather resembles a kinked variant of the normal rhombic form.
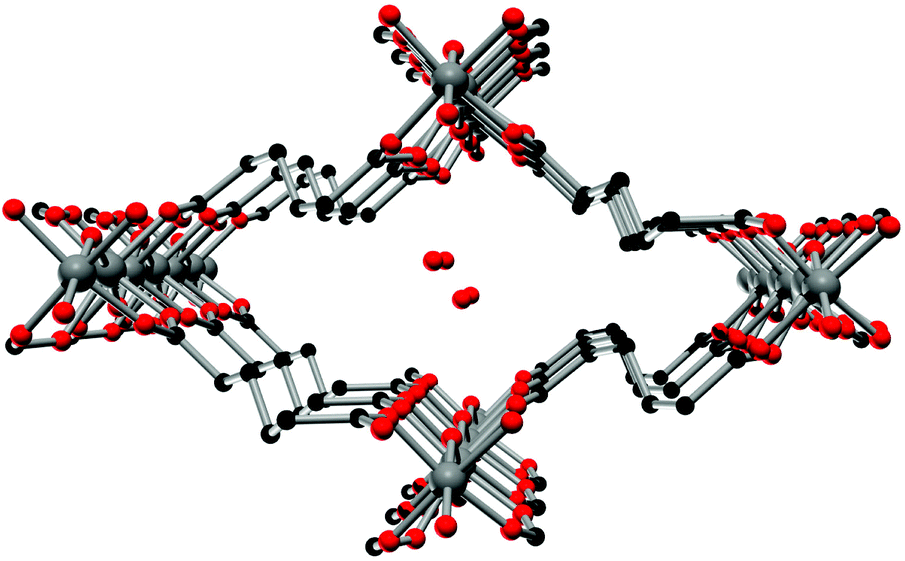 | ||
| Fig. 4 One channel in the structure of Al-MIL-53-ADP-lp as seen along the a axis. Hydrogen atoms are omitted for clarity. Aluminium atoms in grey, oxygen atoms in red and carbon atoms in black. | ||
The hydrated structure reveals that the water molecules are arranged in such a way to form hydrogen bonds with both the μ2-(OH) groups and one oxygen atom of one carboxylate group with characteristic distances between two water/host oxygen atoms ranging from 2.88 to 3.04 Å. In addition to this, the confined water molecules form a relatively strong hydrogen bond network with corresponding Ow_Ow distances (2.92 Å). This spatial distribution and the relevant interactions are very similar to that previously pointed in the MIL-53 series.22,40 Remarkably, the adsorption of water induces a rotation of the AlO-polyhedra chains and a framework expansion of ≈10% (vs. 3–4% and 33% for Al-MIL-53-FUM22 and Al-MIL-53-BDC13 respectively) while the length of the linker molecules is decreased due their conformational changes. The distances between carboxylate groups in the independent linker molecules in Al-MIL-53-ADP-np are 5.79(2) and 5.90(2) Å while they are 5.43 and 5.7 Å in the hydrated form Al-MIL-53-ADP-lp. This “shortening” of the linker molecules could also allow for an easier rotation around the C–C bonds and therefore might be the reason for the observed disordering in the crystal structure. A comparison with the well known highly crystalline layered compound zinc adipate41 pronounces the conformationally compressed character of the linker molecules. Zinc adipate incorporates adipate ions in their longest possible conformation and therefore the average distance between carboxylate groups amounts to 6.432(4) Å (Fig. 5).
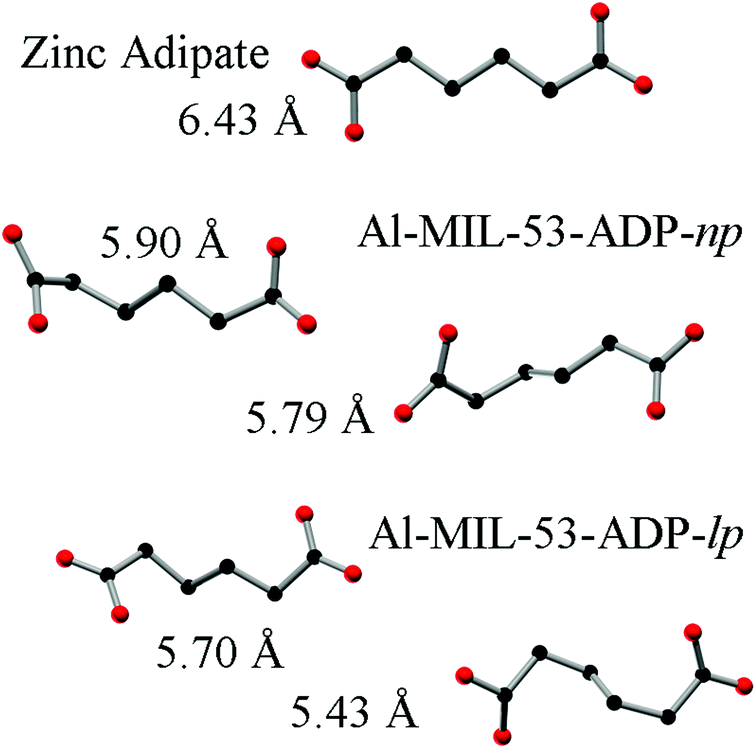 | ||
| Fig. 5 Conformation and lengths (carboxylate to carboxylate) of the adipate molecules in Al-MIL-53-ADP and for comparison also in zinc adipate.41 | ||
This change in conformation also seems to be the dominant mechanism controlling the breathing behaviour. In the aromatic parent compound Al-MIL-5313 the knee-cap mechanism results in profound changes in the angle between adjacently coordinated carboxylate groups. These values amount to 100° and 128° in the large pore and the narrow pore forms of the Al-MIL53-BDC solids, respectively. Here the scenario is different: for the hydrated Al-MIL-53-ADP-lp, the angles between crystallographically independent groups are 97 and 103° and upon dehydration to the narrow pore form they only slightly change to 97 and 107° (Fig. S4†).
It is worth mentioning that we also tried to intercalate several other solvent molecules like ethanol, acetone, toluene or dimethylformamide by dispersion of the dried MOF at elevated temperature, but all attempts proved unsuccessful.
Properties of Al-MIL-53-ADP
The IR-spectrum of Al-MIL-53-ADP-lp is shown in Fig. 6. It is particularly relevant that no signals of residual dimethylformamide molecules or unreacted adipic acid are visible. The most prominent signals are the peaks at 1570 and 1460 cm−1 which can be attributed to the asymmetric carboxylate vibration and the deformation vibration of the CH2-groups, respectively.42 Between 2960 and 2840 cm−1 several signals can be detected which belong to C–H stretching vibrations of the aliphatic linker molecule backbone. The intensity of the broad band around 3500 cm−1 varies, depending on the degree of hydration which can vary during the preparation of the sample.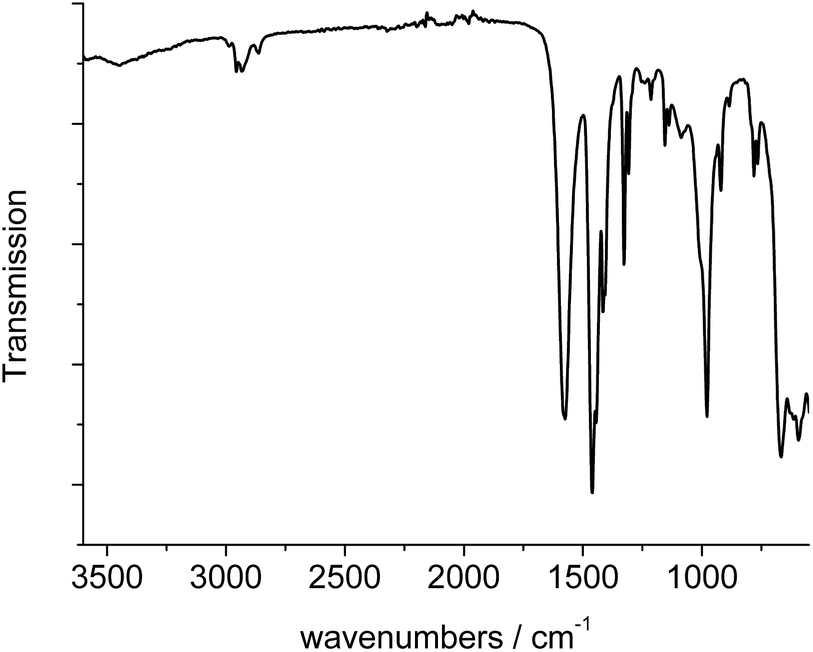 | ||
| Fig. 6 IR-spectrum of Al-MIL-53-ADP-lp. | ||
The thermogravimetric curve (Fig. 7) was measured for a sample prepared directly after filtration from an aqueous dispersion and shows two distinct weight losses. The first step is due to the dehydration of the structure (10 wt%) while the second step of 63.4% above 360 °C is attributed to the decomposition of the framework. The first weight loss of 10 wt% is slightly larger than the expected value (9 wt%) while the second step (63.4%) is slightly smaller than the expected loss (66.5%) based on the deduced composition [Al(OH)(O2C–C4H8–CO2)]·H2O. Taking into account that there are still sulphur containing impurities present, both values are in reasonable agreement with the expected ones.
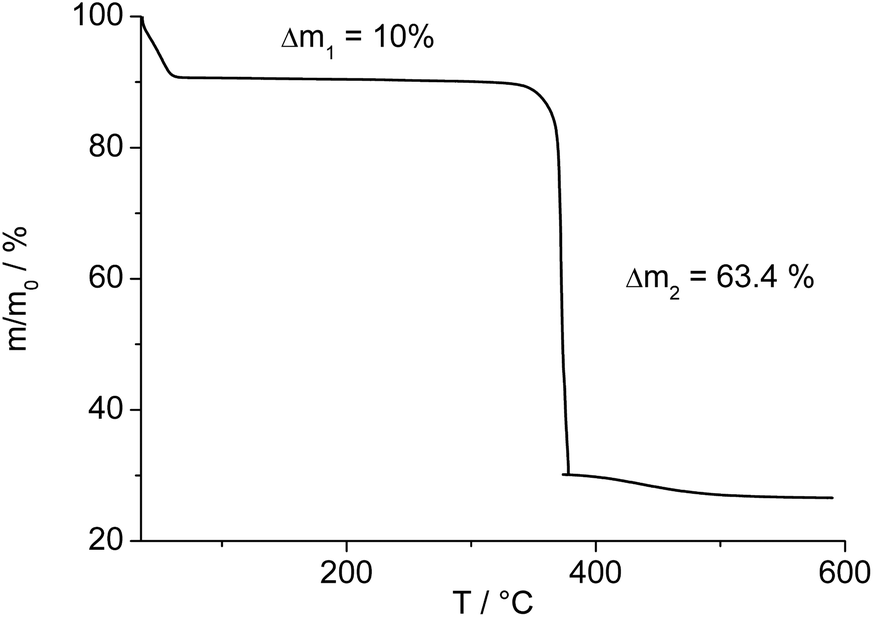 | ||
| Fig. 7 Thermogravimetric curve for Al-MIL-53-ADP-lp. | ||
In order to investigate the sorption properties of Al-MIL-53-ADP, several sorption isotherms were measured. However, no uptake was observed for N2 at −196 °C or for CO2 at 25 °C, regardless of activation conditions (0.1 mbar, between 100 °C and 200 °C). Only for water vapour we observed a considerable adsorption. The isotherm is shown in Fig. 8 and exhibits a strong hysteresis loop. By measuring PXRD patterns depending on relative humidity we were able to identify the respective framework conformation during the sorption process (Fig. 8).
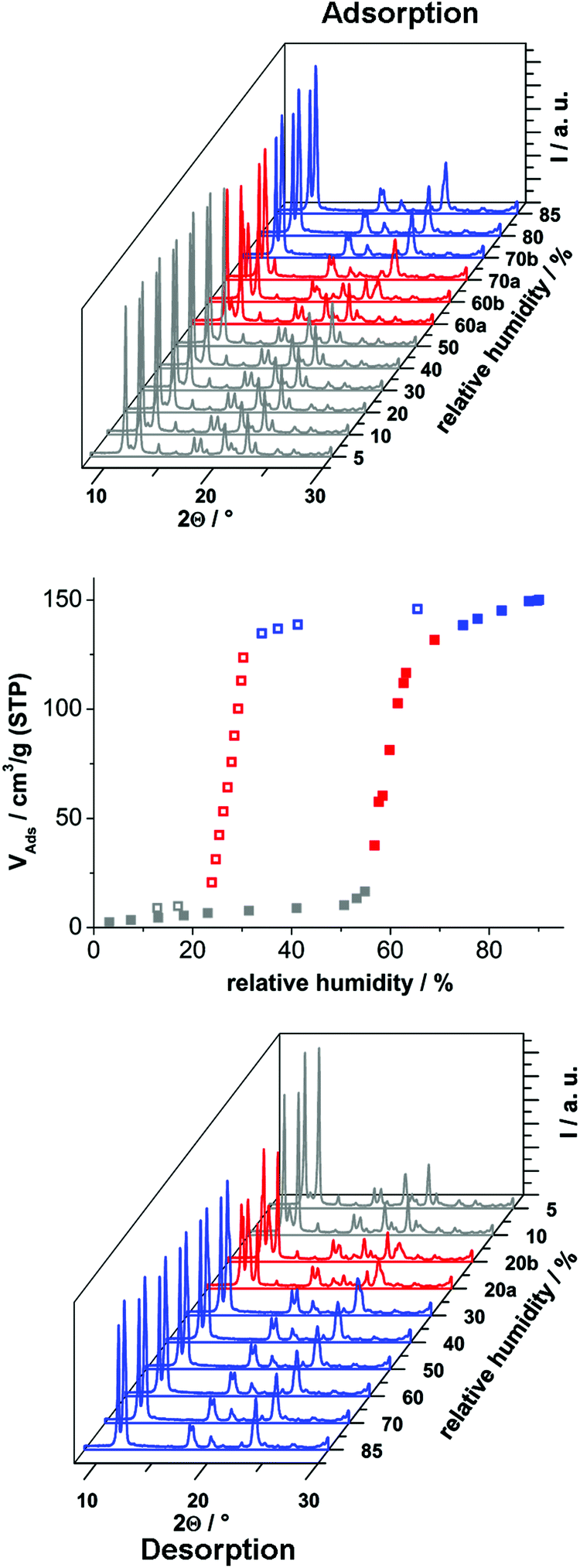 | ||
| Fig. 8 H2O vapour isotherm for Al-MIL-53-ADP and the PXRD patterns depending on relative humidity, measured at 298 K. The colours indicate the respective framework conformation present. Blue indicates the hydrated Al-MIL-53-ADP-lp, grey indicates the dehydrated Al-MIL-53-ADP-np and red indicates a mixture of both conformations. Patterns for 60% and 70% r. h. during adsorption and 20% r. h. during desorption are plotted directly after the humidity value is reached (a) and after 20–45 minutes equilibration (b). | ||
The adsorption of water at 25 °C starts at a relative humidity of ≈55% and proceeds in one step. The adsorbed amount of 1.1 H2O per aluminium atom is in good agreement with the expected amount of 1 molecule. Compared to the adsorption step at ≈55% relative humidity, the desorption starts at a much lower value of ≈25%. Such hysteretic water sorption isotherms are often observed in breathing MOFs, for example for Al-MIL-53-(OH)2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 17 or Al-MIL-53-(1,4-NDC).43 The PXRD data indicate a rather slow equilibration during the uptake and release of H2O molecules. Diffractograms measured at 60% relative humidity during uptake show in general both forms, Al-MIL-53-ADP-np and Al-MIL-53-ADP-lp.
17 or Al-MIL-53-(1,4-NDC).43 The PXRD data indicate a rather slow equilibration during the uptake and release of H2O molecules. Diffractograms measured at 60% relative humidity during uptake show in general both forms, Al-MIL-53-ADP-np and Al-MIL-53-ADP-lp.
Nearly all of the dry form is converted after 20 minutes of equilibration at 70% relative humidity. The same mixture of framework conformers is observed for the desorption curve at 20% relative humidity. Moreover, even after extended equilibration under very moist (relative humidity >70%) or very dry (relative humidity <20%) conditions, there are still traces of the respective counterpart conformation present. A possible explanation could be that the diffusion into and out of the very narrow pores in Al-MIL-53-ADP is very slow and thus impedes fast equilibration times below 30 minutes. In addition the MOF crystals show a strong tendency for intergrowth, indicated by the formation of a thick layer of product at the glass reactor walls during the synthesis. This might also slow down the interparticle diffusion between intergrown crystals and thus delay equilibration.
Solid-state NMR
The 1H NMR spectra of both Al-MIL-53-ADP forms (Fig. 9) exhibit a broad resonance around 1.9 ppm which consists of overlapping peaks of the CH2 and the Al–OH groups. This is confirmed by the first principle DFT calculations performed on the crystal structure of Al-MIL-53-ADP-np which evidence that the chemical isotropic shifts are very similar for both H species (δiso (H(Al–OH) = 2.0 ppm vs. δiso (HCH2) = 1.8 ppm) see Table S2†) Additionally, Al-MIL-53-ADP-lp exhibits two sharper resonances at 2.7 and 3.0 ppm which were attributed to the two crystallographically inequivalent water molecules observed for this structure as also qualitatively shown by first principle calculations (see Table S2†).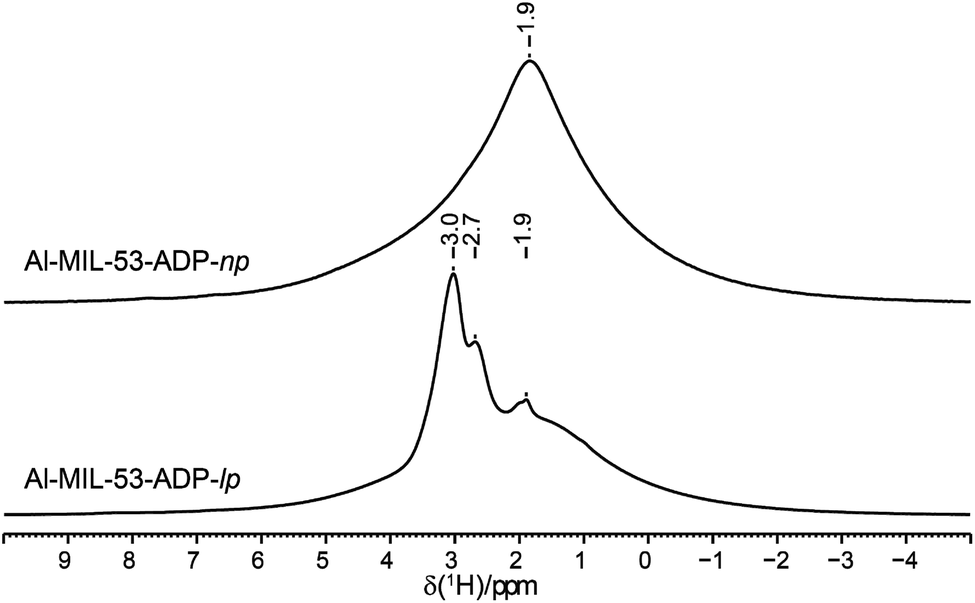 | ||
| Fig. 9 1H MAS NMR spectra of Al-MIL-53-ADP. | ||
The 13C spectra (Fig. 10) show three well-separated peak regions characteristic of the three different carbon types in the structures. While the carboxyl units are observed between 179 and 182 ppm, the outer and inner pairs of CH2 groups resonate in the ranges 37.0–38.2 ppm and 23.7–25.8 ppm, respectively. In the case of Al-MIL-53-ADP-np, six resonances – each two for the inner and outer CH2 groups (23.7/25.8 ppm and 37.1/38.1 ppm) as well as for the CO2 units (179.7/179.8 ppm)) – were observed. These observations are again in very good agreement with our DFT predicted 13C NMR parameters for both CH2 groups (22.5/25.2 ppm and 36.4/37.2 ppm) and CO2 units (183.4 ppm), this accounts for half of the possible resonances with respect to the structural model. Since the two adipate ions in the asymmetric unit are almost inversion symmetric we expect that the intramolecular splitting between resonances of similar chemical units of the same adipate molecule could not be resolved.
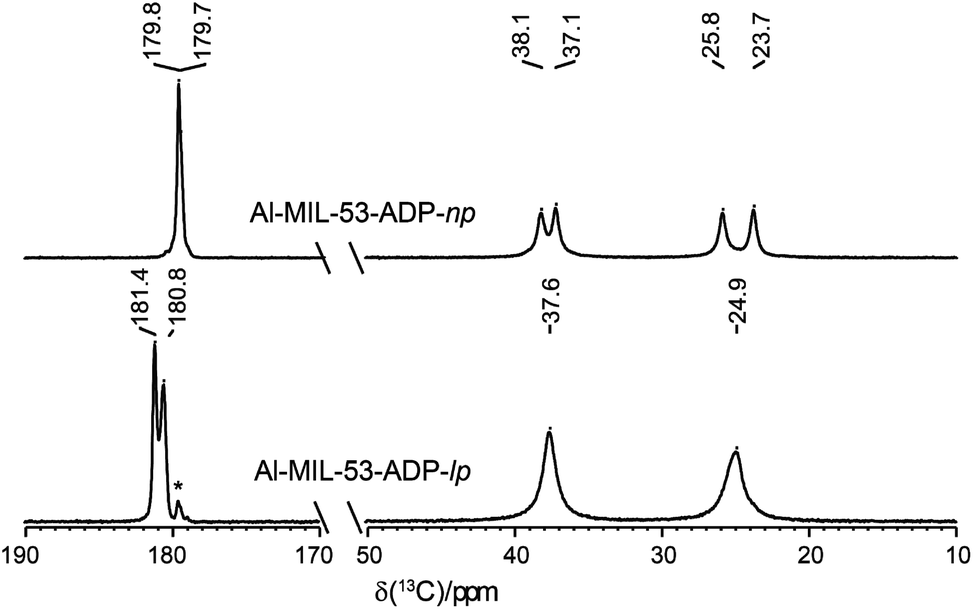 | ||
| Fig. 10 13C CP MAS NMR spectra of Al-MIL-53-ADP. The resonances marked with an asterisk represent a small impurity of the np form in the lp phase. | ||
Due to the pronounced dynamical disorder of the CH2 units of adipate molecules and the water molecules the shift differences between similar inner and outer CH2 units, respectively, are averaged for Al-MIL-53-ADP-lp. This leads to only two resonances at 24.9 and 37.6 ppm in this region. In contrast, for the carboxylic groups of Al-MIL-53-ADP-lp two well resolved resonances at 180.8 and 181.4 ppm with a splitting of 0.6 ppm are visible. These experimental data are consistent with the theoretical findings reported in Table S2.†
The 27Al NMR spectra of both phases are displayed in Fig. 11 along with their simulated spectra including chemical shift and second order quadrupolar interactions (Table 2). For both the np and the lp forms two individual resonances were observed which is in line with the structure solutions of the PXRD data. All resonances exhibit isotropic chemical shifts close to zero reflecting the octahedral coordination of the Al3+ ions. The significantly different quadrupolar coupling constants CQ and asymmetry parameters η for the np phase demonstrate that the local environment of both Al octahedra differs markedly (Table 2). The CQ (9.06 MHz, 7.96 MHz) obtained for the anhydrous form are consistent with the experimental findings previously reported for the dry Al-MIL-53-BDC (CQ = 8.3 MHz) and Al-MIL-53-FUM (8.7 MHz) materials.22 And they also match with the corresponding first-principle calculated NMR parameters (8.60 MHz, 7.90 MHz).22 The quadrupolar interactions of the Al nuclei in the lp phase turn out to be very similar (8.32 and 8.37 MHz see Table 2) and could almost be assimilated into one single resonance. This is in contrast to the first-principles calculations where two types of Al3+, with CQ values of 8.3 MHz and 12.5 MHz respectively have been found (see Table S2†). This deviation probably results from a dynamical disorder, which was experimentally observed for this phase.
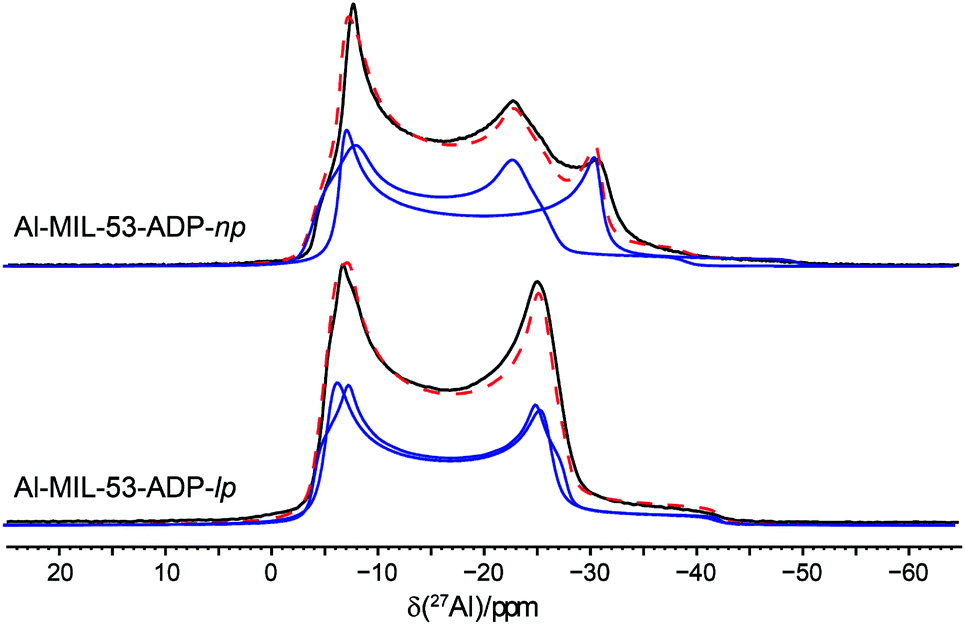 | ||
| Fig. 11 27Al MAS NMR spectra of Al-MIL-53-ADP along with their respective fitted spectra (— individual resonances, -- sum of all resonances). | ||
| [Al(OH)(O2C–C4H8–CO2)] | [Al(OH)(O2C–C4H8–CO2)]·H2O | |
|---|---|---|
| δ iso/ppm | 0.2/−0.5 | 0.2/0.3 |
| C Q/MHz | 9.06/7.96 | 8.32/8.37 |
| η | 0.0/0.19 | 0.0/0.13 |
Overall, the NMR experimental findings are in agreement with the corresponding first principles calculated parameters determined from the resolved crystal structures and support the reliability of the proposed structural models.
Conclusions
Herein we reported the synthesis and detailed characterisation of the first analogue of Al-MIL-53 based on a single chain saturated aliphatic linker molecule. This opens also the opportunity to study other aliphatic carboxylic acids like succinic acid (C4 chain) or suberic acid (C8 chain), which might also form breathing frameworks. The combination of complementary PXRD, solid state NMR and modelling tools was shown to be necessary to elucidate the crystal structures of the anhydrous and hydrated MIL-53-ADP which show pronounced disorder phenomena. Regarding the sorption properties it is especially of interest if functionalisation or extension of the linker molecules would be possible, which might result in similarly selective sorption behaviour towards other guest molecules than water.Acknowledgements
The research leading to these results has received funding from the European Community's Seventh Framework Programme Macademia (FP7/2007-20013) under grant agreement no 228862”. G. M. thanks Institut Universitaire de France for its support. This work has also been supported by the DFG (SE 1417/7-1).Notes and references
- M. P. Suh, H. J. Park, T. K. Prasad and D. Lim, Chem. Rev., 2012, 112, 782 CrossRef CAS PubMed.
- J. Liu, P. K. Thallapally, B. P. McGrail, D. R. Brown and J. Liu, Chem. Soc. Rev., 2012, 41, 2308 RSC.
- F. Vermoortele, M. Maes, P. Modhadam, M. Lennox, F. Ragon, M. Boulhout, S. Biswas, K. Laurier, I. Beurroies, R. Denoyel, M. Roeffaers, N. Stock, T. Düren, C. Serre and D. De Vos, J. Am. Chem. Soc., 2011, 133, 18529 CrossRef PubMed.
- P. Valvekens, F. Vermoortele and D. De Vos, Catal. Sci. Technol., 2013, 13, 1435 Search PubMed.
- O. Shekhah, J. Liu, R. A. Fischer and C. Wöll, Chem. Soc. Rev., 2011, 40, 1081 RSC.
- G. Ferey, Z. Anorg. Allg. Chem., 2012, 638, 1897 CrossRef CAS.
- A. Schneeman, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062 RSC.
- C. Serre, C. Mellot-Draznieks, S. Surble, N. Audebrand, Y. Filinchuk and G. Ferey, Science, 2007, 315, 1828 CrossRef CAS PubMed.
- J. P. S. Mowat, S. R. Miller, A. M. Z. Slawin, V. R. Seymour, S. E. Ashbrook and P. A. Wright, Microporous Mesoporous Mater., 2011, 142, 322 CrossRef CAS.
- K. Barthelet, J. Marrot, D. Riou and G. Ferey, Angew. Chem., Int. Ed., 2002, 114, 291 CrossRef.
- C. Serre, F. Millange, C. Thouvenot, M. Noguès, G. Marsolier, D. Louër and G. Férey, J. Am. Chem. Soc., 2002, 124, 13519 CrossRef CAS PubMed.
- T. R. Whitfield, X. Wang, L. Liu and A. J. Jacobson, Solid State Sci., 2005, 7, 1096 CrossRef CAS.
- T. Loiseau, C. Serre, C. Huguenard, G. Fink, F. Taulelle, M. Henry, T. Bataille and G. Ferey, Chem. – Eur. J., 2004, 10, 1373 CrossRef CAS PubMed.
- C. Volkringer, T. Loiseau, N. Guillou, G. Ferey, E. Elkaim and A. Vimont, Dalton Trans., 2009, 2241 RSC.
- V. Anokhina, M. Vougo-Zanda, X. Wang and A. J. Jacobson, J. Am. Chem. Soc., 2005, 127, 15000–15001 CrossRef PubMed.
- T. Devic, P. Horcajada, C. Serre, F. Salles, G. Maurin, B. Moulin, D. Heurtaux, G. Clet, A. Vimont, J.-M. Greneche, B. Le Ouay, F. Moreau, E. Magnier, Y. Filinchuk, J. Marrot, J.-C. Lavalley, M. Daturi and G. Ferey, J. Am. Chem. Soc., 2010, 132, 1127 CrossRef CAS PubMed.
- S. Biswas, T. Ahnfeldt and N. Stock, Inorg. Chem., 2011, 50, 9518 CrossRef CAS PubMed.
- A. Centrone, T. Harada, S. Speakman and T. A. Hatton, Small, 2010, 6, 1598 CrossRef CAS PubMed.
- I. Senkovska, F. Hoffmann, M. Fröba, J. Getzschmann, W. Böhlmann and S. Kaskel, Microporous Mesoporous Mater., 2009, 122, 93 CrossRef CAS.
- T. Loiseau, C. Mellot-Draznieks, H. Muguerra, G. Ferey, M. Haouas and F. Taulelle, C. R. Chim., 2005, 8, 765 CrossRef CAS.
- Y. Liu, K. Leus, M. Grzywa, D. Weinberger, K. Strubbe, H. Vrielinck, R. Van Deun, D. Volkmer, V. Van Speybroeck and P. Van Ver Voort, Eur. J. Inorg. Chem., 2012, 16, 2819 CrossRef.
- E. Alvarez, N. Guillou, C. Martineau, B. Bueken, B. Van de Voorde, C. Le Guillouzer, P. Fabry, F. Nouar, F. Taulelle, D. de Vos, J. Chang, K. H. Cho, N. Ramsahye, T. Devic, M. Daturi, G. Maurin and C. Serre, Angew. Chem., Int. Ed., 2015, 54, 3664 CrossRef CAS PubMed.
- L. Wang, T. Song, C. Li, J. Xia, S. Wang, L. Wang and J. Xu, J. Solid State Chem., 2012, 190, 208 CrossRef CAS.
- I. H. Kim, X. Wang and A. J. Jacobson, Solid State Sci., 2010, 12, 76 CrossRef CAS.
- F. Niekiel, M. Ackermann, P. Guerrier, A. Rothkirch and N. Stock, Inorg. Chem., 2013, 52, 8699 CrossRef CAS PubMed.
- F. Niekiel, J. Lannoeye, H. Reinsch, A. Munn, A. Heerwig, I. Zizak, S. Kaskel, R. Walton, D. de Vos, P. Llewellyn, A. Lieb, G. Maurin and N. Stock, Inorg. Chem., 2014, 53, 4610 CrossRef CAS PubMed.
- H. Reinsch and D. De Vos, Microporous Mesoporous Mater., 2014, 200, 311 CrossRef CAS.
- Topas Academics 4.2, Coelho Software, 2007 Search PubMed.
- Materials Studio Version 5.0, Accelrys Inc., San Diego, CA, 2009 Search PubMed.
- (a) J. VandeVondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, Comput. Phys. Commun., 2005, 167, 103 CrossRef CAS; (b) J. VandeVondele and J. Hutter, J. Chem. Phys., 2003, 118, 4365 CrossRef CAS; (c) G. Lippert, J. Hutte and M. Parrinello, Theor. Chem. Acc., 1999, 103, 124 CrossRef CAS; (d) B. G. Lippert and J. H. M. Parrinello, Mol. Phys., 1997, 92, 477 CrossRef; (e) J. VandeVondele and J. Hutter, J. Chem. Phys., 2007, 127, 114105 CrossRef PubMed; (f) S. Goedecker, M. Teter and J. Hutter, Phys. Rev. B: Condens. Matter, 1996, 54, 1703 CrossRef CAS; (g) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- M. D. Segall, P. L. D. Lindan, M. J. Probert, C. J. Pickard, P. J. Hasnip, S. J. Clark and M. C. J. Payne, J. Phys.: Condens. Matter, 2002, 14, 2717 CrossRef CAS.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr., 2005, 220, 567 CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- J. R. Yates, C. J. Pickard and F. Mauri, Phys. Rev. B: Condens. Matter, 2007, 76, 024401 CrossRef.
- M. Profeta, F. Mauri and C. J. Pickard, J. Am. Chem. Soc., 2003, 125, 541 CrossRef CAS PubMed.
- C. J. Pickard and F. Mauri, Phys. Rev. B: Condens. Matter, 2001, 63, 245101 CrossRef.
- M. Sanselme, J.-M. Grenèche, M. Riou-Cavelleca and G. Férey, Solid State Sci., 2004, 6, 853 CrossRef CAS.
- W. Kraus and G. Nolze, PowderCell 2.4, 2000 Search PubMed.
- (a) B. Meredig and C. Wolverton, Nat. Mater., 2013, 12, 123 CrossRef CAS PubMed; (b) A. Cadiau, J. S. Lee, D. D. Borges, P. Fabry, T. Devic, M. T. Wharmby, C. Martineau, D. Foucher, F. Taulelle, C. H. Jun, Y. K. Hwang, N. Stock, M. F. De Lange, F. Kapteijn, J. Gascon, G. Maurin, J. S. Chang and C. Serre, Adv. Mater., 2015, 27, 4775 CrossRef CAS PubMed; (c) C. Martineau, Solid State Nucl. Magn. Reson., 2014, 63–64, 1 CrossRef CAS PubMed; (d) S. Devautour-Vinot, G. Maurin, C. Serre, P. Horcajada, D. Cunha, V. Guillerm, E. De Souza Costa, F. Taulelle and C. Martineau, Chem. Mater., 2012, 24, 2168 CrossRef CAS.
- S. Bourrelly, B. Moulin, A. Rivera, G. Maurin, S. Devautour-Vinot, C. Serre, T. Devic, P. Horcajada, A. Vimont, G. Clet, M. Daturi, J.-C. Lavalley, S. Loera-Serna, R. Denoyel, P. L. Llewellyn and G. Férey, J. Am. Chem. Soc., 2010, 132, 9488 CrossRef CAS PubMed.
- S. Klaus, M. W. Lehenmeier, E. Herdtweck, P. Deglmann, A. K. Ott and B. Rieger, J. Am. Chem. Soc., 2011, 133, 13151 CrossRef CAS PubMed.
- G. Socrate, Infrared and Raman Characteristic Group Frequencies: Tables and Charts, John Wiley & Sons, 2004 Search PubMed.
- A. Comotti, S. Bracco, P. Sozzani, S. Horike, R. Matsuda, J. Chen, M. Takata, Y. Kubota and S. Kitagawa, J. Am. Chem. Soc., 2008, 130, 13664 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional XRPD-data, simulated PSD, crystallographic information files, simulated NMR data. See DOI: 10.1039/c5dt03510d |
| This journal is © The Royal Society of Chemistry 2016 |