
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modular solid-phase synthesis, catalytic application and efficient recycling of supported phosphine–phosphite ligand libraries†
Frank J. L.
Heutz
 and
Paul C. J.
Kamer
*
and
Paul C. J.
Kamer
*
EaStCHEM, School of Chemistry, University of St Andrews, North Haugh, St Andrews KY16 9ST, UK. E-mail: pcjk@st-andrews.ac.uk
First published on 13th October 2015
Abstract
In spite of decades of research in the field of homogeneous asymmetric catalysis the discovery of new high performance catalysts still relies heavily on trial-and-error. There is still a lack of efficient combinatorial methods which enable the synthesis and screening of vast ligand libraries, especially for bidentate phosphorus ligands. Here we present a highly modular solid-phase synthetic approach which provides facile access to libraries of phosphine–phosphite ligands in quantitative yield requiring only minimal work-up. The obtained library of supported phosphine–phosphites was successfully applied in rhodium catalyzed asymmetric hydrogenation obtaining high enantioselectivities up to 98%. Also, these polymer supported ligands could be successfully recycled under batch conditions exhibiting only a small decline of activity and no loss of selectivity.
Introduction
Over the last few decades transition metal mediated asymmetric catalysis has evolved into an indispensable tool to both academia and industry, providing access to optically active compounds such as flavors and fragrances, pharmaceuticals and agrochemicals.1 Despite the fact that nowadays homogeneous asymmetric catalysis is a highly evolved field of research, the discovery of high performance ligands for asymmetric transformations remains challenging.2 Although our knowledge of organometallic chemistry and ligand effects in catalysis is ever increasing and computational techniques play an increasingly important role,3 the development of new catalysts still relies heavily on trial-and-error.4Combinatorial synthetic methodologies and high-throughput screening have proven their value in asymmetric catalysis and numerous successful approaches have been reported using both covalent5 and supramolecular chemistry.6 For phosphorus donor ligands the focus has mainly been on standard solution-phase techniques and mainly monodentate ligands.7 Large libraries of bidentate phosphorus ligands and efficient combinatorial methods for synthesizing these remain scarce due to their intrinsically more complicated synthesis and work-up procedures.8 This is even more the case for heterobidentate phosphorus ligands like phosphine–phosphites, which can be attributed to the difficulty of introducing two different phosphorus moieties onto the ligand backbone.
Heterobidentate P-OP ligands such as phosphine–phosphites have received increased attention over the last years and have proven to be very efficient ligands for various asymmetric transformations such as hydrogenation, hydroformylation and allylic substitution.9 BINAPHOS, first reported by the group of Takaya and Nozaki,10 has been highly effective in a wide range of reactions and belongs to the small group of so-called privileged ligands in asymmetric catalysis.2 The highly modular structure of phosphine–phosphites and the fact they possess two electronically different phosphorus moieties, resulting in different trans-labilizing properties, makes them promising candidates for the combinatorial synthesis and high-throughput screening of structurally diverse ligand libraries.11 There have been few reports of modular approaches towards phosphine–phosphite ligand but so far the applications remain fairly limited.12 The preparation of this class of ligands still relies on classical solution-phase methodologies and work-up procedures and suffers from relatively low yields, thus hampering truly high-throughput synthesis and screening.
Solid-phase synthesis (SPS) provides a promising alternative approach towards ligand libraries and has already successfully been employed for many years for the synthesis of large compound libraries such as polypeptides and oligosaccharides.13 Using solid-phase synthesis, ligand structures can be built up step-by-step in a combinatorial fashion while being bound to a resin bead. Using a solid support has as main advantage that it can greatly simplify ligand purification procedures which in turn allows the use of large excesses of reagents to drive reactions to completion. Often the work-up only comprises a simple filtration or decantation step as opposed to the laborious work-up procedures employed in the solution-phase synthesis of phosphorus ligands.14 The facile work-up makes solid-phase synthesis very suitable for automated synthesis using high throughput equipment.
An additional advantage of employing a solid support is that it can greatly facilitate catalyst recovery after the catalytic reaction and potentially even lead to recyclable immobilized catalysts. There are numerous accounts of immobilized (chiral) phosphorus ligands addressing catalyst recovery, which presents one of the major problems in homogeneous catalysis. The vast majority however, are reports of non-modular single ligands or catalysts anchored to a support.15
Surprisingly, the application of solid-phase synthesis for the combinatorial synthesis of supported bidentate phosphorus ligands remains fairly limited.8 However, there have been reports on SPS of a small variety of aminophosphine–phosphine and aminophosphine–phosphite libraries16 and recently we reported on the solid-phase synthesis of diphosphine ligands.17 Using a similar methodology we now report on an efficient combinatorial solid-phase synthetic approach for libraries of highly modular and recyclable phosphine–phosphite ligands.
Results and discussion
Solid-phase synthesis of phosphine–phosphite ligands
A ligand library of 16 supported phosphine–phosphite (P-OP) ligands was prepared of which the general structure is depicted in Fig. 1. These ligands possess a highly modular structure and by varying the three main building blocks in a combinatorial fashion it is possible to quickly generate a ligand library showing large structural diversity. The phosphine moiety can be altered by using phosphines bearing different substituents (R1) and for the ligand backbone cyclic sulfates with a varying backbone length (n) and different substituents (R2) can be used. Finally diversity in the –OP part of the ligand can be created by employing various chlorophosphites to introduce the phosphite moiety giving in total 4 points of diversity.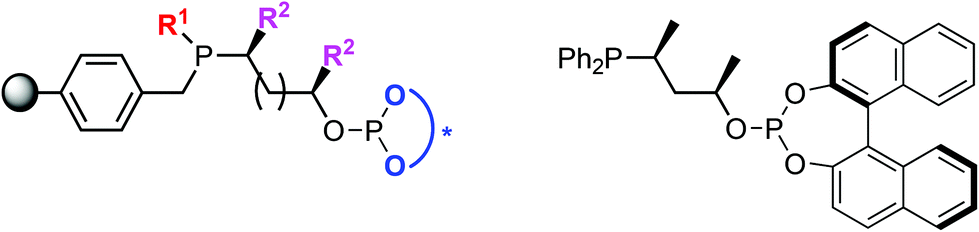 | ||
| Fig. 1 General structure of highly modular supported phosphine–phosphite ligands (left), and a solution-phase analog reported by Bakos et al. (L17, right).18 | ||
We have developed a modular stepwise methodology for the synthesis of P-OP ligands (see Scheme 1). The initial synthetic steps for the preparation of the intermediate supported phosphine–borane sulfates are identical to those reported for supported diphosphines.17 Subsequently, the sulfate group is hydrolyzed and the resulting hydroxyalkyl phosphines are reacted with chlorophosphite reagents yielding supported phosphine–phosphite ligands. A similar solution-phase procedure has been reported by the group of Bakos and one of their homogeneous analogues of our supported P-OP ligands is depicted in Fig. 1.18
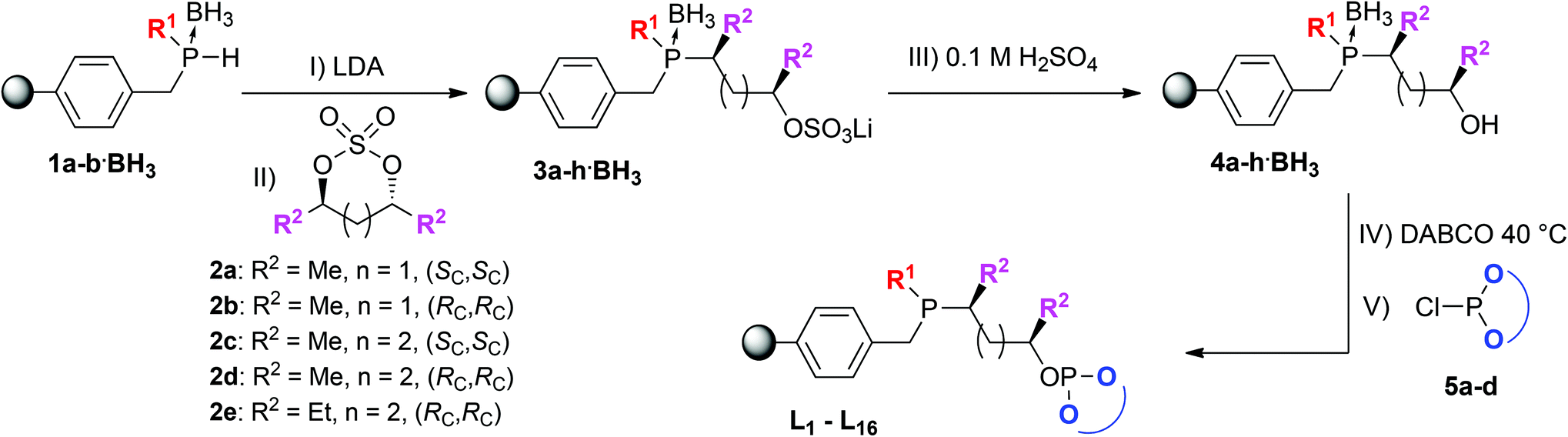 | ||
| Scheme 1 Solid-phase synthetic approach towards supported phosphine–phosphite ligands, all reactions were performed in THF at room temperature unless stated otherwise. | ||
Using our solid-phase synthetic approach this class of ligands is readily accessible under very mild conditions requiring only a very minimal work-up procedure between each reaction step. Moreover, the supported ligands were obtained in high purity with each reaction step proceeding quantitatively as determined by gel-phase 31P NMR (see Fig. 2). Comparatively, when employing traditional solution-phase methodologies laborious purification methods like column chromatography and distillations under inert conditions are often required and the overall yield for similar phosphine–phosphites can be as low as 30%.19
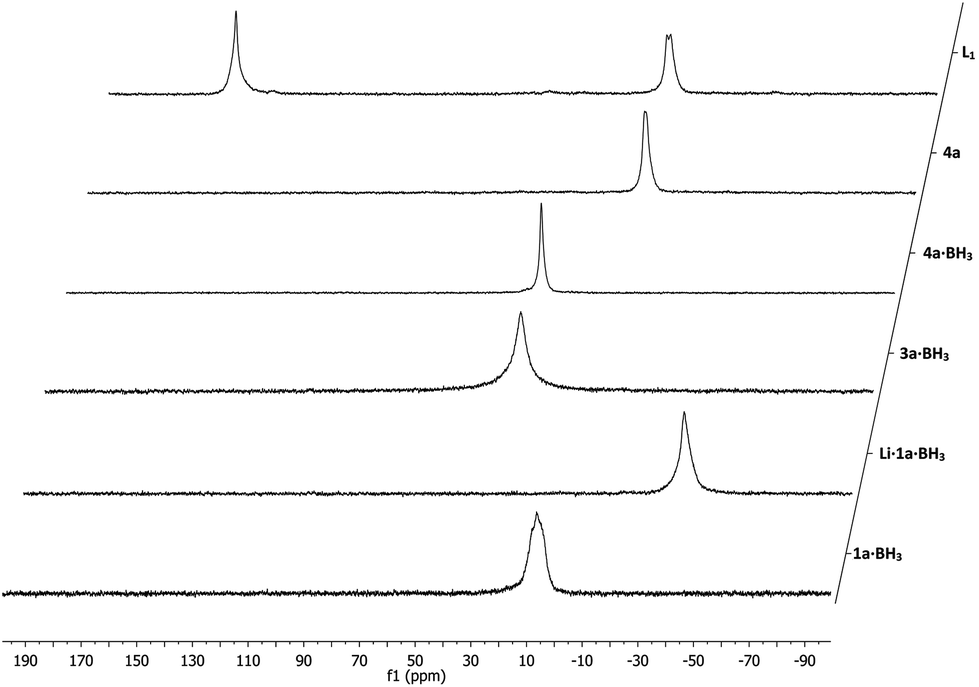 | ||
| Fig. 2 Solid-phase synthesis of representative supported phosphine–phosphite L1 as monitored by gel-phase 31P NMR spectroscopy. | ||
The starting synthon of the solid-phase synthesis are supported phosphine–boranes 1a–b·BH3 which are readily accessible by treating a choromethyl functionalized resin, in this case Merrifield resin, with various primary lithium phosphides having different substituents (R1). The introduction of the phosphine moiety takes place in a non-stereospecific fashion and yields a racemic mixture. Subsequently the obtained supported secondary phosphines can be protected by treatment with BH3·SMe2 to make them less prone to oxidation. BH3·SMe2 was chosen as reagent over the THF adduct due to its higher stability and solubility. Upon deprotonation using lithium diisopropylamide (LDA, step I) the lithiated phosphine–boranes (Li·1a–b·BH3) can be reacted with a cyclic sulfate which after ring opening, with full inversion at one of the stereogenic centers,20 serves as the ligand backbone (step II). Stronger bases such as n-butyl lithium resulted in side products due to transmetallation at the benzylic position. Diversity can be introduced by employing various cyclic sulfates having a varying backbone length (n) and bearing different substituents (R2). All the above mentioned reaction steps could be readily followed by 31P NMR confirming the formation of the desired intermediates (see Fig. 2 for a representative example).
Next step is the hydrolysis of the supported phosphine–borane sulfates (3a–h·BH3) to the corresponding hydroxyalkyl phosphines (4a–h·BH3). Bakos et al. have reported an analogous homogeneous procedure18 but their conditions, 90 °C using 2 M sulfuric acid, were found to be too harsh and led to decomposition of the resin. Instead a very mild hydrolysis at room temperature was applied, using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of THF and 0.1 M sulfuric acid to ensure proper swelling of the resin and to retain its structural integrity. The hydrolysis proceeded relatively slow and proved difficult to monitor by 31P NMR as there was no notable NMR shift observable, although the hydrolysis products did exhibit slightly sharper peaks (Fig. 2). Using 7Li NMR however, it was possible to follow the hydrolysis in time. The consumption of the lithium sulfate group, exhibiting a peak around 1 ppm, could be readily monitored as seen in Fig. 3. Full hydrolysis was on average observed after 3 days and could also be confirmed using FT-IR and elemental analysis, both showing full removal of the sulfate ester group.
:1 mixture of THF and 0.1 M sulfuric acid to ensure proper swelling of the resin and to retain its structural integrity. The hydrolysis proceeded relatively slow and proved difficult to monitor by 31P NMR as there was no notable NMR shift observable, although the hydrolysis products did exhibit slightly sharper peaks (Fig. 2). Using 7Li NMR however, it was possible to follow the hydrolysis in time. The consumption of the lithium sulfate group, exhibiting a peak around 1 ppm, could be readily monitored as seen in Fig. 3. Full hydrolysis was on average observed after 3 days and could also be confirmed using FT-IR and elemental analysis, both showing full removal of the sulfate ester group.
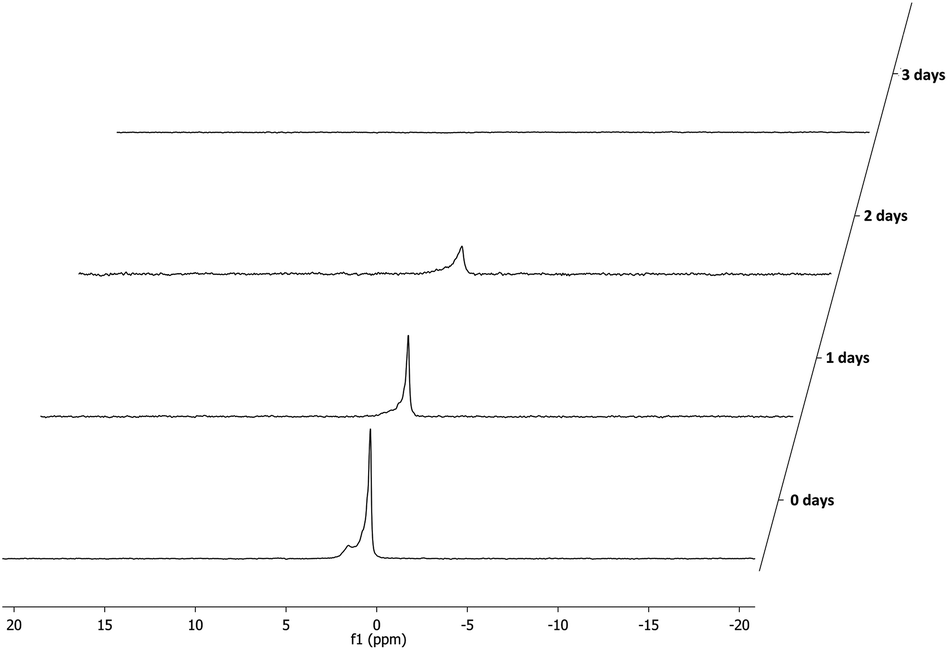 | ||
| Fig. 3 Hydrolysis of representative supported phosphine–borane sulfate 3a·BH3 monitored by 7Li NMR spectroscopy. | ||
It was decided to perform the removal of the BH3 group prior to introduction of the –OP moiety as phosphites are known to be more prone to hydrolysis under basic conditions.21 The borane group was removed by treatment with an excess of 1,4-diazabicyclo[2.2.2]octane (DABCO, 10 eq.) at 40 °C which could be readily followed by 31P NMR (Fig. 2). Next the phosphite moieties were introduced by treating the supported hydroxyalkyl phosphines (4a–h) with various chlorophosphites in the presence of triethylamine. Both (S)-binol and (R)-binol derived chlorophosphites were employed as well as a slightly more bulky trimethylsilyl functionalized binol-PCl. Moreover, also a t-butyl functionalized bisphenol derived chlorophosphite was used, demonstrating the versatility of this solid-phase synthetic approach. In all cases the introduction of the –OP moiety proceeded smoothly although for the more bulky phosphites a slightly larger excess, up to 3 equivalents of reagent, was required.
Fig. 2 shows that this reaction step could be readily monitored using 31P NMR and the appearance of a second peak in a 1:1 ratio could be observed confirming the formation of the desired supported P-OP ligands in high purity. In some cases, like for ligand L8, splitting into two 1:1 signals for the phosphine or phosphite group could be observed (Fig. 4). This is due to the fact that all supported ligands are present as a mixture of two epimers at the phosphine moiety. But for most ligands only single broad peaks could be observed, probably due to overlap of the two epimer signals. Deerenberg et al. reported that for similar phosphine–phosphite systems the P-stereogenic center has little influence on the chiral induction and that the selectivity is mainly determined by the ligand backbone and the phosphite moiety.22 Finally the actual phosphorus loading of the immobilized phosphine–phosphites could be determined by elemental analysis. 1H and 13C NMR of the supported ligands was not very informative due very broad peaks and overlap with signals of the Merrifield resin (see ESI† for representative examples). The complete P-OP ligand library (L1–L16) synthesized using this efficient solid-phase synthetic approach is depicted in Fig. 5.
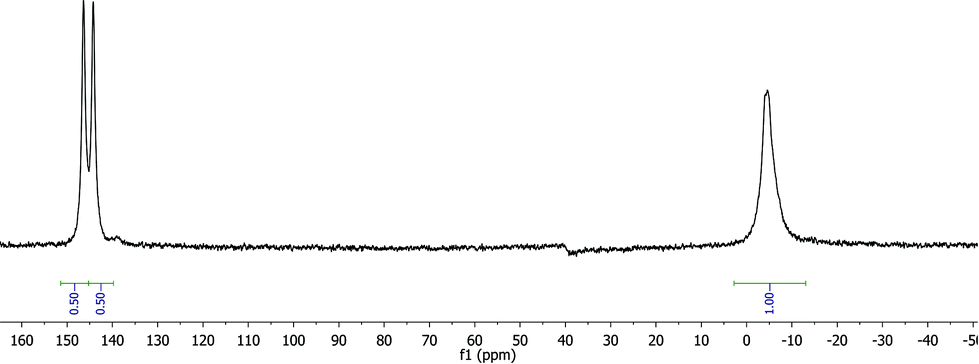 | ||
| Fig. 4
31P NMR spectrum of supported phosphine–phosphite L8 clearly showing the presence of both epimers in a 1:1 ratio. | ||
 | ||
| Fig. 5 Complete Library of supported phosphine–phosphite ligands (L1–L16). | ||
Rh-catalyzed asymmetric hydrogenation
The library of 16 supported phosphine–phosphite ligands was employed in the asymmetric hydrogenation of several benchmark substrates. The complexation was performed prior to catalysis by suspending the resin-bound ligands in dichloromethane in the presence of 0.9 eq. of rhodium precursor ([Rh(COD)2]X). After one hour the white resins had turned bright yellow/orange and the now colorless supernatant was removed. Subsequently the supported rhodium complexes were washed with DCM and THF and filtered off. To confirm full complexation, separate in situ NMR experiments were performed which indicated the expected bidentate coordination to the metal center. Upon complexation very broad NMR signals were observed, this phenomenon was already reported for similar resin-supported systems.17,23Table 1 shows the results for the asymmetric hydrogenation of substrates I–III using supported ligands L1–L8. These 8 ligands all bear the same substituents (R1 and R2) and differ only in the number of carbon atoms in the ligand backbone (n) and the configuration of the backbone and phosphite moiety. In all cases full conversion was achieved and enantioselectivities up to 97% were observed. Interestingly it was found that changing the counterion of the Rh-precursor from BF4− to SbF6− (entries 9 and 10) seemed to have a beneficial effect and led to an increase of ee of up to 9%.This counterion effect has already been reported for similar homogeneous phosphite- and diamidophosphite-based systems.24 Looking at the ligand backbone length it can be seen that for substrate II higher enantioselectivities were obtained for ligands having a C3 backbone, n = 1, (L1 and L4, entries 1 and 4) while for substrates I and III better selectivities were observed with supported phosphine–phosphites having a C4 backbone, n = 2, (L6–L7, entries 6–7).
|
|
||||
|---|---|---|---|---|
| Entry | Ligand | Substrate I | Substrate II | Substrate III |
| eeb | eeb | eeb | ||
|
a Reaction conditions: In a stainless steel autoclave, Rh/substrate = 1:30, p(H2) = 1.2 bar, T = 25 °C, t = 16 h, 0.5 mL of THF, all runs were performed in duplicate, full conversion was obtained in all cases, conversion was determined by GC.
b Enantiomeric excess of product determined by chiral GC (absolute configuration drawn in parenthesis).
c Using [Rh(COD)2]SbF6 as metal precursor.
|
||||
| 1 | L1 | 91 (R) | 93 (R) | 90 (R) |
| 2 | L2 | 92 (R) | 85 (R) | 90 (R) |
| 3 | L3 | 91 (S) | 85 (S) | 90 (S) |
| 4 | L4 | 93 (S) | 93 (S) | 93 (S) |
| 5 | L5 | 32 (R) | 66 (R) | 40 (R) |
| 6 | L6 | 94 (R) | 86 (R) | 95 (R) |
| 7 | L7 | 95 (S) | 87 (S) | 96 (S) |
| 8 | L8 | 34 (S) | 66 (S) | 32 (S) |
| 9c | L2 | 97 (R) | 93 (R) | 95 (R) |
| 10c | L6 | 95 (R) | 95 (R) | 96 (R) |
Also the influence of the configuration of the backbone and phosphite moiety was investigated for L1–L8. The (S)-Binol moiety always leads to the (R) enantiomer and an (R)-Binol group yields the (S) product in the asymmetric hydrogenation of substrates I–III (see Table 1). For the supported ligands bearing a C3 backbone (n = 1, L1–L4) a small matched/mismatched effect could be observed with the matched pairs (RC,SC,Sax) and (SC,RC,Rax) achieving up to 8% higher selectivities in the case of substrate II (entries 1 and 4). Interestingly, for L5–L8 (n = 2) a much larger and inverse matched/mismatched effect was observed. In this case the matched pairs are (RC,SC,Rax) and (SC,RC,Sax) (entries 6 and 7) which exhibited differences in enantioselectivity up to 62% when compared to their mismatched counterparts (entries 5 and 8).
Moreover, the effect of altering the substituents R1 and R2 was studied (Table 2, entries 1–4). It was found that replacing the methyl group on the ligand backbone (R2) with a slightly more bulky ethyl group appeared to have a small positive effect on the selectivity. When comparing L6 (Table 1, entry 6) with L9 (Table 2, entry 1) it can be seen that the latter one bearing an ethyl group achieves up to 5% higher enantioselectivities. Exchanging the phenyl substituent on the phosphine moiety (R1) for a cyclohexyl group on the other hand seemed to have a detrimental effect. While L10 (Table 2, entry 2) exhibits similar selectivity in the hydrogenation of I as its phenyl bearing counterpart L1 (Table 1, entry 1) the selectivity for the other two substrates is up to 26% lower. Similarly, the enantioselectivities achieved by L12 (Table 2, entry 4) are comparable to those of its phenyl-bearing counterpart L9 (Table 2, entry 1) for two of the three substrates but for substrate II the selectivity is much lower. More surprisingly, when comparing L11 (Table 2 entry 3) and L5 (Table 1, entry 5) it was observed that in this case changing R1 from a phenyl group to a cyclohexyl group led to the opposite enantiomer with varying levels of selectivity for all three of the tested substrates. This nicely showcases that small changes in ligand structure can have a huge influence on the outcome of a catalytic reaction and demonstrates the power of this effective modular approach towards the synthesis and screening of large supported P-OP ligand libraries.
| Entry | Ligand | Substrate I | Substrate II | Substrate III |
|---|---|---|---|---|
| eeb | eeb | eeb | ||
|
a Reaction conditions: In a stainless steel autoclave, Rh/substrate = 1:30, p(H2) = 1.2 bar, T = 25 °C, t = 16 h, 0.5 mL of THF, all runs were performed in duplicate, full conversion was obtained in all cases, conversion was determined by GC.
b Enantiomeric excess of product determined by chiral GC (absolute configuration drawn in parenthesis).
c Data taken from ref. 18.
|
||||
| 1 | L9 | 96 (R) | 91 (R) | 98 (R) |
| 2 | L10 | 90 (R) | 67 (R) | 66 (R) |
| 3 | L11 | 41 (S) | 64 (S) | 91 (S) |
| 4 | L12 | 95 (R) | 40 (R) | 97 (R) |
| 5 | L13 | 93 (R) | 87 (R) | 92 (R) |
| 6 | L14 | 88 (R) | 76 (R) | 79 (R) |
| 7 | L15 | 95 (R) | 92 (R) | 94 (R) |
| 8 | L16 | 46 (R) | 2 (R) | 15 (R) |
| 9c | L17 | 97 (R) | — | 96 (R) |
Lastly different phosphite moieties and their influence on the selectivity were investigated. Both a slightly more bulky SiMe3 substituted (S)-binol and an achiral t-butyl substituted bisphenol derived phosphite were employed. From Table 2 it can be concluded that changing to a SiMe3 substituted binol moiety (L13 and L14, entries 5 and 6) in most cases has a small but positive effect on the selectivity when compared to their non-substituted binol counterparts L2 and L6 (Table 1, entries 2 and 6). Going to the achiral t-butyl bisphenol phosphite did seem to have a large influence resulting in lower enantioselectivities. For L15 with a C3 ligand backbone (Table 2, entry 7) enantioselectivities up to 11% lower than for the binol bearing counterpart L2 (Table 1, entry 2) were observed. In the case of L16 bearing a C4 backbone (Table 2, entry 8) the effect of changing to the achiral bisphenol moiety was much larger. For L16 a significant drop in selectivity of respectively 48% and 80% was found for substrates I and III when compared to the parent ligand L6 (Table 1, entry 6). Moreover for substrate II surprisingly, a complete loss of selectivity was observed.
When compared to phosphine–phosphite L17 reported by Bakos et al. (Table 2, entry 9),18 essentially the homogeneous counterpart of resin-bound ligand L2, it was found that some of the supported ligands performed very well. For substrate I it can be seen that comparable enantioselectivities were obtained (Table 1, entry 9) and for substrate III supported P-OP ligand L9 even outperforms it solution-phase analogue by 2% (Table 2, entry 1). This is quite remarkable as in most known cases the immobilization of a homogeneous catalyst has a detrimental effect on the selectivity. This shows that the modular solid-phase synthetic approach demonstrated here not only enables the facile synthesis and screening of large P-OP ligand libraries but can also actually lead to highly selective supported catalysts.
Supported catalysis recycling
To assess the reusability of these supported phosphine–phosphites the best performing member of the ligand library L9 was employed in the asymmetric hydrogenation of acetamidocinnamic acid methyl ester III. The catalyst recycling was performed under batch conditions in a Schlenk vessel under a flow of hydrogen. In between each reaction cycle the resin was washed with substrate solution while maintaining a hydrogen atmosphere to ensure catalyst stability. Finally the reaction time was reduced in the recycling experiments to study recycling at low conversion, which gives a better indication of the actual catalyst stability.The results of the catalyst recycling experiments are summarized in Fig. 6. Merrifield supported ligand L9 could be reused up to 7 times without any loss of activity. After 11 runs only a very marginal drop in conversion of 3.3% was observed. The loss of activity might have been caused by the possible introduction of trace amounts of moisture or air during work-up in between reaction cycles. Moreover the selectivity stayed constant (average 97.2%) over the full extent of the recycling experiments.
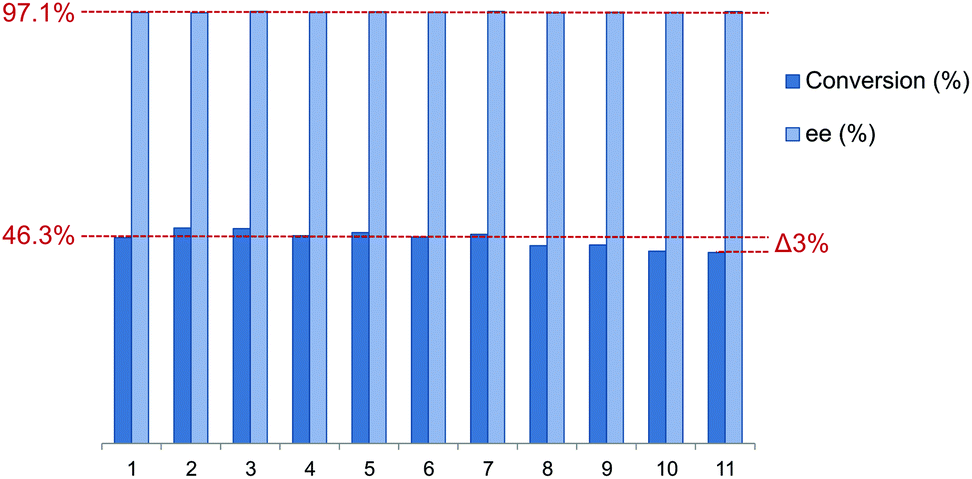 | ||
| Fig. 6 Results of catalyst recycling of supported ligand L9 in the Rh-catalyzed asymmetric hydrogenation of substrate III. Reaction conditions: In a Schlenk vessel under H2 atmosphere, Rh/substrate = 1:30, p(H2) = 1 atm, T = 25 °C, t = 20 min, 1.5 mL of THF, all runs were performed in duplicate. | ||
Also the metal leaching into solution after each reaction cycle was analyzed using ICP-OES. After the first reaction cycle an initial Rh leaching of 1.6 ppm was found and the metal leaching appeared to stay constant after 3 recycling runs and only a minimal leaching of on average 1.3 ppm was detected (for full results see ESI, Table S-I†). The fact that no initial decrease in catalytic activity was observed despite that some metal leaching was detected, seems to indicate the leaching might actually be caused by physically adsorbed rhodium residues residing in the pores of the resin. Moreover no phosphorus leaching could be observed after analyzing the reaction solution with 31P NMR which also supports that the small decrease in catalytic activity is probably caused by other deactivation processes than leaching of active complex into solution. As phosphite-based ligands are prone to hydrolysis and phosphines to oxidation L9 shows remarkable stability for a phosphine–phosphite.25 Potentially this type of supported P-OP ligands could provide promising candidates for catalysis under flow conditions.
Conclusion
A new modular solid-phase synthetic procedure allowing the facile synthesis of large and diverse libraries of phosphine–phosphites was developed. Using this approach a library of 16 phosphine–phosphite ligands could be obtained in very high yields. Moreover only a very minimal work-up was required between reaction steps as opposed to the laborious purification methods employed in the solution-phase synthesis of similar ligands, often resulting in low yields. The supported ligand library was successfully employed in Rh-catalyzed asymmetric hydrogenation and three benchmark substrates were screened. Very high enantioselectivities up to 98% were achieved. Moreover some members of the ligand library even outperformed a homogeneous counterpart showing that the immobilization did not have a detrimental effect on the selectivity. Finally supported ligand L9 was employed in catalyst recycling under batch conditions. The ligand, immobilized on Merrifield resin, could be successfully reused for 11 reaction cycles showing only a very minor loss of activity. Moreover, no decrease in selectivity was observed throughout the recycling experiments. The exhibited recyclability is remarkably high for these types of ligands and might make this system highly suitable for catalysis under flow conditions.Experimental section
General experimental
All reactions and manipulations were carried out under inert conditions using standard Schlenk techniques or in an MBraun glovebox unless stated otherwise. All glassware was dried prior to use to remove traces of water. All chemicals were obtained from commercial suppliers and used as received unless otherwise stated. Diethyl ether and THF were distilled from sodium/benzophenone and triethylamine, dichloromethane and acetonitrile were distilled from calcium hydride. Novabiochem™ Merrifield resin (100–200 mesh, 1.24 mmol g−1 or 1.48 mmol g−1, 1% cross-linked) was obtained from EMD Millipore.General procedure for the synthesis of resin-bound phosphine–boranes (1a–b·BH3)
Step 1. Merrifield resin (2.0 g, 1.24 mmol g−1, 2.48 mmol, 1.0 eq.), was swollen in THF (50 mL) and cooled to −78 °C. A freshly prepared primary lithium phosphide solution (20 mL, 0.15 M, 1.2 eq.), also cooled to −78 °C was added under gentle stirring to avoid mechanical abrasion of the resin. The reaction mixture was allowed to warm up to room temperature and was left overnight without stirring. The supernatant was removed and the resin was washed subsequently with three 20 mL portions of THF followed by three 20 mL portions of Et2O. The product was directly used in the next step without additional purification.Step 2. A resin-bound phosphine, synthesized in the previous step, was swollen in THF (50 mL). Next, BH3·SMe2 (12.5 mL, 2.0 M in toluene, 10 eq.) was added under gentle stirring to avoid mechanical abrasion of the resin. Upon addition the resin colored white and the reaction was stopped when full conversion was observed by 31P NMR. Next, the supernatant was removed and the resin was washed subsequently with three 20 mL portions of THF followed by three 20 mL of Et2O. The product was dried in vacuo yielding a white resin-bound phosphine–borane.
General procedure for the synthesis of resin-bound phosphine–borane sulfates (3a–h·BH3)
Step 1. A resin-bound phosphine–borane (1a–b·BH3, 500 mg, ∼0.6 mmol) was swollen in THF (20 mL). Next, LDA (3 mL, 2.0 M in THF/heptane/ethylbenzene, 10 eq.) was added under gentle stirring to avoid mechanical abrasion of the resin. Upon addition the resin colored dark brown and was allowed to react for 3 hours. Next, the supernatant was removed and the resin was washed subsequently with three 10 mL portions of THF followed by three 10 mL portions of Et2O. The product was used in the next step without additional purification.Step 2. A lithiated resin-bound phosphine–borane synthesized in the previous step was swollen in THF (15 mL). A cyclic sulfate (2a–e, 0.72 mmol, 1.2 eq.) was azeotropically dried with toluene (3 times), dissolved in THF (5 mL) and subsequently added to the resin under gentle stirring to avoid mechanical abrasion. Upon addition the resin turned from dark brown to yellow and was allowed to react overnight. Next, the supernatant was removed and the resin was washed subsequently with three 10 mL portions of THF and three 10 mL portions of Et2O. The product was dried in vacuo yielding a light yellow resin. The product was used in the next step without additional purification.
General procedure for the synthesis of resin-bound hydroxyalkyl phosphine–boranes (4a–h·BH3)
A resin-bound phosphine–borane sulfate (3a–h·BH3, 500 mg, ∼0.5 mmol) was swollen, under gentle stirring, in a 1:1 mixture of THF and 0.1 M of degassed H2SO4 (20 mL). The resin was left overnight without stirring to avoid mechanical abrasion of the resin. Next the resin was washed with three 5 mL portions of THF and resuspended in THF (10 mL). The progress of the hydrolysis was monitored using 7Li NMR. If no full consumption of Li was observed the resin was resuspended in a fresh mixture of THF and H2SO4 and left overnight. This procedure was repeated until no lithium signal could be observed anymore by 7Li NMR (on average 3 days). Next, the supernatant was removed and the resin was washed subsequently with five 10 mL portions of THF followed by three 10 mL portions of Et2O. The product was dried in vacuo affording a white resin which was used in the next step without additional purification.
General procedure for the synthesis of resin-bound phosphine–phosphites (L1–L16)
Step 1. A resin-bound hydroxyalkyl phosphine–borane (4a–h·BH3, 250 mg, ∼0.25 mmol) was suspended in THF (5 mL) and a solution of 1,4-diazabicyclo[2.2.2]octane (5 mL, 0.5 M in THF, 10 eq.) was added. The reaction was heated to 40 °C and was left overnight without stirring. After complete deprotection was confirmed by 31P NMR, the supernatant was removed and the resin was washed subsequently with three 5 mL portions of THF followed by three 5 mL portions of Et2O. The product was used directly in the next step without further purification.Step 2. A deprotected resin-bound hydroxyalkyl phosphine synthesized in the previous step was swollen in THF (10 mL) and triethylamine (2.25 mmol, 9.0 eq.) was added. A chlorophosphite (0.75 mmol, 3.0 eq.) was dissolved in THF (5 mL) and was added to the resin at 0 °C under gentle stirring to avoid mechanical abrasion of the resin. Upon addition a precipitate was formed. The reaction was monitored using 31P NMR and full conversion was reached when a 1:1 ratio of phosphine to phosphite was observed (2–16 hours). Next, the supernatant was removed and the resin was washed subsequently with three 5 mL portions of DCM, three 5 mL portions of THF and three 5 mL portions of Et2O. The product was dried in vacuo yielding a white resin-bound phosphine–phosphite.
General procedure for Rh-catalyzed asymmetric hydrogenation
The hydrogenation experiments were performed in a stainless steel autoclave charged with an insert suitable for 10 reaction vessels including Teflon mini stirring bars. In a typical experiment, a reaction vessel was charged with a resin-bound phosphine–phosphite (5 mg, ∼4.0 μmol) and a solution of [Rh(COD)2]X (0.9 eq.) in CH2Cl2 (1 mL) was added and the heterogeneous mixture was allowed to stir gently for 4 h. The supernatant was removed and the resulting orange resin was washed subsequently with three 1 mL portions of THF followed by three 1 mL portions of Et2O. Next, a solution of substrate (0.5 mL, 0.24 M, 30 eq.) in THF was added to the reaction vessel. Subsequently, the autoclave was purged three times with 5 bar of H2 and then pressurized to 1.2 bar. The reaction mixtures were gently stirred at 25 °C. After 16 h, the autoclave was depressurized and the reaction mixtures were filtered over a plug of silica. Prior to GC measurements substrate II and its products were derivatized using (trimethylsilyl)diazomethane (2 M in diethyl ether), in essence yielding substrate III. The conversion and the enantiomeric excess were determined by chiral GC, see ESI† for column and conditions.Acknowledgements
We thank the European Union (Marie Curie ITN SusPhos, grant agreement no. 317404) for financial support.Notes and references
- (a) H. U. Blaser and E. Schmidt, Asymmetric Catalysis on Industrial Scale, ed. H. U. Blaser and E. Schmidt, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2004 Search PubMed; (b) I. Ojima, Catalytic asymmetric synthesis, John Wiley & Sons, Inc., Hoboken, 3rd edn, 2010 Search PubMed.
- T. P. Yoon and E. N. Jacobsen, Science, 2003, 299, 1691 CrossRef CAS PubMed.
- (a) K. N. Houk and P. H.-Y. Cheong, Nature, 2008, 455, 309 CrossRef CAS PubMed; (b) M. C. Kozlowski and M. Panda, J. Org. Chem., 2003, 68, 2061 CrossRef CAS PubMed.
- (a) M. B. Francis, N. S. Finney and E. N. Jacobsen, J. Am. Chem. Soc., 1996, 118, 8983 CrossRef CAS; (b) R. Kranich, K. Eis, O. Geis, S. Mühle, J. W. Bats and H.-G. Schmalz, Chem. – Eur. J., 2000, 6, 2874 CrossRef CAS PubMed; (c) C. Gennari and U. Piarulli, Chem. Rev., 2003, 103, 3071 CrossRef CAS PubMed.
- (a) M. T. Reetz, Angew. Chem., Int. Ed., 2001, 40, 284 CrossRef CAS; (b) J. G. de Vries and A. H. M. de Vries, Eur. J. Org. Chem., 2003, 799 CrossRef CAS.
- (a) P. E. Goudriaan, X.-B. Jang, M. Kuil, R. Lemmens, P. W. N. M. van Leeuwen and J. N. H. Reek, Eur. J. Org. Chem., 2008, 6079 CrossRef CAS; (b) B. Breit, Angew. Chem., Int. Ed., 2005, 44, 6816 CrossRef CAS PubMed; (c) G. Hattori, T. Hori, Y. Miyake and Y. Nishibayashi, J. Am. Chem. Soc., 2007, 129, 12930 CrossRef CAS PubMed.
- (a) L. Lefort, J. A. F. Boogers, A. H. M. de Vries and J. G. de Vries, Org. Lett., 2004, 6, 1733 CrossRef CAS PubMed; (b) M. T. Reetz and G. Mehler, Tetrahedron Lett., 2003, 44, 4593 CrossRef CAS; (c) M. L. Johansson, S. Berglund, Y. Hu, K. H. O. Andersson and N. Kann, ACS Comb. Sci., 2012, 14, 304 CrossRef CAS PubMed.
- P. E. Goudriaan, P. W. N. M. van Leeuwen, M. N. Birkholz and J. N. H. Reek, Eur. J. Inorg. Chem., 2008, 2939 CrossRef CAS.
- (a) S. Lühr, J. Holz and A. Börner, ChemCatChem, 2011, 3, 1708 CrossRef; (b) S. H. Chikkali, J. I. van der Vlugt and J. N. H. Reek, Coord. Chem. Rev., 2014, 262, 1 CrossRef CAS; (c) H. Fernandez-Perez, P. Etayo, A. Panossian and A. Vidal-Ferran, Chem. Rev., 2011, 111, 2119 CrossRef CAS PubMed.
- N. Sakai, S. Mano, K. Nozaki and H. Takaya, J. Am. Chem. Soc., 1993, 115, 7033 CrossRef CAS.
- (a) A. Suárez, M. A. Méndez-Rojas and A. Pizzano, Organometallics, 2002, 21, 4611 CrossRef; (b) P. W. N. M. van Leeuwen, P. C. J. Kamer, C. Claver, O. Pamies and M. Dieguez, Chem. Rev., 2011, 111, 2077 CrossRef CAS PubMed.
- (a) C. F. Czauderna, D. B. Cordes, A. M. Z. Slawin, C. Müller, J. I. van der Vlugt, D. Vogt and P. C. J. Kamer, Eur. J. Inorg. Chem., 2014, 1797 CrossRef CAS; (b) M. Dindaroğlu, A. Falk and H.-G. Schmalz, Synthesis, 2013, 527 Search PubMed; (c) H. Fernández-Pérez, M. A. Pericàs and A. Vidal-Ferran, Adv. Synth. Catal., 2008, 350, 1984 CrossRef; (d) J. L. Núñez-Rico, P. Etayo, H. Fernández-Pérez and A. Vidal-Ferran, Adv. Synth. Catal., 2012, 354, 3025 CrossRef; (e) J. Velder, T. Robert, I. Weidner, J.-M. Neudörfl, J. Lex and H.-G. Schmalz, Adv. Synth. Catal., 2008, 350, 1309 CrossRef CAS; (f) O. Pàmies, M. Diéguez, G. Net, A. Ruiz and C. Claver, J. Org. Chem., 2001, 66, 8364 CrossRef.
- (a) S. E. Booth, C. M. Dreef-Tromp, P. H. H. Hermkens, J. A. P. A. de Man and H. C. J. Ottenheijm, in Combinatorial Chemistry - Synthesis, Analysis, Screening, ed. G. Jung, Wiley-VCH Verlag GmbH, Weinheim, 1999, pp. 35–76 Search PubMed; (b) D. Obrecht and J. M. Villalgordo, in Solid-Supported Combinatorial and Parallel Synthesis of Small-Molecular-Weight Compound Libraries, Elsevier Science ltd, Oxford, 1998, pp. 1–184 Search PubMed; (c) R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149 CrossRef CAS.
- M. C. Samuels, B. H. G. Swennenhuis and P. C. J. Kamer, in Phosphorus(III) Ligands in Homogeneous Catalysis: Design and Synthesis, ed. P. C. J. Kamer and P. W. N. M. van Leeuwen, John Wiley & Sons, Ltd, 2012, pp. 463–479 Search PubMed.
- (a) J. N. H. Reek, P. W. N. M. van Leeuwen, A. G. J. Ham and A. B. Haan, in Catalyst Separation, Recovery and Recycling, ed. D. J. Cole-Hamilton and R. P. Tooze, Springer, Netherlands, 2006, pp. 39–72 Search PubMed; (b) T. E. Kristensen and T. Hansen, in Catalytic Methods in Asymmetric Synthesis, ed. M. Gruttadauria and F. Giacalone, John Wiley & Sons, Inc., 2011, pp. 209–256 Search PubMed; (c) G. Zhao and Z. Chai, in Recoverable and Recyclable Catalysts, ed. M. Benaglia, John Wiley & Sons, Ltd, 2009, pp. 49–75 Search PubMed; (d) A. F. Trindade, P. M. P. Gois and C. A. M. Afonso, Chem. Rev., 2009, 109, 418 CrossRef CAS PubMed.
- (a) G. Y. Li, P. J. Fagan and P. L. Watson, Angew. Chem., Int. Ed., 2001, 113, 1140 CrossRef; (b) R. den Heeten, B. H. G. Swennenhuis, P. W. N. M. van Leeuwen, J. G. de Vries and P. C. J. Kamer, Angew. Chem., Int. Ed., 2008, 47, 6602 CrossRef CAS PubMed; (c) A. Mansour and M. Portnoy, J. Chem. Soc., Perkin Trans. 1, 2001, 952 RSC; (d) B. Bar-Nir Ben-Aroya and M. Portnoy, Tetrahedron, 2002, 58, 5147 CrossRef CAS; (e) A. Mansour and M. Portnoy, Tetrahedron Lett., 2003, 44, 2195 CrossRef CAS.
- F. J. L. Heutz, M. C. Samuels and P. C. J. Kamer, Catal. Sci. Technol., 2015, 5, 3296 CAS.
- G. Farkas, S. Balogh, Á. Szöllősy, L. Ürge, F. Darvas and J. Bakos, Tetrahedron: Asymmetry, 2011, 22, 2104 CrossRef CAS.
- (a) A. Panossian, H. Fernández-Pérez, D. Popa and A. Vidal-Ferran, Tetrahedron: Asymmetry, 2010, 21, 2281 CrossRef CAS; (b) C. Hegedüs, H. Gulyás, Á. Szöllősy and J. Bakos, Inorg. Chim. Acta, 2009, 362, 1650 CrossRef.
- G. Fries, J. Wolf, K. Ilg, B. Walfort, D. Stalke and H. Werner, Dalton Trans., 2004, 1873 RSC.
- P. W. N. M. van Leeuwen and J. C. Chadwick, in Homogeneous Catalysts: Activity - Stability - Deactivation, Wiley-VCH Verlag GmbH & Co. KGaA, 2011, pp. 1–49 Search PubMed.
- (a) S. Deerenberg, P. C. J. Kamer and P. W. N. M. van Leeuwen, Organometallics, 2000, 19, 2065 CrossRef CAS; (b) S. Deerenberg, H. S. Schrekker, G. P. F. van Strijdonck, P. C. J. Kamer, P. W. N. M. van Leeuwen, J. Fraanje and K. Goubitz, J. Org. Chem., 2000, 65, 4810 CrossRef CAS PubMed.
- T. T. Adint and C. R. Landis, J. Am. Chem. Soc., 2014, 136, 7943 CrossRef CAS PubMed.
- (a) Z. Hua, V. C. Vassar and I. Ojima, Org. Lett., 2003, 5, 3831 CrossRef CAS PubMed; (b) K. N. Gavrilov, S. V. Zheglov, P. A. Vologzhanin, E. A. Rastorguev, A. A. Shiryaev, M. G. Maksimova, S. E. Lyubimov, E. B. Benetsky, A. S. Safronov, P. V. Petrovskii, V. A. Davankov, B. Schäffner and A. Börner, Russ. Chem. Bull., 2008, 57, 2311 CrossRef CAS.
- (a) R. B. Bedford, S. J. Coles, M. B. Hursthouse and V. J. Scordia, Dalton Trans., 2005, 991 RSC; (b) R. Chen, J. Jiang, Y. Wang and Z. Jin, J. Mol. Catal. A: Chem., 1999, 149, 113 CrossRef CAS; (c) B. H. G. Swennenhuis, R. Chen, P. W. N. M. van Leeuwen, J. G. de Vries and P. C. J. Kamer, Eur. J. Org. Chem., 2009, 5796 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and characterisation data. See DOI: 10.1039/c5dt03226a |
| This journal is © The Royal Society of Chemistry 2016 |