
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Spontaneous dehydrocoupling in peri-substituted phosphine–borane adducts†
Laurence J.
Taylor
,
Brian A.
Surgenor
,
Piotr
Wawrzyniak
,
Matthew J.
Ray
,
David B.
Cordes
,
Alexandra M. Z.
Slawin
and
Petr
Kilian
*
School of Chemistry, EastCHEM, University of St Andrews, St Andrews, Fife, KY16 9ST, UK. E-mail: pk7@st-andrews.ac.uk
First published on 21st August 2015
Abstract
Bis(borane) adducts Acenap(PiPr2·BH3)(PRH·BH3) (Acenap = acenaphthene-5,6-diyl; 4a, R = Ph; 4b, R = ferrocenyl, Fc; 4c, R = H) were synthesised by the reaction of excess H3B·SMe2 with either phosphino-phosphonium salts [Acenap(PiPr2)(PR)]+Cl− (1a, R = Ph; 1b, R = Fc), or bis(phosphine) Acenap(PiPr2)(PH2) (3). Bis(borane) adducts 4a–c were found to undergo dihydrogen elimination at room temperature, this spontaneous catalyst-free phosphine-borane dehydrocoupling yields BH2 bridged species Acenap(PiPr2)(μ-BH2)(PR·BH3) (5a, R = Ph; 5b, R = Fc; 5c, R = H). Thermolysis of 5c results in loss of the terminal borane moiety to afford Acenap(PiPr2)(μ-BH2)(PH) (14). Single crystal X-ray structures of 3, 4b and 5a–c are reported.
Introduction
Dehydrocoupling reactions (E–H + E′–H → E–E′ + H2) are an interesting and effective way of generating bonds between main-group elements, with concomitant evolution of H2. Reactions of this type show applications not only in inorganic synthesis, but also in hydrogen storage, transfer hydrogenation and polymer synthesis.1–6Although dehydrocoupling reactions that occur by thermal or autocatalytic routes are known,7,8 the vast majority of recent work has focused on catalysis, particularly with transition metals.6,9–11 In particular, amine–borane adducts have attracted considerable interest as potential hydrogen storage molecules.12,13 However, dehydrocoupling reactions in the chemically related phosphine–boranes have received far less attention.11,14
Dehydrocoupling of phosphine–boranes to form poly(phosphinoboranes) was first reported in the 1950s. Early work in this area is limited, with polymerisations yielding low molecular weight polymers which were often poorly characterised.15,16 In 1999 the Manners’ group pioneered the use of transition metal catalysts in the synthesis of poly(phosphinoboranes)17–20 and more recently B(C6F5)3 has been used as a metal-free dehydrocoupling catalyst.21 Thus formed inorganic polymers have interesting and unusual physical properties, which set them apart from the more traditional carbon-based polymers.17,18
While catalysts are incredibly useful, they are often expensive, especially when they contain precious transition metals such as Rh or Ir. As such, it would be helpful to develop systems which undergo dehydrocoupling without the addition of an external catalyst, but while still under mild conditions. The work of our group has focused on peri-substitution, which is useful in thermodynamically stabilising bonding motifs which are typically unstable at room temperature.22,23 However, lately we have been intrigued by the possibility of using peri-substitution to promote reactivity that would typically require the addition of a catalyst. Due to the unique constraints of the peri-geometry, atoms in the peri-position (E) are forced into close proximity. Strain from the overlap of occupied orbitals can be relieved by, either, the formation of a direct E–E bonding interaction or a bridging motif between the two peri-atoms (E–X–E). As such, it was postulated that if two potentially reactive groups were placed in the peri-positions, the rigid scaffold could lower the kinetic barrier of the coupling reaction, promoting the formation of a direct bond or a bridging motif and hence emulating the role of an external catalyst.
This was indeed found to be the case, as a series of peri-substituted phosphine–borane adducts were synthesised and observed to undergo spontaneous intramolecular dehydrocoupling in solution at room temperature. The results of these investigations are detailed below.
Results and discussion
Bis(borane) Adducts 4a–c
Compounds 1a–b and 2 were used as the starting points for all of the reactions presented in this work. Compound 2 was synthesised according to a previously published procedure,24 while compounds 1a–b were synthesised via a modified version of the literature procedure.25The synthesis and characterisation of the bis(borane) adduct 4a were recently reported by our group.26 In its preparation, treatment of the phosphino-phosphonium salt 1a with excess H3B·SMe2 resulted in borane mediated reduction to afford 4a as a yellow oil in quantitative yield (Scheme 1). An analogous procedure was employed to obtain adduct 4b from the corresponding phosphino-phosphonium salt 1b. The adduct 4b was isolated as an orange solid, which was contaminated with the bridged compound 5b (≈20% as judged by 1H and 31P NMR). Pure 4b was obtained by recrystallisation from acetonitrile.
 | ||
| Scheme 1 Synthesis of bis(borane) adducts 4a–c and BH2 bridged compounds 5a–c. | ||
The 31P{1H} NMR spectrum of 4b exhibits broad singlets at δP 36.3 (iPr2P) and −7.7 (PFcH), and in the 31P NMR spectrum the signal at δP −7.7 is split into a broad doublet (1JPH = 395 Hz). Crystals of 4b suitable for X-ray diffraction were grown from acetonitrile, the structure is shown in Fig. 2 and Tables 1–3. The structure of 4b is similar to the previously reported structure of 4a,26 with a P⋯P distance of 3.521(1) Å and a large positive splay angle of +21.2(7)° (see Fig. 1 for a definition), indicating significant repulsion between the two peri-groups. Additionally, both phosphorus atoms show significant displacement from the mean plane of the acenaphthene ring (0.706 Å for P1, 0.546 Å for P9).
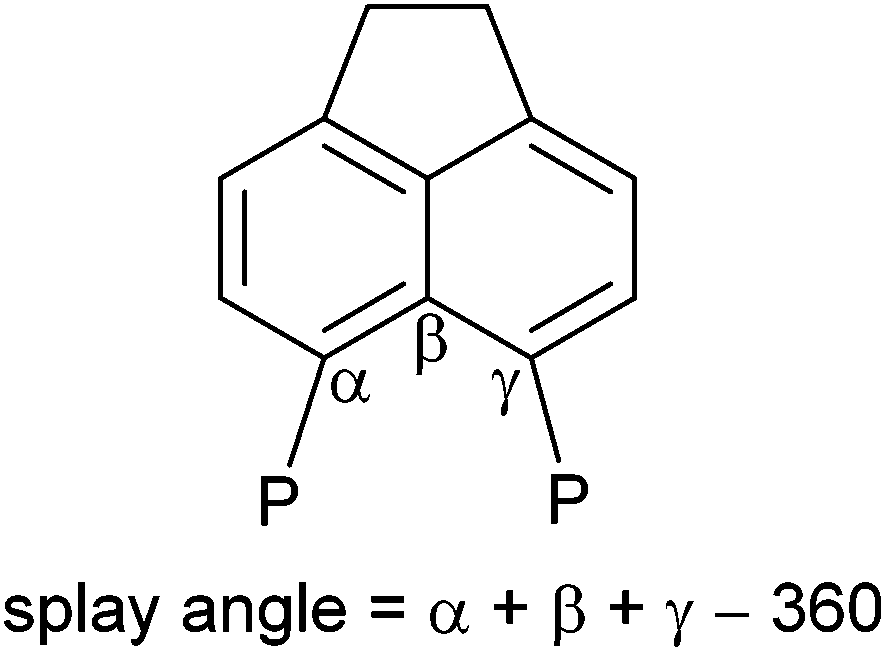 | ||
| Fig. 1 Definition of a splay angle. | ||
 | ||
| Fig. 2 Structures of 3 (top) and 4b (bottom) in the solid state. Carbon-bound hydrogen atoms omitted for clarity. | ||
| a Measurements for second molecule in asymmetric unit shown in square brackets. | |||
|---|---|---|---|
| 3 | |||
| C1–P1 | 1.849(4) | C9–P9 | 1.850(4) |
| C1–P1–H1a | 99(1) | C1–P1–H1b | 94(1) |
| H1a–P1–H1b | 90(2) | ||
| 4b | |||
| C1–P1 | 1.817(3) | C9–P9 | 1.842(3) |
| B1–P1 | 1.929(4) | B9–P9 | 1.980(4) |
| C1–P1–B1 | 109.7(2) | B1–P1–H1 | 117(2) |
| C1–P1–H1 | 105(2) | C9–P9–B9 | 113.6(2) |
| 5a | |||
| C1–P1 | 1.818(2) | C9–P9 | 1.812(2) |
| B1–P1 | 1.932(2) | B9–P9 | 1.927(2) |
| B9–P1 | 1.928(2) | ||
| C1–P1–B1 | 112.21(8) | C9–P9–B9 | 110.95(8) |
| C1–P1–B9 | 106.33(8) | P9–B9–P1 | 108.55(9) |
| B9–P1–B1 | 117.28(9) | ||
| 5b | |||
| C1–P1 | 1.82(1) [1.85(1)] | C9–P9 | 1.82(1) [1.80(2)] |
| B1–P1 | 1.94(2) [1.94(2)] | B9–P9 | 1.92(2) [1.90(2)] |
| B9–P1 | 1.94(2) [1.94(2)] | ||
| C1–P1–B1 | 111.6(7) [108.6(7)] | C9–P9–B9 | 114.1(7) [108.3(7)] |
| C1–P1–B9 | 111.3(7) [107.4(7)] | P9–B9–P1 | 110.9(9) [106.6(8)] |
| B9–P1–B1 | 114.0(9) [118.2(8)] | ||
| 5c | |||
| C1–P1 | 1.820(2) [1.821(2)] | C9–P9 | 1.808(2) [1.807(2)] |
| B1–P1 | 1.937(2) [1.930(2)] | B9–P9 | 1.910(2) [1.911(2)] |
| B9–P1 | 1.922(2) [1.914(2)] | ||
| C1–P1–B1 | 111.90(9) [112.48(9)] | C9–P9–B9 | 110.15(8) [109.62(8)] |
| C1–P1–B9 | 107.56(8) [107.54(8)] | P9–B9–P1 | 109.1(1) [109.0(1)] |
| B9–P1–B1 | 118.16(9) [117.60(9)] | ||
| 3 | 4b | 5a | 5b | 5c | |
|---|---|---|---|---|---|
| a Measurements for second molecule in asymmetric unit shown in square brackets. | |||||
| P1⋯P9 | 3.143(1) | 3.521(1) | 3.1295(8) | 3.181(5) [3.081(5)] | 3.1214(6) [3.1145(6)] |
| splay angle | +16.4(7) | +21.2(7) | +15.1(4) | +16(3) [+15(3)] | +16.5(3) [+16.3(3)] |
| Out-of-plane displacement (P1) | 0.148 | 0.706 | 0.338 | 0.332 [0.213] | 0.050 [0.042] |
| Out-of-plane displacement (P9) | 0.068 | 0.546 | 0.327 | 0.450 [0.296] | 0.068 [0.101] |
| 3 | 4b | 5a | 5b | 5c | |
|---|---|---|---|---|---|
| a I > 2σ(I), R1 = ∑(||Fo| − |Fc||)/∑|Fo|. b wR2 = [∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]]1/2, w = 1/[σ2(Fo2) + [(ap)2 + bp], where p = [(Fo2) + 2Fc2]/3. | |||||
| Chemical formula | C18H24P2 | C28H38B2FeP2 | C24H32B2P2 | C28H36B2FeP2 | C18H28B2P2 |
| Formula weight | 302.34 | 514.02 | 404.08 | 512.01 | 327.99 |
| Crystal dimensions (mm) | 0.12 × 0.10 × 0.03 | 0.10 × 0.10 × 0.01 | 0.10 × 0.06 × 0.06 | 0.20 × 0.03 × 0.01 | 0.18 × 0.12 × 0.08 |
| Crystal system | Triclinic | Monoclinic | Triclinic | Monoclinic | Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c |
P |
P21 | P21/c |
| a (Å) | 7.4543(19) | 15.806(5) | 8.3375(11) | 15.409(5) | 14.0458(16) |
| b (Å) | 8.4923(15) | 13.022(4) | 9.4724(14) | 11.059(3) | 18.841(2) |
| c (Å) | 14.361(5) | 12.835(4) | 16.108(2) | 16.712(6) | 14.4671(14) |
| α (°) | 79.88(3) | 90.0000 | 101.8880(17) | 90.0000 | 90.0000 |
| β (°) | 82.10(3) | 94.825(6) | 93.165(3) | 114.651(5) | 101.176(3) |
| γ (°) | 66.47(2) | 90.0000 | 112.772(3) | 90.0000 | 90.0000 |
| V (Å3) | 818.3(4) | 2632.4(14) | 1134.9(3) | 2588.3(14) | 3768.3(7) |
| Z | 2 | 4 | 2 | 4 | 8 |
| D calc (g cm−3) | 1.227 | 1.297 | 1.182 | 1.314 | 1.156 |
| μ (cm−1) | 2.546 | 7.088 | 1.989 | 7.207 | 2.244 |
| No. rflns measured (unique) | 5176 (2878) | 27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 526 (4765) 526 (4765) |
14052 (4112) |
34241 (9417) |
45236 (6914) |
|
R
1a |
0.0643 | 0.0513 | 0.0362 | 0.0948 | 0.0360 |
| wR2b |
0.1599 | 0.1533 | 0.1041 | 0.2647 | 0.1045 |
The bis(borane) adduct 4c was synthesised from the novel primary phosphine 3 (Scheme 1), which was obtained by clean reduction of the phosphonium-phosphoranide 2 with LiAlH4. The 31P{1H} NMR spectrum of compound 3 displays two doublets at δP −11.3 (iPr2P) and −101.2 (PH2), with a substantial through-space coupling of 4JPP = 205 Hz. In the 31P NMR spectrum, the signal for the PH2 group is split into a pseudo-quartet due to 1JPH = 204 Hz being very similar to that of 4JPP. The 1H NMR spectrum of 3 displays a doublet of doublets for the PH2 protons (δH 4.98, 1JHP = 204 Hz, 5JHP = 48 Hz). This long range 5JHP interaction, in addition to the large 4JPP coupling, indicates a significant through space contribution to coupling operates in this compound.27 Crystals of compound 3 suitable for single crystal X-ray diffraction were grown from THF, the structure is shown in Fig. 2 and Tables 1–3. The structure indicates a clear repulsive interaction between the two phosphorus moieties, with a P⋯P distance of 3.143(1) Å and a positive splay angle of 16.4(7)°. The purity of 3 as obtained from the reaction was established by 31P, 1H and 13C NMR and was found to be sufficient for further syntheses.
Treatment of primary bis(phosphine) 3 with excess H3B·SMe2 afforded bis(borane) adduct 4c as the major product (δP 38.0 (br s, iPr2P), −40.8 (br s, PH2)), although the reaction was not clean. Even with a large excess (12 equivalents) of H3B·SMe2, traces of starting material were found in the 31P{1H} NMR spectra of the crude mixture after the reaction. In addition, a broad singlet at δP 44.0 along with a sharp singlet at δP −101.4 were observed in this spectrum. Rather revealingly, the signal at δP −101.4 splits into a triplet (1JPH = 207 Hz) in the 31P NMR spectrum which, together with the chemical shift values, allowed these signals to be assigned to the monoborane adduct 6 (Scheme 2). In the crude mixture, 4c and 6 were present in a ratio of approximately 5:1. A number of minor, unidentified P containing side products were also formed.
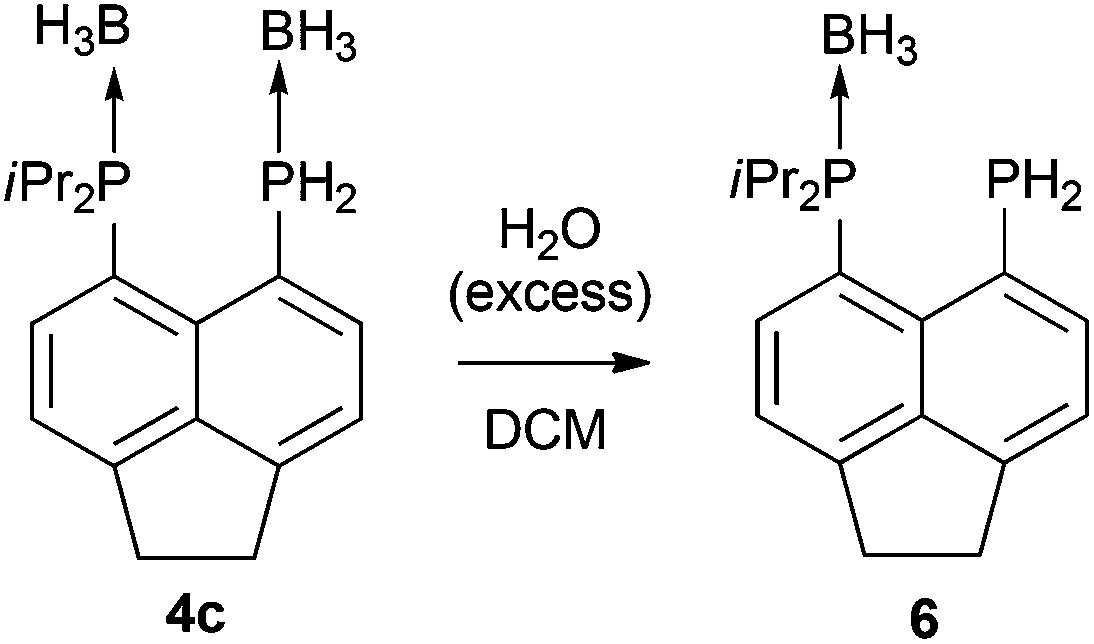 | ||
| Scheme 2 Synthesis of monoborane adduct 6. | ||
In sharp contrast to compounds 4a and 4b, which are stable towards both air and moisture, compound 4c is rather moisture sensitive. On a preparative scale, treatment of a dichloromethane solution of 4c with degassed water afforded 6 as a yellow solid in near quantitative yield (Scheme 2). The new compound was characterised by 1H, 31P, 31P{1H}, 13C{1H}, 11B, and 11B{1H} NMR spectroscopy.
Primary phosphine–borane adducts have been less extensively studied than secondary or tertiary phosphine–boranes, and are known to be generally less stable.28 In addition, steric hindrance arising from the peri-geometry is likely to further destabilise the bis(borane) adduct 4c with respect to the monoborane adduct 6. This corresponds well with our observations of the instability of 4c towards moisture, as well as the difficulty in getting complete conversion to the bis(borane) adduct. Compound 4c could not be isolated in analytically pure form due to its crystallisation being extremely difficult, whilst its sensitivity to air and moisture prevented chromatographic purification.
Compound 4c exhibits two broad singlets in the 31P{1H} NMR spectrum at δP 38.0 (iPr2P) and δP −40.8 (PH2). In the 31P NMR spectrum, the signal at δP −40.8 splits into a broad triplet (1JPH = 379 Hz). One particularly distinctive signal is observed for the PH2 group in the 1H NMR spectrum, which is split into a doublet of quartets (δH 6.14, 1JHP = 377 Hz, 3JHH = 7.1 Hz) due to coupling to the adjacent BH3 hydrogen atoms. This, therefore, provides strong evidence that BH3 is bound to PH2 in this molecule. In contrast, the PH2 signal in the 1H NMR spectrum of compound 6 appears as a sharp doublet (δH 4.48, 1JHP = 207 Hz), indicating the absence of a co-ordinated borane.
It should be noted that, unlike compounds 1a–b, treatment of the phosphonium-phosphoranide 2 with H3B·SMe2 does not result in borane mediated reduction to give 4c, but instead yields the “push–double pull” bis(borane) adduct 7 (Scheme 3).29
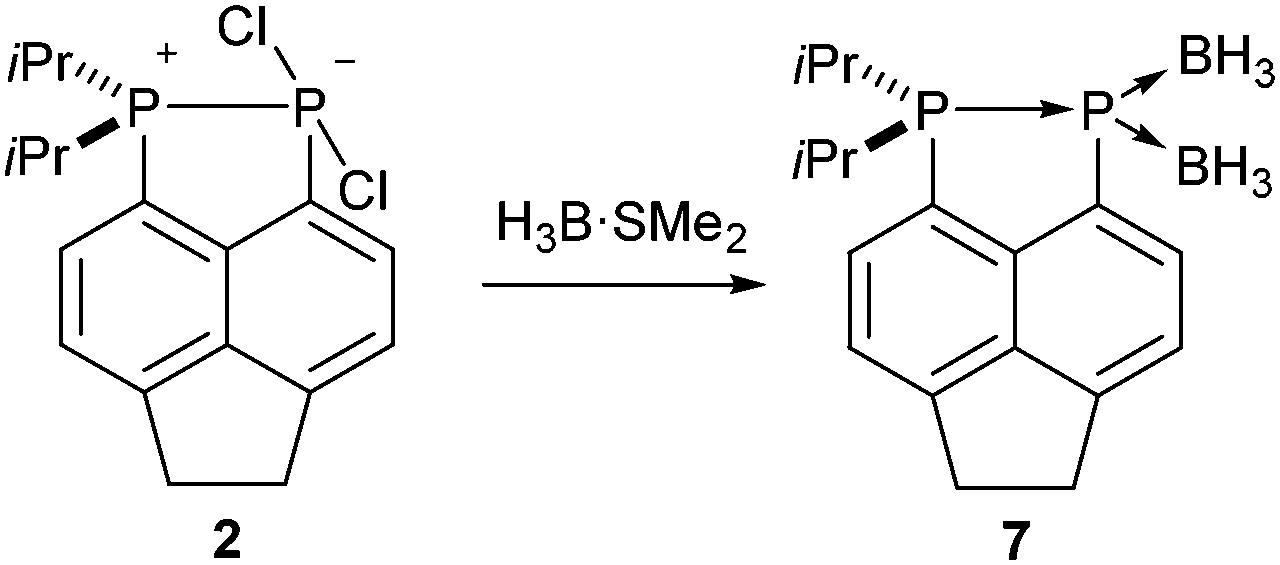 | ||
| Scheme 3 Synthesis of the “push–double pull” bis(borane) adduct 7. | ||
Spontaneous intramolecular dehydrocoupling of 4a–c to give 5a–c
When compound 4a was allowed to stand in solution in DCM, the signals corresponding to the bis(borane) adduct (δP 39.4 (br s, iPr2P) and −6.6 (br s, PhPH))26 were gradually replaced by a broad doublet (δP 13.9, PiPr2, 2JPP ≈ 84.0 Hz) and a very broad signal in which coupling could not be resolved (δP −26.3, PPh),30 corresponding with the formation of 5a (Fig. 3). Complete conversion to 5a was achieved after 8 days at room temperature (Scheme 1). 1H and 31P NMR spectroscopy confirmed that the H atom directly bonded to phosphorus had been lost. Additionally, 11B{1H} NMR spectroscopy revealed a broad pseudo-triplet (δB −39.4, 1JBP ≈ 69 Hz) and a broad doublet (δB −33.6, 1JBP ≈ 46 Hz), consistent with the presence of one bridging P–B–P motif and one terminal B–P motif (Fig. 4).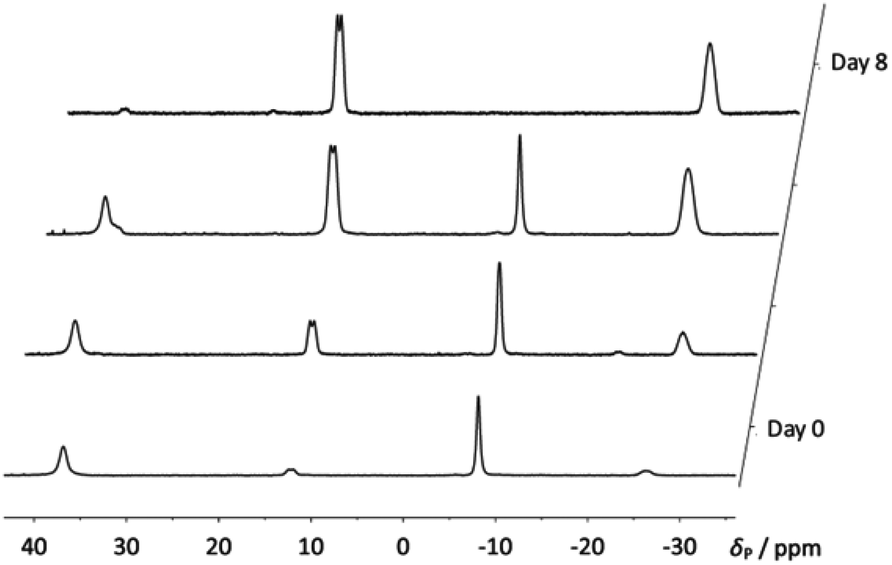 | ||
| Fig. 3 Stacked 31P{1H} NMR spectra showing the gradual formation of compound 5a from 4a over 8 days. | ||
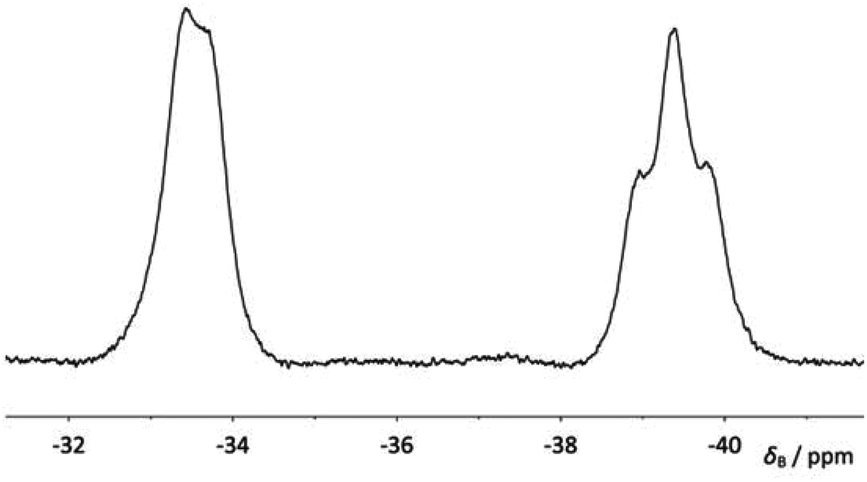 | ||
| Fig. 4 11B{1H} NMR of compound 5a. | ||
Crystals of 5a suitable for single crystal X-ray diffraction were grown from d6-DMSO. The structure confirmed 5a to contain one bridging BH2 and one terminal BH3 motif (Fig. 5, Tables 1–3). A significant reduction of strain is observed in 5a in comparison to 4a, with a reduced P⋯P distance of 3.1295(8) Å and smaller splay angle of +15.1(4)° (cf. 3.61 Å and +24.4(4)° in 4a),26 as well as decreased displacements of the P atoms from the mean plane of the acenaphthene ring (0.338 Å for P1, 0.327 Å for P9; cf. 0.478 and 0.816 Å in 4a).26
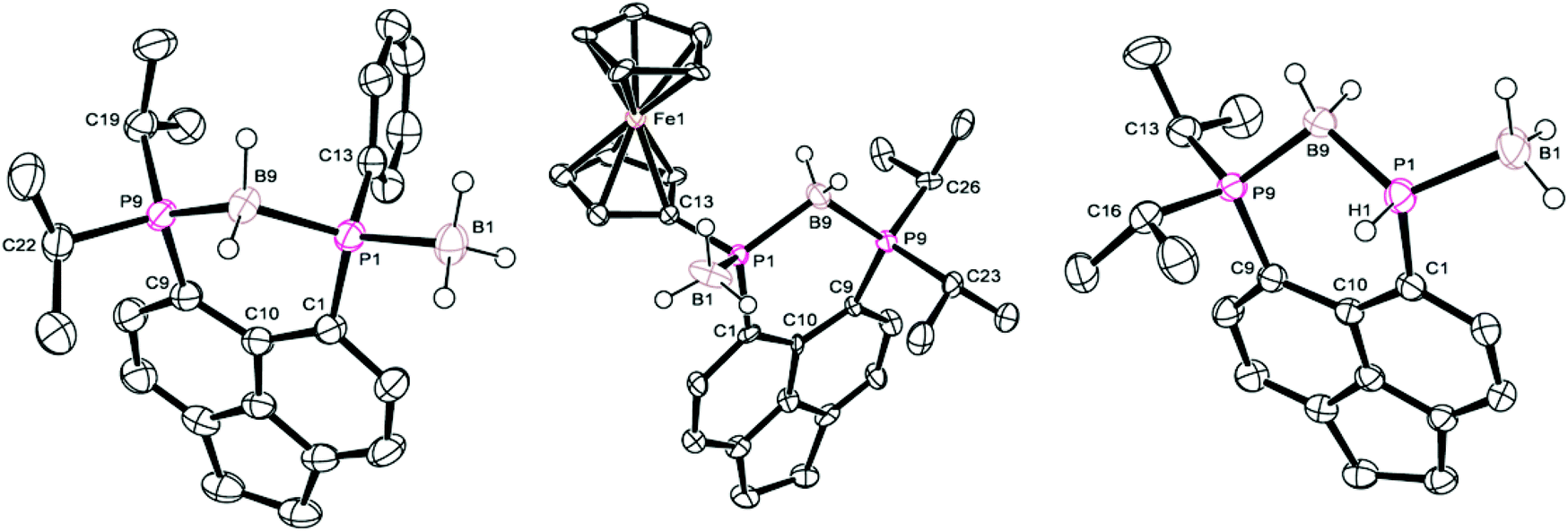 | ||
| Fig. 5 Structures of 5a (left), 5b (centre), and 5c (right) in the solid state. Carbon-bound hydrogen atoms and second molecule in asymmetric unit (for 5b and 5c) omitted for clarity. | ||
Based on the identity of compound 5a, it seemed likely that the bis(borane) adduct 4a had undergone a phosphine–borane dehydrocoupling reaction. In order to confirm the evolution of hydrogen, a solution of 4a in C6D6 was prepared and left to stand in a sealed NMR tube. After 1 day, some conversion to compound 5a was observed by 31P{1H} NMR, and a sharp singlet of dissolved H2 was observed in the 1H NMR (δH 4.47).31 In another experiment, the conversion of 4a to 5a (in CDCl3) was followed over several days at room temperature by 1H NMR spectroscopy.32 The reaction was found to follow simple first order kinetics, with an approximate rate constant of 0.04 h−1. It is likely that the driving force for this reaction is the reduction in strain on going from 4a to 5a, coupled with the entropic gain from hydrogen evolution.
Spontaneous dehydrocoupling reactions occurring at room temperature are rather rare, with a few examples involving very reactive precursors such as primary/secondary stibines or bismuthines.7 In recent work by the Manners’ group, a series of primary arylamine–borane adducts were found to undergo spontaneous dehydrocoupling at room temperature, with the rate of dehydrocoupling increasing with decreasing electron density on the aryl substituent.8 This reactivity was attributed to weak B–N bonding and the increased acidity of the N–H bonds in arylamine–boranes. By contrast, while dehydrocoupling of phosphine–boranes has been observed in the presence of catalysts17–19,33 or at very high temperatures,15,16 spontaneous, room temperature dehydrocoupling of a phosphine–borane adduct is without precedent in the literature.
The compound 5a bears some similarities to two cyclic boronium salts, 10 and 13, reported by Mikołajczyk et al.34 and Costa and Schmidbaur35 (Scheme 4). Compounds 5a, 10 and 13 are all formed by the treatment of peri-substituted precursors with borane, and all consist of two peri-phosphorus atoms bridged by a BH2 unit. However, compounds 10 and 13 are ionic species; 10 is thought to form via the mono(borane) adduct 9, which then reduces the halogenated solvent to form 10.34 Compound 13 exists in equilibrium with the bis(borane) adduct 12 and forms via hydride transfer to give a BH2 bridge and a BH4− counterion.35 Although these reactions are significantly different from the dehydrocoupling observed in 4a, in all cases the driving force for the formation of the BH2 bridge is most likely the same – reduction of strain resulting from the peri-substitution geometry. In compound 4a this is achieved via hydrogen evolution, while for 9 and 12 (which contain no P–H bonds) the formation of the boronium salts is preferred.
 | ||
| Scheme 4 Formation of the cyclic boronium salts 10 and 13. | ||
The ferrocenyl substituted bis(borane) adduct (4b) was also found to undergo spontaneous dehydrocoupling in solution, albeit at a slower rate than 4a. A solution of 4b left standing in CDCl3 achieved approximately 80% conversion to 5b after 2 weeks. Owing to the slow rate of reaction, 5b was more conveniently synthesised by refluxing 4b in THF for 4 days. The observed trend in dehydrocoupling rates (4a > 4b) correlates with the acidity of the P–H hydrogen, which is higher in 4a due to the more electron withdrawing nature of the phenyl substituent as compared to the ferrocenyl substituent.
Compound 5b demonstrates a similar 31P{1H} NMR spectrum to 5a, displaying a broad doublet (δP 16.1, 1JPP = 92.6 Hz, iPr2P) and a very broad unresolved signal (δP −32.0, PFc) located upfield of the corresponding signals for 4b (ΔδP ≈ 20–25). Once again, 1H and 31P NMR spectroscopy confirmed the loss of H directly bonded to phosphorus, and the 11B NMR spectrum displayed two distinct boron environments. Crystals of 5b suitable for single crystal X-ray diffraction were grown from acetonitrile. Obtained data is of somewhat poor quality, but is sufficient to demonstrate the connectivity of the molecule. The crystal structure is shown in Fig. 5 with data in Tables 1–3 and is broadly similar to that seen for 5a.
The dehydrocoupled product of the primary bis(borane) adduct 4c was obtained by treating 3 with excess H3B·SMe2 in DCM and then, without isolating 4c, allowing the reaction mixture to stir at room temperature for 11 days. After this time, no peaks for 4c could be observed in the 31P{1H} NMR spectrum of the reaction mixture. The resultant bridged compound 5c is significantly more inert than the corresponding bis(borane) adduct, and was stable enough to be purified by flash column chromatography. As with the previous compounds, 5c displays peaks in the 31P{1H} NMR spectrum with ΔδP ≈ 25–30 upfield of the corresponding resonances for the parent bis(borane) adduct 4c. Additionally, in the 31P NMR spectrum of 5c, the signal for the PH group (δP −69.5) appears as a doublet (1JPH = 339 Hz) as opposed to the triplet seen for 4c.
Crystals of 5c suitable for single crystal X-ray diffraction were grown from slow diffusion of hexane into its concentrated solution in DCM. The structure is presented in Fig. 5, with data in Tables 1–3. One interesting point of note is that, in contrast to 5a–b, compound 5c displays almost no out-of-plane displacement of the peri-phosphorus atoms (0.050 Å [0.042 Å] for P1, 0.068 Å [0.101 Å] for P8, values in square brackets are for the second molecule in the asymmetric unit). This can be attributed to the significantly reduced steric demands of the hydrogen substituent.
Given the presence of vicinal P–H and B–H bonds in compound 5c, the thermal decomposition of this compound was investigated to verify whether a further molecule of dihydrogen could be eliminated. After refluxing 5c in xylenes for 3 days, partial conversion (≈26% by 31P NMR) to a new compound, compound 14, was observed. Compound 14 shows two resonances in its 31P{1H} NMR spectrum, a broad multiplet (δP 11.0, PiPr2) and a sharp singlet (δP −136.9, PH). The low frequency chemical shift of the singlet suggests that 14 forms by loss of BH3 from 5c (Scheme 5). Furthermore, the 31P NMR spectrum shows a significant reduction in the 1JPH coupling constant (5c, 1JPH = 339 Hz; 14, 1JPH = 185 Hz), consistent with an increase in electron density on phosphorus due to loss of the Lewis acidic BH3.36 Due to the slow rate of the reaction, complete conversion to 14 was not achieved and this compound was not isolated pure. Attempts to drive the reaction to completion by prolonged heating resulted in decomposition.
 | ||
| Scheme 5 Proposed initial product of thermal decomposition of 5c. | ||
Conclusion
Bis(borane) adducts 4a–c were formed by either borane mediated reduction of phosphino-phosphonium salts 1a–b, or by treatment of the bis(phosphine) 3 with excess H3B·SMe2. All three adducts were found to undergo spontaneous intramolecular dehydrocoupling in solution, resulting in the formation of a P–B bond to afford the novel BH2 bridged compounds 5a–c. This reaction is surprisingly facile, occurring at room temperature and in the absence of a catalyst (albeit in some cases at a slow rate). The ease with which the reaction proceeds can be attributed to the unique constraints of the peri-geometry; the two reactive moieties are held in close proximity and the repulsive interaction between them introduces considerable strain into the system, which is reduced on formation of a bridging P–B–P motif.This interesting reaction serves as a demonstration of the utility of peri-substitution for promoting unusual or unexpected reactivity. Furthermore, it highlights how manipulation of the steric properties of a molecule can eliminate the need for a catalyst, which could be a potentially interesting alternative approach to developing compounds for hydrogen storage.
Experimental
General procedures
All experiments were carried out using standard Schlenk technique or glove box unless otherwise stated. Solvents were dried on an MBraun solvent purification system and stored over molecular sieves prior to use. 5-Bromo-6-diisopropylphosphinoacenaphthene and phosphonium-phosphoranide 2 were synthesised according to literature procedures.24 Where possible, new compounds were fully characterized by 31P, 31P{1H}, 1H and 13C{1H} NMR, including measurement of 1H{31P}, H–H DQF COSY, H–P HMQC, H–C HSQC, and H–C HMBC experiments. The NMR numbering scheme for all compounds discussed is shown in Scheme 6. | ||
| Scheme 6 NMR numbering scheme for all compounds discussed. | ||
Instrumentation
All NMR spectra were recorded using a JEOL GSX Delta 270, a Bruker Avance 300, Bruker Avance 400, Bruker Avance 500 or Bruker Avance III 500 spectrometer. 85% H3PO4 was used as an external standard in 31P, BF3·OEt2 in CDCl3 was used as an external standard in 11B, and TMS was used as an internal standard in 1H and 13C NMR. Measurements were performed at 25 °C unless otherwise indicated. All IR and Raman spectra were obtained in the range 4000–300 cm−1 on a Perkin-Elmer System 2000 NIR Fourier transform spectrometer. Mass spectra were acquired by Mrs Caroline Horseburgh at the University of St Andrews on a Micromass LCT. Elemental analysis (C, H and N) was performed by Mr Stephen Boyer at London Metropolitan University.X-ray experimental
Table 3 lists details of data collections and refinements. Data for compound 3 were collected at −180(1) °C by using a Rigaku Mercury70 diffractometer. Data for compounds 4b and 5b were collected at −180(1) °C by using a Rigaku XtaLAB P200 diffractometer. Data for compounds 5a and 5c were collected at −100(1) °C by using a Rigaku XtaLAB P200 diffractometer. All instruments use Mo Kα radiation (λ = 0.71075 Å). Intensities were corrected for Lorentz polarization and for absorption. The structures were solved by direct methods. Refinements were done by full-matrix least-squares based on F2 using SHELXTL.37 CCDC 1410480–1410484 contain the supplementary crystallographic data for this article.[Acenap(PiPr2)(PPh)][Cl] phosphino-phosphonium 1a
1H NMR δH (270 MHz; CDCl3) 8.80 (1H, dd, 3JHP = 9.2 Hz, 3JHH = 7.3 Hz, 2-H), 7.84 (1H, ≈t 3JHH = 6.8 Hz 3JHP = 6.8 Hz, 8-H), 7.72 (1H, dd, 3JHH = 7.3 Hz, 4JHP = 2.9 Hz, 3-H), 7.58 (1H, dd, 3JHH = 7.2 Hz, 4JHP = 2.6 Hz, 7-H), 7.49–7.22 (5H, m, 5 × Ph CH), 3.94–3.75 (1H, m, iPr CH), 3.75–3.61 (1H, m, iPr CH), 3.58 (4H, br s, 11-H, 12-H), 1.39 (3H, dd, 3JHP = 19.3 Hz, 3JHH = 6.9 Hz, iPr CH3), 1.14 (3H, dd, 3JHP = 18.9 Hz, 3JHH = 7.0 Hz, iPr CH3), 1.03 (3H, dd, 3JHH = 7.1 Hz, 3JHP = 3.8 Hz, iPr CH3), 0.95 (3H, dd, 3JHH = 7.1 Hz, 3JHP = 3.8 Hz, iPr CH3).
31P{1H} NMR δP (109 MHz; CDCl3) 61.3 (d, iPr2P), −35.3 (d, PPh), 1JPP = 304 Hz.
[Acenap(PiPr2)(PFc)][Cl] phosphino-phosphonium 1b
1H NMR δH (270 MHz; CDCl3) 8.62 (1H, dd, 3JHP = 9.0 Hz, 3JHH = 7.3 Hz, 2-H), 8.10–8.01 (1H, m, 8-H), 7.67 (1H, dd, 3JHH = 7.2 Hz, 4JHP = 2.7 Hz, 3-H), 7.62 (1H, dd, 3JHH = 7.0 Hz, 4JHP = 2.2 Hz, 7-H), 4.75–4.71 (1H, m, CpH), 4.65–4.55 (1H, m, CpH), 4.34 (5H, s, CpH), 4.30–4.25 (2H, m, 2 × CpH), 3.56 (4H, s, 11-H, 12-H), 3.21–3.19 (2H, m, 2 × iPr CH), 1.35–0.92 (12H, m, 4 × iPr CH3).
31P{1H} NMR δP (109 MHz; CDCl3) 56.1 (d, iPr2P), −37.3 (d, PFc), 1JPP = 313 Hz.
Acenap(PiPr2)(PH2) bis(phosphine) 3
To a stirred suspension of LiAlH4 (0.334 g, 8.8 mmol) in THF (15 mL) cooled to −78 °C, a suspension of 2 (0.50 g, 1.35 mmol) in THF (20 mL) was added slowly via cannula. The resultant bright pink solution was allowed to warm to room temperature, with stirring, overnight. The solution was cooled to 0 °C and degassed water (2.5 mL) was added dropwise with stirring. The mixture was then filtered to remove insoluble impurities. Volatiles were removed in vacuo to give 3 as a pink solid (0.298 g, 0.986 mmol, 73%). The compound is highly soluble in most organic solvents, a small amount of crystals of 3 suitable for single crystal X-ray diffraction were grown from THF.mp 140–144 °C.
IR (nujol mull) νmax/cm−1 2293w, 2240m (PH), 1604w, 840m, 790m.
Raman (glass capillary) νmax/cm−1 3058s (ArH), 2948s and 2929s and 2866s (CH), 2294m and 2241s (PH), 1605m, 1567s, 1331vs, 585s.
1H NMR δH (400 MHz; C6D6) 7.79–7.72 (1H, m, 2-H), 7.60 (1H, dd, 3JHH = 7.1 Hz, 3JHP = 3.3 Hz, 8-H), 7.12 (1H, dt, 3JHH = 7.2 Hz, 4JHH = 1.3 Hz, 7-H), 6.93 (1H, d, 3JHH = 7.1 Hz, 3-H), 4.98 (2H, dd, 1JHP = 204 Hz, 5JHP = 47.8 Hz, PH2), 3.04–2.83 (4H, m, 11-H, 12-H), 2.12–1.99 (2H, m, 2 × iPr CH), 1.17 (6H, dd, 3JHP = 14.3 Hz, 3JHH = 6.9 Hz, 2 × iPr CH3), 1.00 (6H, dd, 3JHP = 12.3 Hz, 3JHH = 7.0 Hz, 2 × iPr CH3).
13C{1H} NMR δC (101 MHz; C6D6) 148.8 (s, qC-6), 147.7 (d, 4JCP = 1.9 Hz, qC-4), 140.4 (m, qC-5, qC-10), 139.7 (s, C-2), 134.5 (d, 2JCP = 2.3 Hz, C-8), 131.0 (dd, 1JCP = 23.9 Hz, 3JCP = 7.4 Hz, qC-9), 125.9 (d, 1JCP = 19.8 Hz, qC-1), 119.8 (s, C-3), 119.4 (s, C-7), 30.3 (s, C-11/C-12), 29.9 (s, C-11/C-12), 26.4 (d, 1JCP = 15.9 Hz, iPr CH), 26.4 (d, 1JCP = 15.8 Hz, iPr CH), 20.4 (s, iPr CH3), 20.3 (s, iPr CH3), 20.2 (s, 2 × iPr CH3).
31P NMR δP (162 MHz; C6D6) −11.3 (dm, 1JPP = 205 Hz, iPr2P), −101.2 (≈q, 1JPP = 205, 1JPH = 204 Hz, PH2).
31P{1H} NMR δP (162 MHz; C6D6) −11.3 (d, iPr2P), −101.2 (d, PH2), 1JPP = 205 Hz.
MS (ES+) m/z 301.1 (100%, M − H).
HRMS (ES+) Found: 301.1278. Calc. for C18H23P2 (M − H): 301.1275.
Acenap(PiPr2·BH3)(PFcH·BH3) bis(borane) 4b
Borane dimethylsulfide (0.10 mL, 94%, 0.99 mmol) was added to a stirred suspension of 1b (120 mg, 0.23 mmol) in THF (30 mL) at −78 °C. The reaction was stirred for 2 hours at −78 °C, then allowed to warm to RT and stirred overnight. Volatiles were removed in vacuo to afford 4b as an orange solid. The crude product contained the bridged compound 5b as a minor byproduct (approximately 20%). Analytically pure material, as well as crystals suitable for single crystal X-ray diffraction, was obtained from acetonitrile at 5 °C (50 mg, 0.10 mmol, 42%).mp 154–155 °C.
Found: C 65.56; H 7.56. Calc. for C28H38FeB2P2: C 65.43; H 7.45.
IR (KBr disk) νmax/cm−1 2966m and 2928m (CH), 2374vs (PH), 2345s (BH), 1638m, 1604m, 1460m, 1414m, 1387m, 1316m, 1256m, 1182m, 1071s, 1028s, 929m, 831s, 671m, 643m, 492m, 446m.
1H NMR δH (400 MHz; CDCl3) 8.09 (1H, dd, 3JHP = 13.2 Hz, 3JHH = 7.4 Hz, 2-H), 7.81 (1H, dd, 3JHP = 16.4 Hz, 3JHH = 7.3 Hz, 8-H), 7.70 (1H, dq, 1JHP = 393 Hz, 3JHH = 6.1 Hz, P–H), 7.39 (1H, d, 3JHH = 7.4 Hz, 3-H), 7.31 (1H, d, 3JHH = 7.3 Hz, 7-H), 4.59–4.55 (2H, m, 2 × CpH), 4.48–4.43 (2H, m, 2 × CpH), 4.32 (5H, s, 5 × CpH), 3.38 (4H, s, 11-H, 12-H), 3.16–3.06 (1H, m, iPr CH), 3.06–2.95 (1H, m, iPr CH), 1.60–0.40 (6H, br m, 2 × BH3), 1.48–1.37 (9H, m, 3 × iPr CH3), 0.99 (3H, dd, 3JHP = 15.2 Hz, 3JHH = 6.8 Hz, iPr CH3).
13C{1H} NMR δC (101 MHz; CDCl3) 152.3 (s, qC-6), 151.9 (s, qC-4), 140.8 (≈t, 3JCP = 7.9 Hz, qC-5), 140.1 (d, 2JCP = 7.5 Hz, C-2), 136.9 (d, 2JCP = 14.3 Hz, C-8), 133.1–132.9 (m, qC-10), 123.5 (d, 1JCP = 53.0 Hz, qC-9), 120.3 (d, 3JCP = 12.9 Hz, C-7), 119.5 (d, 1JCP = 43.4 Hz, qC-1), 119.5 (d, 3JCP = 10.6 Hz, C-3), 74.3 (d, JCP = 15.9 Hz, Cp CH), 72.7 (d, JCP = 6.6 Hz, Cp CH), 72.4 (d, JCP = 3.6 Hz, Cp CH), 71.4 (d, JCP = 9.0 Hz, Cp CH), 70.2 (s, 5 × Cp CH), 66.2 (d, 1JCP = 68.3 Hz, Cp qC), 30.2 (s, C-11/C-12), 30.0 (s, C-11/C-12), 25.5 (dd, 1JCP = 29.1 Hz, 3JCP = 2.1 Hz, iPr CH), 23.7 (d, 1JCP = 33.3 Hz, iPr CH), 19.4 (s, iPr CH3), 18.3 (s, iPr CH3), 17.9–17.3 (m, iPr CH3), 17.5 (s, iPr CH3).
31P NMR δP (162 MHz; CDCl3) 36.2 (br s, iPr2P), −7.7 (d, 1JPH = 395 Hz, PFcH).
31P{1H} NMR δP (162 MHz; CDCl3) 36.3 (br s, iPr2P), −7.7 (br s, PFcH).
11B NMR δB (160 MHz; CDCl3) −39.2 (br m, 2 × BH3).
11B{1H} NMR δB (160 MHz; CDCl3) −38.7 (br m, BH3), −39.4 (br m, BH3).
MS (ES–) m/z 485.1 (12%, M − 2BH3 − H), 499.2 (86, M − BH3 − H), 513.2 (100, M − H).
HRMS (ES−) Found: 513.1919. Calc. for C28H37P2FeB2 (M + H): 513.1906.
Acenap(PiPr2·BH3)(PH2·BH3) Bis(borane) 4c
Borane dimethylsulfide (0.40 mL, 94%, 3.98 mmol) was added to a stirred solution of 3 (100 mg, 0.33 mmol) in DCM (5 mL) at −78 °C. The reaction was allowed to warm to room temperature over 1 h, then stirred for 30 minutes. Volatiles were removed in vacuo to afford crude 4c as an off-white sticky solid (105 mg). Compound was not purified due to its high sensitivity towards moisture and oxygen, which prevented chromatographic separation. NMR data was assigned from the crude product mixture.1H NMR δH (400 MHz; CDCl3) 8.26 (1H, dd, 3JHP = 14.1 Hz, 3JHH = 7.5 Hz, 2-H), 8.19 (1H, dd, 3JHP = 19.3 Hz, 3JHH = 7.3 Hz, 8-H), 7.49–7.42 (2H, m, 3-H, 7-H), 6.14 (2H, dq, 1JHP = 377 Hz, 3JHH = 7.1 Hz, PH2), 3.44 (4H, s, 11-H, 12-H), 2.90–2.77 (2H, m, 2 × iPr CH), 1.50–0.30 (6H, br m, 2 × BH3), 1.38 (6H, dd, 3JHP = 14.6 Hz, 3JHH = 6.9 Hz, 2 × iPr CH3), 1.06 (6H, dd, 3JHP = 15.6 Hz, 3JHH = 7.1 Hz, 2 × iPr CH3).
31P NMR δP (162 MHz; CDCl3) 38.0 (br s, iPr2P), −40.8 (br s, PH2).
31P{1H} NMR δP (162 MHz; CDCl3) 38.0 (br s, iPr2P), −40.8 (t, 1JPH = 379 Hz, PH2).
11B NMR δB (96 MHz; CDCl3) −40.1 (br m, 2 × BH3).
11B{1H} NMR δB (96 MHz; CDCl3) −40.1 (br m, 2 × BH3).
Acenap(PiPr2)(μ-BH2)(PPh·BH3) 5a
Borane dimethylsulfide (0.15 mL, 94%, 1.49 mmol) was added to a stirred solution of 1a (150 mg, 0.363 mmol) in THF (5 mL) at −78 °C. The reaction was stirred for 2 hours at −78 °C, then allowed to warm to room temperature and stirred overnight. Volatiles were removed in vacuo to afford the bis(borane) adduct 4a, which was re-dissolved in DCM (5 mL) and stirred at room temperature for 8 days. Volatiles were removed in vacuo to afford 5a as an off-white solid in near quantitative yield (0.145 g, 0.359 mmol, 99%). Crystals suitable for single crystal X-ray diffraction were grown from d6-DMSO at room temperature.mp 230 °C (decomp).
Found: C 71.23; H 8.05. Calc. for C24H32B2P2: C 71.34; H 7.98.
IR (KBr disk) νmax/cm−1 3030w (ArH), 2970m and 2934m and 2872m (CH), 2449m, 2364vs (BH), 2258m, 1597s, 1488m, 1453s, 1436s, 1388m, 1340m, 1248m, 1139m, 1111m, 1057vs, 882m, 847s, 829m, 739s, 699vs, 666m, 614m, 472m, 402m.
Raman (glass capillary) νmax/cm−1 3060 (s, Ar–H), 2942 (s, νC–H), 2895 (m), 2453 (m), 2388 (m), 2340 (m, νB–H), 1599 (s), 1578 (s), 1444 (s), 1419 (s), 1343 (vs), 1002 (s), 832 (m), 739 (m), 573 (s).
1H NMR δH (500 MHz; CDCl3) 8.01 (1H, dd, 3JHP = 12.2 Hz, 3JHH = 7.2 Hz, 2-H), 7.67 (1H, dd, 3JHP = 10.3 Hz, 3JHH = 7.2 Hz, 8-H), 7.59–7.51 (2H, m, o-Ph CH), 7.42 (1H, d, 3JHH = 7.5 Hz, 7-H), 7.40 (1H, d, 3JHH = 7.3 Hz, 3-H), 7.30–7.23 (3H, m, m/p-Ph CH), 3.50–3.39 (4H, m, 11-H, 12-H), 2.76–2.64 (1H, m, iPr CH), 2.38–2.26 (1H, m, iPr CH), 2.10–0.70 (5H, br m, BH2 and BH3), 1.28 (3H, dd, 3JHP = 14.9 Hz, 3JHH = 7.1 Hz, iPr CH3), 1.25 (3H, dd, 3JHP = 16.0 Hz, 3JHH = 7.2 Hz, iPr CH3), 1.17 (3H, dd, 3JHP = 15.7 Hz, 3JHH = 7.0 Hz, iPr CH3), 0.93 (3H, dd, 3JHP = 15.9 Hz, 3JHH = 7.2 Hz, iPr CH3).
13C{1H} NMR δC (126 MHz; CDCl3) 153.0 (m, qC-6), 149.7 (s, qC-4), 139.8 (dd, 3JCP = 8.6 Hz, 3JCP = 6.5 Hz, qC-5), 138.5 (d, 2JCP = 8.1 Hz, C-2), 137.6 (dd, 1JCP = 42.1 Hz, 3JCP = 7.0 Hz, i-Ph qC), 135.2 (dd, 2JCP = 9.1 Hz, 2JCP = 5.3 Hz, qC-10), 134.2 (s, C-8), 132.5 (d, 2JCP = 8.7 Hz, o-Ph CH), 128.9 (d, 4JCP = 2.1 Hz, p-Ph CH), 128.1 (d, 3JCP = 9.2 Hz, m-Ph CH), 124.6 (dd, 1JCP = 41.4 Hz, 3JCP = 5.0 Hz, qC-1), 121.0 (d, 3JCP = 9.6 Hz, C-3), 118.8 (d, 3JCP = 8.9 Hz, C-7), 114.5 (dd, 1JCP = 56.2 Hz, 3JCP = 3.5 Hz, C-9), 30.6 (s, C-11/C-12), 30.2 (s, C-11/C-12), 24.3 (dd, 1JCP = 35.0 Hz, 3JCP = 1.8 Hz, iPr CH), 23.0 (dd, 1JCP = 35.3 Hz, 3JCP = 4.7 Hz, iPr CH), 18.4 (s, iPr CH3), 17.8 (s, iPr CH3), 16.7 (s, iPr CH3), 16.6 (s, iPr CH3).
31P NMR δP (202 MHz; CDCl3) 13.7 (br s, iPr2P), −26.4 (br s, PPh).
31P{1H} NMR δP (202 MHz; CDCl3) 13.9 (br d, 1JPP = 84.0 Hz, iPr2P), −26.3 (br m, PPh).
11B NMR δB (160 MHz; CDCl3) −33.6 (br m, BH3), −39.4 (br m, BH2).
11B{1H} NMR δB (160 MHz; CDCl3) −33.6 (br d, 1JBP = 46.2 Hz, BH3), −39.4 (br ≈t, 1JBP = 69.3 Hz, BH2).
MS (ES+) m/z 391.2 (28%, M − BH3 + H), 427.2 (100, M + Na).
HRMS (ES+) Found: 427.2052. Calc. for C24H32P2B2Na (M + Na): 427.2058.
Acenap(PiPr2)(μ-BH2)(PFc·BH3) 5b
Borane dimethylsulfide (0.10 mL, 94%, 0.99 mmol) was added to a stirred suspension of 1b (120 mg, 0.23 mmol) in THF (30 mL) at −78 °C. The reaction was stirred for 2 hours at −78 °C, then allowed to warm to RT and stirred overnight to afford 4b, which was not isolated. The reaction mixture was heated under reflux for 4 days. Volatiles were removed in vacuo to afford 5b as an orange oil. Analytically pure material was obtained by filtering through silica gel, eluting with DCM (102 mg, 0.20 mmol, 87%). Crystals of 5b suitable for single crystal X-ray diffraction were grown by slow evaporation from acetonitrile.mp 180 °C (decomp).
Found: C 65.76; H 6.93. Calc. for C28H36FeB2P2: C 65.68; H 7.09.
IR (KBr disk) νmax/cm−1 2967m and 2932m (CH), 2437s, 2362vs (BH), 1598s, 1448m, 1387m, 1332m, 1255m, 1169s, 1105m, 1059s, 1025s, 828s, 670s, 492s, 453m.
Raman (glass capillary) νmax/cm−1 3110s (ArH), 2929s (CH), 2440w, 2381m (BH), 1601s, 1575s, 1447s, 1417s, 1335vs, 1173s, 1107vs, 1060m, 830m, 729m, 552m, 402m, 368m, 321s.
1H NMR δH (500 MHz; CDCl3) 7.71 (1H, dd, 3JHP = 11.5 Hz, 3JHH = 7.2 Hz, 2-H), 7.64 (1H, dd, 3JHP = 10.3 Hz, 3JHH = 7.2 Hz, 8-H), 7.35 (1H, d, 3JHH = 7.1 Hz, 7-H), 7.27–7.23 (1H, m, 3-H), 4.94 (1H, s, CpH), 4.43 (2H, s, 2 × CpH), 4.39 (5H, s, 5 × CpH), 4.32 (1H, s, CpH), 3.43–3.27 (4H, m, 11-H, 12-H), 2.90–2.80 (1H, m, iPr CH), 2.80–2.70 (1H, m, iPr CH), 2.20–0.60 (5H, br m, BH2 and BH3), 1.52 (3H, dd, 3JHP = 16.6 Hz, 3JHH = 7.3 Hz, iPr CH3), 1.42 (3H, dd, 3JHP = 16.3 Hz, 3JHH = 6.9 Hz, iPr CH3), 1.32 (3H, dd, 3JHP = 14.4 Hz, 3JHH = 7.1 Hz, iPr CH3), 1.11 (3H, dd, 3JHP = 15.5 Hz, 3JHH = 7.0 Hz, iPr CH3).
13C{1H} NMR δC (101 MHz; CDCl3) 152.7 (s, qC-6) , 148.1 (s, qC-4) , 139.6 (dd, 3JCP = 9.1 Hz, 3JCP = 6.4 Hz, qC-5), 135.9 (d, 2JCP = 5.7 Hz, C-2), 133.8 (m, qC-10), 133.5 (s, C-8), 130.2 (dd, 1JCP = 38.6 Hz, 3JCP = 3.2 Hz, qC-1), 120.8 (d, 3JCP = 8.5 Hz, C-3), 118.4 (d, 3JCP = 8.8 Hz, C-7), 114.6 (dd, 1JCP = 55.7 Hz, 3JCP = 2.7 Hz, qC-9), 75.1 (dd, 1JCP = 53.3 Hz, 2JCP = 12.2 Hz, Cp qC), 74.2 (d, JCP = 10.4 Hz, Cp CH), 71.7 (d, JCP = 5.9 Hz, Cp CH), 71.0 (d, JCP = 6.1 Hz, Cp CH), 70.6 (d, JCP = 6.8 Hz, Cp CH), 69.8 (s, 5 × Cp CH), 30.6 (s, C-11/C-12), 30.0 (s, C-11/C-12), 25.9 (d, 1JCP = 34.6 Hz, iPr CH), 21.4 (dd, 1JCP = 36.0 Hz, 2JCP = 9.0 Hz, iPr CH), 19.2 (s, iPr CH3), 17.8 (s, iPr CH3), 17.4 (d, 2JCP = 2.7 Hz, iPr CH3), 16.1 (d, 2JCP = 3.8 Hz, iPr CH3).
31P NMR δP (202 MHz; CDCl3) 16.1 (br s, iPr2P), −32.0 (br s, PFc).
31P{1H} NMR δP (202 MHz; CDCl3) 16.1 (br d, 1JPP = 92.6 Hz, iPr2P), −32.0 (br m, PFc).
11B NMR δB (160 MHz; CDCl3) −33.9 (br m, BH3), −41.0 (br m, BH2).
11B{1H} NMR δB (160 MHz; CDCl3) δ −33.7 (br m, BH3), −41.1 (br m, BH2).
MS (ES+) m/z 498.1 (100%, M − BH3), 512.2 (38, M).
HRMS (ES+) Found: 498.1483. Calc. for C28H33FeP2B (M − BH3): 498.1500.
Acenap(PiPr2)(μ-BH2)(PH·BH3) 5c
Borane dimethylsulfide (0.40 mL, 94%, 3.98 mmol) was added to a stirred solution of 3 (100 mg, 0.33 mmol) in DCM (5 mL) at −78 °C. The reaction was allowed to warm to room temperature over 1 h, then left to stir at room temperature for 11 days. Distilled water (10 mL) was added and the reaction stirred for 1 h at room temperature. The product was extracted with DCM (3 × 10 mL) in air and the combined washings were dried over MgSO4. Volatiles were removed in vacuo and the crude product was purified by flash column chromatography on silica, eluting with DCM, to yield 5c as a white crystalline solid (45 mg, 0.137 mmol, 41%). Crystals of 5c suitable for single crystal X-ray diffraction were grown by diffusion of hexane into a concentrated solution of 5c in DCM.mp 140 °C (decomp).
Found: C 65.89; H 8.70. Calc. for C18H28B2P2: C 65.92; H 8.61.
IR (KBr disk) νmax/cm−1 2975m and 2959s (CH), 2930m, 2872m, 2444s (BH), 2368vs (PH), 2259w, 1710w, 1597s, 1492m, 1461s, 1418m, 1387m, 1367w, 1333m, 1257m, 1217w, 1139m, 1103m, 1065vs, 1040m, 909s, 883m, 849s, 715s, 629m, 395w.
Raman (glass capillary) νmax/cm−1 3064m, 2962s, 2934vs (CH), 2901vs, 2462m (BH), 2392m, 2351vs (PH), 1599s, 1575s, 1441s, 1420m, 1337vs, 1054m, 883m, 830m, 735m, 585m, 571s.
1H NMR δH (500 MHz; CDCl3) 8.37 (1H, dd, 3JHP = 13.4 Hz, 3JHH = 7.1 Hz, 2-H), 7.67 (1H, dd, 3JHP = 10.3 Hz, 3JHH = 7.2 Hz, 8-H), 7.46 (1H, d, 3JHH = 7.1 Hz, 3-H), 7.42 (1H, d, 3JHH = 7.2 Hz, 7-H), 4.89 (1H, br d, 1JHP = 328 Hz, PH), 3.45 (4H, s, 11-H, 12-H), 2.78–2.64 (1H, m, iPr CH), 2.55–2.41 (1H, m, iPr CH), 1.90–0.60 (5H, br m, BH2 and BH3), 1.32 (3H, dd, 3JHP = 16.6 Hz, 3JHH = 6.9 Hz, iPr CH3), 1.29–1.23 (6H, m, 2 × iPr CH3), 1.07 (3H, dd, 3JHP = 16.0 Hz, 3JHH = 7.0 Hz, iPr CH3).
13C{1H} NMR δC (126 MHz; CDCl3) 153.1 (d, 4JCP = 2.0 Hz, qC-4), 150.1 (s, qC-6), 139.5 (dd, 3JCP = 8.2 Hz, 3JCP = 5.9 Hz, qC-5), 138.4 (d, 2JCP = 10.9 Hz, C-2), 135.8 (dd, 2JCP = 8.6, 2JCP = 2.2 Hz, qC-10), 134.4 (s, C-8), 120.6 (d, 3JCP = 10.7 Hz, C-3), 119.1 (d, 3JCP = 8.9 Hz, C-7), 118.8 (dd, 1JCP = 41.0 Hz, 3JCP = 6.8 Hz, qC-1), 112.6 (dd, 1JCP = 55.9 Hz, 3JCP = 4.1 Hz, qC-9), 30.5 (s, C-11/C-12), 30.0 (s, C-11/C-12), 25.1 (d, 1JCP = 36.1 Hz, iPr CH), 22.4 (dd, 1JCP = 36.4, 3JCP = 4.5 Hz, iPr CH), 17.7 (d, 2JCP = 2.2 Hz, iPr CH3), 17.2 (s, iPr CH3), 17.1 (s, iPr CH3), 16.6 (d, 2JCP = 1.8 Hz, iPr CH3).
31P NMR δP (202 MHz; CDCl3) 12.8 (br s, iPr2P), −69.5 (br d, 1JPH = 339 Hz, PH).
31P{1H} NMR δP (202 MHz; CDCl3) 12.8 (br d, 1JPP = 79.9 Hz, iPr2P), −69.5 (br m, PH).
11B NMR δB (160 MHz; CDCl3) −37.2 (br m, BH3), −41.9 (br m, BH2).
11B{1H} NMR δB (160 MHz; CDCl3) −37.1 (br m, BH3), −41.9 (br ≈t, 1JBP = 63.0 Hz, BH2).
MS (ES+) m/z 351.2 (100%, M + Na).
HRMS (ES+) Found: 351.1753. Calc. for C18H28P2B2Na (M + Na): 351.1750.
Acenap(PiPr2·BH3)(PH2) 6
Borane dimethylsulfide (0.72 mL, 94%, 7.6 mmol) was added to a stirred solution of 3 (180 mg, 0.60 mmol) in DCM (10 mL) at 0 °C. The reaction was stirred for 2 hours at room temperature, then cooled to 0 °C and degassed water (10 mL) was added cautiously. The reaction mixture was stirred for a further 2 hours at room temperature, the organic layer was separated and volatiles removed in vacuo to afford 6 as a yellow solid in quantitative yield (188 mg, 0.59 mmol, 99%).1H NMR δH (400 MHz; CDCl3) 8.54 (1H, dd, 3JHP = 17.4 Hz, 3JHH = 7.4 Hz, 8-H), 8.04–8.00 (1H, m, 2-H), 7.33 (1H, d, 3JHH = 7.3 Hz, 7-H), 7.27 (1H, d, 3JHH = 7.2 Hz, 3-H), 4.48 (2H, d, 1JHP = 207 Hz, PH2), 3.63–3.49 (2H, m, 2 × iPr CH), 3.38 (4H, s, 11-H, 12-H), 1.70–0.30 (3H, br m, BH3), 1.42 (6H, dd, 3JHP = 15.0 Hz, 3JHH = 7.0 Hz, 2 × iPr CH3), 0.88 (6H, dd, 3JHP = 16.2 Hz, 3JHH = 7.1 Hz, 2 × iPr CH3).
13C{1H} NMR δC (75 MHz; CDCl3) 152.3 (s, qC-6), 150.1 (s, qC-4), 143.8 (d, 2JCP = 4.6 Hz, C-8), 143.5 (br d, 2JCP = 20.0 Hz, C-2), 140.9 (≈t, 3JCP = 7.1 Hz, 3JCP = 7.1 Hz, qC-5), 137.0 (d, 2JCP = 26.6 Hz, qC-10), 120.5 (d, 1JCP = 43.7, qC-9), 119.8 (s, C-3), 119.2 (d, 3JCP = 14.7 Hz, C-7), 117.8 (d, 1JCP = 16.0 Hz, qC-1), 30.0 (s, C-11/C-12), 29.9 (s, C-11/C-12), 25.2 (d, 1JCP = 28.4 Hz, iPr CH), 24.8 (d, 1JCP = 28.3 Hz, iPr CH), 19.1 (s, 2 × iPr CH3), 18.9 (s, 2 × iPr CH3).
31P NMR δP (109 MHz; CDCl3) 44.5 (m, iPr2P), −101.3 (t, 1JPH = 207 Hz, PH2).
31P{1H} NMR δP (109 MHz; CDCl3) 44.0 (m, iPr2P), −101.4 (s, PH2).
11B NMR δB (96 MHz; CDCl3) −41.9 (br m, BH3).
11B{1H} NMR δB (96 MHz; CDCl3) −41.9 (br d, 1JBP = 66.4 Hz, BH3).
Acenap(PiPr2)(μ-BH2)(PH) 14
A suspension of 5c (0.100 g, 0.304 mmol) in xylenes (20 mL) was heated under reflux. At high temperatures, all solid dissolved to give a yellow solution. After 3 days volatiles were removed in vacuo to give a yellow solid (0.098 g) containing ≈74% 5c and ≈26% 14. Compound 14 was not isolated pure due to its instability to air and moisture and NMR data was assigned from the mixture. Attempts to bring the reaction to completion via prolonged heating under reflux resulted in decomposition to a complex mixture of products.1H NMR δH (400 MHz; CDCl3) 7.98 (1H, dd, 3JHP = 12.1 Hz, 3JHH = 7.0 Hz, 2-H), 7.61 (1H, dd, 3JHP = 9.9 Hz, 3JHH = 7.2 Hz, 8-H), 7.35 (1H, d, 3JHH = 7.2 Hz, 7-H), 7.25 (1H, d, 3JHH = 7.0 Hz, 3-H), 3.40–3.33 (4H, m, 11-H, 12-H). Signals for H directly bound to phosphorus/boron and iPr groups were obscured by signals from 5c or were too weak to be seen.
31P NMR δP (162 MHz; CDCl3) 11.1 (br s, iPr2P), −137.0 (br d, 1JPH = 185 Hz, PH).
31P{1H} NMR δP (162 MHz; CDCl3) 11.0 (m, iPr2P), −136.9 (s, PH).
Acknowledgements
This work was financially supported by the EPSRC and COST action CM1302 SIPs. The authors would also like to thank the University of St Andrews NMR Service and to Mrs Caroline Horsburgh for running the MS spectra.References
- E. M. Leitao, T. Jurca and I. Manners, Nat. Chem., 2013, 5, 817–829 CrossRef CAS PubMed.
- R. Waterman, Chem. Soc. Rev., 2013, 42, 5629–5641 RSC.
- R. J. Less, R. L. Melen and D. S. Wright, RSC Adv., 2012, 2, 2191–2199 RSC.
- R. J. Less, R. L. Melen, V. Naseri and D. S. Wright, Chem. Commun., 2009, 4929–4937 RSC.
- S. Greenberg and D. W. Stephan, Chem. Soc. Rev., 2008, 37, 1482–1489 RSC.
- T. J. Clark, K. Lee and I. Manners, Chem. – Eur. J., 2006, 12, 8634–8648 CrossRef CAS PubMed.
- G. Balázs, H. J. Breunig and E. Lork, Organometallics, 2002, 21, 2584–2586 CrossRef.
- H. Helten, A. P. M. Robertson, A. Staubitz, J. R. Vance, M. F. Haddow and I. Manners, Chem. – Eur. J., 2012, 18, 4665–4680 CrossRef CAS PubMed.
- R. Waterman, Dalton Trans., 2009, 18–26 RSC.
- F. Gauvin, J. F. Harrod and H. G. Woo, Adv. Organomet. Chem., 1998, 42, 363–405 CrossRef CAS.
- R. Waterman, Curr. Org. Chem., 2008, 12, 1322–1339 CrossRef CAS.
- C. W. Hamilton, R. T. Baker, A. Staubitz and I. Manners, Chem. Soc. Rev., 2009, 38, 279–293 RSC.
- A. Staubitz, A. P. M. Robertson and I. Manners, Chem. Rev., 2010, 110, 4079–4124 CrossRef PubMed.
- A. Staubitz, A. P. M. Robertson, M. E. Sloan and I. Manners, Chem. Rev., 2010, 110, 4023–4078 CrossRef CAS PubMed.
- R. I. Wagner and F. F. Caserio, J. Inorg. Nucl. Chem., 1959, 11, 259 CrossRef CAS.
- A. B. Burg, J. Inorg. Nucl. Chem., 1959, 11, 258 CrossRef CAS.
- H. Dorn, R. A. Singh, J. A. Massey, A. J. Lough and I. Manners, Angew. Chem., Int. Ed., 1999, 38, 3321–3323 CrossRef CAS PubMed.
- H. Dorn, R. A. Singh, J. A. Massey, J. M. Nelson, C. A. Jaska, A. J. Lough and I. Manners, J. Am. Chem. Soc., 2000, 122, 6669–6678 CrossRef CAS.
- C. A. Jaska, H. Dorn, A. J. Lough and I. Manners, Chem. – Eur. J., 2003, 9, 271–281 CrossRef CAS PubMed.
- A. Schäfer, T. Jurca, J. Turner, J. R. Vance, K. Lee, V. A. Du, M. F. Haddow, G. R. Whittell and I. Manners, Angew. Chem., Int. Ed., 2015, 54, 4836–4841 CrossRef PubMed.
- J.-M. Denis, H. Forintos, H. Szelke, L. Toupet, T.-N. Pham, P.-J. Madec and A.-C. Gaumont, Chem. Commun., 2003, 54–55 RSC.
- P. Kilian, F. R. Knight and J. D. Woollins, Chem. – Eur. J., 2011, 17, 2302–2328 CrossRef CAS PubMed.
- B. A. Chalmers, M. Bühl, K. S. A. Arachchige, A. M. Z. Slawin and P. Kilian, J. Am. Chem. Soc., 2014, 136, 6247–6250 CrossRef CAS PubMed.
- P. Wawrzyniak, A. L. Fuller, A. M. Z. Slawin and P. Kilian, Inorg. Chem., 2009, 48, 2500–2506 CrossRef CAS PubMed.
- M. J. Ray, A. M. Z. Slawin, M. Bühl and P. Kilian, Organometallics, 2013, 32, 3481–3492 CrossRef CAS.
- M. J. Ray, M. Bühl, L. J. Taylor, K. S. A. Arachchige, A. M. Z. Slawin and P. Kilian, Inorg. Chem., 2014, 53, 8538–8547 CrossRef CAS PubMed.
- J.-C. Hierso, Chem. Rev., 2014, 114, 4838–4867 CrossRef CAS PubMed.
- B. Németh, B. Khater, T. Veszprémi and J.-C. Guillemin, Dalton Trans., 2009, 3526–3535 RSC.
- B. A. Surgenor, M. Bühl, A. M. Z. Slawin, J. D. Woollins and P. Kilian, Angew. Chem., Int. Ed., 2012, 51, 10150–10153 CrossRef CAS PubMed.
- The signal at δP −26.3 appears as a very broad signal due to extensive quadrupolar line broadening arising from the two directly bound B atoms. As such, the 2JPP coupling constant is only observed on the signal at δP 13.9, as this P atom is bound to only one B atom and gives a less broad signal.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- A solution of 4a (approximately 100 mg) in CDCl3 containing tetramethylsilane (TMS) was prepared under an inert atmosphere and flame sealed. A measure of relative concentration was obtained by integrating the iPr CH signal of 4a relative to the TMS signal.
- S. Pandey, P. Lönnecke and E. Hey-Hawkins, Inorg. Chem., 2014, 53, 8242–8249 CrossRef CAS PubMed.
- K. Owsianik, R. Chauvin, A. Balińska, M. Wieczorek, M. Cypryk and M. Mikołajczyk, Organometallics, 2009, 28, 4929–4937 CrossRef CAS.
- T. Costa and H. Schmidbaur, Chem. Ber., 1982, 115, 1374–1378 CrossRef CAS.
- O. Kühl, Phosphorus-31 NMR Spectroscopy, Springer, Berlin, 1st edn, 2008 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
Footnote |
| † CCDC 1410480–1410484. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt02539g |
| This journal is © The Royal Society of Chemistry 2016 |