
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFormation of the spirocyclic, Si-centered cage cations [ClP(NSiMe3)2Si(NSiMe3)2P5]+ and [P5(NSiMe3)2Si(NSiMe3)2P5]2+†
M. H.
Holthausen
and
J. J.
Weigand
*
TU Dresden, Fachrichtung Chemie und Lebensmittelchemie, Professur für Anorganische Molekülchemie, 01062 Dresden, Germany. E-mail: jan.weigand@tu-dresden.de
First published on 15th June 2015
Abstract
On account of our interest in P4 activation by phosphenium ion insertion into P–P bonds we have developed synthetic routes to bicyclic N–P–Si-heterocycle 7 and probed its reactivity towards GaCl3 and P4. A GaCl3-induced rearrangement of 7 leads to the in situ formation of spirocyclic, Si-centered phosphenium ions. Their insertion into P–P bonds of one or two P4 tetrahedra yields polyphosphorus cages [ClP(NSiMe3)2Si(NSiMe3)2P5]+ (19+) and [P5(NSiMe3)2Si(NSiMe3)2P5]2+ (132+).
Introduction
White phosphorus (P4) is an archetypal building block for the syntheses of polyphosphorus cations featuring a high P to substituent ratio.1 In contrast to highly substituted cations RnPm (n > m), which are obtained via synthetic methods based on catena or cyclic polyphosphanes,2 the targeted preparation of P-rich cations RnPm (n < m) is achieved by taking advantage of the tetrahedral P4 framework.1,3 In a seminal paper, Krossing et al. reported that dicoordinated phosphenium ions, like other predominantly electrophilic ambiphiles,4 are able to insert into a P–P bond of the P4 tetrahedron.5 This was exploited for the preparation of a series of P5X2+-cages (X = Cl, Br, I).5,6 We expanded this methodology and prepared a series of symmetrically and unsymmetrically-substituted R2P5+- and RP5Cl+-cations (R = aryl, alkyl, R2N).7 The additional stability added by organo-substituents allowed for the stepwise insertion of up to three [Ph2P]+ phosphenium ions into P–P bonds of one P4 molecule yielding mono- to tri-cationic [Ph2P5]+-, [Ph4P6]2+- and [Ph6P7]3+-cages.8 A complementary study exploited 1,3-dichloro-cyclo-1,3-diphosphadiazane ClP(NDipp)2PCl (Dipp = 2,6-diisopropylphenyl), as a twofold phosphenium ion source for the stepwise activation of two P4 tetrahedra which yielded the mono- and dicationic species [ClP(NDipp)2P5]+ (1+) and [P5(NDipp)2P5]2+ (22+, Fig. 1).9 The related P5+-species 3[GaCl4] was obtained by the reaction of P4 with the four-membered heterocycle Cl2Si(NSiMe3)2PCl and GaCl3 while the isolobale Al-species 4 is the result of the reaction of phosphenium zwitterion Cl2Al(NSiMe3)2P with P4.10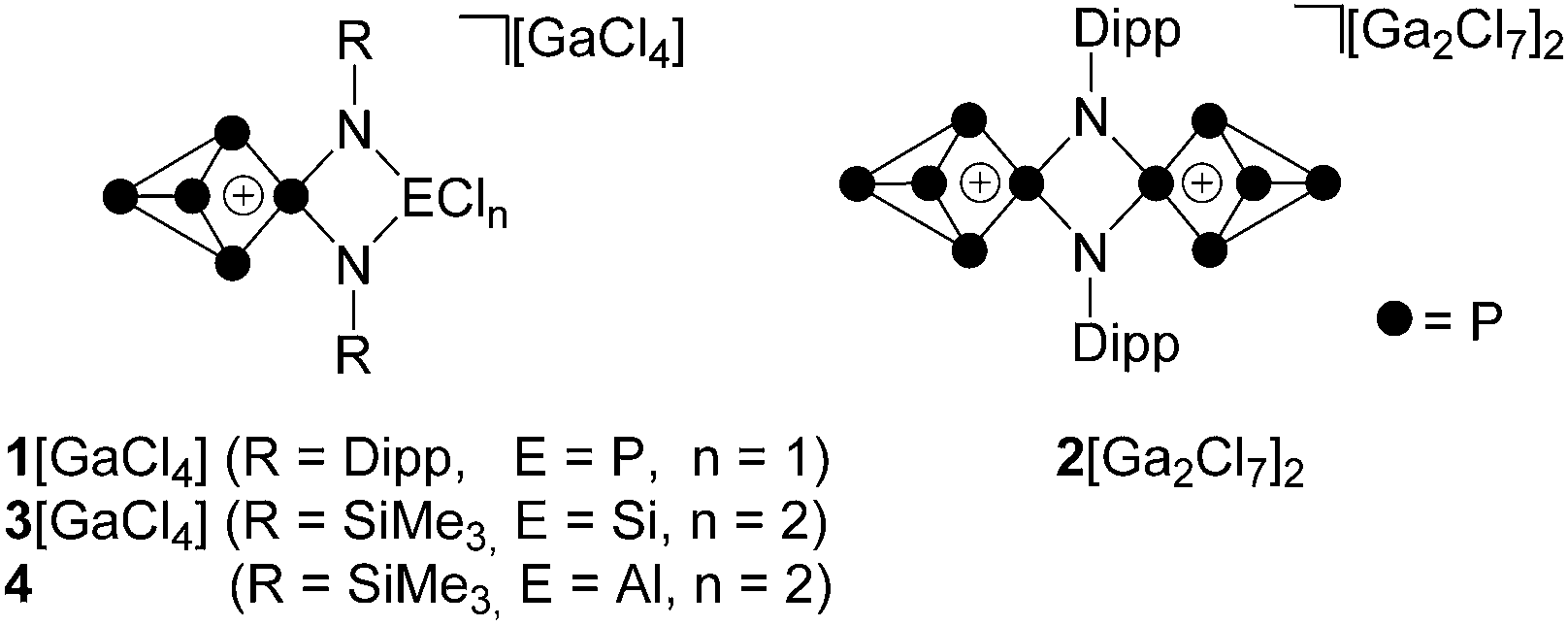 | ||
| Fig. 1 P5+-cages 1+, 22+, 3+ and 4 featuring a fused four-membered heterocycle. | ||
Only recently, cationic polyphosphorus cages have emerged as valuable synthetic building blocks for three purposes. First, they can be selectively fragmented in reactions with carbenes which results in the formation of Pn-species (e.g. a P5+-cage cation yields a P2- and cationic P3+-species).11 Second, they can be oxidized with selenium or sulphur which allows for the targeted preparation of cationic phosphorus–chalcogen cages.7c Third, they can be used for the controlled release of P4 due to the reversibility of the phosphenium ion insertion.10b
With the intention to further expand the range of methods for the in situ generation of phosphenium ions for P4 activation, we investigated the reactivity of bicyclic phosphorus–nitrogen–silicon heterocycle 7 with GaCl3 and P4. A GaCl3-induced rearrangement reaction was observed which formally gives access to the spirocyclic, Si-centered compound ClP(NSiMe3)2Si(NSiMe3)2PCl. Chloride abstraction by GaCl3 and insertion of the respective phosphenium ion in P–P bonds of one or two P4 molecules yields the Si-centered polyphosphorus cages [ClP(NSiMe3)2Si(NSiMe3)2P5]+ (19+) and [P5(NSiMe3)2Si (NSiMe3)2P5]2+ (132+).
Results and discussion
Synthetic routes towards and characterization of 7
As part of our ongoing interest in four-membered phosphorus–nitrogen-element heterocycles as precursors for phosphenium ions12 that can insert into a P–P bond of P4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10a we revisited the synthesis of heterocycle 6. This compound is synthesized by the reaction of iminophosphane 5 with SiCl4 according to a procedure reported by Niecke and co-workers (Scheme 1).13 Compound 6 was obtained from the reaction mixture by distillation (40 °C, 8 × 10−2 mbar) and isolated in 41% yield in accordance with the literature report. Surprisingly, however, a second fraction was obtained at higher temperatures (105 °C, 2 × 10−3 mbar) and identified as bicyclic compound 7 (18% yield).14 The 31P{1H} NMR spectrum of 7 dissolved in C6D6 shows a singlet resonance at δ(P) = 211.8 ppm which is in the typical chemical shift region of diphosphadiazanes featuring amino-groups on P.15
10a we revisited the synthesis of heterocycle 6. This compound is synthesized by the reaction of iminophosphane 5 with SiCl4 according to a procedure reported by Niecke and co-workers (Scheme 1).13 Compound 6 was obtained from the reaction mixture by distillation (40 °C, 8 × 10−2 mbar) and isolated in 41% yield in accordance with the literature report. Surprisingly, however, a second fraction was obtained at higher temperatures (105 °C, 2 × 10−3 mbar) and identified as bicyclic compound 7 (18% yield).14 The 31P{1H} NMR spectrum of 7 dissolved in C6D6 shows a singlet resonance at δ(P) = 211.8 ppm which is in the typical chemical shift region of diphosphadiazanes featuring amino-groups on P.15
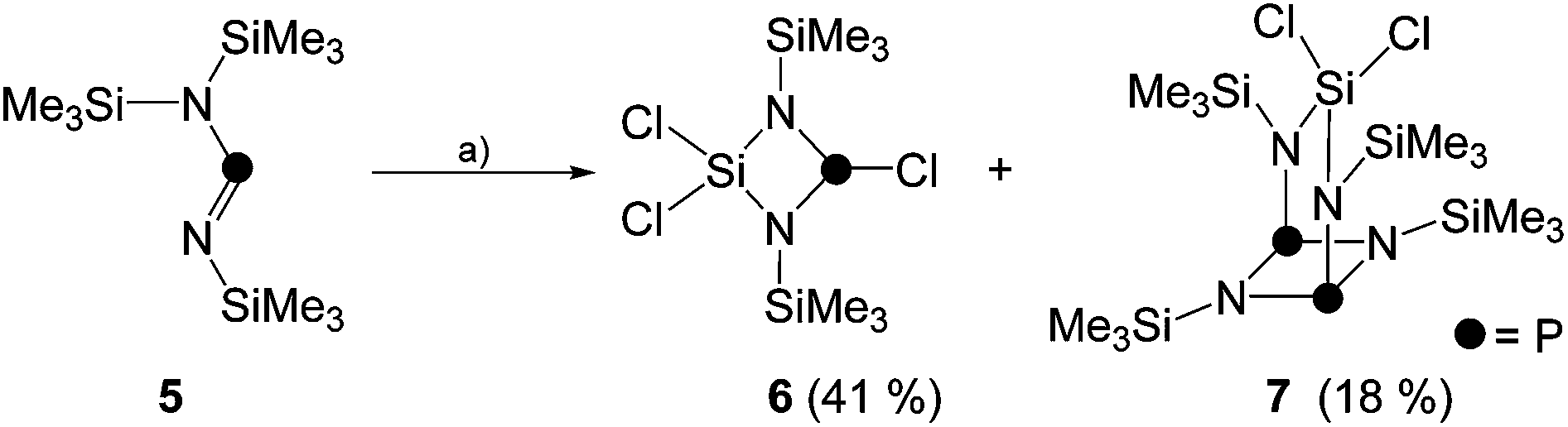 | ||
| Scheme 1 Preparation of compounds 6 and 7; (a) +SiCl4, −Me3SiCl, neat, 80 °C, 3 d. | ||
The molecular structure of 7 is C2V-symmetric and features an distorted planar four-membered [NP]2-ring (largest deviation from the plane 0.195 Å, Fig. 2). All four N atoms exhibit a trigonal planar arrangement (angular sums range from 358.8(3)° to 360.0(3)°) whereas the P atoms are involved in pyramidal bonding environments (angular sums P1: 286(3)° and P2: 286.2(2)°). The P1–N1 and P2–N2 bonds are almost orthogonal to the [NP]2-plane (N1–P1⋯P2: 95.84(4)°, N2–P2⋯P1: 95.84(4)°). The C2V-symmetric arrangement of the N2SiCl2-moiety in 7 is rare16 and contrasts other known diamido-cyclodiphosphazane compounds compounds coordinating main group fragments.15 Compounds of type (PNR)2NR2EXn typically exhibit CS-symmetric, seco-heterocube type structures in which the main group element fragment (e.g. EXn = Mg, BPh, AlCl, GaCl, Ge, Sn, SnCl2, SiCl2, AsCl, SbCl, BiCl, R = t-Bu, Ph) is coordinated by three N atoms and occupies the edge of a distorted cube.15
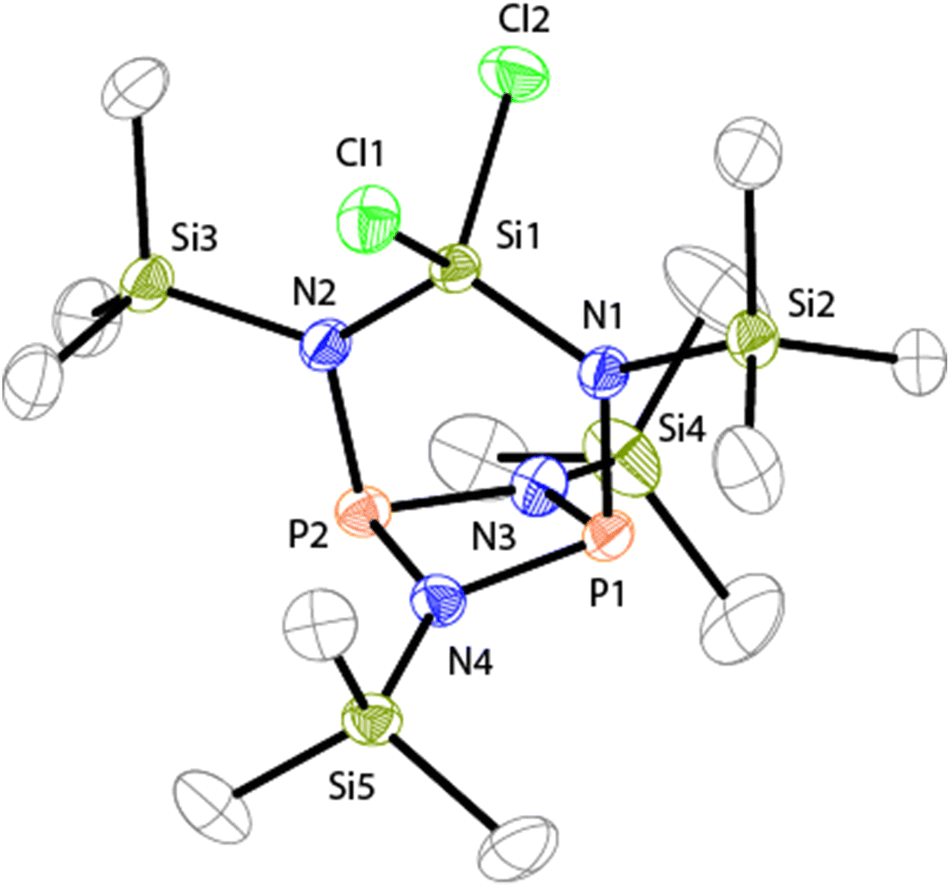 | ||
| Fig. 2 Molecular structure of 7 (hydrogen atoms are omitted for clarity and thermal ellipsoids are displayed at 50% probability); selected bond lengths [Å] and angles [°]: N1–P1 1.734(1), N3–P1 1.721(1), N4–P1 1.719(4), N2–P2 1.738(1), N3–P2 1.724(1), N4–P2 1.720(1), P1⋯P2 2.5235(6), N1–Si1 1.708(1), N1–Si2 1.773(1), N2–Si1 1.710(1), N2–Si3 1.774(1), N3–Si4 1.728(1), N4–Si5 1.740(1), Si1–Cl2 2.0554(6), Si2–Cl1 2.5235(6); P1–N4–P2 94.39(6), P1–N3–P2 94.19(7), N3–P1–N4 84.06(7), N3–P2–N4 83.96(7), N1–P1–N3 100.48(6), N1–P1–N4 102.10(6), N2–P2–N3 100.29(7), N2–P2–N4 101.91(6), N1–P1⋯P2 95.84(4), N2–P2⋯P1 95.58(4), Si1–N1–P1 117.14(7), Si1–N2–P2 117.18(8), N1–Si1–N2 114.19(6), Cl1–Si1–Cl2 104.35(2). | ||
The molecular arrangement observed for 7 in the solid state also persists in solution. The observation of two sets of resonances for its chemically inequivalent Me3Si-groups in the 1H NMR spectrum (δ(H) = 0.21 and 0.40 ppm) confirm the C2V-symmetry. The 13C{1H} NMR spectrum shows a triplet resonance which is assigned to the carbon atoms of the Me3Si-groups attached to the [PN]2-ring (δ(C) = −0.1 ppm, 3J(CP) = 3.6 Hz). A pseudo-triplet is observed for the Me3Si-groups adjacent to the N2SiCl2-moiety (δ(C) = 2.3 ppm, |3J(CP) + 5J(CP)| = 5.0 Hz) and is a result of an AA′X3X′3 spin system (A = 31P, X = 13C) with a comparatively large 2J(PAPA′) coupling constant.17 The same arguments account for the pseudo-triplet resonance in the 29Si{1H} NMR spectrum of 7 (δ(Si) = 7.7 ppm) which is assigned to the Me3Si-groups adjacent to the N2SiCl2-moiety (|2J(SiP) + 4J(SiP)| = 12.5 Hz). The triplet resonance corresponding to the Me3Si-groups attached to the [PN]2-ring (δ(Si) = 1.8 ppm, 3J(SiP) = 11.7 Hz) and the resonance of the SiCl2 moiety (δ(Si) = −47.4 ppm) are observed in the expected regions.18
Due to the low isolated yield of 7, it was of interest to develop an alternative synthetic approach. Compound 8 is the head to tail dimer of iminophosphane 5 and was obtained according to a literature known procedure.17 The solvent free reaction of 8 with an excess of SiCl4 gave selectively and quantitatively compound 9via Me3SiCl elimination (Scheme 2).19 Compound 9 is the trans-conformer of an unsymmetrically-substituted diphosphadiazane derivative and, thus, shows an AX spin system in its 31P{1H} NMR spectrum (δ(PA) = 219.5 ppm, δ(PX) = 232.3 ppm, 2J(PAPX) = 12.0 Hz). The 1H NMR spectrum of 9 shows singlet resonances for the SiMe3-groups on the [NP]2-ring and for both pointing towards the plane of the four-membered ring. The SiMe3 group pointing away from the ring, however, shows a relatively large 4J(HP) coupling constant (δ(H) = 0.27 ppm, 4J(PAH) = 3.7 Hz). For structurally related compounds, this was rationalized by the close proximity of the CH3-groups to the lone pair of electrons on the adjacent P atom.20 Due to the same reason, two relatively large 4J(PH) coupling constants (3.4 and 3.7 Hz) were observed for the isomer of 9 in which the SiCl3-group points towards the face of the four-membered ring (9′).19 The chloro-substituted Si atom in 9 appears as doublet resonance in the typical range in the 29Si NMR spectrum (δ(Si) = −27.3 ppm)21 with a remarkably large coupling constant of 2J(SiPX) = 26.0 Hz. A coupling constant of similar magnitude is observed for the Me3Si-group pointing away from the four-membered ring (δ(Si) = 7.8 ppm, 2J(SiPA) = 32.4 Hz), whereas those pointing to the faces of the [NP]2-ring exhibit rather small coupling constants (δ(Si) = 4.9 and 0.2 ppm; 2J(SiP) = 4.2 and 3.0 Hz). Similarly the 13C{1H} NMR spectrum reveals rather small coupling constants for the latter Me3Si-groups (3J(CP) = 8.0 and 10.5 Hz) and a large coupling constant for the Me3Si-group pointing away from the [NP]2-ring plane (3J(CPA) = 19.8 Hz).
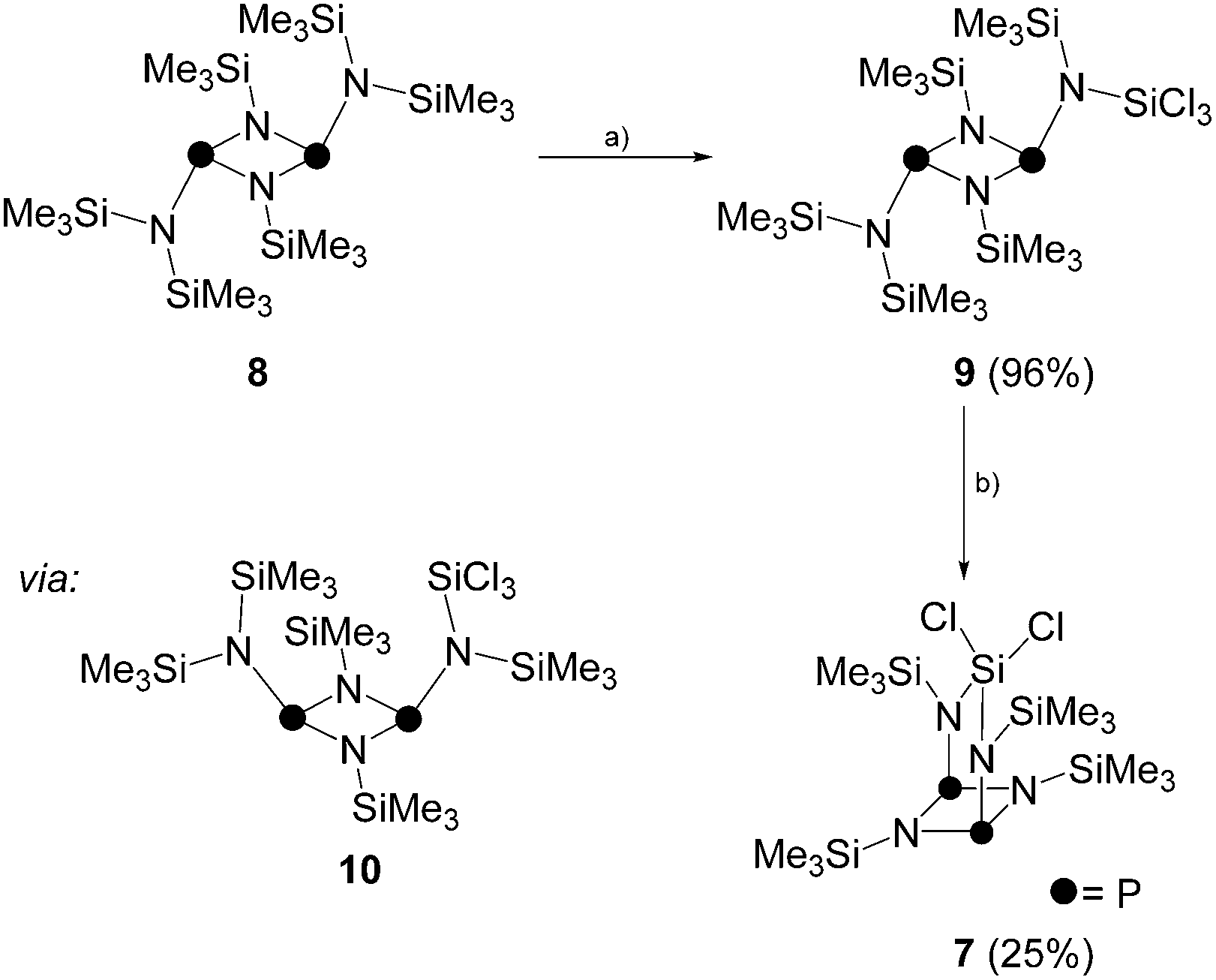 | ||
| Scheme 2 Synthesis of 9via reaction of 8 and SiCl4; (a) +SiCl4, −Me3SiCl, rt, 12 h, 96%; and subsequent transformation to 7; (b) neat, 185 °C, 10 min, 25%. | ||
It is assumed, that 7 is obtained from 9via an intermediate of type 10 which forms upon rotation22 of the P–N bond involving the SiCl3-substituted N atom and inversion23 of one of the P centers in 9 (Scheme 2). The arrangement of the SiCl3-group and one of the SiMe3-groups in intermediate 10 allows for Me3SiCl elimination yielding 7. Indeed, heating 9 for ten minutes to 185 °C results in conversion to 7 in 25% yield.24
Reactivity of 7 towards GaCl3 and P4
Diphosphadiazanes and related compounds bearing chloro- and Me3Si-substituents are known to undergo a variety of distinct reactions with Lewis acids. Next to halide abstraction,10 also elimination of Me3SiCl,25 migration of Me3Si-,26 chloro- or methyl-substituents27 and P–N bond cleavage reactions are reported.28 Especially the latter reaction is of interest, since it is assumed to proceed via phosphenium ion intermediates. Thus, bicyclic compound 7 is a promising substrate for the generation of phosphenium ions and, subsequently, insertion of the latter into P–P bonds of P4.Thus, the reaction of 7 with the Lewis acid GaCl3 was probed (Scheme 3). The addition of one equivalent of GaCl3 to a solution of 7 in C6H5F gave a reddish colored reaction mixture which was investigated by means of 31P{1H} NMR spectroscopy. The formation of the Lewis-acid/base adduct 11 is proposed on the basis of the observation of two broad resonances in an approximate ratio of 1:1 (δ(PA) = 115 ppm, Δν1/2 = 1200 Hz; δ(PX) = 179.6 ppm, Δν1/2 = 600 Hz). The coordination of GaCl3 to a P atom is very likely, since they have been identified as the most basic sites in related compounds.29 The resonance at low field is tentatively assigned to the tri-coordinated P atom. Accordingly, the resonance at high field corresponds to the tetra-coordinated P atom and exhibits a stronger line broadening due to the coordination of GaCl3.
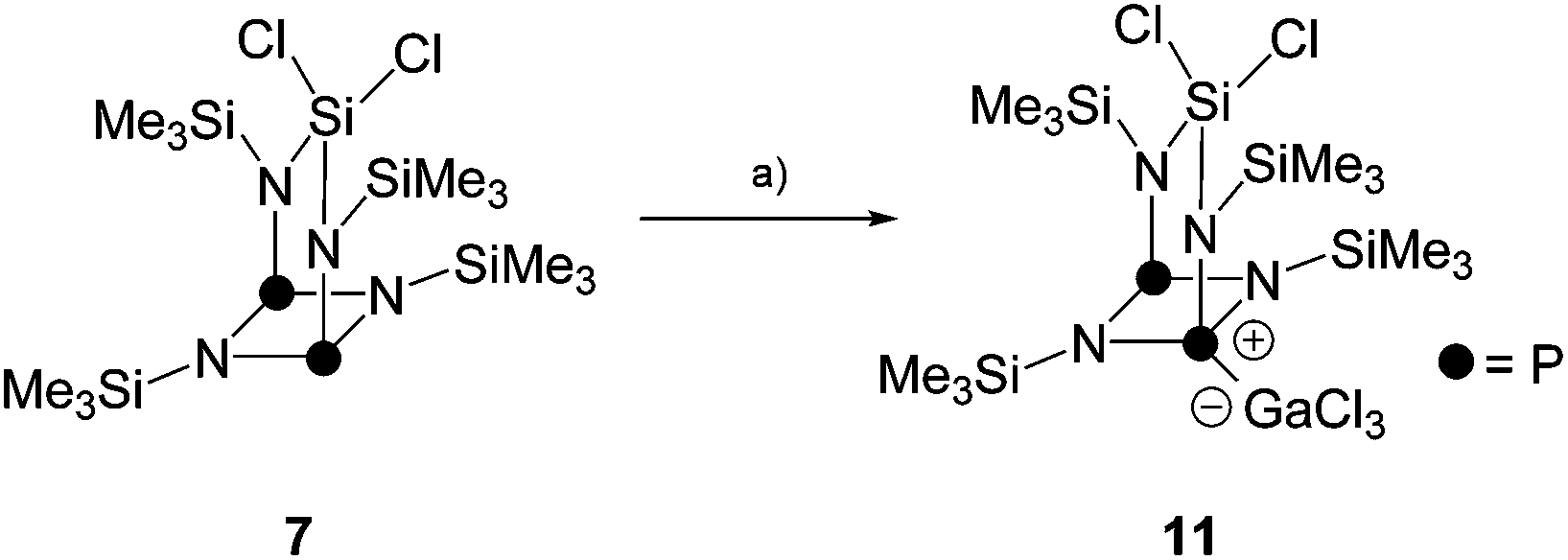 | ||
| Scheme 3 Formation of Lewis-acid/base adduct 11via the reaction of 7 and GaCl3; (a) +GaCl3, C6H5F, r.t., 1 h. | ||
Interestingly, P4 does not react with 11 which indicates that the latter is not a suitable phosphenium ion source. Adding two equivalents of GaCl3 to a solution of 7 in C6H5F, however, results in the rapid consumption of in situ generated 11. This reaction yields a complex mixture of not identified products and bodes well for the generation of reactive intermediates that might be able to activate P4. Indeed, mixtures of 7, P4 and GaCl3 in 1:1:2 and 1:2:4 stoichiometries were bright red and the consumption of P4 accompanied by a color change to brown was observed. Subsequent investigation by means of 31P{1H} NMR spectroscopy revealed the formation of P5+ cage cation 12+ and bridged bis(P5+)-cage dication 132+ by their characteristic A2MOX2 and A2MX2 spin systems (Scheme 4).
 | ||
| Scheme 4 Reaction of 7 with P4 and GaCl3 in various stoichiometries; (a) +P4, +2, GaCl3, C6H5F, r.t., 24 h; (b) +2, P4, +4, GaCl3, r.t., C6H5F, 24 h; the equation depicts the major products and is not balanced; anions of the products are not depicted. | ||
In addition, both spectra reveal significant amounts of P4 but neither remaining 7 nor the corresponding adduct 11 were observed which is attributed to unidentified side reactions. The products 12+ and 132+ are formally obtained by the insertion of phosphenium ions based on ClP(NSiMe3)2Si(NSiMe3)2PCl which features a Si-centered spiro[3.3]heptane-motif, into P–P bonds of P4. Interestingly, both reactions yield 12+ and 132+ in comparable ratios (1:1.8 for (a) and 1:1.5 for (b), Scheme 4). This contrasts the anticipated increase of the amount of dication 132+ in the presence of excess P4 and GaCl3. In addition, the formation of large quantities of 132+ in the reaction of 1:1:2 stoichiometry indicates that the reaction rate of a GaCl3-induced rearrangement of 7 yielding the spiro-motif is slow compared to that of the subsequent phosphenium ion insertion.
A tentative mechanism for the formation of such a Si-centered spiro[3.3]heptane-type species from 7 is illustrated in Scheme 5. While the reaction of 7 with one equivalent GaCl3 gives the Lewis-acid/base adduct 11 (vide supra), an excess of GaCl3 is assumed to initiate a chloride abstraction from the SiCl2-moiety. This is facilitated by the tendency of bicyclic compounds of type 7 to form a seco-heterocube-type structure.15 Thus, compound 7 might be in equilibrium with the related derivative 7I. Compound 7I features a hypervalent penta-coordinated Si-moiety which might favor the sequestering of a chloride anion. Thus, it is assumed that the reaction with GaCl3 proceeds via chloride abstraction and formation of the cationic seco-heterocube 14+ featuring a tetra-coordinated N atom. The next two steps constitute P–N bond cleavage and formation reactions yielding intermediates 15+ and 16+via formal retention of the seco-heterocube-type structure. Similar P–N bond ruptures were reported as decomposition pathways of diamino-cyclo-diphosphadiazanes.26 Intermediate 16+ features a diphosphadiazane [NP]2-ring which is assumed to react via a cyclo-reversion reaction to intermediate 17+. Cation 17+ features an aminoiminophosphane moiety similar to 5 which is tethered to a four-membered SiN2P-ring. Intramolecular nucleophilic attack of the imino-N atom on the chloro-substituted Si atom initiates a GaCl3-mediated transfer of a chloride anion from the Si atom to a di-coordinated P atom to give formally the phosphenium ion 18+. Cation 18+ features the Si-centered spiro[3.3]heptane-motif and, thus, is assumed to be accountable for the formation of cages cations 12+ and 132+via insertion into a P–P bond of P4, chloride abstraction by GaCl3 and subsequent insertion into a P–P bond of a second P4 tetrahedron.
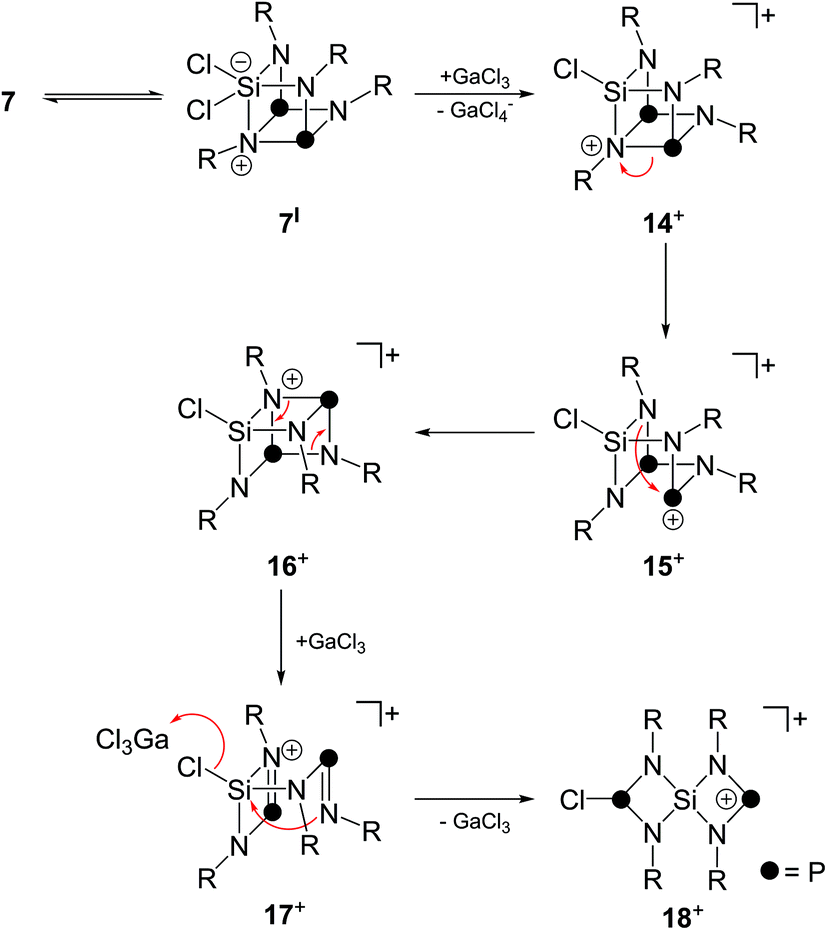 | ||
| Scheme 5 Suggested GaCl3-induced rearrangement mechanism of 7 to phosphenium ion 18+ featuring a Si-centered spiro[3.3]heptane-type motif; R denotes Me3Si-substituents; for reasons of simplification the reactions of intermediates or products possessing a di-coordinated P atom with P4 as well as retransfer of a chloride ion to a di-coordinated P atom are not considered. | ||
Attempts to isolate a gallate salt of 12+ from both reaction mixtures were unsuccessful, possibly due to the fluxional coordination behavior of the GaCl3-molecule to the PCl-function. However, slow diffusion of n-hexane into the reaction mixture of 1:1:2 stoichiometry yielded compound 19[GaCl4] in low yields (10%, Scheme 6). The 31P{1H} NMR spectrum of 19[GaCl4] dissolved in CD2Cl2 shows an ABMX2Y spin system which is in accordance with the Cs symmetry of the molecule (Fig. 3). The mirror plane is defined by the tetra-coordinated P atom and the two adjacent P atoms. The P–Cl unit is included in the plane and exhibits a spatial proximity to one of the two bridge-head P atoms. The chemical shifts and coupling constants involving the P5+-cage motif are similar to those observed for the cage cation 3+.10a The chloro-substituted P atom in 19+ exhibits a singlet resonance (δ(PY) = 166.8 ppm) in the typical range of silyl-substituted diamino-chlorophosphanes.13,30 A 4J(PP) coupling between this P atom and the tetra-coordinated P atom of the P5+-cage is not resolved. The 1H NMR spectrum of 19+ reveals three singlet resonances assigned to the chemically different Me3Si-groups which integrate in a 2:1:1 ratio. The high-field resonance (δ(H) = 0.31 ppm) exhibits the highest intensity and is assigned to the two Me3Si-groups bonded to the four-membered ring which incorporates the P–Cl moiety. The chemical shift is comparable to related four-membered ring compounds (6: δ(H) = 0.18 ppm,137: δ(H) = 0.21 ppm). Both resonances assigned to the Me3Si-groups bonded to the N atoms adjacent to the P5+-moiety exhibit a low field shift (δ(H) = 0.67 ppm and 0.76 ppm) and are comparable to the corresponding resonance of 3+ (δ(H) = 0.68 ppm).10a The 29Si{1H} NMR spectrum exhibits a resonance at δ(Si) = −58.0 ppm which is assigned to the Si spiro-atom. This resonance reveals a doublet of doublet splitting caused by 2J(SiP)-couplings to the tetra-coordinated P atom (2J(SiPM) = 8.5 Hz) and the chloro-substituted P atom (2J(SiPY) = 18.5 Hz).
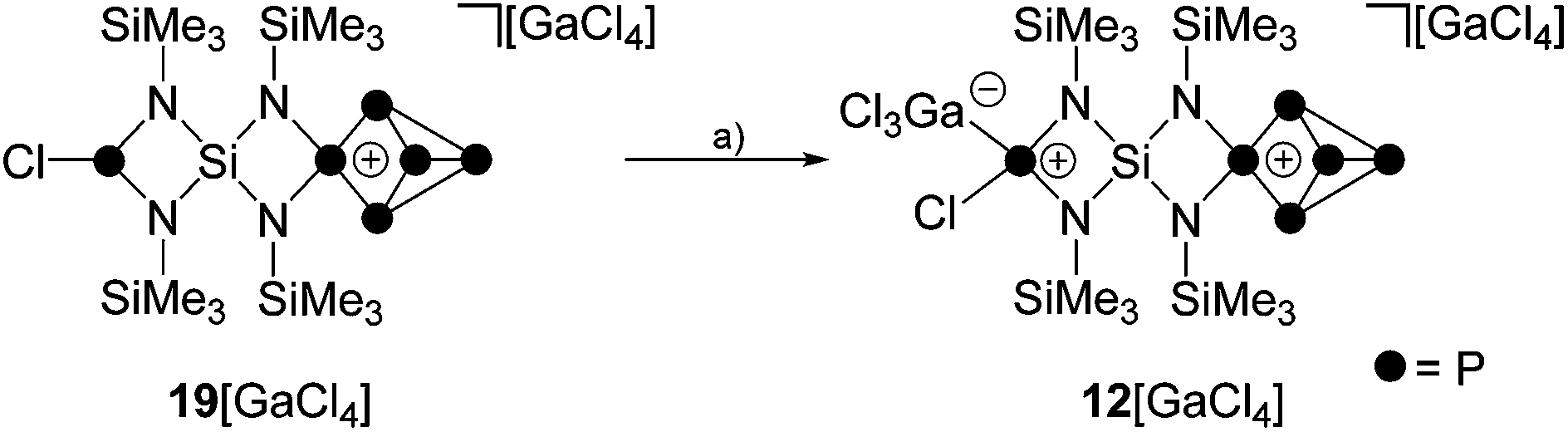 | ||
| Scheme 6 Reaction of 19[GaCl4] with GaCl3; (a) +GaCl3, CD2Cl2, r.t., 30 min. | ||
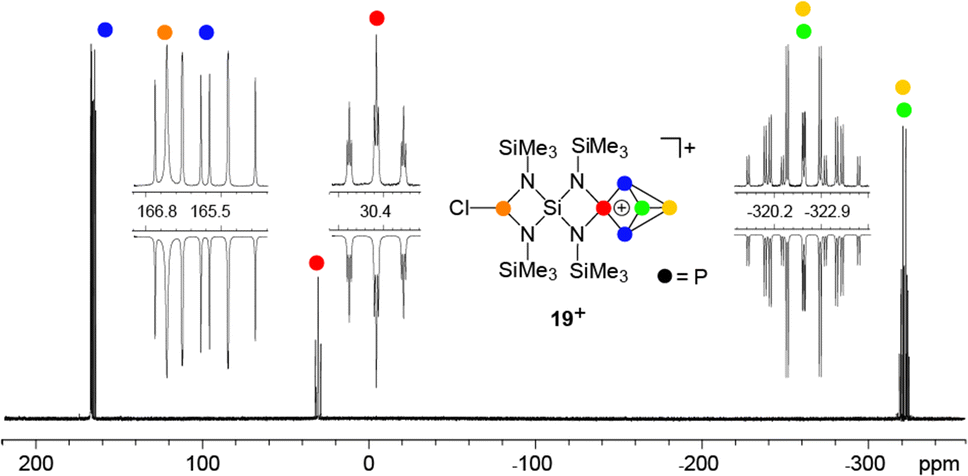 | ||
| Fig. 3 31P{1H} NMR spectrum of 19[GaCl4] (CD2Cl2, r.t.); insets show experimental (upwards) and fitted spectra (downwards); ABMX2Y spin system of 19+: δ(PA) = −322.9 ppm, δ(PB) = −320.2 ppm, δ(PM) = 30.4 ppm, δ(PX) = 165.5 ppm, δ(PY) = 166.8 ppm, 1J(PAPB) = −189.8 Hz, 1J(PAPX) = −143.4 Hz, 1J(PBPX) = −147.7 Hz, 1J(PMPX) = −245.1 Hz, 2J(PAPM) = 18.8 Hz, 2J(PBPM) = 18.0 Hz. | ||
The molecular structure of 19+ is depicted in Fig. 4 and the P–P bond lengths and angles in the P5+-moiety are comparable to those of 3+.10a Both four-membered rings are almost planar (largest deviation from the planes N1: 0.026 Å and N3: 0.022 Å) and exhibit a perpendicular arrangement (angle between both planes: 89.79(9)°). Due to the steric limitations of the four-membered heterocycles the spiro-Si atom exhibits a distorted tetrahedral arrangement with two rather small (N1–Si3–N2: 85.4(1)°, N3–Si3–N4: 87.7(1)°) and two widened (N1–Si3–N3: 122.9(1)°, N2–Si3–N4: 118.8(1)°) N–Si–N angles. Alternating bond lengths are observed within the two four-membered rings. The P–N bonds involving the tetra-coordinated P atom are shorter (N2–P1: 1.656(3) Å, N1–P1: 1.661(3) Å) than those involving the tri-coordinated P atom (N3–P6: 1.716(3) Å, N4–P6: 1.711(3) Å) and both magnitudes of bond lengths are also observed in the related cages 3+ and 2+.9,10a The Si–N bonds in the [SiN2P]-ring fused to the P5+-cage (N1–Si3: 1.755(3) Å, N2–Si3: 1.751(3) Å) are of similar lengths as observed for 3+.10a In contrast, the Si–N bonds in the second [SiN2P]-ring are shorter (N3–Si3: 1.711(3) Å, N4–Si3: 1.704(3) Å).
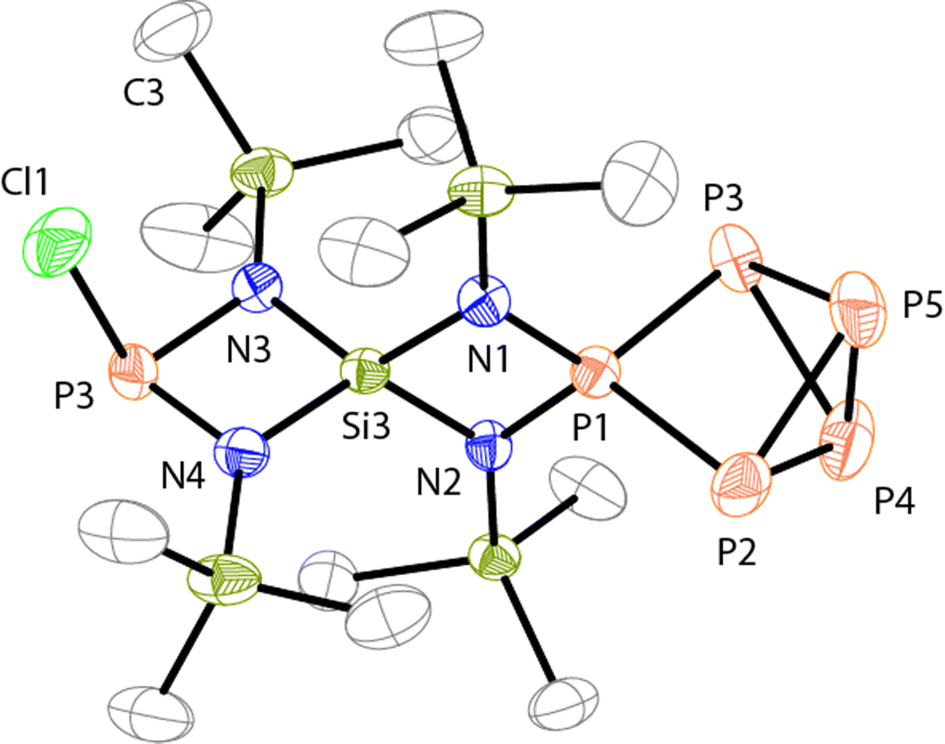 | ||
| Fig. 4 Molecular structure of 19+ in compound 19[GaCl4]·C6H5F (hydrogen atoms are omitted for clarity and thermal ellipsoids are displayed at 50% probability); selected bond lengths [Å] and angles [°]: N2–P1 1.656(3), N1–P1 1.661(3), N1–Si3 1.755(3), N2–Si3 1.751(3), N3–Si3 1.711(3), N4–Si3 1.704(3), N3–P6 1.716(3), N4–P6 1.711(3), Cl1–P6 2.238(2), P1⋯Si3 2.445(1), P6⋯Si3 2.472(1), P1–P2 2.167(1), P1–P3 2.165(1), P2–P4 2.249(2), P2–P5 2.238(2), P3–P4 2.251(2), P3–P5 2.229(2), P4–P5 2.169(2); N1–P1–N2 91.5(1), P1–N1–Si3 91.4(1), P1–N2–Si3 91.7(1), N1–Si3–N2 85.4(1), N3–Si3–N4 87.7(1), N1–Si3–N3 122.9(1), N2–Si3–N4 118.8(1), N3–P6–N4 87.2(1), N3–P6–Cl1 102.2(1), N4–P6–Cl1 101.7(1), P3–P1–P2 91.15(5), P1–P2–P5 83.95(5), P1–P2–P4 82.53(5), P5–P2–P4 57.82(5), P5–P4–P2 60.83(5), P2–P5–P3 87.67(5). | ||
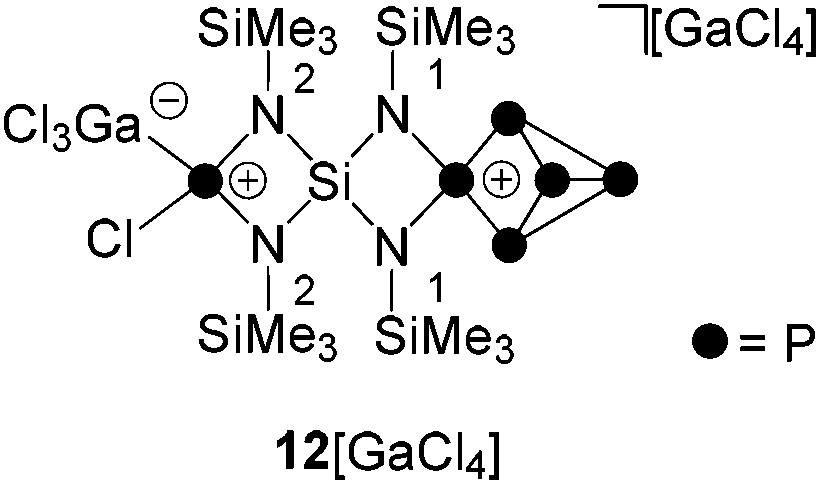
The addition of GaCl3 to a solution of 19[GaCl4] in CD2Cl2 yields the previously mentioned Lewis-acid base adduct 12+ (Scheme 6). The A2MOX2 spin system observed in the 31P{1H} NMR spectrum of 12+ suggests a time averaged C2v-symmetry of the molecule in solution which can be explained by a fluxional behavior of the coordinated GaCl3 molecule (Fig. 5). The A2MX2 part of the A2MOX2 spin system corresponds to the P5+-cage moiety and reveals comparable chemical shifts and coupling constants as observed for 3+.10a The resonance corresponding to the O part of the spin system is very broad (δ(PO) = 144 ppm, Δν1/2 = ∼1200 Hz) which is caused by the fluxional behavior of the coordinated GaCl3 molecule and its quadrupole moment.
 | ||
| Fig. 5 31P{1H} NMR spectrum of 12[GaCl4] (CD2Cl2, r.t.); A2MOX2 spin system of 12+: δ(PA) = −318.3 ppm, δ(PM) = 33.4 ppm, δ(PO) = 144 ppm (Δν1/2 = ∼1200 Hz), δ(PX) = 170.8 ppm, 1J(PAPX) = −144.8 Hz, 1J(PMPX) = −248.6 Hz, 2J(PAPM) = 19.0 Hz. | ||
Dication 132+ was isolated as a [Ga2Cl7]− salt from the reaction of 7, P4 and GaCl3 in a 1:2:4 stoichiometry (Scheme 4) by the addition of n-hexane. This gave a brown oil which was isolated by decanting the supernatant and upon addition of small amounts of 1,2-C6H4F2 yielded a suspension containing a yellow microcrystalline material of 13[Ga2Cl7]2. This compound was isolated by filtration in low yields (20%) in an approximate purity of 75%. Further purification by recrystallization from CH2Cl2/n-hexane leads to significantly decreased yields (7% yield in >90% purity). The 31P{1H} NMR spectrum of 13[Ga2Cl7]2 dissolved in CD2Cl2 shows an A2MX2-spin system in accordance with the C2v-symmetry of the molecule (Fig. 6). The C2-axis includes both tetra-coordinated P atoms and the spiro-Si atom and the mirror planes are defined by the four-membered [SiN2P]-rings. The resonances and coupling constants of 132+ are similar to those observed for the related compounds 19+ and 3+.10a
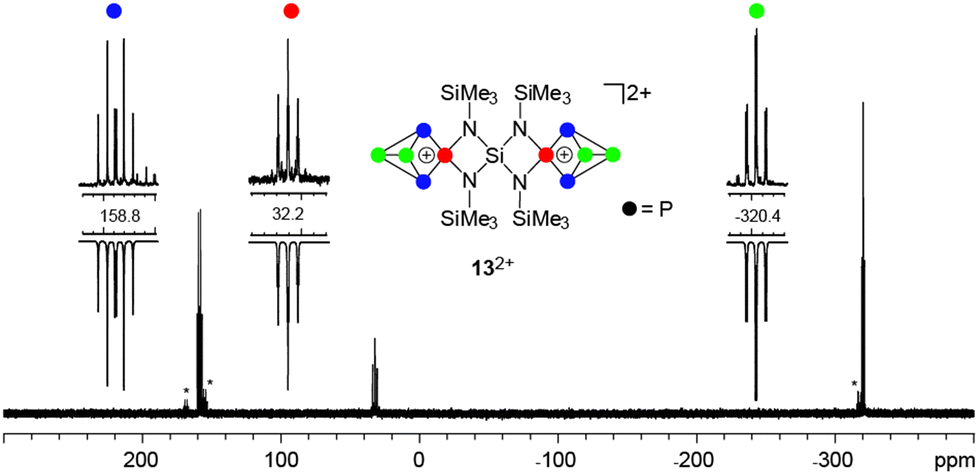 | ||
| Fig. 6 31P{1H} NMR spectrum of 13[Ga2Cl7]2 (CD2Cl2, r.t.); unidentified side products are marked with asterisks; A2MX2 spin system of 132+: δ(PA) = −320.4 ppm, δ(PM) = 32.2 ppm, δ(PX) = 158.8 ppm, 1J(PAPX) = −142.7 Hz, 1J(PMPX) = −259.1 Hz, 2J(PAPM) = 20.0 Hz. | ||
Single crystals of compound 13[Ga2Cl7]2 were obtained by diffusion of n-hexane in a CH2Cl2 solution of 13[Ga2Cl7]2 at −35 °C. The compound crystallizes with two independent formula units in the asymmetric unit. Two of the four Ga2Cl7− anions are highly disordered exhibiting unusually high thermal displacement parameters (see ESI† for details). Single crystals of 13[Ga2Cl7][GaCl4] were obtained by layering the supernatant solution of the reaction mixture of the synthesis of 13[Ga2Cl7]2 with n-hexane at −35 °C. The data obtained by X-ray single crystal structure determination was of higher quality and, thus, the molecular structure of 13[Ga2Cl7][GaCl4] is discussed (Fig. 7). The P–P bond lengths and angles in the P5+-moieties are comparable to those of related P5+-cage compounds.9,10a The P–N bonds in 132+ are rather short (av. P–N: 1.666(8) Å) which is a typical feature of P–N bonds involving phosphonium moieties. The Si–N bond lengths in the four-membered rings are nearly identical (av. Si–N: 1.732(8) Å) and are between the two types of bond lengths observed for 19+ (av. Si–N: 1.753(6) Å and Si–N: 1.7007(6) Å).
 | ||
| Fig. 7 Molecular structure of 132+ in compound 13[Ga2Cl7][GaCl4](hydrogen atoms are omitted for clarity and thermal ellipsoids are displayed at 50% probability); selected bond lengths [Å] and angles [°]: N1–P1 1.667(2), N2–P1 1.671(2), N3–P6 1.663(2), N4–P6 1.665(2), Si1–N1 1.731(2), Si1–N2 1.732(2), S1–N3 1.736(2), Si1–N4 1.728(2), P1⋯Si1 2.4452(8), P6⋯Si1 2.4277(8), P1–P2 2.1540(8), P1–P3 2.1499(8), P2–P4 2.245(1), P2–P5 2.245(1), P3–P4 2.246(1), P3–P5 2.2464(9), P4–P5 2.168(1), P6–P7 2.1511(8), P6–P8 2.1572(2), P7–P9 2.240(1), P7–P10 2.252(1), P8–P9 2.2415(9), P8–P10 2.242(1), P9–P10 2.172(1); N1–P1–N2 90.81(9), N3–P6–N4 90.99(9), N1–Si1–N2 86.73(9), N3–Si1–N4 86.49(9), P1–N1–Si1 91.26(9), P1–N2–Si1 91.12(9), P6–N3–Si1 91.14(9), P6–N4–Si1 91.34(9), P3–P1–P2 92.59(3), P1–P2–P5 81.73(3), P1–P2–P4 83.19(3), P5–P2–P4 57.69(3), P5–P4–P2 61.25(3), P2–P5–P3 87.60(3), P7–P6–P8 92.30(3), P6–P7–P10 82.39(3), P6–P7–P9 82.89(3), P10–P7–P9 57.83(3), P10–P9–P7 61.36(3), P7–P10–P8 87.48(3). | ||
Conclusions
The bicyclic P–N–Si heterocycle 7 was targeted as source for the in situ generation of phosphenium cations for P4 activation. In this context, two distinct synthetic protocols for its preparation were thoroughly investigated and gave insights into the formation of the bicycle. The reaction of 7 with GaCl3 initially yields adduct 11. This adduct is not stable and subsequently rearranges to give in situ spirocyclic, Si-centered compound ClP(NSiMe3)2Si(NSiMe3)2PCl. The latter species gives access to polyphosphorus cage cations [ClP(NSiMe3)2Si(NSiMe3)2P5]+ (19+) and [P5(NSiMe3)2Si(NSiMe3)2P5]2+ (132+) in the presence of GaCl3 and P4. We are continuing to investigate the Lewis-acid mediated generation of phosphenium ions for P4 activation from related phosphorus–nitrogen-element bicycles. Furthermore, studies directed towards the utilization of 12+, 19+ and 132+ as synthetic building blocks will be the target of future efforts.Experimental
General
General information on materials and methods as well as 31P{1H} NMR spectra of reaction mixtures are given in the ESI.†Synthesis of Cl2Si(NSiMe3)2(PNSiMe3)2 (7)
Method A: The literature reported synthesis of 5 was performed on a 20 mmol scale.13 Compound 5 was removed by distillation from the reaction mixture (40 °C, 8 × 10−2 mbar). The remaining colorless, slushy residue was dissolved in C6H5F (5 mL) yielding a turbid suspension. The solvent was removed in vacuo yielding a sludgy residue which was redistilled employing a short Vigreux column (5 cm). Compound 7 was obtained as colorless oil (1.789 g, 3.51 mmol, 18%, 105 °C, 2 × 10−3 mbar) which solidified shortly after distillation. Method B: 9 (305 mg, 0.50 mmol, 1.0 eq.) was heated to 185 °C for 10 min. In the course of the reaction a colorless liquid is formed accompanied by the condensation of Me3SiCl on colder parts of the reaction vessel. After cooling to ambient temperature the reaction mixture remains a liquid. Isolation of 7 from this mixture proceeds as described in method A.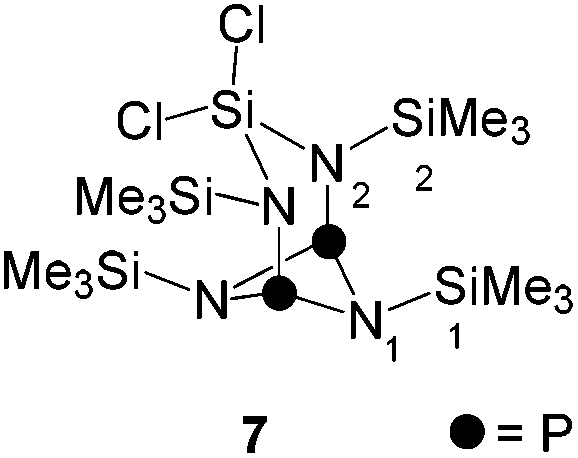
m.p.: 55.6–57.8 °C; Raman (300 mW, [cm−1]): ν = 2959 (390), 2899 (100), 1410 (11), 690 (10), 645 (22), 613 (51), 562 (4), 349 (34), 185 (30), 141 (6), 75 (10); IR (ATR, [cm−1]): ν = 2956 (w), 1408 (vw), 1249 (s), 1098 (vw), 973 (vw), 942 (w), 883 (w), 830 (vs), 776 (vw), 754 (vw), 713 (w), 682 (w), 643 (vw), 556 (s), 450 (m); 1H NMR (C6D6, [ppm]): δ = 0.21 (18H, s, H1), 0.39 (18H, s, H2); 13C{1H} NMR (C6D6, [ppm]): δ = −0.08 (6C, t, C1, 3J(CP) = 3.6 Hz), 2.7 (6C, pseudo-t, C2, 3J(CP) = 5.0 Hz); 29Si{1H} NMR (C6D6, [ppm]): δ = −47.4 (1Si, s, Si3), 1.8 (2Si, t, Si1, 2J(SiP) = 11.7 Hz), 7.7 (2Si, pseudo-t, Si2, 2J(SiP) = 11.8 Hz); 15N NMR (C6D6, [ppm]): δ = −397 (t, N1, 1J(NP) = 55 Hz), −374 (d, N2, 1J(NP) = 75 Hz); 31P{1H} NMR (C6D6, [ppm]): δ = 211.8 (s); elemental analysis for C12H36Cl2N4P2Si5: calcd: C 28.3, H 7.1, N 11.0; found: C 28.5, H 7.3, N 10.6; MS-ESI-EM: 473.0943 [M-Cl−], calcd: for C12H36Cl1N4P2Si5: 473.0945.
Synthesis of (SiMe3)2N(PNSiMe3)2N(SiMe3)(SiCl3) (9)
Compound 8 (1.114 g, 2.00 mmol, 1.0 eq.) was suspended in SiCl4 (6.796 g, 40.0 mmol, 40.0 eq.) and stirred for 12 h at ambient temperature. After removal of all volatiles in vacuo9 was isolated in quantitative yields as colorless solid (1.187 g, 1.92 mmol, 96%).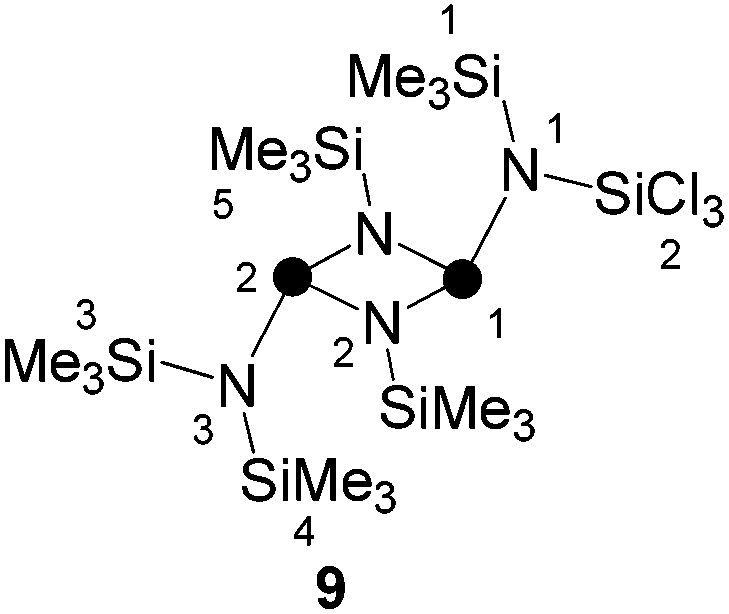
m.p.: 180.2–182.5 °C; Raman (300 mW, [cm−1]): ν = 2957 (37), 2906 (100), 1410 (15), 686 (13), 651 (4), 641 (64), 586 (21), 488 (4), 436 (38), 351 (21), 206 (37), 107 (27), 78 (18); IR (ATR, [cm−1]): ν = 3139 (vw), 3047 (m), 2958 (vw), 1406 (m), 1251 (s), 1060 (m), 923 (m), 834 (vs), 778 (vw), 755 (w), 712 (w), 677 (w), 572 (vw), 559 (m), 506 (w), 432 (vw); 1H NMR (C6D6, [ppm]): δ = 0.17 (18H, s, H5), 0.27 (9H, d, H3, 4J(HP) = 3.7 Hz), 0.52 (9H, s, H4), 0.64 (9H, s, H1); 13C{1H} NMR (C6D6, [ppm]): δ = 1.4 (6C, t, C5, 3J(CP) = 2.5 Hz), 4.7 (3C, d, C1, 3J(CP) = 10.5 Hz), 4.8 (3C, d, C3, 3J(CP) = 19.8 Hz), 5.0 (3C, d, C4, 3J(CP) = 8.0 Hz); 29Si{1H} NMR (C6D6, [ppm]): δ = −27.3 (1Si, d, Si2, 2J(SiP) = 26.0 Hz), 0.2 (1Si, d, Si4, 2J(SiP) = 3.0 Hz), 1.5 (2Si, t, Si5, 2J(SiP) = 7.4 Hz), 4.5 (1Si, d, Si2, 2J(SiP) = 4.2 Hz), 7.6 (1Si, d, Si3, 2J(SiP) = 32.4 Hz); 15N NMR (C6D6, [ppm]): δ = 67.1 (t, N2, 1J(NP) = 50 Hz), 93.8 (d, N1, 1J(NPX) = 90 Hz), 113.9 (d, N3, 1J(NPA) = 90 Hz); 31P{1H} NMR (C6D6, [ppm]): AX spin system: δ(PA) = 218.6 (d, P1, Δν1/2 = 42 Hz, 2J(PAPX) = 12 Hz), δ(PX) = 232.2 (d, P2, Δν1/2 = 37 Hz, 2J(PAPX) = 12 Hz); elemental analysis for C15H45Cl3N4P2Si6: calcd: C 29.1, H 7.3, N 9.1; found: C 28.8, H 7.3, N 8.6.
Reaction of 7, P4 and GaCl3 in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :1:2 and 1:2:4 stoichiometries
:1:2 and 1:2:4 stoichiometries
1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :1:2: Compound 7 (256 mg, 0.50 mmol, 1.0 eq.) was added to a suspension of P4 (62 mg, 0.50 mmol, 1.0 eq.) in C6H5F (5 mL). A solution of GaCl3 (176 mg, 1.00 mmol, 2.0 eq.) in C6H5F (2 mL) was added dropwise to the suspension giving a red colored reaction mixture which was stirred for 12 h at ambient temperature. In the course of the reaction the color of the reaction mixture changed to yellow and the dissolving of P4 was observed. The reaction mixture was investigated by means of 31P{1H} NMR spectroscopy (see ESI†). 1:2:4: Compound 7 (256 mg, 0.50 mmol, 1.0 eq.) was added to a suspension of P4 (124 mg, 1.00 mmol, 2.0 eq.) in C6H5F (5 mL). A solution of GaCl3 (352 mg, 2.00 mmol, 4 eq.) in C6H5F (4 mL) was added dropwise to the suspension giving a red colored reaction mixture which was stirred for 12 h at ambient temperature. In the course of the reaction the color of the reaction mixture changed to brown and the dissolving of P4 was observed. The reaction mixture was investigated by means of 31P{1H} NMR spectroscopy (see ESI†). n-Hexane (2 mL) was added leading to the formation of a brown oil. The supernatant was removed, diluted with C6H5F (6 mL) and layered with n-hexane (3 mL) at −35 °C. Small amounts of crystalline material of 19[GaCl4] (41 mg, 10%), suitable for X-ray single crystal structure determination, were obtained within a few days. The remaining oil was washed with n-hexane (3 × 3 mL) transforming it into a brown sludge. All volatiles were removed in vacuo and the sludge was suspended in 1,2-C6H4F2 (2 mL) leading to the formation of a yellow, microcrystalline solid. The supernatant was removed and the yellow powder was washed with 1,2-C6H4F2/n-hexane (1:1 mixture, 2 × 2 mL). The obtained yellow powder consisting of 13[Ga2Cl7]2 in an approximate purity of 75% (determined by 31P{1H} NMR spectroscopy, 20% yield, 145 mg) was isolated by filtration and dried in vacuo. Recrystallization from a CH2Cl2 solution by slow diffusion of n-hexane yielded crystalline material of 19[Ga2Cl7]2 (purity > 90%) which was suitable for single crystal structure determination. Isolation was conducted via filtration and removal of all volatiles in vacuo (7% yield, 55 mg). Single crystals of 13[Ga2Cl7][GaCl4] were obtained by layering the diluted supernatant of the reaction mixture with n-hexane at −35 °C.
:1:2: Compound 7 (256 mg, 0.50 mmol, 1.0 eq.) was added to a suspension of P4 (62 mg, 0.50 mmol, 1.0 eq.) in C6H5F (5 mL). A solution of GaCl3 (176 mg, 1.00 mmol, 2.0 eq.) in C6H5F (2 mL) was added dropwise to the suspension giving a red colored reaction mixture which was stirred for 12 h at ambient temperature. In the course of the reaction the color of the reaction mixture changed to yellow and the dissolving of P4 was observed. The reaction mixture was investigated by means of 31P{1H} NMR spectroscopy (see ESI†). 1:2:4: Compound 7 (256 mg, 0.50 mmol, 1.0 eq.) was added to a suspension of P4 (124 mg, 1.00 mmol, 2.0 eq.) in C6H5F (5 mL). A solution of GaCl3 (352 mg, 2.00 mmol, 4 eq.) in C6H5F (4 mL) was added dropwise to the suspension giving a red colored reaction mixture which was stirred for 12 h at ambient temperature. In the course of the reaction the color of the reaction mixture changed to brown and the dissolving of P4 was observed. The reaction mixture was investigated by means of 31P{1H} NMR spectroscopy (see ESI†). n-Hexane (2 mL) was added leading to the formation of a brown oil. The supernatant was removed, diluted with C6H5F (6 mL) and layered with n-hexane (3 mL) at −35 °C. Small amounts of crystalline material of 19[GaCl4] (41 mg, 10%), suitable for X-ray single crystal structure determination, were obtained within a few days. The remaining oil was washed with n-hexane (3 × 3 mL) transforming it into a brown sludge. All volatiles were removed in vacuo and the sludge was suspended in 1,2-C6H4F2 (2 mL) leading to the formation of a yellow, microcrystalline solid. The supernatant was removed and the yellow powder was washed with 1,2-C6H4F2/n-hexane (1:1 mixture, 2 × 2 mL). The obtained yellow powder consisting of 13[Ga2Cl7]2 in an approximate purity of 75% (determined by 31P{1H} NMR spectroscopy, 20% yield, 145 mg) was isolated by filtration and dried in vacuo. Recrystallization from a CH2Cl2 solution by slow diffusion of n-hexane yielded crystalline material of 19[Ga2Cl7]2 (purity > 90%) which was suitable for single crystal structure determination. Isolation was conducted via filtration and removal of all volatiles in vacuo (7% yield, 55 mg). Single crystals of 13[Ga2Cl7][GaCl4] were obtained by layering the diluted supernatant of the reaction mixture with n-hexane at −35 °C.
m.p.: 115.0–116.9 °C; Raman (300 mW, [cm−1]): ν = 2961 (37), 2998 (100), 1412 (24), 636 (37), 552 (63), 442 (22), 398 (24), 376 (14), 344 (43), 152 (44), 121 (13); IR (ATR, [cm−1]): ν = 2956 (vw), 2897 (vw), 1411 (vw), 1254 (m), 1123 (vw), 993 (s), 905 (s), 822 (vs), 756 (vw), 724 (s), 690 (w), 639 (w), 548 (vw), 509 (w), 462 (m); 1H NMR (CD2Cl2, [ppm]): δ = 0.31 (18H, s, H1), 0.67 (9H, s, H2), 0.76 (9H, s, H3); 13C{1H} NMR (CD2Cl2, [ppm]): δ = 1.4 (6C, d, C1, 3J(CP) = 3.1 Hz), 2.2 (3C, m, C2), 3.1 (3C, m, C3); 29Si{1H} NMR (CD2Cl2, [ppm]): δ = −58.0 (dd, Si4, 2J(SiPY) = 18.5 Hz, 2J(SiPM) = 8.5 Hz), 7.4 (d, Si1, 2J(SiP) = 8.4 Hz), 10.9 (s, Si2), 11.7 (s, Si3); 15N NMR (CD2Cl2, [ppm]): δ = 96 (s, N1), 115 (s, N2), 125 (s(br), N3); 71Ga{1H} NMR (CD2Cl2, [ppm]): δ = 249.6 (s); 31P{1H} NMR (CD2Cl2, [ppm]): ABMX2Y spin system: δ(PA) = −322.9, δ(PB) = −320.2, δ(PM) = 30.4, δ(PX) = 165.5, δ(PY) = 166.8, 1J(PAPB) = −189.8 Hz, 1J(PAPX) = −143.4 Hz, 1J(PBPX) = −147.7 Hz, 1J(PMPX) = −245.1 Hz, 2J(PAPM) = 18.8 Hz, 2J(PBPM) = 18.0 Hz; elemental analysis for C12H36GaCl5P6N4Si5: calcd: C 17.8, H 4.5, N 6.9; found: C 17.1, H 4.4, N 5.5.
1
H NMR (CD
2
Cl
2
, [ppm]): δ = 0.37 (18H, s, H2), 0.75 (18H, s, H1); 31P{1H} NMR (CD2Cl2, [ppm]): ABMOX2 spin system: δ(PA) = −318.3, δ(PM) = 33.4, δ(PO) = 144 (Δν1/2 = ∼1200 Hz), δ(PX) = 170.8, 1J(PAPX) = −144.8 Hz, 1J(PMPX) = −248.6 Hz, 2J(PAPM) = 19.0 Hz. Compound 12[GaCl4] was independently synthesized by addition of GaCl3 (18 mg, 0.10 mmol, 1.0 eq.) to a solution of 19[GaCl4] (64 mg, 0.10 mmol, 1.0 eq.) in CD2Cl2 (1 mL). The obtained colorless solution was stirred for 30 min at ambient temperature and subsequently investigated by 1H and 31P{1H} NMR spectroscopy.

m.p.: 164.5–167.5 °C (decomposition); Raman (250 mW, [cm−1]): ν = 2962 (10), 2898 (17), 1095 (10), 633 (13), 549 (100), 441 (11), 397 (17), 384 (10), 354 (10), 138 (81) the Raman measurement was hampered by strong fluorescence effects; IR (ATR, [cm−1]): ν = 2956 (vw), 2898 (vw), 1409 (w), 1257 (s), 994 (vs), 899 (m), 814 (vs), 759 (vw), 729 (w), 692 (vw), 637 (w), 544 (w), 490 (w), 441 (vw), 409 (w); 1H NMR (CD2Cl2, [ppm]): δ = 0.72 (36H, s, CH3); 13C{1H} NMR (CD2Cl2, [ppm]): δ = 2.7 (12C, s, CH3); 29Si{1H} NMR (CD2Cl2, [ppm]): δ = 13.7 (s, Si(CH3)3), the Si atom of the SiN4-moiety was not detected; 15N NMR (CD2Cl2, [ppm]): δ = 115 (s); 31P{1H} NMR (CD2Cl2, [ppm]): A2MX2 spin system: δ(PA) = −320.4, δ(PM) = 32.2, δ(PX) = 158.8, 1J(PAPX) = −142.7 Hz, 1J(PMPX) = −259.1 Hz, 2J(PAPM) = 20.0 Hz; elemental analysis for C12H36Ga4Cl14P10N4Si5: calcd: C 10.8, H 2.5, N 3.8; found: C 10.9, H 2.5, N 4.0.
Acknowledgements
We gratefully acknowledge financial support from the Fonds der Chemischen Industrie (fellowship to M.H.H.) and the DFG (WE4621/2-1).Notes and references
- M. H. Holthausen and J. J. Weigand, Chem. Soc. Rev., 2014, 43, 6639 RSC.
- C. A. Dyker and N. Burford, Chem. – Asian J., 2008, 3, 28 CrossRef CAS PubMed.
- P-rich cations RnPm (n < m) were also prepared from P1-sources, however in most cases the reaction outcome is unpredictable, see: (a) M. Donath, E. Conrad, P. Jerabek, G. Frenking, R. Fröhlich, N. Burford and J. J. Weigand, Angew. Chem., Int. Ed., 2012, 51, 2964 CrossRef CAS PubMed; (b) K.-O. Feldmann and J. J. Weigand, Angew. Chem., Int. Ed., 2012, 51, 7545 CrossRef CAS PubMed; (c) M. Donath, M. Bodensteiner and J. J. Weigand, Chem. – Eur. J., 2014, 20, 17306 CrossRef CAS PubMed.
- (a) Y. Peng, H. Fan, H. Zhu, H. W. Roesky, J. Magull and C. E. Hughes, Angew. Chem., Int. Ed., 2004, 43, 3443 CrossRef CAS PubMed; (b) G. Prabusankar, A. Doddi, C. Gemel, M. Winter and R. A. Fischer, Inorg. Chem., 2010, 49, 7976 CrossRef CAS PubMed; (c) W. Uhl and M. Benter, Chem. Commun., 1999, 771 RSC; (d) Y. Xiong, S. Yao, M. Brym and M. Driess, Angew. Chem., Int. Ed., 2007, 46, 4511 CrossRef CAS PubMed; (e) S. Khan, R. Michel, S. S. Sen, H.-W. Roesky and D. Stalke, Angew. Chem., Int. Ed., 2011, 50, 11786 CrossRef CAS PubMed; (f) S. Khan, R. Michel, J. M. Dieterich, R. A. Mata, H. W. Roesky, J.-P. Demers, A. Lange and D. Stalke, J. Am. Chem. Soc., 2011, 133, 17889 CrossRef CAS PubMed; (g) C. D. Martin, C. M. Weinstein, C. E. Moore, A. L. Rheingold and G. Bertrand, Chem. Commun., 2013, 49, 4486 RSC; For overviews of P4 activation by nucleophiles, electrophiles and predominantly nucleophilic ambiphiles see: (h) M. Scheer, G. Balázs and A. Seitz, Chem. Rev., 2010, 110, 4236 CrossRef CAS PubMed; (i) N. A. Giffin and J. D. Masuda, Coord. Chem. Rev., 2011, 255, 1342 CrossRef CAS; (j) S. Khan, S. S. Sen and H. W. Roesky, Chem. Commun., 2012, 48, 2169 RSC.
- I. Krossing and I. Raabe, Angew. Chem., Int. Ed., 2001, 40, 4406 CrossRef CAS PubMed ; For insertion of a related NO+ species into a P–P bond of P4 see: (a) T. Köchner, S. Riedel, A. J. Lehner, H. Scherer, I. Raabe, T. A. Engesser, F. W. Scholz, U. Gellrich, P. Eiden, R. A. Paz Schmidt, D. A. Plattner and I. Krossing, Angew. Chem., Int. Ed., 2010, 49, 8139 CrossRef PubMed; (b) T. Köchner, T. A. Engesser, H. Scherer, D. A. Plattner, A. Steffani and I. Krossing, Angew. Chem., Int. Ed., 2012, 51, 6529 CrossRef PubMed.
- (a) M. Gonsior, I. Krossing, L. Müller, I. Raabe, M. Jansen and L. van Wüllen, Chem. – Eur. J., 2002, 8, 4475 CrossRef CAS PubMed; (b) I. Krossing, J. Chem. Soc., Dalton Trans., 2002, 500 RSC.
- (a) M. H. Holthausen, K.-O. Feldmann, S. Schulz, A. Hepp and J. J. Weigand, Inorg. Chem., 2012, 51, 3374 CrossRef CAS PubMed; (b) M. H. Holthausen and J. J. Weigand, Z. Anorg. Allg. Chem., 2012, 638, 1103 CrossRef CAS; (c) M. H. Holthausen, A. Hepp and J. J. Weigand, Chem. – Eur. J., 2013, 19, 9895 CrossRef CAS PubMed.
- J. J. Weigand, M. H. Holthausen and R. Fröhlich, Angew. Chem., Int. Ed., 2009, 48, 295 CrossRef CAS PubMed.
- M. H. Holthausen and J. J. Weigand, J. Am. Chem. Soc., 2009, 131, 14210 CrossRef CAS PubMed.
- (a) M. H. Holthausen, C. Richter, A. Hepp and J. J. Weigand, Chem. Commun., 2010, 46, 6921 RSC; (b) J. W. Dube, C. M. E. Graham, C. L. B. Macdonald, Z. D. Brown, P. P. Power and P. J. Ragogna, Chem. – Eur. J., 2014, 20, 6739 CrossRef CAS PubMed.
- M. H. Holthausen, S. K. Surmiak, P. Jerabek, G. Frenking and J. J. Weigand, Angew. Chem., Int. Ed., 2013, 52, 11078 CrossRef CAS PubMed.
- E. Niecke and R. Kröher, Angew. Chem., Int. Ed. Engl., 1976, 15, 692 CrossRef.
- E. Niecke and W. Bitter, Chem. Ber., 1976, 109, 415 CrossRef CAS ; performing the reaction in 2:1 stoichiometry of 5 and SiCl4 gave 6 and 7 in similar ratios.
- Ref. 13 reports that after removal of 6 the residue consists mainly of polymeric material of unknown constitution and minor amounts of spirocyclic compound ClP(NSiMe3)2Si(NSiMe3)2PCl. Investigation of the residue by 31P{1H} NMR (C6H5F solvent), however, revealed 7 is the main compound (∼60%) next to several minor species, see ESI† for more information.
- L. Stahl, Coord. Chem. Rev., 2000, 210, 203 CrossRef CAS.
- (a) N. Burford and D. J. LeBlanc, Inorg. Chem., 1999, 38, 2248 CrossRef CAS; (b) I. Schranz, L. P. Grocholl and L. Stahl, Inorg. Chem., 2000, 39, 3037 CrossRef CAS PubMed.
- E. Niecke, W. Flick and S. Pohl, Angew. Chem., Int. Ed. Engl., 1976, 15, 309 CrossRef.
- J. Heinicke, S. Mantey, A. Oprea, M. K. Kindermann and P. G. Jones, Heteroat. Chem., 1999, 10, 605 CrossRef CAS.
- Next to main species 7, also minor amounts of compounds 8, 9 and 9′ were observed in the 1H and 31P{1H} NMR spectra of the residue described in ref. 14, see ESI† for more information.
- P. Paetzgold, C. von Plotho, E. Niecke and R. Rüger, Chem. Ber., 1983, 116, 1678 CrossRef.
- M. L. Filleus-Blanchard, Org. Mag. Res., 1979, 12, 12 CrossRef.
- This rotation is not observed at ambient temperature due to the high barrier of rotation reported for exocyclic P–N bonds in amino-substituted diphosphadiazanes (e.g.8: ΔG > 27 kcal mol−1, ref. 17).
- Several isomerization pathways are conceivable: (a) L. Horner and H. Winkler, Tetrahedron Lett., 1964, 461 CrossRef CAS; (b) A. Rauk, L. C. Allen and K. Miskow, Angew. Chem., Int. Ed. Engl., 1970, 9, 400 CrossRef CAS; (c) J. Silaghi-Dumitrescu, F. Lara-Ochoa and I. Haiduc, Main Group Chem., 1998, 2, 309 CrossRef; (d) I. Schranz, D. F. Moser, L. Stahl and R. J. Staples, Inorg. Chem., 1999, 38, 5814 CrossRef CAS; (e) U. Wirringa, H. Voelker, H. W. Roesky, Y. Shermolovich, L. Markovski, I. Uson, M. Noltemeyer and H.-G. Schmidt, J. Chem. Soc., Dalton Trans., 1995, 1951 RSC; (f) H.-J. Chen, R. C. Haltiwanger, T. G. Hill, M. L. Thompson, D. E. Coons and A. D. Norman, Inorg. Chem., 1985, 24, 4725 CrossRef CAS.
- Next to 7 (25%), compounds 5 (33%) and 6 (42%) are main products of this reaction. It is assumed, that 5 and Me3SiN
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) PN(SiCl3)(SiMe3) form upon cyclo-reversion of 9. Compound Me3SiNPN(SiCl3)(SiMe3) is assumed to eliminate Me3SiCl yielding four-membered heterocycle 6.
PN(SiCl3)(SiMe3) form upon cyclo-reversion of 9. Compound Me3SiNPN(SiCl3)(SiMe3) is assumed to eliminate Me3SiCl yielding four-membered heterocycle 6. - A. Villinger, P. Mayer and A. Schulz, Chem. Commun., 2006, 1236 RSC.
- A. Villinger, A. Westenkirchner, R. Wustrack and A. Schulz, Inorg. Chem., 2008, 47, 9140 CrossRef CAS PubMed.
- (a) B. Luo, V. G. Young and W. L. Gladfelter, J. Organomet. Chem., 2002, 268 CrossRef CAS; (b) H. Schmidtbauer and W. Findeiss, Angew. Chem., Int. Ed. Engl., 1964, 3, 696 Search PubMed.
- M. Rastätter, P. W. Roesky, D. Gudat, G. B. Deacon and P. C. Junk, Chem. – Eur. J., 2007, 13, 7410 CrossRef PubMed.
- (a) R. W. Shaw, Phosphorus, Sulfur Silicon Relat. Elem., 1978, 4, 101 CrossRef CAS; (b) S. S. Krishnamurthy, Phosphorus, Sulfur Silicon Relat. Elem., 1994, 87, 101 CrossRef CAS.
- A. H. Cowley, M. Lattman and J. C. Wilburn, Inorg. Chem., 1981, 20, 2916 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data, 31P NMR spectra of reaction mixtures and general experimental information. CCDC 1060461–1060464. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt01512j |
| This journal is © The Royal Society of Chemistry 2016 |