
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHeterogeneous chemistry and reaction dynamics of the atmospheric oxidants, O3, NO3, and OH, on organic surfaces
Robert C.
Chapleski
Jr.
,
Yafen
Zhang
,
Diego
Troya
and
John R.
Morris
*
Department of Chemistry, Virginia Tech, Blacksburg, Virginia 24061, USA. E-mail: jrmorris@vt.edu; Tel: +1-540-231-2471
First published on 11th November 2015
Abstract
Heterogeneous chemistry of the most important atmospheric oxidants, O3, NO3, and OH, plays a central role in regulating atmospheric gas concentrations, processing aerosols, and aging materials. Recent experimental and computational studies have begun to reveal the detailed reaction mechanisms and kinetics for gas-phase O3, NO3, and OH when they impinge on organic surfaces. Through new research approaches that merge the fields of traditional surface science with atmospheric chemistry, researchers are developing an understanding for how surface structure and functionality affect interfacial chemistry with this class of highly oxidizing pollutants. Together with future research initiatives, these studies will provide a more complete description of atmospheric chemistry and help others more accurately predict the properties of aerosols, the environmental impact of interfacial oxidation, and the concentrations of tropospheric gases.
I. Introduction
O3, OH, and NO3 are the most important atmospheric oxidants due to their high chemical potentials, abundance, and negative effects on human health. While the importance of these oxidants has led many scientists to investigate their gas-phase chemistry,1–4 detailed studies into the reactions of O3, OH, and NO3 at the gas–surface interface have only recently been reported. Interfacial reactions between organic particles and oxidative gases result in changes in particulate composition, size, and physical properties. These changes affect human health and visibility, climate, and the global carbon cycle.5–7 Organic particles in the atmosphere form or grow through the following four mechanisms (Fig. 1): (i) biogenic emissions in remote regions and anthropogenic emissions in urban areas, which result in particles described as soot or primary organic aerosols;8–10 (ii) adsorption of volatile organic compounds onto liquid or solid surfaces, which results in aerosols coated by organic films;11,12 (iii) coagulation of smaller carbonaceous nanoparticles;13 and (iv) reactions of atmospheric oxidants on the surfaces of existing organic particles, which produce so-called secondary organic aerosols (SOAs).14,15 The concentrations of these types of organic aerosols typically range from 1 to 10 μg m−3 and can reach levels exceeding 15 μg m−3 in heavily industrialized environments.16,17 Once formed, they nearly immediately begin contributing to the heterogeneous chemistry of the lower atmosphere.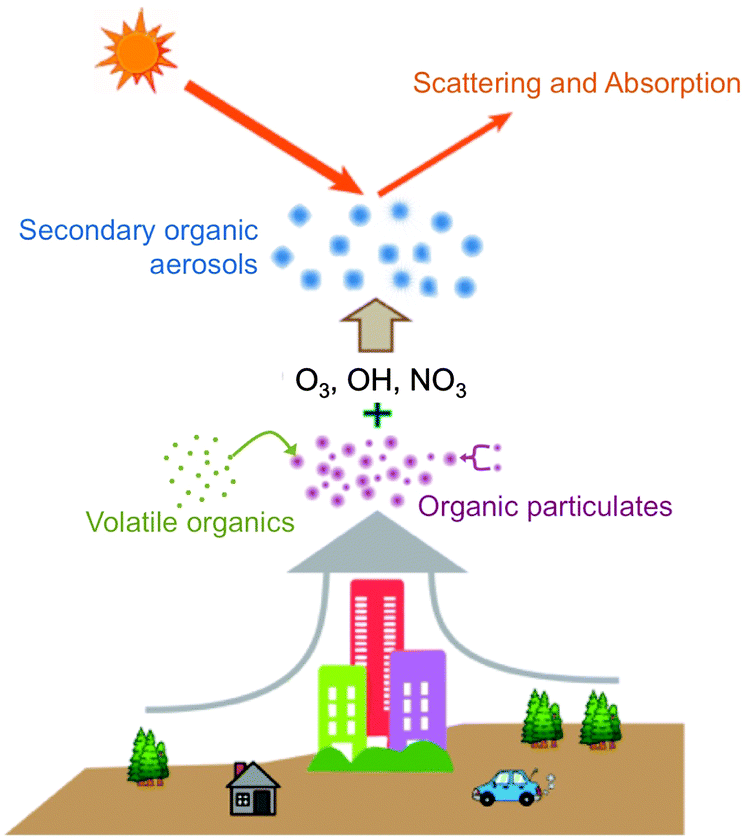 | ||
| Fig. 1 Schematic illustration of the generation and transformation of organic particles in the atmosphere. Particles can form and grow from biogenic and anthropogenic emission products, adsorption of volatile organics onto surfaces, coagulation of carbonaceous nanoparticles, and reactions of atmospheric oxidants on the surfaces of existing organic particles. These particles can affect the balance of incoming and outgoing solar radiation. | ||
Surface reactions involving O3, NO3, and OH are known to alter the properties and fate of organic particulates, often in unexpected ways. For example, Fig. 2 shows scanning-electron microscope images of SOAs generated through the oxidation of volatile organic emissions from pine seedlings.18 When exposed to OH in the presence of SO2, particles (shown as white spots in the figure) with an average radius of 100 nm are formed. Conversely, exposure to O3 results in the formation of significantly smaller particles (28 nm). Such modifications in particulate properties due to atmospheric oxidation most likely affect the scattering and absorption of light by the particles, thereby altering the balance between incoming and outgoing solar radiation5,19 (Fig. 1). Further, the overall surface area of each individual particle is altered, thereby affecting the subsequent adsorption and reaction rates on these surfaces. These findings have motivated scientists to investigate the role of organic surfaces in atmospheric interfacial oxidative reactions—the chemistry contributing to atmospheric organic particle transformation and growth.20–23 Such chemistry is particularly important in polluted regions of the atmosphere where elevated concentrations of both particulates and oxidative gases often coincide.
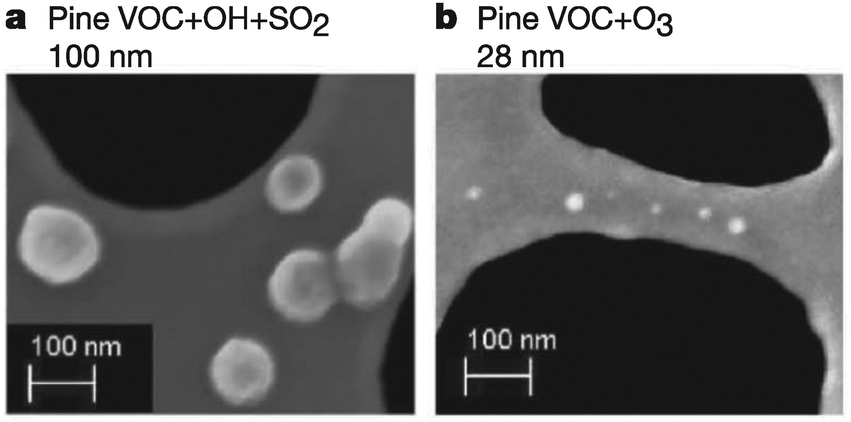 | ||
| Fig. 2 SEM images of SOA particles of different sizes resulting from the oxidation of pine-seedling volatile organic compound (VOC) emissions following (a) OH-initiated oxidation in the presence of SO2, and (b) O3-initiated oxidation. The white spots are SOAs supported on a lacey carbon grid (black regions). Reprinted with permission from Macmillan Publishers Ltd: Nature, 2010, 467, 824. Copyright 2010. | ||
Tropospheric O3 has been recognized as a worldwide environmental problem, hallmarked by the presence of NO and NO2 in the lower atmosphere. Anthropogenic O3 first became of concern as an atmospheric pollutant in the late 1940s,24 when high concentrations were reported in the Los Angeles area. Since then, dangerously elevated tropospheric concentrations have been measured in Greece, Japan, Sydney, Jerusalem, Mexico City, and many other locations.19,25–27 Today, tropospheric O3 concentrations are monitored to characterize the extent of air pollution in urban areas, where emissions from transportation and industrial plants are substantial. Anthropogenic O3 is primarily generated from the reaction of atmospheric O2 with ground-state O(3P) radicals that result from the photolytic dissociation of ambient NO219,28 (Scheme 1).
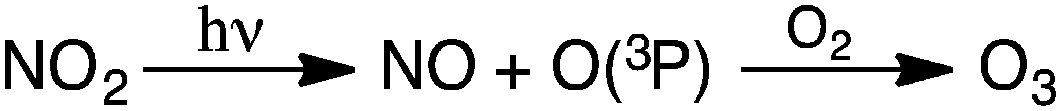 | ||
| Scheme 1 Generation of O3 in the atmosphere during the daytime. | ||
Though air pollution is often evaluated using O3 concentrations, hydroxyl (OH) radicals also play a critical role in daytime atmospheric chemistry. In fact, concentrations of OH radicals are difficult to quantify precisely because of their high reactivity and consequent short atmospheric lifetime (≤1 s). Techniques for quantifying OH have been reviewed by Heard and Pilling.29 One important reaction of OH involves volatile organic compounds (VOCs) and the conversion of two molecules of NO to NO2 (Scheme 2).30 This conversion is primarily responsible for the formation of NO2, a precursor to O3 in the atmosphere (Scheme 1).30,31 Further, the atmospheric concentration of OH is dependent on the photodissociation of O3 by UV radiation, which produces an electronically excited oxygen atom (O(1D)), able to undergo rapid reactions with water vapor in the air to form two OH radicals (Scheme 3).32,33
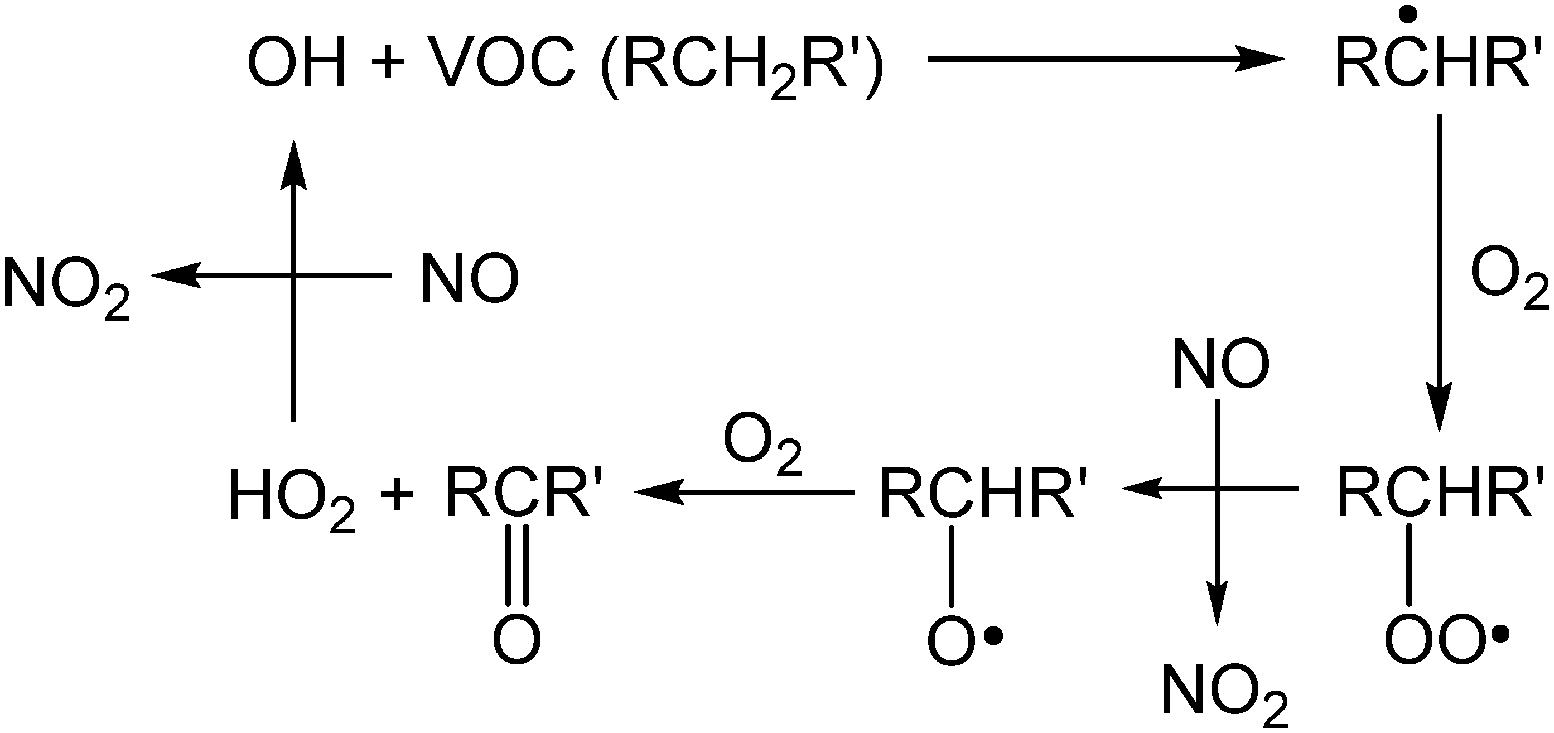 | ||
| Scheme 2 Generation of NO2 during typical oxidation of volatile organic compounds (VOC) by OH. | ||
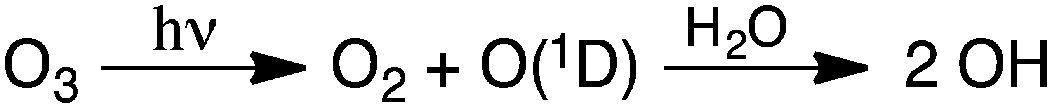 | ||
| Scheme 3 Formation of OH radicals in the atmosphere during the daytime. | ||
Because the primary mechanisms of O3 and OH generation involve the photodissociation of other atmospheric molecules, reactions initiated by these two oxidants are of highest importance during the daytime.28,32 At night, in the absence of photochemical reactions, the consumption of O3 and OH is much faster than the formation of these two oxidants, and their atmospheric concentrations diminish.22,34 However, oxidation of NO2 by O3 generates NO3, which drives a great deal of tropospheric nighttime chemistry.26,35 Interestingly, NO3 chemistry is most relevant to the atmosphere after sunset because NO3 photodissociates rapidly in daylight.26,36 NO3 initially reacts with certain VOCs through addition or hydrogen abstraction reactions, ultimately yielding peroxyacetyl nitrate (PAN).37–40 PAN (due to its eventual photo- and thermal-decomposition into NO2) acts as a reservoir and a transportation medium for NO2.41 In addition, reactions of NO3 and NO2 result in the production of dinitrogen pentoxide (N2O5), which is an important source of nitric acid in the atmosphere.36
While the rich interplay amongst O3, OH, and NO3 and their reactions in the gas phase have key implications in tropospheric chemistry, this review focuses on reactions of each of these gases with organic surfaces. These heterogeneous reactions alter the size and composition of atmospheric particulates and modify the surfaces of other anthropogenic and natural materials as well, such as metals, metal oxides, and polymers. The importance of these processes has provided ample motivation for detailed laboratory investigation. Below, we highlight a subset of key studies into the chemistry of organic materials with O3, OH, and NO3. For each gas, the review begins with a focus on reactions at highly relevant, yet relatively uncharacterized, surfaces and particles (including soot), followed by studies performed with model aerosols composed of a single type of molecule or surface-adsorbate combination. Finally, each section concludes with a discussion of fundamental research involving highly ordered synthetic surfaces that helps provide insight into specific details of the gas–surface reaction dynamics and kinetics, while serving as a valuable benchmark for theoretical studies.
II. O3 reactions with organic surfaces
Reactions between ozone and organic surfaces have long been recognized as critical to the overall chemistry of the atmosphere. It is therefore not surprising that, relative to NO3 and OH radicals, there have been many studies into the interfacial chemistry of ozone. In the gas phase, O3 initiates reactions with vinyl-containing organics and polycyclic aromatics via addition across double bonds to form an unstable primary ozonide, which triggers a series of subsequent reactions.42–44 Analogous chemistry may occur on surfaces;45–47 however, scientists are only beginning to decipher how surface structure and functionality affect ozone accommodation, diffusion, and reaction pathways. A quintessential result for these processes is the reactive uptake coefficient: the probability that a gas-phase molecule that collides with the surface will react with the surface. For a thorough explanation of this coefficient in terms of reactions of oxidative gases with organic surfaces, the reader is referred to work by Houle et al.48In an investigation of the reaction kinetics of ozone with several different polycyclic aromatic hydrocarbon (PAH) compounds on laboratory-generated soot, Bedjanian and Nguyen49 found that fast initial consumption of ozone was followed by rapid irreversible changes in the molecules at the interface, rendering the surface unreactive to further ozone exposure. In complementary work, Disselkamp and coworkers22 investigated the reaction between soot and ozone in a static aerosol chamber. By interpreting ozone and CO2 infrared signals over the course of ozone exposure, they obtained a stoichiometry of two O3 molecules for every molecule of CO2 product formed and reported a very small pseudo first-order reaction probability of 10−8 for ozone with “aged” soot. The interfacial reactivity of ozone on soot has also been reported in a recent study by Browne et al., which yielded an uptake coefficient of 2 × 10−7.50 The He group further augmented these findings by performing in situ Raman and infrared studies51,52 of ozone reactions with soot. Their work suggested that amorphous carbon and disordered graphitic sites were responsible for initiating the reactions. They also revealed that some of the reaction products included surface-bound ketone, lactone, and anhydride groups. Other research teams extended this work to specific surface-bound compounds (including polycyclic aromatics,43,53–58 biogenic volatile organics,59 biomass burning products,60 and fungicides61) and functionalities at well-characterized aerosol surfaces.62–66
Within the body of literature on O3 reactions with aerosols, investigations of the ozonolysis of oleic acid aerosols are of particular interest because these particulates appear in relative high abundance in certain regions of the atmosphere.67–70 From these studies, reactive uptake coefficients have been determined to be generally on the order of 10−3.67–69,71 Variations in this coefficient have been attributed to the effects of particle size on diffusion of O3 into the bulk68 and the likelihood of secondary reaction pathways beyond ozonolysis.69 Further, in an experiment in which the substrate was not uniformly covered by oleic acid, a reactive uptake coefficient on the order of 10−5 was found.71 Importantly, the use of fresh surfaces in these studies contributed to a higher reactive uptake coefficient than in experiments employing passivated surfaces.22
Motivated by the challenge of building a highly fundamental understanding of interfacial ozone chemistry, several groups have explored ozone reactions with organic particles adsorbed onto extended (planar) solid surfaces. In their work, Ham and Wells72 identified products formed during exposure of alpha-terpineol on glass and vinyl flooring tile to ozone. Kahan et al. implemented fluorescence spectroscopy in a Teflon reaction chamber to investigate the degradation kinetics of PAHs adsorbed on a microscope slide,73 and Kwamena et al.74 investigated the role of relative humidity and ozone concentration in the kinetics of ozonolysis of anthracene deposited on a pyrex flow tube. In research highlighted in these two works, the Langmuir–Hinshelwood mechanism, whereby the impinging ozone molecule thermally accommodates on the surface before reacting, adequately describes the time-resolved data.
To add insight to the role of the interaction between organic molecules and the underlying surfaces on which they are supported on oxidation reactions, Chu et al.21 used density functional theory methods to model the ozonolysis of surface-bound planar PAHs. They found an energetic expense to reaction resulting from the “lift-off” of the molecules from the surface as the planar PAH reactants form nonplanar intermediates or products. Fig. 3 shows structures and energies for the ozone reaction with one of the available double bonds in pyrene. The first step in the reaction is an exoergic addition to form a primary ozonide, which retains planarity. Decomposition of the primary ozonide in step 2 leads to a non-planar Criegee intermediate. The reaction Gibbs energy (ΔGrxn) for this step is +14.8 kcal mol−1, and the energetic penalty resulting from loss of interactions with the underlying surface increases the reaction Gibbs energy by an additional ∼14 kcal mol−1, with variations depending on the nature of the surface (e.g., fly ash vs. NaCl). The last step of the reaction is the thermodynamically favorable formation of a secondary ozonide (step 3), which results in a planar species.
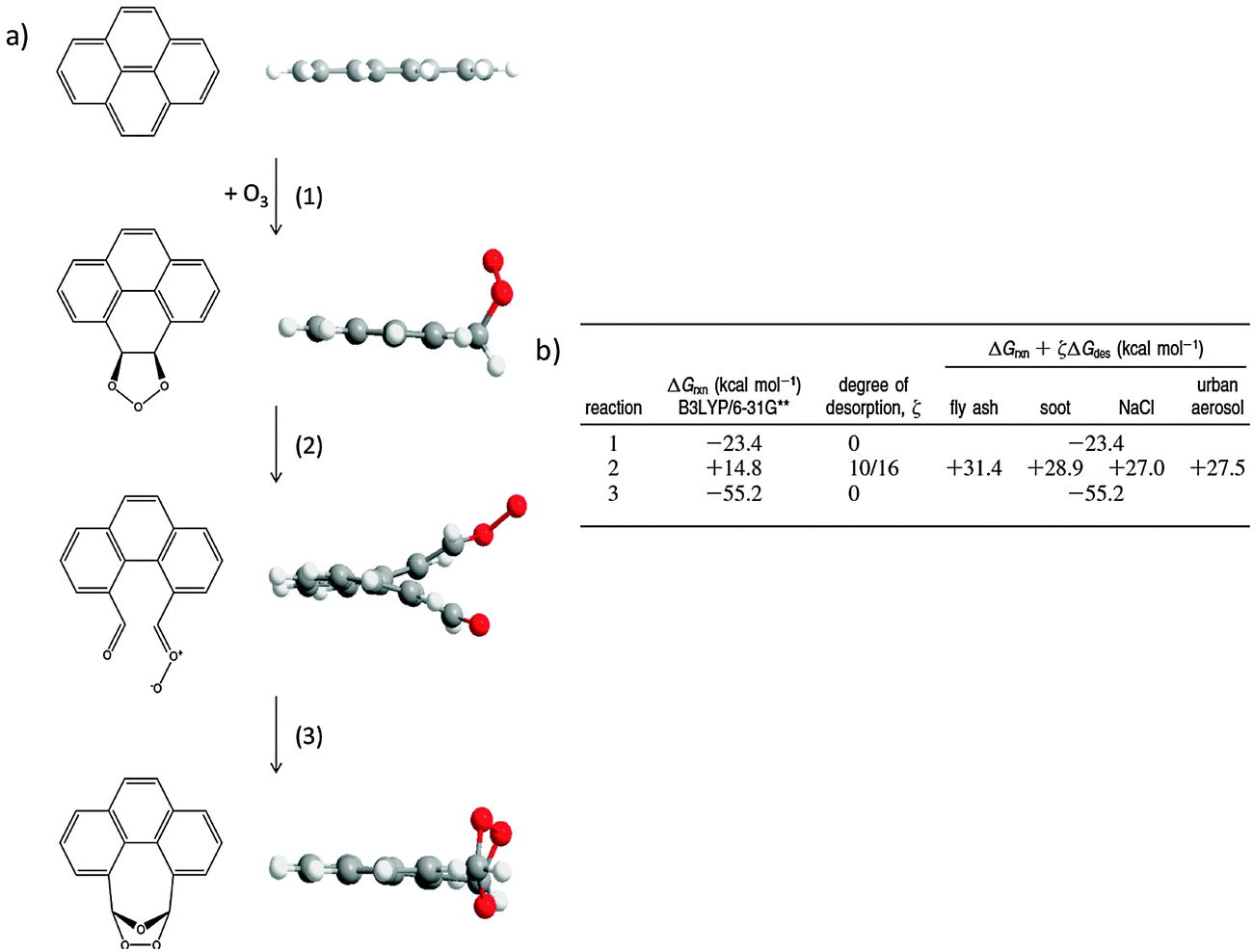 | ||
| Fig. 3 (a) Structures of minima in the oxidation of pyrene by ozone show a diversion from planarity in an intermediate. (b) Energetics of reaction steps in (a) show an energetic penalty for surface liftoff as a result of loss of planarity. ΔGrxn is determined with gas-phase molecules (surface-absent), ς is defined as the fraction of carbon atoms that leave the plane of the molecule, and ΔGdes is the energy of complete desorption from the surface. Reprinted (adapted) with permission from J. Am. Chem. Soc., 2010, 132, 15968. Copyright 2010 American Chemical Society. | ||
Additionally, attenuated total reflectance infrared (ATR-IR) experiments75–78 have been used to investigate the effects of temperature,75–77 relative humidity,75–77 and film thickness77 on reaction mechanisms,76 product yields,76,78 product hygroscopicity,75,77,78 and redox activity.75 As ATR-IR allows for in situ collection of infrared spectra, the change in the IR intensity of specific vibrational modes during exposure can be used to obtain kinetic information such as ozone's reactive uptake coefficient, which has been determined to be in the range of 1.0 × 10−5,76 to 5.1 × 10−4.75 The measured variations in ozone uptake coefficients are likely related to differences in the reaction conditions, surface properties, and the particular infrared band analyzed to extract kinetic information.
Beyond solid surfaces, the ozonolysis of organic compounds at the air–water interface has also been studied in thin films. Raja and Valsaraj79 investigated the impact of ozone exposure on the rate of naphthalene vapor uptake onto single droplets of water. They reported that reactions between ozone and naphthalene at the air–water interface increased the rate of naphthalene uptake onto the droplet surface. Oxidation products with higher water solubility than naphthalene more readily diffused into the bulk, thus allowing for greater mass transfer of naphthalene from the gas phase to the surface of the droplet. Moreover, the rate of the heterogeneous ozone-naphthalene reaction—15 times greater than that of the homogeneous gas-phase reaction—was found to be dependent upon the size of the droplet. Wadia and coworkers80 used an experimental flow chamber and molecular dynamics simulations to investigate the ozonolysis of saturated and terminally unsaturated phospholipid molecules at the air–water interface. No reaction with the saturated compound was observed, yet reaction with the unsaturated compound yielded an aldehydic product. This reaction was facilitated by the availability of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond at the interface, which was observed to be invariant to surface film compression. Recently, Mmereki et al.81 provided additional insight into the ozone-anthracene reaction in organic films on water via laser-induced fluorescence. Interestingly, the presence of organic acids in the aqueous phase reduced the reaction rate with the organic film, and the presence of alcohols enhanced the overall rate of reaction.
C double bond at the interface, which was observed to be invariant to surface film compression. Recently, Mmereki et al.81 provided additional insight into the ozone-anthracene reaction in organic films on water via laser-induced fluorescence. Interestingly, the presence of organic acids in the aqueous phase reduced the reaction rate with the organic film, and the presence of alcohols enhanced the overall rate of reaction.
The dependence of the ozone reaction rate with organic surfaces on the physical properties of the surface has received additional scrutiny. Of particular interest, Moise and Rudich82 noted an order-of-magnitude increase in the reactive uptake coefficient for ozone at liquid organic surfaces relative to solid samples (Fig. 4). This result was attributed to the participation of subsurface layers in liquid uptake, which is not as prevalent when the organic surface is frozen. In the figure, dashed lines mark differences in coefficients of the same compound resulting from a liquid/solid phase change. The coefficient for linoleic acid decreased from (1.2 ± 0.2) × 10−3 to (1.4 ± 0.1) × 10−4 upon freezing. For oleic acid, a decrease from (8.3 ± 0.2) × 10−4 to (5.2 ± 0.1) × 10−5 was observed. This change of phase also caused a decrease in the coefficient of 1-hexadecene from (3.8 ± 0.6) × 10−4 to (2.5 ± 0.4) × 10−5. These differences are similar in magnitude to those observed for liquid-phase uptake compared to a well-ordered thin film.83 In particular, 1-octene exhibited a decrease in the uptake coefficient from 1 × 10−3 for liquid to 1 × 10−4 for a monolayer in the same temperature range.
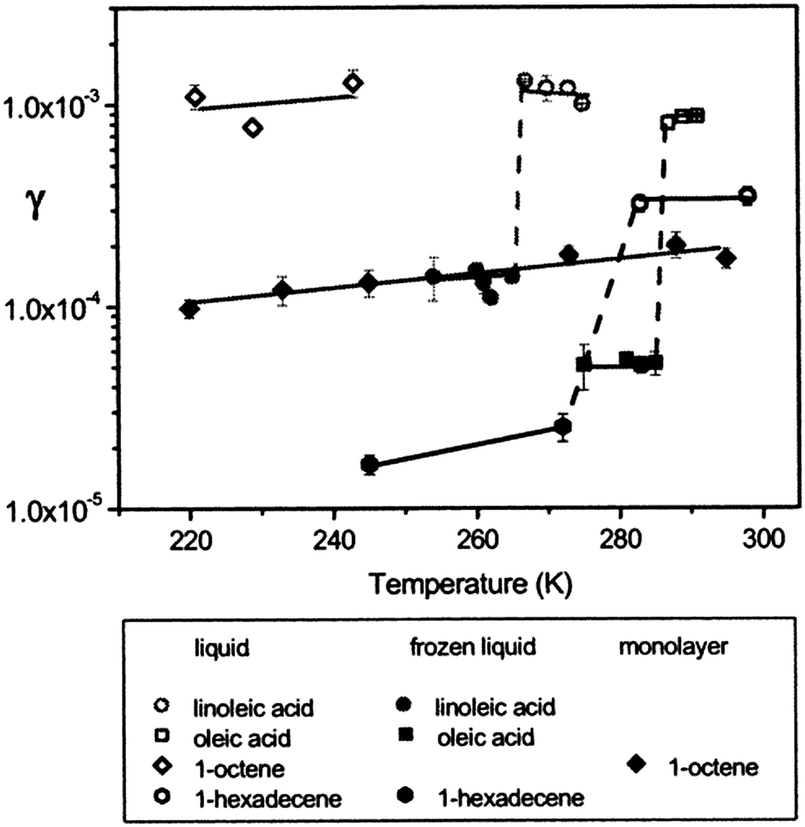 | ||
| Fig. 4 Ozone reactive uptake coefficients, γ, vs. temperature (K) for several organic liquids, frozen liquids, and a monolayer.82,83 The dashed lines show an order of magnitude difference in uptake coefficient between liquid and frozen samples. Reprinted (adapted) with permission from J. Phys. Chem. A, 2002, 106, 6469–6476. Copyright 2002 American Chemical Society. | ||
As with reaction rate, the mechanism of ozonolysis has been shown to depend on the characteristics of the surface. Enami et al. performed a series of studies that employed electrospray mass spectrometry to investigate ozonolysis of aqueous microdroplets of several organic compounds including uric acid,84 ascorbic acid,85 sulfonic acid,86 phenol and α-tocopherol,87 cysteine,88 and β-caryophyllene.89 After identifying unique products from these reactions that were distinct from those in the bulk solution, they concluded that an air–water shell a few nanometers thick presents a reaction environment that is fundamentally different from that found in the bulk. They showed, specifically in β-caryophyllene ozonolysis, that different products were formed at different depths within this shell, in correlation with varying water densities. As the water density changes, the rate of vibrational relaxation changes as well, allowing for the activation of different processes.89 To further elucidate the role of water molecules at the interface, Beauchamp and coworkers90 investigated the ozonolysis of phospholipid surfactant mixtures using field-induced droplet ionization mass spectrometry. They found known metastable species in the bulk as major products of ozonolysis at the air–water interface. Lower water density at the interface relative to the bulk may play a key role, as proton transfer from water leads to rapid decomposition of these products. Also, as found in the study of ozone exposure to naphthalene on water droplets by Raja and Valsaraj,79 ozonolysis of unsaturated reactants resulted in increased molecular hydrophilicity, leading to dissolution of products into the aqueous phase. Most recently, Beauchamp and coworkers have developed a method for generating controlled nanoliter-sized droplets for mass-spectrometric sampling using bursting bubble ionization and interfacial sampling with an acoustic transducer.91 This method has been used to investigate the time-dependent ozonolysis of oleic acid.
Investigations of the heterogeneous ozonolysis of oleic acid have more recently been expanded to include macroscopic surface samples.82,92 For example, flow-tube methods have been used to examine interfacial reactions of oleic acid on a variety of substrates.71,82,92 Further, ATR-IR has been utilized to monitor oxidation at the interface of large and small oleic acid droplets.93 The reactive uptake coefficients measured for extended surfaces were found to range from 10−5 to 10−3, depending on experimental technique, sample geometry, the influence of secondary reactions,93 and whether the coefficient was calculated using changes in O392 or in oleic acid concentrations or properties.82,93 Additionally, Reid and coworkers investigated oxidative aging of aerosol particles containing environmentally relevant organic acids using optical tweezers and cavity enhanced ringdown spectroscopy.94,95 For mixed NaCl/oleic acid particles, they report an uptake coefficient of 2.3 × 10−4, which is consistent with measurements cited above that employed ATR-IR experiments.82,92,93 In another droplet-based study, O3 reactions with oleic acid surfaces were investigated through the use of a pendant drop of water coated with a monolayer of oleic acid.96 The authors attributed a relatively low uptake coefficient ((2.6 ± 0.1) × 10−6) to the decrease in accessibility of ozone to the CC bond, which was submerged within the oleic acid monolayer (Fig. 5).42
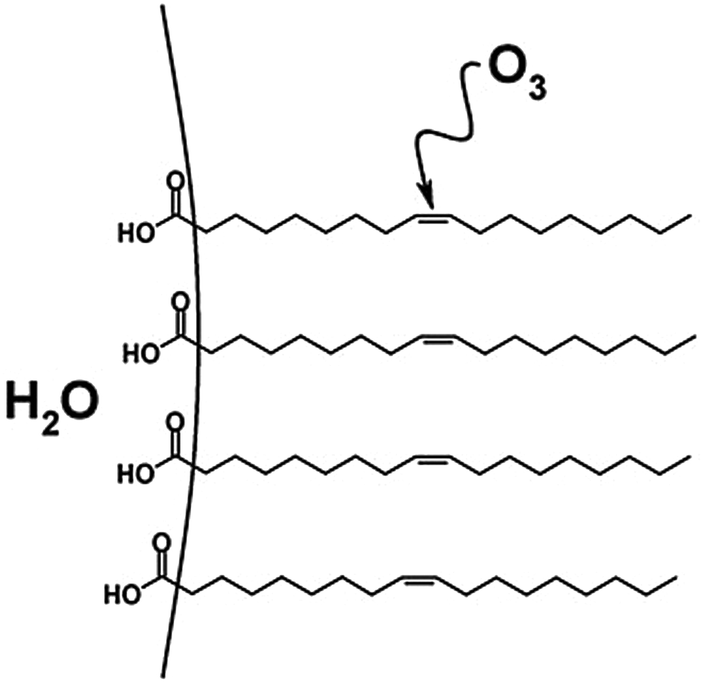 | ||
| Fig. 5 An oleic acid monolayer on a pendant drop of water. The non-terminal positioning of the CC double bond hinders reaction with O3, resulting in a decreased reactive uptake relative to a monolayer with a terminal CC bond. Reproduced from ref. 42 with permission from the PCCP Owner Societies. | ||
From a much more fundamental perspective, studies employing self-assembled monolayers (SAMs) as model surfaces have provided detailed insight into the dynamics of gas–surface collisions involving ozone. Specifically, dynamic properties including energy transfer and thermal accommodation coefficients, as well as reaction probability of a single surface-bound functional group, have been revealed using SAMs. These surfaces present many advantages for fundamental work: they can be synthesized in a highly reproducible manner, they can be characterized in situ with infrared spectroscopic probes, and they enable one to locate a specific functional group precisely at the gas–surface interface. With this strategy, Dubkowski et al.97 and Vieceli et al.98 employed time-resolved changes in atmospheric-pressure ATR-IR band intensities as well as molecular dynamics simulations of alkene-terminated SAMs to describe the interaction of O3 with a SAM. Their experimental results suggested a surprisingly long surface residence time for ozone: ∼7 s, which was many orders of magnitude longer that that shown by their simulations: ∼17 ps. As nonreactive molecular dynamics were used in the simulations, the vast difference in residence time was attributed to the formation of covalent bonds initially leading to the primary ozonide in the experiment, which was not modeled in the simulations. The simulations also showed that (possibly reactive) collisions of O3 with CC double bonds in organic systems were dependent upon the residence time of ozone on the organic surface before desorption, as well as the accessibility of unsaturated sites within the organic surface to ozone.98 Specifically, the ozone collision probability with CC bonds was found to be a factor of two lower for a vinyl-terminated SAM than for a liquid slab of 1-tetradecene. In the SAM, the CC bonds are located at the gas–surface interface, allowing access to ozone without uptake into the organic phase. However, a shorter residence time on the SAM (17 ps) than in the liquid (53 ps) leads to an overall lower collision probability. Further, for a phospholipid monolayer surface with submerged CC bonds, this probability decreased by a factor of five relative to the SAM. Though ozone had a similar residence time on the phospholipid surface (51 ps) as on the liquid, the localization of the CC double bonds beneath the gas–surface interface hindered accessibility to ozone as compared to the SAM, or to the liquid, which had CC bonds dispersed evenly throughout.
To gain insight into the fate of vinyl-functionalized organic surfaces upon reaction with ozone, McIntire et al.99 obtained AFM and SEM images, as well as Auger spectra of a model surface (vinyl-terminated SAM on silicon) following exposure to ozone in a Teflon reaction chamber. Their AFM data, shown in Fig. 6, displays images of a silicon substrate before and after deposition of the SAM, as well as after ozone oxidation, at both low and high relative humidity. At low relative humidity, Criegee intermediates (CI) decompose or react with adjacent organic species (Fig. 6e). Subsequent reactions of these products ultimately lead to radical-induced polymerization of the SAM chains into the aggregates shown in Fig. 6c (white). In contrast to these interesting structures, high relative humidity appears to provide competing reaction pathways that limit the degree of agglomeration (Fig. 6d).
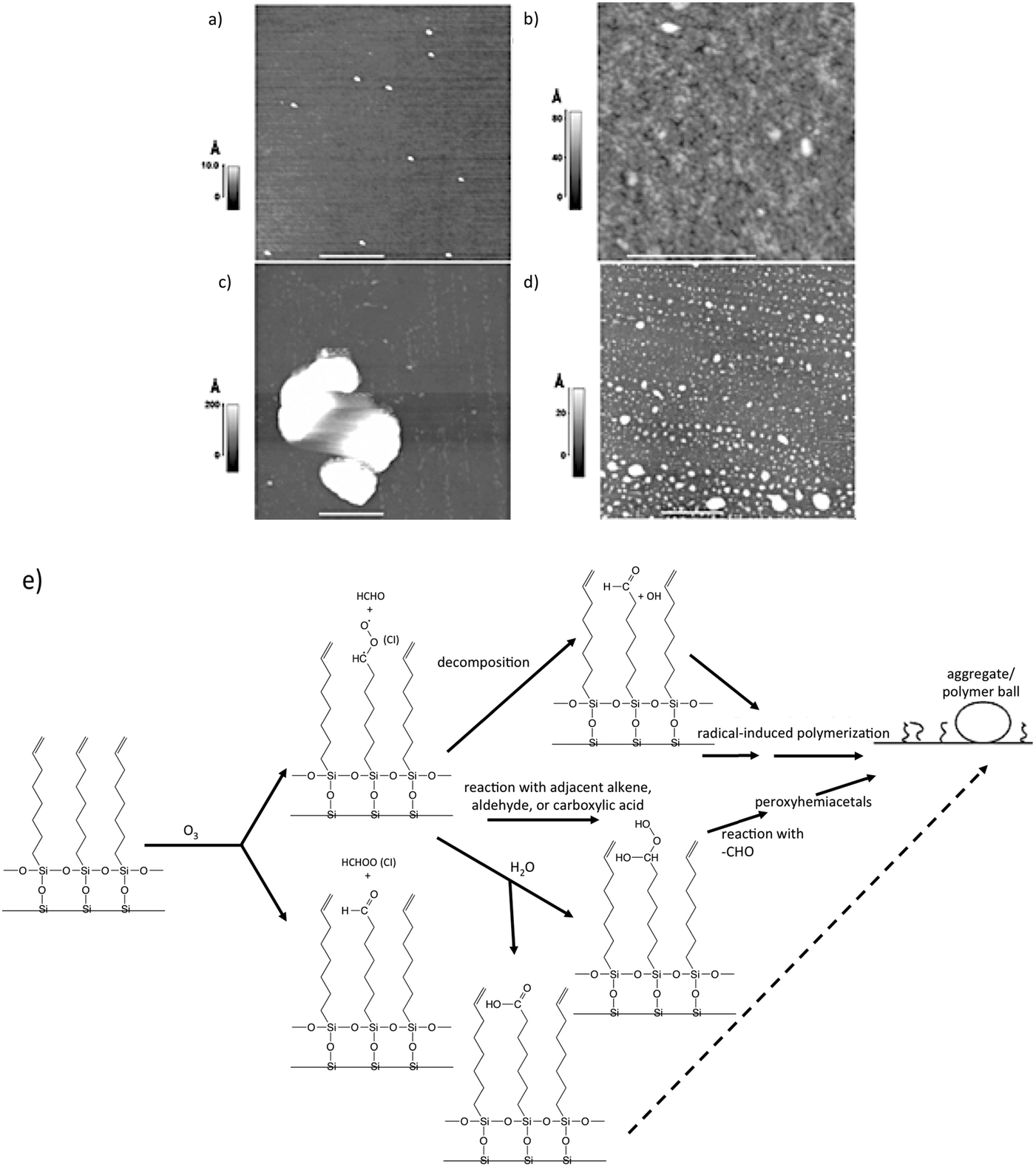 | ||
| Fig. 6 AFM images of a clean Si substrate (a) before deposition of a vinyl-terminated SAM. Small particles of silica shown result from surface “chipping” during handling; (b) following deposition of SAM; (c) following exposure to ozone for 40 min at <5% relative humidity; (d) same as in (c), but at 60% relative humidity; (e) scheme showing reactions of Criegee Intermediates leading to radical-induced polymerization. Reproduced and adapted from ref. 99 with permission from the PCCP Owner Societies. | ||
While SAMs provide well-characterized model organic surfaces for fundamental studies of interfacial ozone chemistry, vacuum-based molecular beam methods enable one to also precisely control the properties of the impinging gas molecules. Ultra-high vacuum (UHV) approaches, in particular, virtually eliminate the participation of background gases in the surface chemistry. Although the UHV-based measurements are not immediately relevant to reactions that take place under atmospheric conditions, they do provide valuable benchmarks for theoretical studies of these reactions, aid in spectral assignments of interfacial functional groups, and reveal how O3 reacts with organic surfaces under pristine conditions. Such experiments are the first steps in building a more comprehensive understanding of these reactions.
Lu, Fiegland, and coworkers100–102 have combined SAMs, UHV, and molecular beam scattering methods to explore the dynamics of surface-bound functional group oxidation during collisions with ozone molecules. Using time-of-flight methods, they found bimodal energy distributions for ozone scattered from methyl-, hydroxyl-, and perfluoro-terminated SAMs.102 One mode of the energy distribution was attributed to those molecules that transferred sufficient energy to the surface upon collision to become transiently trapped where they are unable to immediately surmount the energy barrier for desorption. Eventually, the thermal energy of the surface drives desorption of this fraction of ozone molecules, which exhibit a Boltzmann distribution of final velocities. The remaining component of the scattering distribution was attributed to those molecules with sufficient energy to scatter impulsively from the surface upon collision without becoming fully accommodated. By comparing the incident and final energies of molecules, the extent of energy transfer to the surface upon collision was deduced. For the methyl-terminated SAM, extensive energy transfer evidenced by a dominant Boltzmann component was attributed to the large number of low-energy degrees of freedom that efficiently absorb ozone's translational energy. Interestingly, ozone scattering from the hydroxyl-terminated SAM, which displays increased rigidity due to hydrogen bonding amongst terminal groups, resulted in a Boltzmann component similar to the one obtained with the methyl-terminated SAM. The authors suggested that a relatively strong gas–surface attractive potential plays a major role in bringing ozone to thermal equilibration on the more rigid hydroxyl-terminated SAM. Conversely, the perfluoro-terminated SAM possesses both high rigidity and weak gas–surface interactions that lead to a much smaller Boltzmann component to the final energy distribution and relatively low overall energy transfer from the gas to the surface.
More recently, molecular beam scattering methods, together with in situ monitoring of surface functional groups via RAIRS, have been employed to explore the reaction dynamics of ozone during exposure to a vinyl-terminated SAM in UHV. Results from the molecular beam scattering experiments support a mechanism that involves extensive thermal accommodation, especially at low beam energies, followed by inter-chain anhydride group formation.101 Importantly, the initial reaction probability for ozone incident energies near room temperature was found to be 1.1 × 10−5.100 Finally, the effect of ozone translational energy on the reaction probability was also investigated in this experimental study. The results suggest that accommodation of ozone on the surface is required prior to reaction for room-temperature translational energies; however, at higher incident energies, a direct reaction driven by the beam energy appears to occur.100 As shown in Fig. 7, this change of mechanism is evidenced by an increase in the initial reaction probability with incident energy.
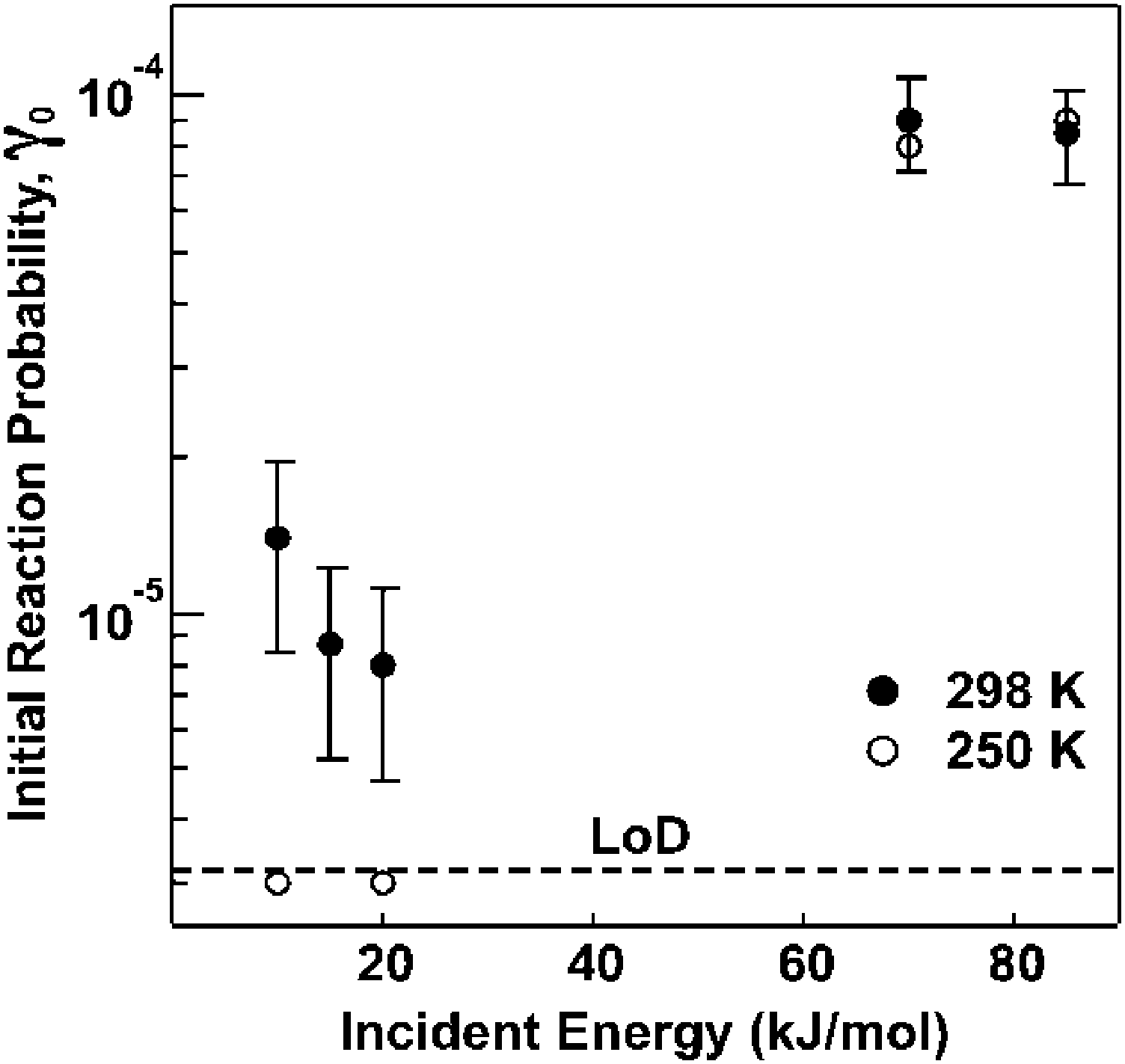 | ||
| Fig. 7 Initial reaction probability for O3 impinging on a vinyl-terminated self-assembled monolayer as a function of collision energy and surface temperature. Error bars represent one standard deviation. Reprinted with permission from J. Phys. Chem. C, 2011, 115(51), 25348. Copyright 2011 American Chemical Society. | ||
An interesting result stemming from these UHV experiments is that the reaction probability measured in the UHV-based molecular beam experiments (for thermal energies) is surprisingly similar to those found in an ambient environment, even though the reaction conditions are significantly different. To reconcile these findings, a comparison of the microscopic mechanisms of the gas–surface collisions in both types of experiments is required. When ozone collides with the SAM, it can impulsively scatter from the surface or become trapped onto the surface. Once trapped, an accommodated ozone molecule can diffuse along the surface until it either reacts or desorbs. In an ultra-high vacuum environment, once an adsorbed molecule surmounts the trapping potential, it desorbs irreversibly, as the likelihood of encountering another gas-phase molecule and becoming deflected back to the surface is very low. At atmospheric pressures, the desorbing gas molecule might be reflected back to the surface by other gases present in the environment, which would make the reaction probability higher than in UHV conditions. The reaction probability is similar under both sets of conditions likely because the ordered nature of the SAM used in UHV environment facilitates reactions in a manner that does not occur within other, less ordered surfaces used in the investigations under atmospheric conditions. Thus, an opportunity for further work in this area exists, as one may explore similar systems under atmospheric conditions or investigate the role of monolayer order, chain tilt, and packing density on the overall chemistry.
III. NO3 reactions with organic surfaces
As with ozone, many of the studies of heterogeneous reactions of organics with nitrate radicals have involved the deposition of liquid37 and solid37,39,103–105 organic substrates onto flow tube reactors. Subsequent product analysis typically employs mass spectrometry, though alternative methods of detection have been used.37One representative study by Knopf et al. investigated the effect of relative humidity on the uptake of NO3 radicals on aerosols generated during biomass burning.103 By varying the length of the flow tube and therefore the exposure area of the substrate, they determined first-order NO3 loss rate constants, which they subsequently used to calculate NO3 uptake coefficients. Further, using insights from proposed reaction mechanisms and physical properties of substrate molecules such as vapor pressure, they explained differences in NO3 uptake behavior as a function of NO3 exposure. In the presence of O2, reactive uptake coefficients were found to be in the range of 1–26 × 10−3. Other groups have investigated the reactive uptake coefficients of NO3 (both in the presence and absence of O2) on organic aerosols using flow tube reactors, and the reported uptake coefficients were also within this range.37,104 Conversely, additional work, using similar methods, have measured NO3 uptake coefficients up to two orders of magnitude larger.39,105 Variances in the measured uptake coefficients may be attributed to differences in the linear flow velocities employed in different laboratories or differences in surface composition and preparation conditions.
In a comparison of liquid-phase and solid-phase deposition, Moise and coworkers37 reported a decrease of the uptake coefficients by as much as a factor of five upon freezing for some organics (i.e., n-hexadecane and n-octanoic acid) and no change for others (e.g., 1-hexadecene, 2,2,4,4,6,8,8 heptamethyl nonane, and 10,12 octadecadienoic acid). For the reactions in which the coefficient did not change upon freezing, evaporation or mobility of the surface molecules was thought to provide a constantly renewed surface. The authors related key differences in these surface mechanisms to the solubility constant and diffuso-reactive length (“the approximate distance within the liquid over which reaction is taking place”,106 calculated using the ratio of the diffusion coefficient to the first-order loss rate) of NO3 in each liquid organic.
To more completely describe the reactive uptake coefficient of NO3 at substrates investigated by Knopf et al.,103 Shiraiwa et al. used computational kinetic flux models to quantify the relative role of surface and bulk reactions for highly functionalized organic molecules relevant to biomass burning.107,108 More specifically, by relating uptake coefficients to bulk accommodation coefficients (i.e., the probability of a gas molecule colliding with the surface to enter the bulk of the particle), the contribution of surface reaction to the overall reaction was revealed to be dependent on the mechanism of reaction. For example, surface contributions were minor for levoglucosan, which reacts with NO3via an initial hydrogen-abstraction step—a relatively slow process. On the other hand, for abietic acid, which can react with NO3via relatively rapid addition across a double bond, surface contributions dominate. Further, desorption lifetimes and bulk diffusion coefficients of NO3 into these organic surfaces were also determined.38
To complement the studies of NO3 reaction with organic molecules deposited on surfaces, Schütze and Hermann109 investigated the uptake of nitrate radicals into single drops of an aqueous solution of an organic dye inside a flow-tube reactor. A change in absorbance of the dye solution brought about by reaction with nitrate radicals allowed for the in situ determination of the reactive uptake coefficient of NO3 radicals using UV-Vis spectroscopy. Falling within the range of values reported in studies by Knopf et al.103 and others,37,104 the uptake coefficient was found to be (2.7 ± 0.8) × 10−3.109
Aside from experimental approaches that involve the direct deposition of substrates onto the surface of a flow tube, heterogeneous reactions of NO3 with organic surfaces have been examined using particles/aerosols with organic compounds adsorbed on them. Organic molecules ranging from simple polycyclic aromatic compounds110 to more complex organophosphorus pesticide molecules111,112 have been deposited at the surface of azelaic acid for the study of nitrate radical reaction rates and mechanisms. Many of these studies have employed mass spectrometry coupled with gas chromatography to help characterize gas-phase product and reactant concentrations. Typical uptake coefficients obtained in this manner lie within the same range determined from the afore-described flow-tube deposition studies, thereby helping to validate the flow-tube approach as a viable means by which to explore the type of chemistry that occurs on aerosol particles.
Beyond aerosol and liquid flow-tube experiments, model organic molecular substrates have proven to be valuable tools in the study of nitrate radical reaction pathways. Bertram and coworkers, for example, studied the reactions of nitrate radicals with alkane- and alkenethiol self-assembled monolayers on gold substrates in a flow-tube reactor arrangement.113,114 The reactive uptake coefficients they determined were 8.8 × 10−4 for an alkane monolayer and 3.4 × 10−2 for an alkene-terminated monolayer. A similar variation in coefficients was found when they compared other alkane and alkene systems. Differences in the reactive uptake coefficients found by the Bertram group from those obtained in previous studies of NO3 chemistry37,103,104 may be due to the greater accessibility of the double bond to NO3 radicals in the monolayer studies. In a separate study,105 Bertram and coworkers suggest that NO3, like O3, interacts with atmospheric surfaces through the Langmuir–Hinshelwood mechanism, though a complete description of the overall reaction mechanism has yet to be developed.
In addition to the measurements of reaction rates by Bertram and coworkers, the mechanism of reaction between these monolayers and nitrate radicals was explored using time-of-flight mass spectrometry to detect gas-phase products, while XPS and infrared spectroscopy were employed to analyze the surface-bound molecules following exposure.114 The reaction was conducted in the presence of atmospheric gases that may have interfered with the reaction of pure NO3 with the surface. In fact, background O2 and NO2 are critical co-reactants in the mechanisms proposed by Bertram et al. As shown in Fig. 8, nitrate initially abstracts a hydrogen atom from alkanes (Fig. 8a), or adds to one side of the CC bond in alkenes (Fig. 8b), leaving in a radical site for subsequent O2 addition and further reactions with other oxidative species.
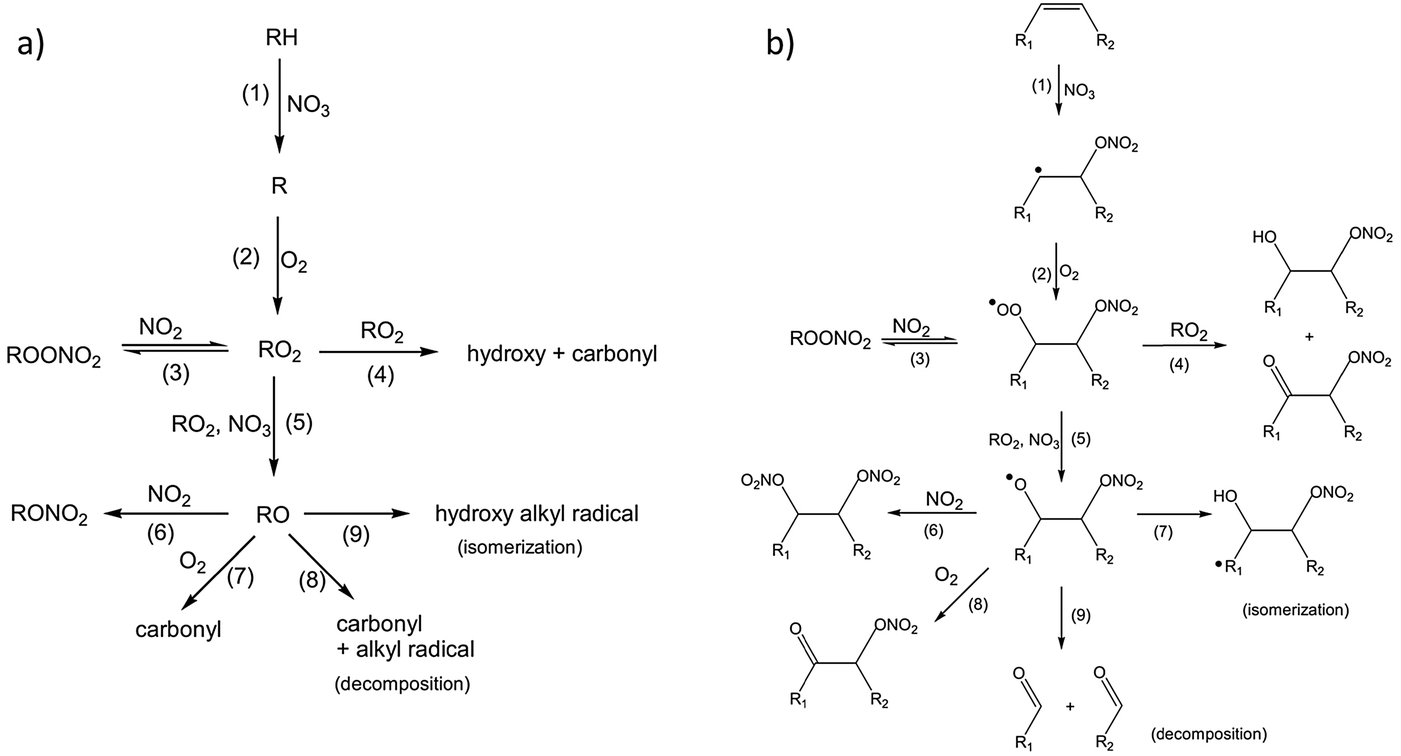 | ||
| Fig. 8 Proposed mechanism for addition of NO3 to an (a) alkane and (b) alkene surface in the presence of NO2 and O2. Copyright 2009 Wiley. Used with permission from S. Gross and A. K. Bertram, Products and kinetics of the reactions of an alkane monolayer and a terminal alkene monolayer with NO3radicals, J. Geophys. Res.: Atmos., 2009, 114, D02307. | ||
In an effort to explore the interfacial chemistry of nitrate radicals at organic surfaces in the absence of background gases and other possible contaminants, Zhang et al.115 explored these reactions at the surface of an alkenethiol self-assembled monolayer in the highly regulated environment of a UHV chamber. Additionally, the use of in situ reflection–absorption infrared spectroscopy (RAIRS) in their work allowed for the characterization of the surface during the course of nitrate radical exposure, thereby enabling an in-depth study of the reaction kinetics. The majority of studies prior to this work presented reactive uptake coefficients with little-to-no direct tracking of chemical bond breaking and formation at the surface. By directly monitoring changes in the infrared band intensity of the stretching mode associated with the surface carbon–carbon double bonds, Zhang et al. reported an initial reaction probability of (2.3 ± 0.5) × 10−3. This investigation was complemented by a quantum/molecular mechanical computational study that facilitated the identification of intermediates and products as well as the elucidation of a reaction mechanism from vibrational spectra. As shown in Fig. 9, a mechanism was proposed wherein NO3 adds to the CC double bond resulting in the formation of an alkyl nitrate radical. Electronic structure calculations revealed that hydrogen abstraction from the terminal carbon atom could be deemed unlikely under the thermal conditions of the experiment, as only a very small fraction of NO3 molecules had enough energy to overcome the calculated abstraction barrier. Notably, this is the first study of the reactions involving NO3 and organic surfaces to incorporate gas collision energy into a discussion of reaction mechanisms.
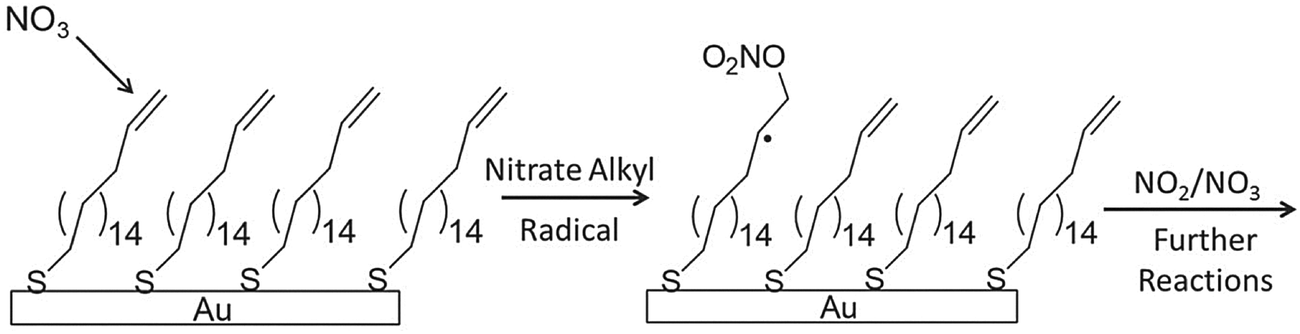 | ||
| Fig. 9 A scheme for the reaction of nitrate radical with a vinyl-terminated self-assembled monolayer. Positioning of the CC bond at the gas–surface interface affords a higher reaction probability than in experiments with a submerged CC bond (as shown in Fig. 5 for O3 reactions). Reproduced from ref. 115 with permission from the PCCP Owner Societies. | ||
The synergistic link between the work performed under atmospheric pressure by Bertram and coworkers and that of Zhang et al. is that both studies suggest that the first step in the reaction is the generation of a surface-bound radical. This step defines the initial reaction probability for NO3 at vinyl-terminated surfaces and appears to be uninfluenced by background gases. Therefore, computational studies that approach this problem in the absence of background contamination are likely appropriate approaches for exploring the fundamental energetics of the first steps in the reaction. Once the surface-bound radicals are formed, background gases likely play a major role in scavenging the radicals and further oxidizing the surface. The next step in both types of studies must be to increase the complexity of the systems to better model heterogeneous atmospheric chemistry by including highly defective surfaces and the presence of critical co-reactants within the surfaces as well as within the gas phase.
IV. OH reactions with organic surfaces
Reactions with hydroxyl radicals alter the fate and properties of atmospheric organic particles. Moreover, heterogeneous OH chemistry affects the atmospheric concentrations of ozone and nitrate. A distinctive aspect of this oxidant is that its high reactivity makes it difficult to quantify its concentration, and therefore, quantitative experiments are not as well developed as for ozone and nitrate. Nonetheless, as with ozone and nitrate gases, studies of hydroxyl radical heterogeneous chemistry have been performed largely with the use of flow reactor tubes coupled to mass spectrometric product identification methods. In many of these studies, squalane (a long, branched liquid alkane at ambient conditions20) has been employed as the liquid of choice to model the surface of ambient aerosol particles.In one such study, Smith et al.116 generated hydroxyl radicals from O3 photolysis in the presence of H2O in a flow tube containing deposited particles of squalane, and they monitored the composition of aerosol particles leaving the tube with a custom-built aerosol mass spectrometer. A relative-rate method employing the loss of particle-phase squalane over the course of OH exposure was used to determine a reactive uptake coefficient of 0.3 ± 0.07. As expected from previous work,1,117,118 the reactive uptake coefficient was found to be several orders of magnitude above that for either ozone or nitrate radicals—a consequence of the relatively high chemical potential of the hydroxyl radical and small (or nonexistent) barrier for hydrogen abstraction.119,120 Interestingly, the coefficient is still over a factor of three lower than unity, which indicates that 70% of the OH molecules escape the gas-squalane collision without reaction. Those molecules either impulsively scatter without mass accommodation or trap on the surface but desorb prior to achieving a particular orientation leading to reaction. The authors proposed reaction mechanisms that depend on OH exposure. At low exposures, sequential oxidation predominates, yet at higher, atmospherically relevant exposures, volatilization into various gas-phase functionalized organic products prevails. In other words, there is a competition between oxidation and surface mass loss that changes with time.
In another experiment, aimed at correlating the structure and phase of an organic aerosol to its reactivity, Ruehl et al.121 also used a photo-oxidation flow-tube method coupled to GC/MS analytical methods. They measured a reactive uptake coefficient similar to that described above for squalane (0.36 ± 0.11), which they found to be higher than that for octacosane (0.18 ± 0.11), a normal alkane with a slightly smaller molecular weight. The difference in coefficients between the two molecules is not only due to differences in structure (i.e., branched vs. straight chain), but also to differences in phase. Octacosane is a solid at room temperature; therefore, the diffusion of unreacted organic molecules from the bulk to the surface is much slower than in a liquid. Moreover, the authors confirmed a “surface freezing” phenomenon in which linear alkane molecules were preferentially oriented normal to the surface. They substantiated this claim with a positional distribution of functionalized octacosane products, showing a higher probability for functionalized products near the terminus of the octacosane backbone (Fig. 10), as this region is most accessible to OH exposure. As with the study by Smith et al.,116 Ruehl et al. propose a higher likelihood of functionalization at shorter oxidation lifetimes and fragmentation leading to carbon loss at longer lifetimes.
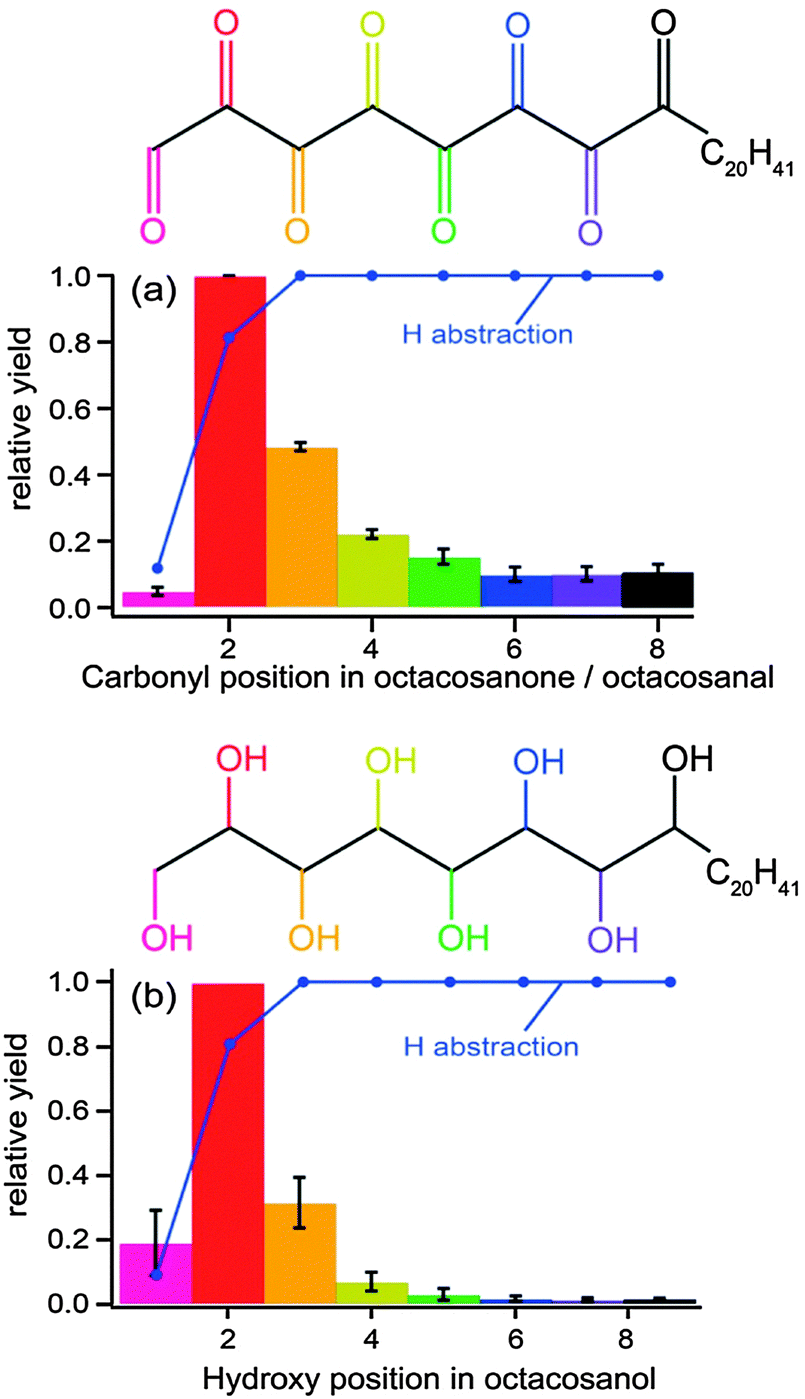 | ||
| Fig. 10 Preferential positioning of ketone and alcohol groups resulting from the functionalization of octacosane by OH exposure suggests a “surface freezing” phenomenon. Reprinted (adapted) with permission from J. Phys. Chem. A, 2013, 117, 3990. Copyright 2013 American Chemical Society. | ||
Che et al.20 also investigated the reaction between hydroxyl radicals and squalane aerosols in an experiment that showcased a novel method in which the heterogeneous chemistry of organic aerosols was studied in a continuous-flow stirred tank reactor in place of a stationary flow-tube reactor. This modification affords long-term stable reaction conditions. With this method, a larger reactive uptake coefficient (0.51 ± 0.10) for squalane exposure to OH radicals was reported. The authors attribute the difference between this uptake coefficient and that obtained in the study by Smith et al.116 to the influence of secondary chemistry (more than one squalane molecule may react per OH), which is a pathway that their approach may be more sensitive to than the stationary flow-tube method.
To gain more insight into the extent of secondary chemistry of OH with organic aerosols, Kolesar et al. investigated the effect of exposing a squalane seed coated with viscous SOA material (a product of α-pinene ozonolysis) to OH in a flow tube.122 The presence of the SOA coating enhanced squalane loss. The authors hypothesized the formation of radicals at the particle surface that are able to migrate through the SOA matrix to the squalane support where further reactions occur.
Flow-tube studies of heterogeneous reactions of hydroxyl radicals with organic aerosols ranging from those present in soot50,117,123–125 and biomass-burning aerosols,126–128 to those from herbicides,129 pesticides,130,131 and flame retardants,132,133 have been performed to obtain information such as product structure,117,123,125,130,134 yield and volatilization,123,126 changes in hygroscopic properties124 and particle size,124,131 as well as kinetic information such as reactive and degradative rate constants50,126,127,129–134 and reactive uptake coefficients.50,117,125,126,132,135 These reactive uptake coefficients are generally large, but vary significantly between 0.19 and 2.0,50,117,132,135 although even higher values have been reported.125 It should be noted that values above 1.0 are only possible for those coefficients determined by measuring particle loss117,125,132,135 as opposed to OH loss.126 That is, secondary reactions, such as propagation steps in radical polymerization may result in the elimination of multiple particles for a single OH radical.117,125,135,136 Small variations in uptake coefficient have also been attributed to differences in parametric fit of loss data, presence of O2 in the reaction environment, and variations in the kinetic energy of OH.116
Recently, flow-tube studies by Wilson and coworkers have added insight into the role of molecular structure and O2 concentration on the mechanism of organic aerosol oxidation by OH.137,138 Using mass-spectrometric data, this group compared the reaction mechanisms for both branched squalene and linear linonenic acid (Fig. 11). Both reactions were found to be initiated by the addition of an OH radical to a CC double bond to yield a hydroxyalkyl radical. O2 was found to add to this radical, which results in the formation of a hydroxyperoxy moiety. In reactions with squalene (Fig. 11a), the OH radical adds to the less substituted carbon, leading to a tertiary hydroxyperoxy radical. This radical has no adjacent hydrogens with which to self-react, which preferentially leads to the production of a hydroxyalkoxy radical (A6) over a diol (A5). The hydroxyalkoxy radical may then dissociate (A7) to form a variety of fragmentation products. In the absence of O2, hydrogen abstraction (A2) or subsequent OH radical addition (A3) after the initiation step leads to a variety of products. Thus, at lower O2 concentrations (∼1% in this study), “functionalization products” dominate, and at higher O2 concentrations (∼10% in this study), “fragmentation products” dominate. Conversely, in reactions with linolenic acid (Fig. 11b), a secondary hydroxyperoxy is available to react with an adjacent hydrogen atom (B6), favoring functionalized product formation over hydroxyalkoxy formation (B7). At both O2 concentrations used in this study, functionalization products dominate for the linolenic acid reaction. The Wilson group has also developed and experimentally validated a theoretical model for the reaction of squalane with OH radicals.48 As such, they related the uptake coefficient to the gas-phase OH density and the size and materials properties of the aerosol.
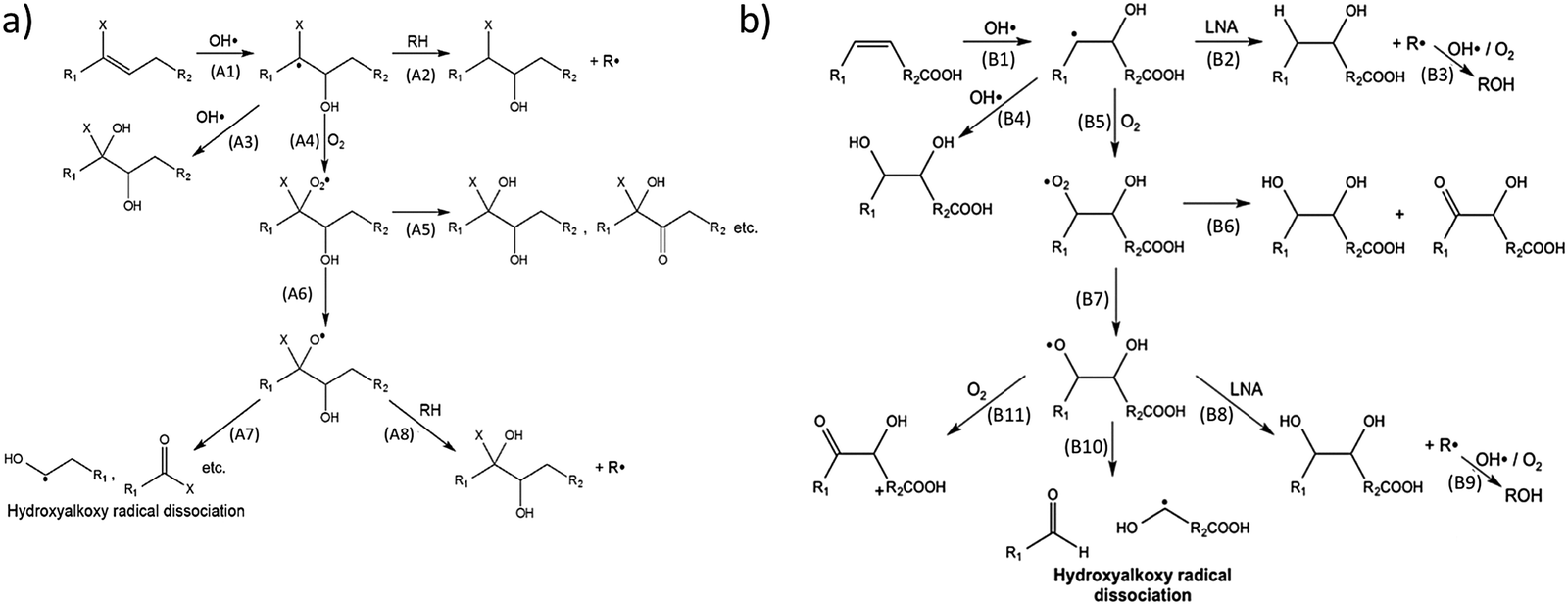 | ||
| Fig. 11 Proposed mechanism for addition of OH to (a) squalene, a branched alkane, and (b) linolenic acid, a linear, unsaturated carboxylic acid, in the presence of O2. Reprinted (adapted) with permission from (a) J. Phys. Chem. A, 2014, 118, 4106. (b) J. Phys. Chem. A, 2014, 118, 11555. Copyright 2014 American Chemical Society. | ||
Beyond aerosol studies, larger-scale surfaces have provided insight into the heterogeneous chemistry of OH. In a laboratory study by Bertram et al.,139 several surfaces including waxes, methyl- and vinyl- self-assembled monolayers, pyrene, and soot were deposited on the inside wall of a Pyrex flow tube as a proxy for suspended particles in the atmosphere. The reactive uptake coefficient in the Bertram work (determined via hydroxyl loss, as in the study by Slade et al.126), ranged from (6 ± 3) × 10−4 for halocarbon wax to 0.88 ± 0.38 (with an upper limit of 1) for soot.
In an effort to provide insight into the role of surface properties in the interaction between hydroxyl radicals and surface-adsorbed organics, Iuga et al. used density functional theory methods to explore the potential energy of the reaction between a hydroxyl radical and formaldehyde bound to the monomer Si(OH)4140 and small silicate polymers.141 In these studies, reaction pathways, along with rate constants and energy profiles, were generated for several possible mechanisms. A notable result of this theoretical work is that the OH—formaldehyde reaction yielded a lower rate constant in the presence of dust (i.e., silicates) than in the gas phase, thus suggesting that surface-adsorbed molecules may not be as reactive to OH as they are in the gas phase. This difference likely stems from the obstruction of particular reactive approach geometries for the OH + formaldehyde pair on the surface relative to the gas phase.
Further key information about OH surface chemistry emerged from the work of Dilbeck and Finlayson-Pitts,142 who studied the reaction between hydroxyl radicals and both saturated and unsaturated phospholipid molecules adsorbed onto an NaCl surface in the presence of O2. Infrared-spectroscopic measurements recorded during OH exposure, in conjunction with MALDI-ToF mass spectrometric measurements, were used to identify product functional groups. Further, changes in IR bands were used to determine the loss rate of specific vibrational modes during the gas–surface reaction. Consequently, probabilities for reaction at specific sites on the organic molecule could be obtained. With these methods, the scientists determined a reaction probability of (4 ± 1) × 10−3 for OH radical addition to the CC double bond of the unsaturated phospholipid surface molecules and a hydrogen abstraction probability of (8 ± 1) × 10−4 from the methylene groups of the saturated surface. The authors suggest that the surprisingly low measured probabilities may be attributed to phospholipid aggregation during sample preparation, thus restricting the availability of surface molecules for reaction. Hence, the values reported here are likely only lower limits.
The importance of functional group accessibility was exemplified in follow-up work by Moussa and Finlayson-Pitts143 that utilized ATR-IR to investigate the reaction of OH radicals with unsaturated self-assembled monolayers (in the presence of O2). In that work, infrared spectra were recorded during OH exposure and analyzed to provide a reaction probability of 1.1 ± 0.9 for addition of OH to the CC double bond. These measurements clearly showed that vinyl groups positioned precisely at the gas–surface interface are extremely reactive toward hydroxyl radicals and that their lifetime is directly related to the gas-phase OH concentration. Furthermore, the unit reaction probability indicates that every OH molecule, regardless of impact parameter, orientation, or translational energy, reacts with the surface, which is fundamentally different from interfacial reactions involving the nitrate radical or ozone (vide supra).
In another thin film study, Mysak et al.144 used XPS to investigate the reaction of hydroxyl radicals with coronene on a single-crystal metallic substrate in ultra-high vacuum. These measurements allowed for the examination of changes in film thickness as well as functional group distribution as a function of reaction time. The authors found that there is an increase in the surface oxygen-to-carbon ratio over the course of a heterogeneous OH reaction that is controlled by the formation of oxygenated functional groups as well as through carbon loss, as described in prior work.20,116,138 To help explain the increase in oxygen-to-carbon surface ratio with reaction time, Mysak et al. proposed that the initial transformation of reduced organic aerosols occurs via a different pathway (functionalization) than that of the highly oxidized aerosols (carbon loss), which is consistent with chemistry described in previous flow-tube type studies.116,121
Highly fundamental investigations have helped to shed significantly more light on these reaction pathways. Two key studies by the McKendrick group have implemented laser-induced fluorescence techniques in vacuo to probe the collision dynamics between hydroxyl radicals and liquid organic thin films of squalane. In the first published study,145 the objective was to examine a novel photolytic source of hydroxyl radicals and determine the contributions of impulsive scattering and thermal desorption mechanisms to the final energy distribution of OH radicals scattered from the surface. Separate trajectory calculations complemented this work by revealing the importance of the impulsive scattering and thermal desorption in collisions of OH with a perfluorinated SAM surface.146 In the second study,147 a broader range of liquids (squalane, squalene, and oleic acid) was examined with the same techniques. The reactive uptake coefficients (determined via OH loss) were found to be between 0.24 ± 0.10 (oleic acid) and 0.39 ± 0.10 (squalane). Interestingly, these values are similar to those determined for the analogous reactions at particulates.121
In other key studies, McKendrick and coworkers scattered OH radicals from a non-reactive fluorinated surface and determined the contributions of impulsive scattering and thermal-desorption mechamisms in the desorbing OH flux.148 The thermal-desorption fraction of the OH flux was then assumed to react following a Langmuir–Hinshelwood mechanism in experiments in which OH was aimed at reactive surfaces. Enami and coworkers further explored the idea of OH reactions undergoing a Langmuir–Hinshelwood mechanism using electrospray ionization mass spectrometry. Results from dosing aqueous carboxylic acid microjets with a stream of hydroxyl radicals led to a description of the mechanism that involves initial reactions precisely at the gas–surface interface. Importantly, they inferred from the extent of acid depletion during reaction, as well as from previous knowledge that OH radicals prefer interfacial layers over the bulk,149 that there is always an excess of OH radicals available to react in the outermost interfacial layers. This trapped layer of radicals is consistent with the Langmuir–Hinshelwood mechanism.150,151
The fundamental nature of the OH + –CC and –CH3 surface reaction was further investigated by D'Andrea et al.136 when they examined the reaction of OH radicals with alkane- and alkene-thiol self-assembled monolayers deposited on gold using molecular beam scattering methods in an UHV environment. Coupled with RAIRS, this experiment showed real-time changes in surface functionalization over the entire course of OH exposure (as opposed to after different, long-exposure times143), thus providing a more detailed spectroscopic picture of the progression of the reaction. Following examination of spectral changes during exposure, reaction mechanisms of OH with both self-assembled monolayers were proposed. Both mechanisms, as shown schematically in Fig. 12, highlight the possibility for radical-induced polymerization of the organic chains in the monolayer, thereby providing insight into the secondary chemistry alluded to in previous studies (i.e., where researchers reported reactive uptake coefficients greater than unity117,125,132,135).
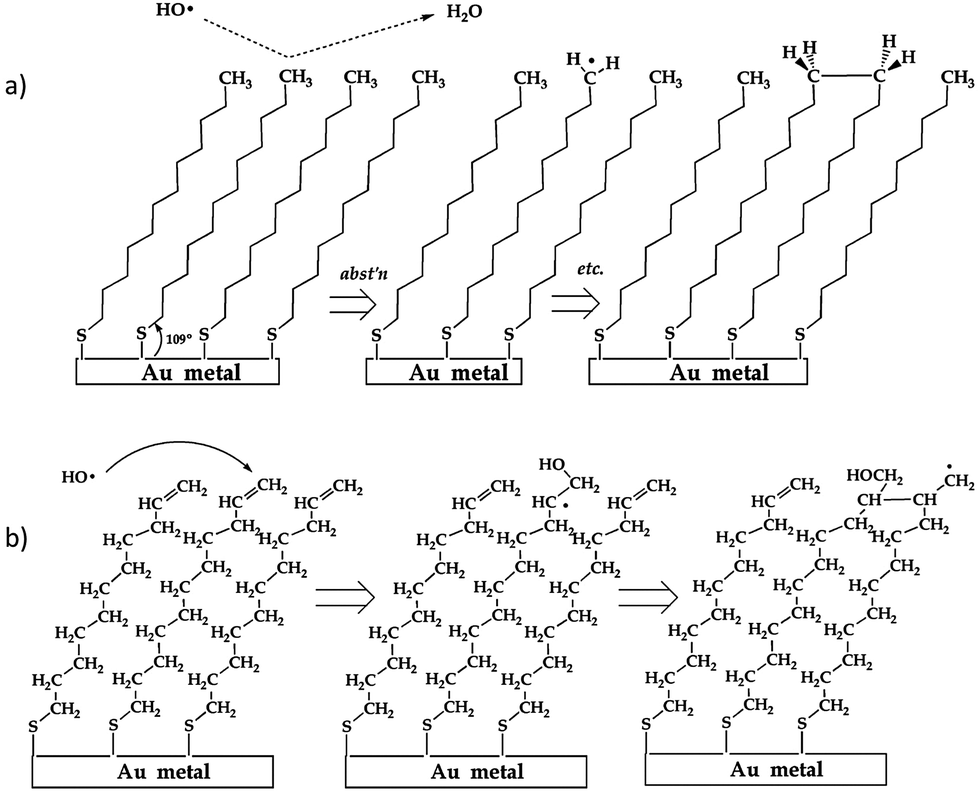 | ||
| Fig. 12 Mechanisms of OH + (a) alkane and (b) alkene-terminated SAM surfaces proposed by D'Andrea et al. In (a), isolated alkyl radicals cannot diffuse due to SAM stability. Subsequent OH collisions can result in further radical generation and cross-linking dimerization. In (b), polymerization results in a cross-linked alcohol. Reprinted (adapted) with permission from J. Phys. Chem. B, 2008, 112, 535. Copyright 2008 American Chemical Society. | ||
V. Summary and perspectives
Oxidation by O3, NO3, and OH results in changes in the physical and chemical properties of organic particles and surfaces in the atmosphere. Importantly, these molecules functionalize initially hydrophobic organic particles rendering them hydrophilic, which dramatically affects their solubility in rain droplets, role as cloud condensation nuclei, and environmental transport. Moreover, changes in particulate size and composition affect the absorption and scattering of incoming sunlight. Consequences such as these have motivated scientists to study the interaction between highly oxidative pollutants and organic surfaces.This review described the progress of research surrounding heterogeneous reactions of atmospheric oxidative gases and organic surfaces of atmospheric importance. A wide range of methods from flow tubes and mass spectrometry to molecular beam scattering and infrared spectroscopy have been used to inform scientists about kinetics and reaction mechanisms. Most recently, researchers have been challenged by the possibilities of polymerization initiated by radical-containing pollutants and further promoted by secondary mechanisms that do not directly involve the atmospheric initiator.
In addition to highly practical field measurements152 and atmospheric sampling,153 future research will continue to focus on deconstructing complex systems into their fundamental components and investigating real-time changes to organic surfaces during exposure to a controlled flux of gas-phase oxidants. In this way, scientists will independently study how key properties, such as gas-surface polarity, molecular structure, and density, affect the outcome of interfacial collisions.
Modern computational techniques will also continue to find a role in adding insight into the fundamental aspects of atmospheric heterogeneous chemistry. The same experimental systems that allow laboratory researchers to investigate individual components of complex atmospheric reactions readily lend themselves to in silico modeling. The uniform structure of model organic surfaces such as self-assembled monolayers allows for their facile computational modeling using quantum/molecular mechanical hybrid models as well as molecular dynamics techniques, which will provide great theoretical insight into oxidative processes on both the molecular and surface nanoscales.
As a result of investigations into heterogeneous chemistry and reaction dynamics of the atmospheric oxidants, O3, NO3, and OH, researchers are gaining insight into the role of surface structure and functionality in the reactions of these gases. While research in this field continues to progress, more will be learned about the effects of this chemistry on organic particulates and the balance of oxidative gas concentrations in the troposphere. Armed with this information, researchers will be able to make well-informed predictions about the fate of organics in the atmosphere as well as the impact of these oxidants on the environment.
Acknowledgements
The authors are grateful to Dr. Linsey Marr from the Virginia Tech Department of Civil and Environmental Engineering for her constructive comments on this work. This material is based upon work supported by, or in part by, the U. S. Army Research Laboratory and the U. S. Army Research Office under contract/grant number W911NF1410159.References
- R. Atkinson, Atmos. Chem. Phys., 2003, 3, 2233–2307 CrossRef CAS.
- D. Johnson and G. Marston, Chem. Soc. Rev., 2008, 37, 699–716 RSC.
- R. Atkinson, J. Phys. Chem. Ref. Data, 1997, 26, 215–290 CrossRef CAS.
- S. S. Brown and J. Stutz, Chem. Soc. Rev., 2012, 41, 6405–6447 RSC.
- D. M. Etheridge, L. P. Steele, R. J. Francey and R. L. Langenfelds, J. Geophys. Res.: Atmos., 1998, 103, 15979–15993 CrossRef CAS.
- L. W. Richards, J. Air Pollut. Control Assoc., 1988, 38, 784–791 CrossRef.
- M. C. Jacobson, H. C. Hansson, K. J. Noone and R. J. Charlson, Rev. Geophys., 2000, 38, 267–294 CrossRef CAS.
- B. T. Simoneit, J. Atmos. Chem., 1989, 8, 251–275 CrossRef CAS.
- L. J. Standley and B. R. T. Simoneit, Environ. Sci. Technol., 1987, 21, 163–169 CrossRef CAS.
- T. E. Graedel, D. T. Hawkins and L. D. Claxton, Atmospheric chemical compounds: sources, occurrence and bioassay, Elsevier, 1986 Search PubMed.
- D. J. Donaldson and D. Anderson, J. Phys. Chem. A, 1999, 103, 871–876 CrossRef CAS.
- P. S. Gill, Rev. Geophys., 1983, 21, 903 CrossRef CAS.
- A. J. Tiwari and L. C. Marr, J. Environ. Qual., 2010, 39, 1883–1895 CrossRef CAS PubMed.
- M. Hallquist, J. C. Wenger, U. Baltensperger, Y. Rudich, D. Simpson, M. Claeys, J. Dommen, N. M. Donahue, C. George, A. H. Goldstein, J. F. Hamilton, H. Herrmann, T. Hoffmann, Y. Iinuma, M. Jang, M. E. Jenkin, J. L. Jimenez, A. Kiendler-Scharr, W. Maenhaut, G. McFiggans, T. F. Mentel, A. Monod, A. S. H. Prévôt, J. H. Seinfeld, J. D. Surratt, R. Szmigielski and J. Wildt, Atmos. Chem. Phys., 2009, 9, 5155–5236 CrossRef CAS.
- D. Schuetzle, D. Cronn, A. L. Crittenden and R. J. Charlson, Environ. Sci. Technol., 1975, 9, 838–845 CrossRef CAS.
- C. Fountoukis, A. G. Megaritis, K. Skyllakou, P. E. Charalampidis, C. Pilinis, H. A. C. Denier van der Gon, M. Crippa, F. Canonaco, C. Mohr, A. S. H. Prévôt, J. D. Allan, L. Poulain, T. Petäjä, P. Tiitta, S. Carbone, A. Kiendler-Scharr, E. Nemitz, C. O'Dowd, E. Swietlicki and S. N. Pandis, Atmos. Chem. Phys., 2014, 14, 9061–9076 CrossRef CAS.
- J. Jimenez, M. R. Canagaratna, N. M. Donahue, A. S. H. Prevot and Q. Zhang, Science, 2009, 326, 1525–1529 CrossRef CAS PubMed.
- A. Virtanen, J. Joutsensaari, T. Koop, J. Kannosto, P. Yli-Pirila, J. Leskinen, J. M. Makela, J. K. Holopainen, U. Poschl, M. Kulmala, D. R. Worsnop and A. Laaksonen, Nature, 2010, 467, 824–827 CrossRef CAS PubMed.
- B. J. Finlayson-Pitts and J. N. Pitts Jr, Chemistry of the upper and lower atmosphere: theory, experiments, and applications, Academic press, 1999 Search PubMed.
- D. L. Che, J. D. Smith, S. R. Leone, M. Ahmed and K. R. Wilson, Phys. Chem. Chem. Phys., 2009, 11, 7885–7895 RSC.
- S. N. Chu, S. Sands, M. R. Tomasik, P. S. Lee and V. F. McNeill, J. Am. Chem. Soc., 2010, 132, 15968–15975 CrossRef CAS PubMed.
- R. S. Disselkamp, M. A. Carpenter, J. P. Cowin, C. M. Berkowitz, E. G. Chapman, R. A. Zaveri and N. S. Laulainen, J. Geophys. Res.: Atmos., 2000, 105, 9767–9771 CrossRef CAS.
- J. L. Fry and K. Sackinger, Atmos. Chem. Phys., 2012, 12, 8797–8811 CAS.
- J. T. Middleton, et al., Plant Dis. Rep., 1950, 34, 245–252 CAS.
- N. L. Bernard, M. J. Gerber, C. M. Astre and M. J. Saintot, Environ. Sci. Technol., 1998, 33, 217–222 CrossRef.
- D. Asaf, E. Tas, D. Pedersen, M. Peleg and M. Luria, Environ. Sci. Technol., 2010, 44, 5901–5907 CrossRef CAS PubMed.
- Y. Tsutsumi, Y. Zaizen and Y. Makino, Geophys. Res. Lett., 1994, 21, 1727–1730 CrossRef CAS.
- F. E. Blacet, Ind. Eng. Chem., 1952, 44, 1339–1342 CrossRef CAS.
- D. E. Heard and M. J. Pilling, Chem. Rev., 2003, 103, 5163–5198 CrossRef CAS PubMed.
- K. R. Darnall, A. C. Lloyd, A. M. Winer and J. N. Pitts, Environ. Sci. Technol., 1976, 10, 692–696 CrossRef CAS.
- H. Levy, Science, 1971, 173, 141–143 CAS.
- M. R. Kurpius and A. H. Goldstein, Geophys. Res. Lett., 2003, 30, 1371 CrossRef.
- S. Li, J. Matthews and A. Sinha, Science, 2008, 319, 1657–1660 CrossRef CAS PubMed.
- K. Salo, M. Hallquist, A. M. Jonsson, H. Saathoff, K. H. Naumann, C. Spindler, R. Tillmann, H. Fuchs, B. Bohn, F. Rubach, T. F. Mentel, L. Muller, M. Reinnig, T. Hoffmann and N. M. Donahue, Atmos. Chem. Phys., 2011, 11, 11055–11067 CrossRef CAS.
- J. L. Ambrose, H. Mao, H. R. Mayne, J. Stutz, R. Talbot and B. C. Sive, J. Geophys. Res.: Atmos., 2007, 112, D21302 CrossRef.
- R. P. Wayne, I. Barnes, P. Biggs, J. P. Burrows, C. E. Canosamas, J. Hjorth, G. Lebras, G. K. Moortgat, D. Perner, G. Poulet, G. Restelli and H. Sidebottom, Atmos. Environ., Part A, 1991, 25, 1–203 CrossRef.
- T. Moise, R. K. Talukdar, G. J. Frost, R. W. Fox and Y. Rudich, J. Geophys. Res.: Atmos., 2002, 107, AAC 6-1 CrossRef.
- M. Shiraiwa, U. Poschl and D. A. Knopf, Environ. Sci. Technol., 2012, 46, 6630–6636 CrossRef CAS PubMed.
- Z. Zhao, S. Husainy, C. T. Stoudemayer and G. D. Smith, Phys. Chem. Chem. Phys., 2011, 13, 17809–17817 RSC.
- A. M. Winer, R. Atkinson and J. N. Pitts, Science, 1984, 224, 156–159 CAS.
- J. H. Seinfeld and S. N. Pandis, Atmospheric chemistry and physics: from air pollution to climate change, John Wiley & Sons, 2012 Search PubMed.
- E. Gonzalez-Labrada, R. Schmidt and C. E. DeWolf, Phys. Chem. Chem. Phys., 2007, 9, 5814–5821 RSC.
- K. Miet, K. Le Menach, P. M. Flaud, H. Budzinski and E. Villenave, Atmos. Environ., 2009, 43, 3699–3707 CrossRef CAS.
- R. C. Chapleski, Jr., J. R. Morris and D. Troya, Phys. Chem. Chem. Phys., 2014, 16, 5977–5986 RSC.
- E. D. Davis, A. Wagner, M. McEntee, M. Kaur, D. Troya and J. R. Morris, J. Phys. Chem. Lett., 2012, 3, 3193–3198 CrossRef CAS PubMed.
- Y. Liu, J. Liggio, S.-M. Li, D. Breznan, R. Vincent, E. M. Thomson, P. Kumarathasan, D. Das, J. Abbatt, M. Antiñolo and L. Russell, Environ. Sci. Technol., 2015, 49, 2806–2814 CrossRef CAS PubMed.
- A. J. Tiwari, J. R. Morris, E. P. Vejerano, M. F. Hochella and L. C. Marr, Environ. Sci. Technol., 2014, 48, 2706–2714 CrossRef CAS PubMed.
- F. A. Houle, W. D. Hinsberg and K. R. Wilson, Phys. Chem. Chem. Phys., 2015, 17, 4412–4423 RSC.
- Y. Bedjanian and M. L. Nguyen, Chemosphere, 2010, 79, 387–393 CrossRef CAS PubMed.
- E. C. Browne, J. P. Franklin, M. R. Canagaratna, P. Massoli, T. W. Kirchstetter, D. R. Worsnop, K. R. Wilson and J. H. Kroll, J. Phys. Chem. A, 2015, 119, 1154–1163 CrossRef CAS PubMed.
- Y. C. Liu, C. Liu, J. Z. Ma, Q. X. Ma and H. He, Phys. Chem. Chem. Phys., 2010, 12, 10896–10903 RSC.
- C. Han, Y. C. Liu, J. Z. Ma and H. He, J. Chem. Phys., 2012, 137, 084507 CrossRef PubMed.
- N. O. A. Kwamena, M. G. Staikova, D. J. Donaldson, I. J. George and J. P. D. Abbatt, J. Phys. Chem. A, 2007, 111, 11050–11058 CrossRef CAS PubMed.
- N. O. A. Kwamena, J. A. Thornton and J. P. D. Abbatt, J. Phys. Chem. A, 2004, 108, 11626–11634 CrossRef CAS.
- J. Z. Ma, Y. C. Liu and H. He, Atmos. Environ., 2010, 44, 4446–4453 CrossRef CAS.
- J. Z. Ma, Y. C. Liu, Q. X. Ma, C. Liu and H. He, Atmos. Environ., 2013, 72, 165–170 CrossRef CAS.
- J. J. Nájera, R. Wamsley, D. J. Last, K. E. Leather, C. J. Percival and A. B. Horn, Int. J. Chem. Kinet., 2011, 43, 694–707 CrossRef.
- E. Gloaguen, E. R. Mysak, S. R. Leone, M. Ahmed and K. R. Wilson, Int. J. Mass Spectrom., 2006, 258, 74–85 CrossRef CAS.
- Y. B. Gai, W. G. Wang, M. F. Ge, H. G. Kjaergaard, S. Jorgensen and L. Du, Atmos. Environ., 2013, 77, 696–702 CrossRef CAS.
- S. M. Forrester and D. A. Knopf, Atmos. Chem. Phys., 2013, 13, 6507–6522 CrossRef CAS.
- J. Gan, B. Yang, Y. Zhang, X. Shu, C. G. Liu and J. N. A. Shu, J. Phys. Chem. A, 2010, 114, 12231–12236 CrossRef CAS PubMed.
- F. D. Pope, P. J. Gallimore, S. J. Fuller, R. A. Cox and M. Kalberer, Environ. Sci. Technol., 2010, 44, 6656–6660 CrossRef CAS PubMed.
- J. J. Najera, C. J. Percival and A. B. Horn, Phys. Chem. Chem. Phys., 2009, 11, 9093–9103 RSC.
- T. L. Eliason, S. Aloisio, D. J. Donaldson, D. J. Cziczo and V. Vaida, Atmos. Environ., 2003, 37, 2207–2219 CrossRef CAS.
- P. J. Gallimore, P. Achakulwisut, F. D. Pope, J. F. Davies, D. R. Spring and M. Kalberer, Atmos. Chem. Phys., 2011, 11, 12181–12195 CrossRef CAS.
- D. Ray, J. K. Malongwe and P. Klan, Environ. Sci. Technol., 2013, 47, 6773–6780 CrossRef CAS PubMed.
- J. W. Morris, P. Davidovits, J. T. Jayne, J. L. Jimenez, Q. Shi, C. E. Kolb, D. R. Worsnop, W. S. Barney and G. Cass, Geophys. Res. Lett., 2002, 29, 71 Search PubMed.
- G. D. Smith, E. Woods, C. L. DeForest, T. Baer and R. E. Miller, J. Phys. Chem. A, 2002, 106, 8085–8095 CrossRef CAS.
- J. D. Hearn, A. J. Lovett and G. D. Smith, Phys. Chem. Chem. Phys., 2005, 7, 501–511 RSC.
- B. J. Dennis-Smither, K. L. Hanford, N. O. A. Kwamena, R. E. H. Miles and J. P. Reid, J. Phys. Chem. A, 2012, 116, 6159–6168 CrossRef CAS PubMed.
- E. P. Rosen, E. R. Garland and T. Baer, J. Phys. Chem. A, 2008, 112, 10315–10324 CrossRef CAS PubMed.
- J. E. Ham and J. R. Wells, Indoor Air, 2008, 18, 394–407 CrossRef CAS PubMed.
- T. F. Kahan, N. O. A. Kwamena and D. J. Donaldson, Atmos. Environ., 2006, 40, 3448–3459 CrossRef CAS.
- N.-O. A. Kwamena, M. E. Earp, C. J. Young and J. P. D. Abbatt, J. Phys. Chem. A, 2006, 110, 3638–3646 CrossRef CAS PubMed.
- D. Fu, C. B. Leng, J. Kelley, G. Zeng, Y. H. Zhang and Y. Liu, Environ. Sci. Technol., 2013, 47, 10611–10618 CAS.
- L. Petrick and Y. Dubowski, Indoor Air, 2009, 19, 381–391 CrossRef CAS PubMed.
- C. B. Leng, J. Hiltner, H. Pham, J. Kelley, M. Mach, Y. H. Zhang and Y. Liu, Phys. Chem. Chem. Phys., 2014, 16, 4350–4360 RSC.
- G. Zeng, S. Holladay, D. Langlois, Y. H. Zhang and Y. Liu, J. Phys. Chem. A, 2013, 117, 1963–1974 CrossRef CAS PubMed.
- S. Raja and K. T. Valsaraj, J. Air Waste Manage. Assoc., 2005, 55, 1345–1355 CAS.
- Y. Wadia, D. J. Tobias, R. Stafford and B. J. Finlayson-Pitts, Langmuir, 2000, 16, 9321–9330 CrossRef CAS.
- B. T. Mmereki, D. J. Donaldson, J. B. Gilman, T. L. Eliason and V. Vaida, Atmos. Environ., 2004, 38, 6091–6103 CrossRef CAS.
- T. Moise and Y. Rudich, J. Phys. Chem. A, 2002, 106, 6469–6476 CrossRef CAS.
- T. Moise and Y. Rudich, J. Geophys. Res.: Atmos., 2000, 105, 14667–14676 CrossRef CAS.
- S. Enami, M. R. Hoffmann and A. J. Colussi, J. Phys. Chem. B, 2008, 112, 4153–4156 CrossRef CAS PubMed.
- S. Enami, M. R. Hoffmann and A. J. Colussi, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 7365–7369 CrossRef CAS PubMed.
- S. Enami, M. R. Hoffmann and A. J. Colussi, Chem. Res. Toxicol., 2009, 22, 35–40 CrossRef CAS PubMed.
- S. Enami, M. R. Hoffmann and A. J. Colussi, J. Phys. Chem. A, 2009, 113, 7002–7010 CrossRef CAS PubMed.
- S. Enami, M. R. Hoffmann and A. J. Colussi, J. Phys. Chem. B, 2009, 113, 9356–9358 CrossRef CAS PubMed.
- S. Enami, M. R. Hoffmann and A. J. Colussi, J. Phys. Chem. Lett., 2010, 1, 2374–2379 CrossRef CAS.
- H. I. Kim, H. Kim, Y. S. Shin, L. W. Beegle, W. A. Goddard, J. R. Heath, I. Kanik and J. L. Beauchamp, J. Phys. Chem. B, 2010, 114, 9496–9503 CrossRef CAS PubMed.
- D. A. Thomas, L. Wang, B. Goh, E. S. Kim and J. L. Beauchamp, Anal. Chem., 2015, 87, 3336–3344 CrossRef CAS PubMed.
- T. Thornberry and J. P. D. Abbatt, Phys. Chem. Chem. Phys., 2004, 6, 84–93 RSC.
- H. M. Hung and C. W. Tang, J. Phys. Chem. A, 2010, 114, 13104–13112 CrossRef CAS PubMed.
- B. J. Dennis-Smither, R. E. H. Miles and J. P. Reid, J. Geophys. Res.: Atmos., 2012, 117, D20204 CrossRef.
- B. J. Dennis-Smither, F. H. Marshall, R. E. H. Miles, T. C. Preston and J. P. Reid, J. Phys. Chem. A, 2014, 118, 5680–5691 CrossRef CAS PubMed.
- E. Gonzalez-Labrada, R. Schmidt and C. E. DeWolf, Chem. Commun., 2006, 2471–2473 RSC.
- Y. Dubowski, J. Vieceli, D. J. Tobias, A. Gomez, A. Lin, S. A. Nizkorodov, T. M. McIntire and B. J. Finlayson-Pitts, J. Phys. Chem. A, 2004, 108, 10473–10485 CrossRef CAS.
- J. Vieceli, O. L. Ma and D. J. Tobias, J. Phys. Chem. A, 2004, 108, 5806–5814 CrossRef CAS.
- T. M. McIntire, A. S. Lea, D. J. Gaspar, N. Jaitly, Y. Dubowski, Q. Q. Li and B. J. Finlayson-Pitts, Phys. Chem. Chem. Phys., 2005, 7, 3605–3609 RSC.
- J. W. Lu, L. R. Fiegland, E. D. Davis, W. A. Alexander, A. Wagner, R. D. Gandour and J. R. Morris, J. Phys. Chem. C, 2011, 115, 25343–25350 CAS.
- L. R. Fiegland, M. McCorn Saint Fleur and J. R. Morris, Langmuir, 2005, 21, 2660–2661 CrossRef CAS PubMed.
- J. W. Lu and J. R. Morris, J. Phys. Chem. A, 2011, 115, 6194–6201 CrossRef CAS PubMed.
- D. A. Knopf, S. M. Forrester and J. H. Slade, Phys. Chem. Chem. Phys., 2011, 13, 21050–21062 RSC.
- L. Lee, P. Wooldridge, T. Nah, K. Wilson and R. Cohen, Phys. Chem. Chem. Phys., 2013, 15, 882–892 RSC.
- S. Gross, R. Iannone, S. Xiao and A. K. Bertram, Phys. Chem. Chem. Phys., 2009, 11, 7792–7803 RSC.
- D. R. Hanson, A. R. Ravishankara and S. Solomon, J. Geophys. Res.: Space Phys., 1994, 99, 3615–3629 CrossRef CAS.
- M. Shiraiwa, C. Pfrang and U. Poeschl, Atmos. Chem. Phys., 2010, 10, 3673–3691 CrossRef CAS.
- M. Shiraiwa, R. M. Garland and U. Poschl, Atmos. Chem. Phys., 2009, 9, 9571–9586 CrossRef CAS.
- M. Schutze and H. Herrmann, J. Atmos. Chem., 2005, 52, 1–18 CrossRef.
- Y. Zhang, B. Yang, J. Gan, C. G. Liu, X. Shu and J. N. Shu, Atmos. Environ., 2011, 45, 2515–2521 CrossRef CAS.
- Y. F. Wang, P. Zhang, B. Yang, C. G. Liu and J. N. Shu, Chemosphere, 2013, 90, 848–855 CrossRef CAS PubMed.
- C. G. Liu, B. Yang, J. Gan, Y. Zhang, M. Liang, X. Shu and J. N. Shu, Chemosphere, 2012, 87, 470–476 CrossRef CAS PubMed.
- D. A. Knopf, J. Mak, S. Gross and A. K. Bertram, Geophys. Res. Lett., 2006, 33, L17816 CrossRef.
- S. Gross and A. K. Bertram, J. Geophys. Res.: Atmos., 2009, 114, D02307 CrossRef.
- Y. Zhang, R. C. Chapleski, J. W. Lu, T. H. Rockhold, D. Troya and J. R. Morris, Phys. Chem. Chem. Phys., 2014, 16, 16659–16670 RSC.
- J. D. Smith, J. H. Kroll, C. D. Cappa, D. L. Che, C. L. Liu, M. Ahmed, S. R. Leone, D. R. Worsnop and K. R. Wilson, Atmos. Chem. Phys., 2009, 9, 3209–3222 CrossRef CAS.
- I. J. George, A. Vlasenko, J. G. Slowik, K. Broekhuizen and J. P. D. Abbatt, Atmos. Chem. Phys., 2007, 7, 4187–4201 CrossRef CAS.
- B. D'Anna, O. Andresen, Z. Gefen and C. J. Nielsen, Phys. Chem. Chem. Phys., 2001, 3, 3057–3063 RSC.
- S. Mitroka, S. Zimmeck, D. Troya and J. M. Tanko, J. Am. Chem. Soc., 2010, 132, 2907–2913 CrossRef CAS PubMed.
- J. X. Zhang, L. Yang and D. Troya, Chin. J. Chem. Phys., 2013, 26, 765–773 CrossRef CAS.
- C. R. Ruehl, T. Nah, G. Isaacman, D. R. Worton, A. W. H. Chan, K. R. Kolesar, C. D. Cappa, A. H. Goldstein and K. R. Wilson, J. Phys. Chem. A, 2013, 117, 3990–4000 CrossRef CAS PubMed.
- K. R. Kolesar, G. Buffaloe, K. R. Wilson and C. D. Cappa, Environ. Sci. Technol., 2014, 48, 3196–3202 CrossRef CAS PubMed.
- A. Vlasenko, I. J. George and J. P. D. Abbatt, J. Phys. Chem. A, 2008, 112, 1552–1560 CrossRef CAS PubMed.
- I. J. George, R. Y. W. Chang, V. Danov, A. Vlasenko and J. P. D. Abbatt, Atmos. Environ., 2009, 43, 5038–5045 CrossRef CAS.
- T. Nah, S. H. Kessler, K. E. Daumit, J. H. Kroll, S. R. Leone and K. R. Wilson, Phys. Chem. Chem. Phys., 2013, 15, 18649–18663 RSC.
- J. H. Slade and D. A. Knopf, Phys. Chem. Chem. Phys., 2013, 15, 5898–5915 RSC.
- C. Lai, Y. Liu, J. Ma, Q. Ma and H. He, Phys. Chem. Chem. Phys., 2015, 17, 10953–10962 RSC.
- C. Lai, Y. Liu, J. Ma, Q. Ma and H. He, Atmos. Environ., 2014, 91, 32–39 CrossRef CAS.
- M. Pflieger, A. Monod and H. Wortham, Environ. Sci. Technol., 2013, 47, 6239–6246 CAS.
- M. Al Rashidi, A. Chakir and E. Roth, Atmos. Environ., 2014, 82, 164–171 CrossRef CAS.
- M. Segal-Rosenheimer, R. Linker and Y. Dubowski, Phys. Chem. Chem. Phys., 2011, 13, 506–517 RSC.
- Y. C. Liu, J. Liggio, T. Harner, L. Jantunen, M. Shoeib and S. M. Li, Environ. Sci. Technol., 2014, 48, 1041–1048 CrossRef CAS PubMed.
- Y. Liu, L. Huang, S. M. Li, T. Harner and J. Liggio, Atmos. Chem. Phys., 2014, 14, 12195–12207 Search PubMed.
- Y. Liu, S. M. Li and J. Liggio, Atmos. Chem. Phys., 2014, 14, 9201–9211 CAS.
- V. F. McNeill, R. L. N. Yatavelli, J. A. Thornton, C. B. Stipe and O. Landgrebe, Atmos. Chem. Phys., 2008, 8, 5465–5476 CrossRef CAS.
- T. M. D'Andrea, X. Zhang, E. B. Jochnowitz, T. G. Lindeman, C. Simpson, D. E. David, T. J. Curtiss, J. R. Morris and G. B. Ellison, J. Phys. Chem. B, 2008, 112, 535–544 CrossRef PubMed.
- T. Nah, S. H. Kessler, K. E. Daumit, J. H. Kroll, S. R. Leone and K. R. Wilson, J. Phys. Chem. A, 2014, 118, 4106–4119 CrossRef CAS PubMed.
- T. Nah, H. Zhang, D. R. Worton, C. R. Ruehl, B. B. Kirk, A. H. Goldstein, S. R. Leone and K. R. Wilson, J. Phys. Chem. A, 2014, 118, 11555–11571 CrossRef CAS PubMed.
- A. K. Bertram, A. V. Ivanov, M. Hunter, L. T. Molina and M. J. Molina, J. Phys. Chem. A, 2001, 105, 9415–9421 CrossRef CAS.
- C. Iuga, R. E. Olea and A. Vivier-Bunge, J. Mex. Chem. Soc., 2008, 52, 36–46 CAS.
- C. Iuga, A. Vivier-Bunge, A. Hernandez-Laguna and C. I. Sainz-Diaz, J. Phys. Chem. C, 2008, 112, 4590–4600 CAS.
- C. W. Dilbeck and B. J. Finlayson-Pitts, Phys. Chem. Chem. Phys., 2013, 15, 9833–9844 RSC.
- S. G. Moussa and B. J. Finlayson-Pitts, Phys. Chem. Chem. Phys., 2010, 12, 9419–9428 RSC.
- E. R. Mysak, J. D. Smith, P. D. Ashby, J. T. Newberg, K. R. Wilson and H. Bluhm, Phys. Chem. Chem. Phys., 2011, 13, 7554–7564 RSC.
- K. L. King, G. Paterson, G. E. Rossi, M. Iljina, R. E. Westacott, M. L. Costen and K. G. McKendrick, Phys. Chem. Chem. Phys., 2013, 15, 12852–12863 RSC.
- D. Troya, Theor. Chem. Acc., 2012, 131, 1072 CrossRef.
- C. Waring, K. L. King, P. A. J. Bagot, M. L. Costen and K. G. McKendrick, Phys. Chem. Chem. Phys., 2011, 13, 8457–8469 RSC.
- P. A. J. Bagot, C. Waring, M. L. Costen and K. G. McKendrick, J. Phys. Chem. C, 2008, 112, 10868–10877 CAS.
- M. Roeselová, J. Vieceli, L. X. Dang, B. C. Garrett and D. J. Tobias, J. Am. Chem. Soc., 2004, 126, 16308–16309 CrossRef PubMed.
- S. Enami, M. R. Hoffmann and A. J. Colussi, J. Phys. Chem. Lett., 2015, 6, 527–534 CrossRef CAS PubMed.
- S. Enami, M. R. Hoffmann and A. J. Colussi, J. Phys. Chem. A, 2014, 118, 4130–4137 CrossRef CAS PubMed.
- L. E. Hatch, J. M. Creamean, A. P. Ault, J. D. Surratt, M. N. Chan, J. H. Seinfeld, E. S. Edgerton, Y. X. Su and K. A. Prather, Environ. Sci. Technol., 2011, 45, 5105–5111 CrossRef CAS PubMed.
- P. Q. Fu, K. Kawamura, Y. F. Cheng, S. Hatakeyama, A. Takami, H. Li and W. Wang, Atmos. Chem. Phys., 2014, 14, 4185–4199 CrossRef.
| This journal is © The Royal Society of Chemistry 2016 |