
Comprehensive evaluation of ten docking programs on a diverse set of protein–ligand complexes: the prediction accuracy of sampling power and scoring power†
Zhe
Wang
a,
Huiyong
Sun
a,
Xiaojun
Yao
b,
Dan
Li
a,
Lei
Xu
c,
Youyong
Li
d,
Sheng
Tian
d and
Tingjun
Hou
*ae
aCollege of Pharmaceutical Sciences, Zhejiang University, Hangzhou, Zhejiang 310058, China. E-mail: tingjunhou@zju.edu.cn; tingjunhou@hotmail.com; Tel: +86-571-88208412
bState Key Laboratory of Quality Research in Chinese Medicine, Macau Institute For Applied Research in Medicine and Health, Macau University of Science and Technology, Avenida Wai Long, Taipa, Macau (SAR), China
cInstitute of Bioinformatics and Medical Engineering, School of Electrical and Information Engineering, Jiangsu University of Technology, Changzhou 213001, China
dInstitute of Functional Nano and Soft Materials (FUNSOM), Soochow University, Suzhou, Jiangsu 215123, China
eState Key Lab of CAD&CG, Zhejiang University, Hangzhou, Zhejiang 310058, China
First published on 7th April 2016
Abstract
As one of the most popular computational approaches in modern structure-based drug design, molecular docking can be used not only to identify the correct conformation of a ligand within the target binding pocket but also to estimate the strength of the interaction between a target and a ligand. Nowadays, as a variety of docking programs are available for the scientific community, a comprehensive understanding of the advantages and limitations of each docking program is fundamentally important to conduct more reasonable docking studies and docking-based virtual screening. In the present study, based on an extensive dataset of 2002 protein–ligand complexes from the PDBbind database (version 2014), the performance of ten docking programs, including five commercial programs (LigandFit, Glide, GOLD, MOE Dock, and Surflex-Dock) and five academic programs (AutoDock, AutoDock Vina, LeDock, rDock, and UCSF DOCK), was systematically evaluated by examining the accuracies of binding pose prediction (sampling power) and binding affinity estimation (scoring power). Our results showed that GOLD and LeDock had the best sampling power (GOLD: 59.8% accuracy for the top scored poses; LeDock: 80.8% accuracy for the best poses) and AutoDock Vina had the best scoring power (rp/rs of 0.564/0.580 and 0.569/0.584 for the top scored poses and best poses), suggesting that the commercial programs did not show the expected better performance than the academic ones. Overall, the ligand binding poses could be identified in most cases by the evaluated docking programs but the ranks of the binding affinities for the entire dataset could not be well predicted by most docking programs. However, for some types of protein families, relatively high linear correlations between docking scores and experimental binding affinities could be achieved. To our knowledge, this study has been the most extensive evaluation of popular molecular docking programs in the last five years. It is expected that our work can offer useful information for the successful application of these docking tools to different requirements and targets.
Introduction
As we know, lead identification is one of the most important and difficult steps in modern drug design and discovery.1,2 Thus, numerous methods and strategies have been developed and used to identify promising lead candidates for targets of interest.3–5 Among them, experimental high-throughput screening (HTS), the large-scale approach widely used since the early 1990s, has been out of favor due to its high cost and low hit rate.6,7 But, on the other hand, computational virtual high-throughput screening (vHTS) has attracted increasing attention in lead identification due to the advantages of high performance computing (HPC), highly optimized software and publicly accessible libraries of purchasable compounds.8–10 As the most important approach used in structure-based virtual screening, molecular docking can predict the binding mode and affinity of a ligand (usually a small organic molecule) within the binding pocket of the target of interest.11–16During the past two decades, a great variety of docking tools and programs, such as AutoDock,17 AutoDock Vina,18 LeDock,19 rDock,20 UCSF DOCK,21 LigandFit,22 Glide,23 GOLD,24 MOE Dock,25 and Surflex-Dock26, have been developed for both commercial and academic use.27–29 For a docking program, the two most critical components are the sampling algorithm and the scoring function, which determine its sampling power and scoring power, respectively. As far as we know, the popular sampling algorithms can be roughly divided into three categories: shape matching, systematic search (such as exhaustive search, fragmentation, and conformational ensemble), and stochastic search algorithms (such as Monte Carlo methods, genetic algorithms, Tabu search methods, and swarm optimization methods);30 and the popular scoring functions can be roughly grouped into three major classes: force field, empirical, and knowledge-based scoring functions.31–33 More recently, some quantum mechanical (QM) and semi-empirical QM (SQM) based scoring functions have been designed to capture the binding affinity trend and native pose identification.34,35 With the rapid development of computer hardware, the problem of sampling efficiency can be effectively or at least partially overcome, but it is still a huge challenge for available scoring functions to predict the binding affinities of diverse small molecules with high accuracy.36
Because different sampling strategies and scoring functions are employed by different docking programs, it is important to evaluate and compare the performance of these programs. The evaluation results can reveal the advantages and limitations of each docking program, which may help users to make reasonable choices among different docking programs. To date, a number of evaluation studies with the purpose of assessing the accuracy of different molecular docking programs and workflows have been reported.25,37–42 However, most important comparative studies providing evaluation benchmarks were published before 2011, and similar comparative studies in recent five years were quite limited. Previously in 2013, Damm-Ganamet et al. published a paper on the benchmark exercise from the Community Structure-Activity Resource (CSAR), which documented the evaluation of the results for binding pose, enrichment and relative ranking of blinded congeneric series submitted by 20 different research groups.43 Undoubtedly, the result of this work is very meaningful for the developer community, but it may be not quite useful for users as the software comparison is anonymous and most of the evaluated programs are customized versions or in-house programs that are not easily accessible. More recently, Tuccinardi and colleagues reported an extensive consensus docking reliability analysis by considering the consensus predictions of ten different docking procedures, and they found that consensus docking was able to not only predict the ligand binding pose better than any single docking program but also give hints concerning the reliability of the docking poses.44 With the rapid development of docking algorithms, many traditional docking programs have been updated and some new docking engines have been developed. However, the corresponding evaluation studies are antiquated and insufficient. Generally speaking, although a large number of comparative studies have been reported in the past two decades, it still remains difficult to determine which docking program is more suitable for specific targets. Therefore, extensive evaluation studies on the performance of popular docking programs and tools are still quite demanding.
In this study, we evaluated the capabilities of ten molecular docking programs to predict the ligand binding poses (sampling power) and rank the binding affinities (scoring power). The evaluated docking programs include five academic programs (AutoDock, AutoDock Vina, LeDock, rDock, and UCSF DOCK) and five commercial programs (LigandFit, Glide, GOLD, MOE Dock, and Surflex-Dock). The features of the evaluated docking programs are outlined in Table 1. Most commercial docking programs are quite expensive, and therefore it is expected that the commercial docking programs with stronger funding support may show better performance than the academic ones. According to our evaluation study, we want to answer the following question: do the commercial docking programs more dominant advantages than the academic ones? Among the evaluated programs, AutoDock, GOLD and Glide are the most commonly used docking programs by analyzing all docking publications from 1990 to 2013.27 Certainly, this does not mean that these three programs are more accurate than the other evaluated programs. According to our evaluation study, we want to answer the second question: do the more popular docking programs show better performance than the less popular ones? Meanwhile, two newly released docking programs, LeDock and rDock, were included in the evaluation study. Actually, we have tested a variety of new freely available programs in our routine docking test work, and the reason we selected LeDock and rDock rather than others is that they have relatively better accuracy and speed. Certainly, compared with other more traditional programs, these new programs may not be well validated and their performance is questionable. Therefore, according to our evaluation study, we want to answer the third question: do the traditional docking programs show better performance than the newly released ones?
| Program | Feature | Website |
|---|---|---|
| AutoDock17 | LGA-based docking software. Free for academic use. Maintained by the Molecular Graphics Laboratory, Scripps Research Institute, La Jolla. | http://autodock.scripps.edu/ |
| AutoDock Vina18 | AutoDock Vina employs an iterated local search global optimizer. Free for academic use. Maintained by the Molecular Graphics Laboratory, The Scripps Research Institute, La Jolla. | http://vina.scripps.edu/ |
| LeDock19 | LeDock is based on a combination of simulated annealing and evolutionary optimization of the ligand pose (position and orientation) and its rotatable bonds, using a physics/knowledge hybrid scoring scheme derived from prospective virtual screening campaigns. Free for academic use. Maintained by Lephar Research. | http://lephar.com/ |
| rDock20 | rDock is based on a combination of stochastic and deterministic search techniques (GA and MC) to generate low energy ligand poses. Free for academic use. Maintained by rDock Development Team | http://rdock.sourceforge.net/ |
| UCSF DOCK21 | Anchor-and-grow based docking program. Free for academic use. Maintained by the Shoichet group at the University of California San Francisco. | http://dock.compbio.ucsf.edu/ |
| LigandFit22 | Ligand conformations generated using Monte Carlo techniques are initially docked into an active site based on the shape, followed by further CHARMm minimization. Provided by Accelrys. | http://accelrys.com/ |
| Glide23 | Exhaustive search-based docking program. It has extra precision (XP), standard precision (SP) and high-throughput virtual screening (HTVS) scoring modes. Provided by Schrödinger. | http://www.schrodinger.com/ |
| GOLD24 | GA-based docking program. Product of collaboration between the University of Sheffield, GlaxoSmithKline, and the Cambridge Crystallographic Data Centre. | http://www.ccdc.cam.ac.uk/ |
| MOE Dock25 | MOE Dock supplies a database of conformations or generates conformations on the fly, and then refines the poses using a force field based method with MM/GBVI. Distributed by Chemical Computing Group. | http://www.chemcomp.com/ |
| Surflex-Dock26 | Docking program based on a “protomol” that can be automatically generated and/or user-defined. Poses are scored using an updated and re-parameterized empirical scoring function (based on the Hammerhead docking system). Distributed by Tripos. | http://www.tripos.com/ |
Materials and methods
Benchmark dataset
The benchmark dataset contains 2002 protein–ligand complexes with high resolution crystal structures and experimental binding affinity data that were selected from the refined set of PDBbind.45,46 The latest version of the PDBbind refined set (version 2014) has more than 3400 complexes. In order to avoid the failure of processing proteins with non-standard residues during docking calculations, all protein structures with any other hetero atoms, such as cofactors and metal ions, were filtered out automatically by an in-house script. Finally, a collection of 2002 protein–ligand complexes chosen from the PDBbind refined set was used for our evaluation study. The distributions of the experimental binding affinities and five molecular properties of the ligands included in the 2002 complexes are illustrated in Fig. 1. | ||
Fig. 1 Distributions of (A) experimental binding affinities and (B–F) five molecular properties, including the molecular weight, log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P, logS, topological polar surface area (TPSA) and the number of rotatable bonds, of the 2002 ligands in the dataset for the evaluation study. P, logS, topological polar surface area (TPSA) and the number of rotatable bonds, of the 2002 ligands in the dataset for the evaluation study. | ||
Structure preparation
In order to examine the robustness of the sampling algorithm implemented in each docking program, three different starting conformations (referred to as original, rotated and optimized, respectively) of each ligand were subsequently docked into the binding pocket of the corresponding target. The original conformation of each ligand is identical to that in the crystal structure of the complex, the rotated conformation was generated by rotating the original conformation on the Z-axis by 180°, and the optimized conformation was generated by optimizing the rotated conformation with the OPLS-2005 force field.47 The rotated and optimized conformations of each ligand were generated automatically by using an in-house python script developed based on the Python API available in Schrödinger Suite 2015 (Schrodinger, LLC, New York). The partial atomic charges of each original ligand structure were generated by using the AM1-BCC method implemented in Antechamber.48 For docking a program, which needs partial atomic charges but not support for calculating by itself, these types of charges should be used; otherwise, partial atomic charges were reassigned by the corresponding tool of the docking program. Hydrogens of each protein were added and the AMBER ff14SB partial charges were assigned by Chimera.49 For each complex, the processed protein structure was saved as a mol2-format file, and the processed ligand structure was saved as mol2-format and mol-format files.Molecular docking calculations
Ten docking programs employed in our benchmark study can be categorized into five academic programs, including AutoDock (version 4.2.6),17 AutoDock Vina (version 1.1.2),18 LeDock19 (version 1.0), rDock (version 2013.1)20 and UCSF DOCK (version 6.7),21 and five commercial programs, including LigandFit (version 2.4),22 Glide (version 67011),23 GOLD (version 5.2),24 MOE Dock (version 2014.0901)25 and Surflex-Dock (version 2.706.13302).26 In our benchmark study, the docking site of each target was determined on the basis of the position of the co-crystalized ligand within the active pocket. The maximum number of docking conformations for each ligand was set to 20 and a clustering distance cutoff was set to 0.5 Å in all docking programs. Other key parameters and scoring functions used in our study for individual software packages are described as follows:000 for LGA and PSO, respectively. In the LGA runs, the number of energy evaluations was set to 2500000, and that for the PSO runs was set to 250000. Docking scores were calculated by the default scoring function.
500120, 41200300, 61500350, 102000500, and 253000750”. LigScore1, LigScore2, PLP1, PLP2, Jain, and PMF scoring functions were selected for the docking score calculation.
000 GA operations. In our benchmark, “early termination” was turned on that means that GOLD will terminate docking runs on a given ligand as soon as a specified number of runs have given essentially the same answer. Docking poses were scored using only the ChemPLP function.
Assessment methods
The sampling algorithm and scoring function are two most important components for a docking program. In this study, the capability of each docking program to predict the ligand binding poses (sampling power) and rank the binding affinities (scoring power) was evaluated. In our study, RMSD (root mean square deviation) was used as the main parameter to evaluate the sampling power of each program. When the RMSD between the docked binding pose and the native binding pose is below 2.0 Å, the prediction was regarded to be successful. We checked not only the conformation with the highest docking score (referred to as the top scored pose) but also the conformation that is the closest to the native binding pose (referred to as the best pose). All RMSD values were calculated by using the “rmsd.py” script in Schrödinger.The scoring power, which represents the ability of a scoring function to rank the binding capabilities of studied molecules, was quantitatively evaluated by Pearson's correlation coefficient (rp) and Spearman's ranking coefficient (rs) between the docking scores and experimental binding data.
Protein classification
Due to the fact that any scoring function used in molecular docking cannot give reliable predictions for all protein families, the tested proteins were clustered into different protein families and the prediction accuracy for individual protein families were assessed. Here, we utilized SCOPe50 (Structural Classification of Proteins extended), an extended database of SCOP51 (Structural Classification of Proteins) that used a structural similarity-based algorithm to classify proteins into divers scaffolds, to achieve the clustering task. Finally, 1705 out of the 2002 tested proteins were successfully assigned to different protein families based on the indices of SCOPe (version 2.05).Results and discussion
Evaluation of sampling power on the entire dataset
In order to give a more realistic evaluation for the tested docking programs, the optimized conformations of all ligands were used as the starting structures for molecular docking. As a simple and effective method to evaluate the sampling power of a docking tool,8 the fraction of the complexes with the RMSD values between the predicted ligand binding poses and the native ligand binding poses lower than predefined thresholds was plotted and is shown in Fig. 2. The success rates (RMSD between the top scored pose and the native pose less than 2 Å) of all ten docking programs for the top scored and best poses are illustrated in Fig. 3A. Overall, if the optimized conformations of ligands were used as the input for molecular docking, the success rate for the top scored poses is about from 40% to 60%, and that for the best poses is about from 60% to 80%. On the basis of the results for the top scored poses, the performance of the academic programs follows the following order: LeDock (57.4%) > rDock (50.3%) ≈ AutoDock Vina (49.0%) > AutoDock (PSO) (47.3%) > UCSF DOCK (44.0%) > AutoDock (LGA) (37.4%), and that of the commercial programs follows the following order: GOLD (59.8%) > Glide (XP) (57.8%) > Glide (SP) (53.8%) > Surflex-Dock (53.2%) > LigandFit (46.1%) > MOE Dock (45.6%). The averaged success rates of the commercial docking programs for the top scored poses and best poses are 54.0% and 67.8%, respectively, and those of the academic programs for the top scored poses and best poses are 47.4% and 68.4%, respectively. That is to say, the capability of the commercial programs to predict ligand binding poses is slightly better than that of the academic programs from a global perspective, but the difference is not obvious. | ||
| Fig. 2 Cumulative distribution plots for docking of protein–ligand complexes. Top scored poses when the optimized ligands were used as the input from freely available programs (A) and commercially available programs (B), best poses when the optimized ligands were used as the input from freely available programs (C) and commercially available programs (D). Dotted lines indicate a 2.0 Å RMSD cutoff. | ||
 | ||
| Fig. 3 Success rates (A) and consistent rates (B) of ten docking programs in the docking power test. Optimized ligands were used as the input and 2.0 Å was used as the RMSD cutoff. | ||
Among the free docking tools, LeDock and rDock exhibited an eye-catching performance on ligand pose prediction, and LeDock is even better than most commercial programs. As the authors mentioned, a combination of simulated annealing (SA) and genetic algorithm (GA) is used by LeDock to optimize the position, orientation, and rotatable bonds of the docked ligand.52 SA and GA are two popular machine learning algorithms that have been widely employed by many docking programs. However, integrating such two algorithms in one tool is still very rare. Our results suggest that employing blended sampling algorithms may be an expedient strategy to improve the sampling power of a docking program. LeDock is a new molecular docking program, and we even could not find enough technical details of this docking algorithm. But from the results of the present study, it exhibits a high accuracy with good speed (slightly faster than AutoDock Vina) and is a recommended program for the virtual screening task.
In AutoDock, two sampling methods, including the Lamarckian Genetic Algorithm (LGA) and Particle Swarm Optimization (PSO), were utilized to optimize the binding poses of each ligand within the protein binding pocket. As illustrated in Fig. 2A and C, both the success rates for the top scored poses and best poses predicted by AutoDock (PSO) are obviously higher than those predicted by AutoDock (LGA). In addition, we also found that the speed of the PSO version is much faster than that of the LGA version. AutoDock Vina, the new generation of AutoDock, was also included in our evaluation. As shown in Table 1, the predictions of AutoDock Vina are slightly better than those of AutoDock (PSO), but substantially better than those of AutoDock (LGA). Compared with the report of AutoDock Vina developers, we could find that our evaluation results of AutoDock (LGA) and AutoDock Vina were consistent with their findings (only the LGA version of AutoDock was compared), i.e., AutoDock Vina significantly improved the average accuracy of the binding mode predictions compared to AutoDock.18
By comparing Fig. 2B and D, we found that the success rate of Surflex-Dock for the best poses was 80.0% but that for the top scored poses was much lower (53.2%). The huge gap between the prediction accuracies for the top scored poses and best poses reveals that the pose ranking capability of Surflex-Dock may be unsatisfactory and needs to be improved. Another unforeseen outcome is that the performance of Glide with the XP scoring mode on the best poses is even worse than that of Glide with the SP scoring mode. In our previous studies, we also observed that the XP scoring did not always perform better than the SP scoring for many systems.53,54 By analyzing the binding conformations generated by Glide (SP) and Glide (XP), we found that the cluster number of binding conformations provided by XP was generally less than that provided by SP; in other words, the docking poses from SP have more diversity than those from XP, which may partially account for its better performance on the best poses.
Although the overall success rates of the top scored poses and best poses (Fig. 3A) can help us to distinguish the sampling power of the tested programs, it is still not comprehensive enough. In real cases, e.g., in virtual screening studies, the top scored poses are generally considered to be the most reasonable binding structures. However, we found that there was a big difference between the success rates for the top scored poses and best poses, suggesting that the top scored poses are usually not the best (or native) poses, which is mainly caused by the drawbacks of scoring functions. It is reported that some SQM-based scoring function may be employed in the late stages of virtual screening to overcome this unbalance.34 Here, the consistent rate was used to assess the consistency between the predictions for the top scored poses and best poses. The consistent rate is defined as SRtsp/SRbp, where SRtsp and SRbp are the success rates for the top scored poses and best poses, respectively. As shown in Fig. 3B, the consistent rate of Glide (XP) and GOLD are up to 87.7% and 82.5%, respectively. To some extent, these two programs may be more suitable for a virtual screening study.
Then, we analyzed the failure cases with large prediction errors. We found that a total of 72 crystal structures could not be well predicted by any docking program (Table S1, ESI†). We believe that the following two reasons account for the unsuccessful docking. First, it can be found in Table S2 (ESI†) that about 82.0% (59/72) of ligands in failure cases are not neutral, which means nowadays docking methodologies are still not accurate enough to predict charged systems. Second, the large flexibility of ligands is another key factor leading to failure. As listed in Table S2 (ESI†), more than half (40/72) of ligands contain over than 10 rotatable bonds.
Influence of ligand flexibility on sampling power
The number of rotatable bonds of a ligand is directly related to the flexibility of this ligand, which has a critical influence on the conformation sampling performance of a docking program. As far as we know, there are still no similar benchmarking studies on the sampling power according to the number of rotatable bonds of ligands based on such an extensive dataset. Fig. 4 shows that whether for the top scored poses or best poses, the success rates of most docking programs dropped significantly when the numbers of rotatable bonds of ligands are higher than 20. On the other hand, it has been reported that the rotatable bond counts of most drugs and drug-like compounds were less than 10.55 The data shown in Fig. S1 (ESI†) illustrate that more than 90.0% of 1790 drugs approved by FDA possess fewer than 10 rotatable bonds. Therefore, it is more valuable to assess the performance of the tested docking programs on the ligands with the rotatable bond counts less than 10. As shown in Fig. 4, LeDock, rDock, Glide (SP), Glide (XP) and GOLD exhibit better performance on the top scored poses, and LeDock, rDock, Glide (SP) and Surflex-Dock have relatively better performance on the best poses. | ||
| Fig. 4 Heat map of the success rates of docking for ligands with different numbers of rotatable bonds. (A) Top scored poses and (B) best poses. A docking pose is considered successful if RMSD between the docking pose and the experimentally conformation of the ligand is less than 2.0 Å. | ||
In the PBDbind refined set database some ligands are small peptides or peptide mimics. The properties of peptides or peptide mimics are more similar to those of proteins, e.g., higher molecular weights and more rotatable bonds. Generally, a peptide or peptide mimic ligand is more difficult to be docked successfully. In order to conduct further investigation on the performances of the tested programs for peptides or peptide mimics, the whole dataset was separated into two groups: regular organic molecule ligands and peptide or peptide mimic ligands. The numbers of regular organic molecule ligands and peptide or peptide mimic ligands are 1843 and 159, respectively. The success rates of the two types of ligands are summarized in Table 2. As we expected, the predictions for organic ligands are significantly better than those for peptides or peptide mimic ligands for all docking programs. It is notable that for peptides or peptide mimic ligands Surflex-Dock achieves the success rates of 44.0% and 67.3% for the top scored poses and best poses, respectively.
| Docking program | Regular organic molecule | Peptide or peptide mimic | ||
|---|---|---|---|---|
| Top scored pose | Best pose | Top scored pose | Best pose | |
| AutoDock (LGA) | 0.378 | 0.559 | 0.216 | 0.324 |
| AutoDock (PSO) | 0.477 | 0.686 | 0.331 | 0.439 |
| AutoDock Vina | 0.485 | 0.726 | 0.384 | 0.597 |
| LeDock | 0.574 | 0.808 | 0.352 | 0.465 |
| rDock | 0.503 | 0.763 | 0.283 | 0.465 |
| UCSF DOCK | 0.445 | 0.591 | 0.340 | 0.415 |
| LigandFit | 0.479 | 0.689 | 0.267 | 0.504 |
| Glide (SP) | 0.544 | 0.754 | 0.403 | 0.547 |
| Glide (XP) | 0.584 | 0.666 | 0.403 | 0.484 |
| GOLD | 0.599 | 0.726 | 0.371 | 0.472 |
| MOE Dock | 0.457 | 0.612 | 0.195 | 0.245 |
| Surflex-Dock | 0.533 | 0.800 | 0.440 | 0.673 |
Impact of ligand starting conformations on sampling power
It has been reported that the results of docking calculations are very sensitive to the starting geometries of ligands.56 When the starting geometry of the input ligand is similar to the native binding structure of the ligand, the better binding poses may be predicted by molecular docking.57 However, as a robust docking program, the docked binding poses produced from different input conformations of a ligand should be similar. In order to examine the sensitivity of each docking program to starting conformations, three different starting conformations (original, rotated and optimized conformations) of each ligand were subsequently docked. The corresponding dockings were referred to as original docking, rotated docking and optimized docking, respectively.As shown in Fig. 5, the cumulative distributions based on different starting conformations of the docking results of AutoDock, AutoDock Vina, LigandFit and GOLD have relatively large fluctuations, suggesting that these several docking programs are relatively more sensitive to the initial structures of ligands. We found that the pose prediction accuracies of original docking and rotated docking (input ligand structure has memory of the crystallized conformation) were generally better than those of optimized docking (input ligand structure ‘forgets’ the crystallized conformation) for most docking programs. This is consistent with the results reported by Onodera and colleagues, i.e., if the input ligand structure is similar to the native one, the better poses are usually predicted by docking programs.57 Among the tested docking programs, LeDock, rDock, UCSF DOCK, Glide, MOE Dock and Surflex-Dock are not sensitive to the starting conformations of ligands, and that is to say, the sampling algorithms implemented in these several docking programs are quite robust.
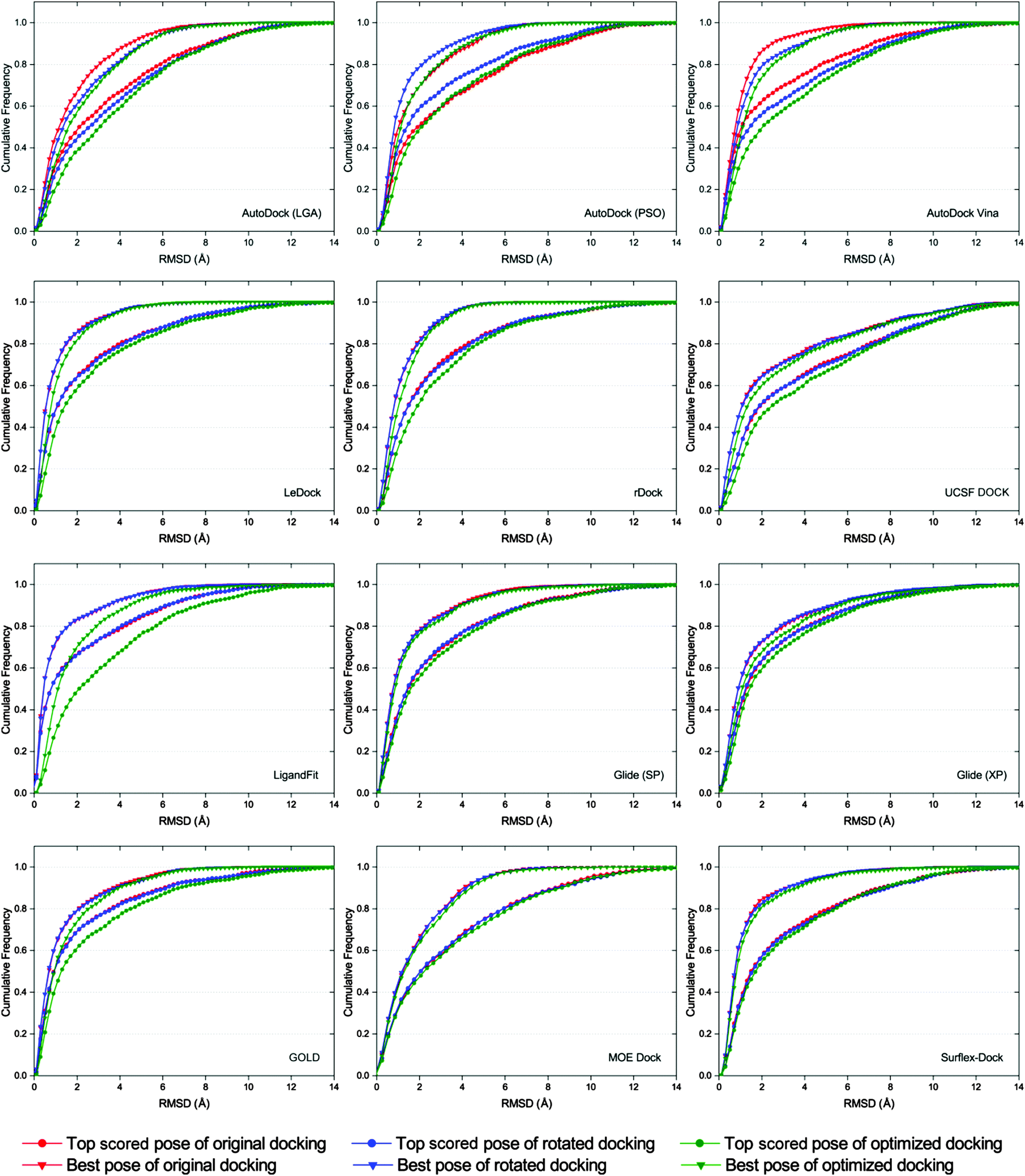 | ||
| Fig. 5 Comparison of the top scored pose and best pose cumulative distribution of docking using different starting conformations. | ||
Evaluation of scoring power on the entire dataset
Besides sampling power, the capability of a docking program to rank the binding affinities of different ligands (scoring power) is also another important issue for a docking program because the appraisal and ranking of predicted ligand conformations is a decisive step for docking-based virtual screening. Generally, scoring power is defined as the prediction accuracy of a scoring function implemented in a docking program to rank the binding capabilities of a series of protein–ligand complexes. Usually, multiple scoring functions are integrated in the same docking program to meet the requirement of different precisions and computational cost. The scoring function we evaluated for each program is described in the Method section. Top scored poses and best poses are two different types of “correct” conformations under different circumstances, and thus, the scoring power of each program was evaluated based on both of them.The Pearson correlation coefficient (rp) and Spearman ranking coefficient (rs) between the docking scores and experimental binding affinities for the entire test set are summarized in Table 3. The docking program with the best scoring power is AutoDock Vina, which produced rp (rs) of 0.564 (0.580) and 0.569 (0.584) for the top scored poses and best poses, respectively. The next two top-ranked docking tools are MOE Dock and GOLD, which gave rp (rs) of 0.564 (0.589) and 0.500 (0.511) for the top scored poses, respectively, and rp (rs) of 0.411 (0.457) and 0.494 (0.513) for the best poses, respectively. Unexpectedly, we found that there is no obvious difference in the scoring powers between the top scored poses and best poses for most docking programs, except MOE Dock. On the whole, the scoring powers of the tested docking programs on the entire test set are not quite satisfactory. Based on the Spearman ranking coefficients for the top scored poses, the performance of the academic programs can be ordered in the following way: AutoDock Vina (0.580) > AutoDock (PSO) (0.534) > LeDock (0.462) > UCSF DOCK (0.331) > rDock (0.017) and that of the commercial programs can be ordered in the following way: MOE Dock (0.589) > GOLD (0.515) > Glide (0.473) > Surflex-Dock (0.370) > LigandFit (0.221). Overall, compared with the academic programs, the commercial programs do not have improved capability to rank the binding affinities for a diverse dataset. Moreover, it seems that the good performance of a scoring function to identify correct binding poses cannot guarantee the good performance of this function to rank binding affinities. For example, rDock has relatively good sampling power, but its ranking power is quite low; GOLD has the best sampling power for the top scored poses, but its ranking power for the top scored poses is not the best. Apparently, there was no single docking program that outperformed all others in both sampling power and scoring power. Therefore, the best solution for docking-based virtual screening would be the combination of different docking tools into a single platform, which could be benefited from the advantages of different approaches. For example, we can use LeDock to virtually screen the chemical database, and then use AutoDock Vina or MOE Dock to rescore the top scored poses predicted by LeDock.
| Docking program | Correlation coefficient | Top scored pose | Best pose |
|---|---|---|---|
| a r p represents Pearson's correlation coefficient. b r s represents Spearman's ranking coefficient. c The standard error was estimated by randomly sampling 80% of the tested dataset 100 repeats. | |||
| AutoDock (LGA) | r p | 0.433 ± 0.009c | 0.404 ± 0.009 |
| r s | 0.477 ± 0.008 | 0.450 ± 0.009 | |
| AutoDock (PSO) | r p | 0.492 ± 0.008 | 0.466 ± 0.008 |
| r s | 0.534 ± 0.007 | 0.513 ± 0.008 | |
| AutoDock Vina | r p | 0.564 ± 0.008 | 0.569 ± 0.008 |
| r s | 0.580 ± 0.008 | 0.584 ± 0.008 | |
| LeDock | r p | 0.442 ± 0.009 | 0.463 ± 0.009 |
| r s | 0.462 ± 0.010 | 0.486 ± 0.009 | |
| rDock | r p | −0.015 ± 0.011 | −0.021 ± 0.011 |
| r s | −0.017 ± 0.011 | −0.005 ± 0.011 | |
| UCSF DOCK | r p | 0.291 ± 0.010 | 0.276 ± 0.011 |
| r s | 0.331 ± 0.011 | 0.323 ± 0.011 | |
| LigandFit | r p | −0.132 ± 0.011 | −0.105 ± 0.011 |
| r s | −0.221 ± 0.012 | −0.192 ± 0.012 | |
| Glide (SP) | r p | 0.444 ± 0.008 | 0.402 ± 0.009 |
| r s | 0.473 ± 0.009 | 0.419 ± 0.010 | |
| Glide (XP) | r p | 0.367 ± 0.010 | 0.356 ± 0.010 |
| r s | 0.389 ± 0.010 | 0.374 ± 0.010 | |
| GOLD | r p | −0.500 ± 0.008 | −0.494 ± 0.008 |
| r s | −0.515 ± 0.008 | −0.513 ± 0.008 | |
| MOE Dock | r p | 0.564 ± 0.008 | 0.411 ± 0.009 |
| r s | 0.589 ± 0.009 | 0.457 ± 0.009 | |
| Surflex-Dock | r p | −0.340 ± 0.009 | −0.350 ± 0.009 |
| r s | −0.370 ± 0.009 | −0.382 ± 0.009 | |
Performance of scoring power on different protein families
Given the uneven performances of scoring functions on different kinds of protein targets, we classified the proteins into different families based on the indices of SCOPe and then the scoring powers of the tested programs on different protein families were evaluated. In order to ensure the statistical significance of the test, only six protein families with over 50 members were selected for the further study. The correlation coefficients (rp and rs) of the docking scores and experimental binding affinities on different protein families, which are a.123.1.1 (nuclear receptor ligand-binding domain), b.47.1.2 (eukaryotic proteases), b.50.1.1 (retroviral protease), b.50.1.2 (pepsin-like), c.94.1.1 (phosphate binding protein-like) and d.144.1.7 (protein kinases, catalytic subunit), are illustrated in Fig. 6. It can be found that the scoring powers for most families are much better than those for the whole dataset (Table 3).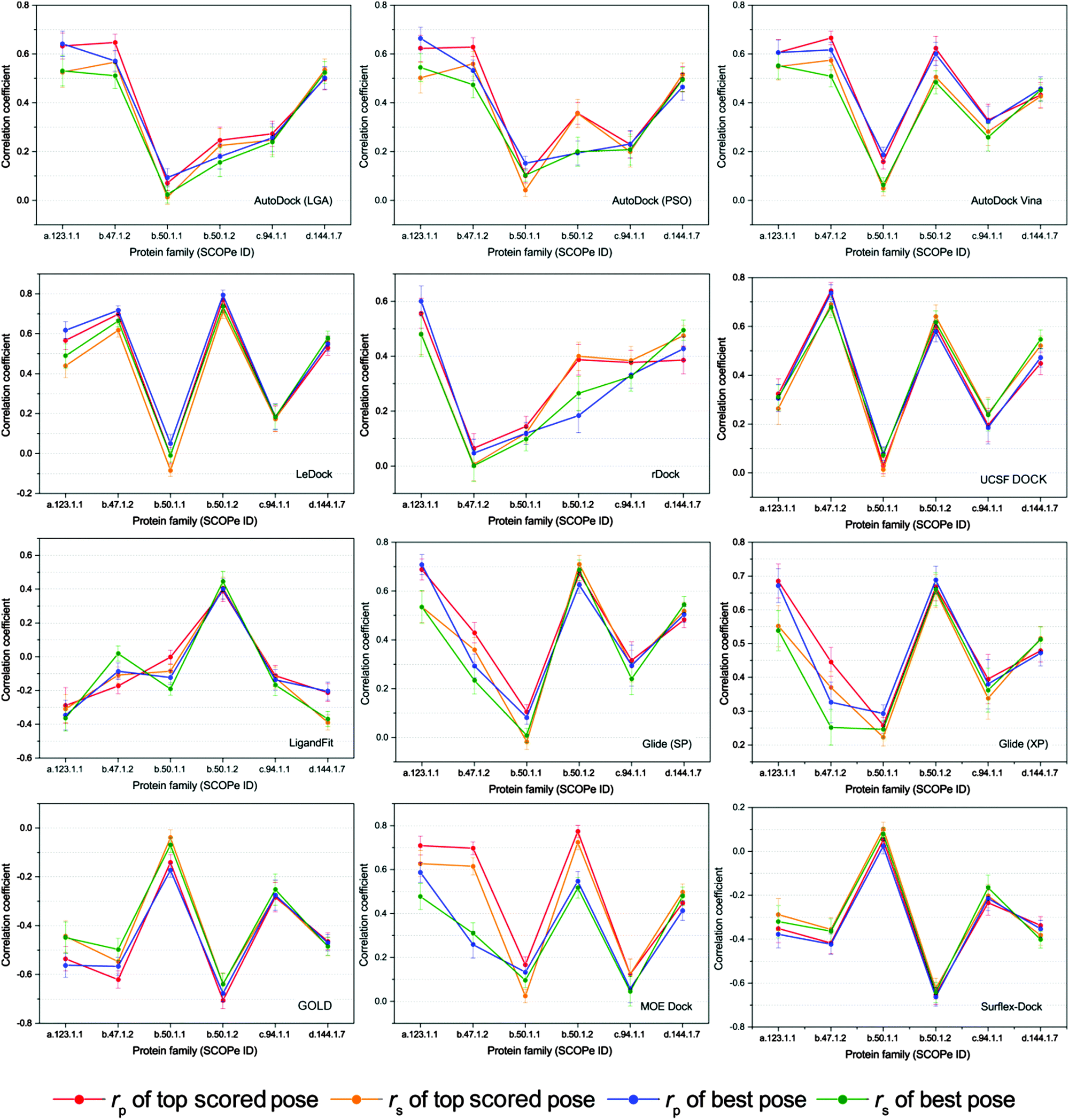 | ||
| Fig. 6 Scoring power of all tested docking tools for the protein families with more than 50 members. The total number of a.123.1.1, b.47.1.2, b.50.1.1, b.50.1.2, c.94.1.1, and d.144.1.7 is 51, 86, 263, 61, 68, and 113, respectively. rp represents Pearson's correlation coefficient and rs represents Spearman's ranking coefficient. The standard error was estimated by randomly sampling 80% of the tested dataset 50 times. | ||
The scoring power of the same docking tool on different protein families is widely divergent, for example, the Pearson correlation coefficients of LeDock to the groups of b.47.1.2 and b.50.1.2 for the top scored poses are 0.698 and 0.770, respectively, while those to b.50.1.1 and c.94.1.1 are only −0.010 and 0.176, respectively. On the other hand, the performances of different docking programs on the same protein family are also intermingled. As shown in Fig. 6, the Pearson correlation coefficients of AutoDock, rDock, and LigandFit to the group of b.50.1.2 for both top scored poses and best poses are less than 0.500, while those of LeDock, Glide (XP), GOLD, and Surflex-Dock are around 0.700 or −0.700. This result fully explains the importance of selecting the right program. Exceptionally, we found that the performances of all investigated programs to the subset of b.50.1.1, the largest group, were much worse than the others. Actually, the proteins in the family b.50.1.1 are HIV proteases, and several reports have pointed out that both the lack of consideration of the entropic term in the scoring function and the narrow distribution of the experimental binding free energies may contribute to the low correlation.58–60
Conclusions
In this study, based on an extensive benchmark dataset with 2002 complexes, the sampling power and scoring power of ten docking programs were evaluated. GOLD and LeDock had the best capabilities to identify the correct ligand binding poses (GOLD: 59.8% accuracy for the top scored poses; LeDock: 80.8% prediction accuracy for the best poses). Among the ten tested programs, five of them can achieve 50.0–60.0% accuracies for pose predictions. It is notable that Glide (XP) and GOLD are the two most robust programs on pose predictions and they possess the consistent rates of nearly 90.0%. Therefore, overall, the ligand binding poses could be identified in most cases by the evaluated docking programs.Among the tested programs, three of them, including AutoDock Vina, GOLD and MOE Dock, achieved the best scoring powers, with rp/rs of 0.564/0.580, 0.500/0.515 and 0.569/0.589 for the top scored poses, respectively. However, the relatively weak correlation between the docking scores and experimental binding affinities for the entire dataset indicates that current scoring functions are still not reliable and universal enough. Evaluation of the scoring powers on different protein families illustrates that the scoring powers of the same docking tool on different protein families are quite different (rp from 0.000 to 0.800) and therefore different docking programs may be used for different protein families.
Our evaluation results illustrate that no single docking program has dominative advantages than other programs. The combination of different docking tools into a single platform may be a practical method to achieve better predictions for docking-based virtual screening. To sum up, we made an updated comprehensive docking benchmark with emphasis on sampling power and scoring power, and we expect our work could provide new useful reference for people to select the most appropriate docking program for their projects.
Acknowledgements
Hongtao Zhao (Lephar Research, Rindögatan 21, 11558 Stockholm, Sweden) is thanked for his helpful discussion and critical reading of the manuscript. This study was supported by the National Science Foundation of China (21575128).References
- A. M. Davis, D. J. Keeling, J. Steele, N. P. Tomkinson and A. C. Tinker, Curr. Top. Med. Chem., 2005, 5, 421–439 CrossRef CAS PubMed.
- V. Schnecke and J. Bostrom, Drug Discovery Today, 2006, 11, 43–50 CrossRef CAS PubMed.
- A. Golebiowski, S. R. Klopfenstein and D. E. Portlock, Curr. Opin. Chem. Biol., 2001, 5, 273–284 CrossRef CAS PubMed.
- A. Golebiowski, S. R. Klopfenstein and D. E. Portlock, Curr. Opin. Chem. Biol., 2003, 7, 308–325 CrossRef CAS PubMed.
- T. Honma, Med. Res. Rev., 2003, 23, 606–632 CrossRef CAS PubMed.
- R. Lahana, Drug Discovery Today, 1999, 4, 447–448 CrossRef PubMed.
- W. L. Jorgensen, Science, 2004, 303, 1813–1818 CrossRef CAS PubMed.
- J. B. Cross, D. C. Thompson, B. K. Rai, J. C. Baber, K. Y. Fan, Y. B. Hu and C. Humblet, J. Chem. Inf. Model., 2009, 49, 1455–1474 CrossRef CAS PubMed.
- J. J. Irwin and B. K. Shoichet, J. Chem. Inf. Model., 2005, 45, 177–182 CrossRef CAS PubMed.
- A. Lavecchia and C. Di Giovanni, Curr. Med. Chem., 2013, 20, 2839–2860 CrossRef CAS PubMed.
- I. D. Kuntz, Science, 1992, 257, 1078–1082 CAS.
- B. K. Shoichet, Nature, 2004, 432, 862–865 CrossRef CAS PubMed.
- A. R. Leach, B. K. Shoichet and C. E. Peishoff, J. Med. Chem., 2006, 49, 5851–5855 CrossRef CAS PubMed.
- E. Yuriev, J. Holien and P. A. Ramsland, J. Mol. Recognit., 2015, 28, 581–604 CrossRef CAS PubMed.
- E. Yuriev and P. A. Ramsland, J. Mol. Recognit., 2013, 26, 215–239 CrossRef CAS PubMed.
- L. S. Azevedo, F. P. Moraes, M. M. Xavier, E. O. Pantoja, B. Villavicencio, J. A. Finck, A. M. Proenca, K. B. Rocha and W. F. de Azevedo, Curr. Bioinf., 2012, 7, 352–365 CrossRef CAS.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- H. Zhao and A. Caflisch, Bioorg. Med. Chem. Lett., 2013, 23, 5721–5726 CrossRef CAS PubMed.
- S. Ruiz-Carmona, D. Alvarez-Garcia, N. Foloppe, A. B. Garmendia-Doval, S. Juhos, P. Schmidtke, X. Barril, R. E. Hubbard and S. D. Morley, PLoS Comput. Biol., 2014, 10, e1003571 Search PubMed.
- W. J. Allen, T. E. Balius, S. Mukherjee, S. R. Brozell, D. T. Moustakas, P. T. Lang, D. A. Case, I. D. Kuntz and R. C. Rizzo, J. Comput. Chem., 2015, 36, 1132–1156 CrossRef CAS PubMed.
- C. M. Venkatachalam, X. Jiang, T. Oldfield and M. Waldman, J. Mol. Graphics Modell., 2003, 21, 289–307 CrossRef CAS PubMed.
- R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic, D. T. Mainz, M. P. Repasky, E. H. Knoll, M. Shelley, J. K. Perry, D. E. Shaw, P. Francis and P. S. Shenkin, J. Med. Chem., 2004, 47, 1739–1749 CrossRef CAS PubMed.
- G. Jones, P. Willett, R. C. Glen, A. R. Leach and R. Taylor, J. Mol. Biol., 1997, 267, 727–748 CrossRef CAS PubMed.
- C. R. Corbeil, C. I. Williams and P. Labute, J. Comput.-Aided Mol. Des., 2012, 26, 775–786 CrossRef CAS PubMed.
- A. N. Jain, J. Med. Chem., 2003, 46, 499–511 CrossRef CAS PubMed.
- Y. C. Chen, Trends Pharmacol. Sci., 2015, 36, 78–95 CrossRef CAS PubMed.
- M. Bello, M. Martinez-Archundia and J. Correa-Basurto, Expert Opin. Drug Discovery, 2013, 8, 821–834 CrossRef CAS PubMed.
- S. F. Sousa, A. J. Ribeiro, J. T. Coimbra, R. P. Neves, S. A. Martins, N. S. Moorthy, P. A. Fernandes and M. J. Ramos, Curr. Med. Chem., 2013, 20, 2296–2314 CrossRef CAS PubMed.
- S. Y. Huang and X. Zou, Int. J. Mol. Sci., 2010, 11, 3016–3034 CrossRef CAS PubMed.
- S. Y. Huang, S. Z. Grinter and X. Q. Zou, Phys. Chem. Chem. Phys., 2010, 12, 12899–12908 RSC.
- H. Gohlke, M. Hendlich and G. Klebe, J. Mol. Biol., 2000, 295, 337–356 CrossRef CAS PubMed.
- T. Schulz-Gasch and M. Stahl, Drug Discovery Today: Technol., 2004, 1, 231–239 CrossRef CAS PubMed.
- A. Pecina, R. Meier, J. Fanfrlik, M. Lepsik, J. Rezac, P. Hobza and C. Baldauf, Chem. Commun., 2016, 52, 3312–3315 RSC.
- K. Raha and K. M. Merz, Jr., J. Med. Chem., 2005, 48, 4558–4575 CrossRef CAS PubMed.
- N. Moitessier, P. Englebienne, D. Lee, J. Lawandi and C. R. Corbeil, Br. J. Pharmacol., 2008, 153, S7–S26 CrossRef CAS PubMed.
- C. Bissantz, G. Folkers and D. Rognan, J. Med. Chem., 2000, 43, 4759–4767 CrossRef CAS PubMed.
- E. Kellenberger, J. Rodrigo, P. Muller and D. Rognan, Proteins, 2004, 57, 225–242 CrossRef CAS PubMed.
- S. M. Vogel, M. R. Bauer and F. M. Boeckler, J. Chem. Inf. Model., 2011, 51, 2650–2665 CrossRef CAS PubMed.
- Y. Li, L. Han, Z. Liu and R. Wang, J. Chem. Inf. Model., 2014, 54, 1717–1736 CrossRef CAS PubMed.
- Y. Li, Z. Liu, J. Li, L. Han, J. Liu, Z. Zhao and R. Wang, J. Chem. Inf. Model., 2014, 54, 1700–1716 CrossRef CAS PubMed.
- G. M. Sastry, M. Adzhigirey, T. Day, R. Annabhimoju and W. Sherman, J. Comput.-Aided Mol. Des., 2013, 27, 221–234 CrossRef PubMed.
- K. L. Damm-Ganamet, R. D. Smith, J. B. Dunbar, J. A. Stuckey and H. A. Carlson, J. Chem. Inf. Model., 2013, 53, 1853–1870 CrossRef CAS PubMed.
- T. Tuccinardi, G. Poli, V. Romboli, A. Giordano and A. Martinelli, J. Chem. Inf. Model., 2014, 54, 2980–2986 CrossRef CAS PubMed.
- Z. Liu, Y. Li, L. Han, J. Li, J. Liu, Z. Zhao, W. Nie, Y. Liu and R. Wang, Bioinformatics, 2015, 31, 405–412 CrossRef PubMed.
- R. Wang, X. Fang, Y. Lu and S. Wang, J. Med. Chem., 2004, 47, 2977–2980 CrossRef CAS PubMed.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- J. Wang, W. Wang, P. A. Kollman and D. A. Case, J. Mol. Graphics Modell., 2006, 25, 247–260 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- N. K. Fox, S. E. Brenner and J. M. Chandonia, Nucleic Acids Res., 2014, 42, D304–D309 CrossRef CAS PubMed.
- A. G. Murzin, S. E. Brenner, T. Hubbard and C. Chothia, J. Mol. Biol., 1995, 247, 536–540 CAS.
- H. T. Zhao and A. Caflisch, Bioorg. Med. Chem. Lett., 2013, 23, 5721–5726 CrossRef CAS PubMed.
- S. Tian, H. Sun, Y. Li, P. Pan, D. Li and T. Hou, J. Chem. Inf. Model., 2013, 53, 2743–2756 CrossRef CAS PubMed.
- S. Tian, H. Sun, P. Pan, D. Li, X. Zhen, Y. Li and T. Hou, J. Chem. Inf. Model., 2014, 54, 2664–2679 CrossRef CAS PubMed.
- S. Ekins, J. Bradford, K. Dole, A. Spektor, K. Gregory, D. Blondeau, M. Hohman and B. A. Bunin, Mol. BioSyst., 2010, 6, 840–851 RSC.
- M. Feher and C. I. Williams, J. Chem. Inf. Model., 2009, 49, 1704–1714 CrossRef CAS PubMed.
- K. Onodera, K. Satou and H. Hirota, J. Chem. Inf. Model., 2007, 47, 1609–1618 CrossRef CAS PubMed.
- V. Lafont, A. A. Armstrong, H. Ohtaka, Y. Kiso, L. Mario Amzel and E. Freire, Chem. Biol. Drug Des., 2007, 69, 413–422 CAS.
- H. Sun, Y. Li, S. Tian, L. Xu and T. Hou, Phys. Chem. Chem. Phys., 2014, 16, 16719–16729 RSC.
- A. Weis, K. Katebzadeh, P. Soderhjelm, I. Nilsson and U. Ryde, J. Med. Chem., 2006, 49, 6596–6606 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1: distributions of five properties of 1790 FDA approved drugs; Table S1: the unsuccessful docking instances of the individual tested docking program in the benchmark; and Table S2: the formal charge and the number of rotatable bonds of ligands for the 72 failure cases that could not be successfully predicted by any docking program. See DOI: 10.1039/c6cp01555g |
| This journal is © the Owner Societies 2016 |