
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design and characterisation of bodipy sensitizers for dye-sensitized NiO solar cells†
Gareth H.
Summers‡
ab,
Jean-François
Lefebvre‡
a,
Fiona A.
Black
b,
E.
Stephen Davies
a,
Elizabeth A.
Gibson
*ab,
Tönu
Pullerits
c,
Christopher J.
Wood
a and
Karel
Zidek
c
aSchool of Inorganic Chemistry, The University of Nottingham, University Park, Nottingham, NG7 2RD, UK
bSchool of Chemistry, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK. E-mail: Elizabeth.gibson@ncl.ac.uk
cDepartment of Chemical Physics, Lund University, Box 124, 22241, Lund, Sweden. E-mail: Tonu.Pullerits@chemphys.lu.se
First published on 27th November 2015
Abstract
A series of photosensitizers for NiO-based dye-sensitized solar cells is presented. Three model compounds containing a triphenylamine donor appended to a boron dipyrromethene (bodipy) chromophore have been successfully prepared and characterised using emission spectroscopy, electrochemistry and spectroelectrochemistry, to ultimately direct the design of dyes with more complex structures. Carboxylic acid anchoring groups and thiophene spacers were appended to the model compounds to provide five dyes which were adsorbed onto NiO and integrated into dye-sensitized solar cells. Solar cells incorporating the simple Bodipy-CO2H dye were surprisingly promising relative to the more complex dye 4. Cell performances were improved with dyes which had increased electronic communication between the donor and acceptor, achieved by incorporating a less hindered bodipy moiety. Further increases in performances were obtained from dyes which contained a thiophene spacer. Thus, the best performance was obtained for 7 which generated a very promising photocurrent density of 5.87 mA cm−2 and an IPCE of 53%. Spectroelectrochemistry combined with time-resolved transient absorption spectroscopy were used to determine the identity and lifetime of excited state species. Short-lived (ps) transients were recorded for 4, 5 and 7 which are consistent with previous studies. Despite a longer lived (25 ns) charge-separated state for 6/NiO, there was no increase in the photocurrent generated by the corresponding solar cell.
Introduction
Dye-sensitized photocathodes offer a versatile means of converting sunlight into electricity or fuel by reducing the redox couple in tandem dye-sensitized solar cells, or protons in water to produce hydrogen in photoelectrochemical devices.1–4 These systems have garnered less interest compared to photoanodic systems, such as the conventional dye-sensitized solar cell pioneered by Grätzel. However, substituting the platinum electrode in a TiO2-based solar cell with a photocathode could provide a route to increasing the efficiency.5 The theoretical maximum efficiency of a single junction solar cell has been calculated to be 33.4% under AM1.5 conditions but this can be increased to 45.7% by stacking two photoelectrodes in one device.6 A more realistic maximum efficiency for DSCs of 20% has been proposed, but after 25 years of research, only 12% has been achieved.7,8 Matching the output from a state-of-the art TiO2 photoanode with a dye-sensitized photocathode could lead to a step change in efficiency that would make tandem DSCs competitive with thin film devices. In order to assemble a working tandem cell, the currents on both photoelectrodes must be matched and the spectral properties of the dyes must complement each other. Typically the photocathodic current densities produced from sensitized NiO electrodes are less than half of the photoanodic currents produced by a dye-sensitized TiO2 electrode.8–10 To improve the performance of the working cell, dye regeneration must be able to compete with charge recombination. Therefore, the photoinduced charge-separated state must be sufficiently long-lived (μs) and unfortunately for most organic dyes adsorbed on NiO the charge-separated state lifetime is generally shorter (ca. 10−7 s). This makes the photocathodes unsuitable for use with iodine-free electrolytes such as cobalt-based systems, which avoid the overpotential wasted in the dye regeneration step that limits the efficiency of devices using the iodide/triiodide redox couple.11,12 Thus, there are a number of important considerations regarding the design of photosensitizers for NiO and the best used to date contain a combination of key features of those dyes which have generated either high photovoltages or high currents in NiO-based dye-sensitized solar cells. For example, Spiccia et al. reported record efficiency NiO solar cells based on Co(en)33+/2+ (1.3%) and Fe(acac)33+/2+ (2.51%) using a perylene-thiophene-triphenylamine sensitizer (“PMI-6T-TPA”).13,14 Detailed reviews on the performance and limitations of p-type DSCs can be found in ref. 1, 2 and 33.Based on the features of the best performing dyes, we have designed new molecular sensitizers which exhibit high extinction coefficients, donor–acceptor character and strong anchoring to the NiO surface (based on the general structure in Fig. 1). This article describes the electrochemical and photochemical properties of the model structures (1–3) and corresponding dyes (Bodipy-CO2H, 4–7) (Fig. 2). We have chosen bodipy-based acceptor groups due to the ease of functionality and typically high extinction coefficients of bodipy compounds.15–17 A triphenylamine motif with a carboxylic acid group as the electron donating anchoring group has been used since these have been shown to bind to NiO and perform well in push–pull photosensitizers.18–21 In order to assess the impact of the distance between the bodipy and the NiO surface on the lifetime of the photoreduced bodipy and the device performance π-spacers were incorporated into the design. Previously we reported the solar cell efficiency of dyes 6 and 7 which contain thiophene groups between the amine and the bodipy. In this study we have removed the thiophene and introduced phenyl groups between the benzoic acid and the amine in 4 and 5.8,22 To determine whether the rotation of the bodipy around the C–C bond between the bodipy motif and the aniline affected the electronic properties of the dye and the device performance, methyl substituents were incorporated into dyes 4 and 6 but not into dyes 5 and 7. To assess the effect of these modifications on the charge transfer dynamics at the dye/NiO interface we carried out transient absorption spectroscopy and emission experiments on the dyed films and compared them to our previous results for 6.22
 | ||
| Fig. 1 Illustration of the principle components of the dyes designed and investigated in this work. | ||
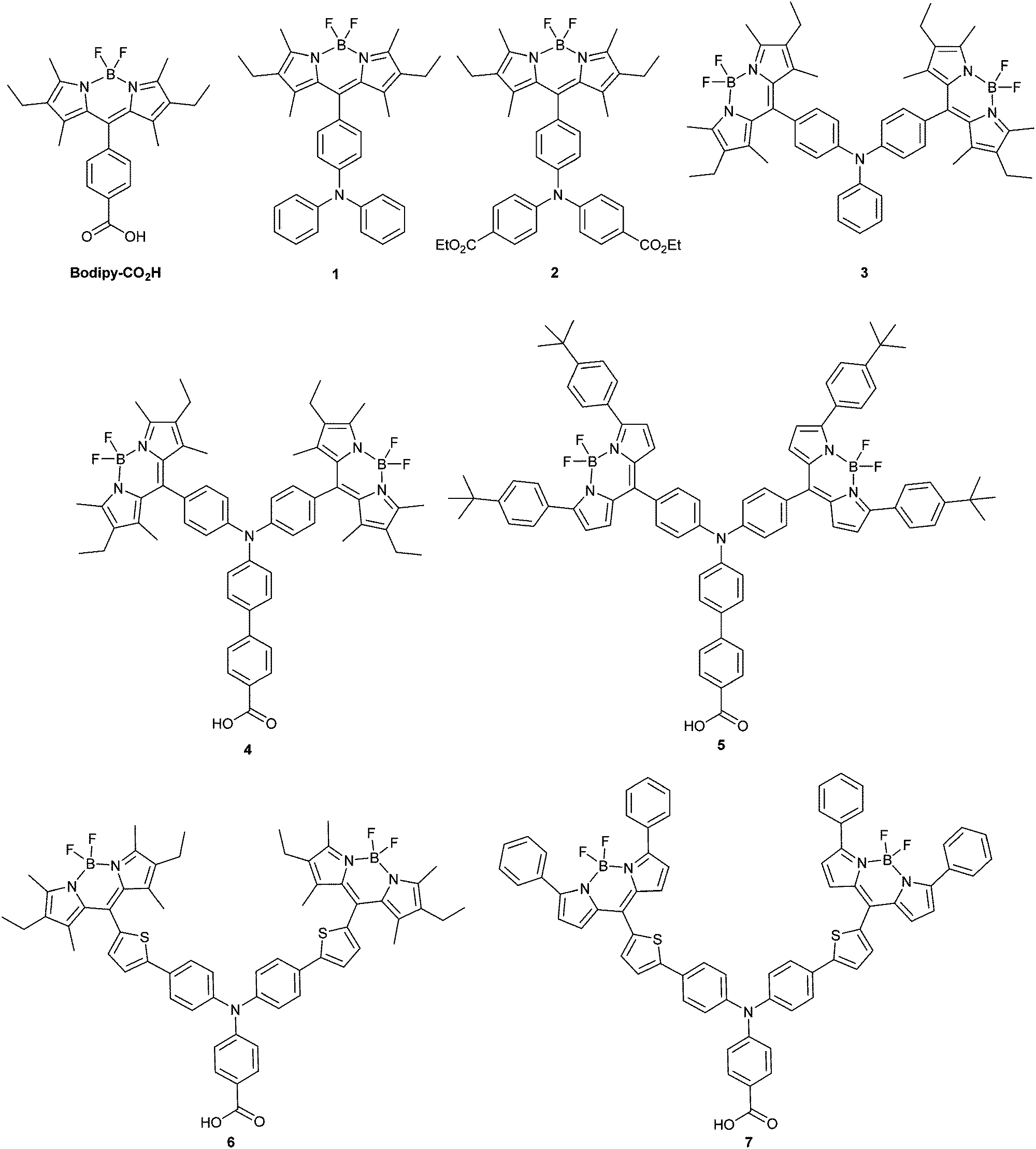 | ||
| Fig. 2 Dyes 1–7 and 4,4-difluoro-8-(4-carboxyphenyl)-1,3,5,7-tetramethyl-2,6-diethyl-3a,4a-diaza-4-bora-s-indacene (Bodipy-CO2H) investigated as NiO photosensitizers in this work. | ||
Results
Synthesis
Details regarding the synthesis of the bodipy molecules is provided in the ESI.†1–4 (Fig. 2) were prepared by reacting 2,4-dimethyl-3-ethylpyrrole with the formyltriphenylamine derivatives, followed by the coordination to –BF2. 5 was prepared similarly by reacting 2-(4-(tert-butyl)phenyl)-pyrrole with the formyltriphenylamine derivatives, followed by the coordination to –BF2. The identity of the products were determined by NMR spectroscopy (see ESI† for assigned spectra), HR ESI-MS and elemental analysis. The synthesis of 6 and 7 has been reported previously.8,224-Formyltriphenylamine and 4,4′-diformyltriphenylamine were prepared using the Vilsmeier–Haack reaction in 79% and 11% yield respectively, by tuning the ratio of triphenylamine![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :POCl3:DMF and the reaction time. The products were separated using column chromatography and reacted with 2,4-dimethyl-3-ethylpyrrole to give the mono and bis-dipyrromethane derivatives. These were then converted to 1 and 3 by the one-pot sequential oxidation with p-chloranil followed by reaction with boron trifluoride etherate in the presence of a base (N,N-diisopropylethylamine) with reasonable yields (50% and 23%).
:POCl3:DMF and the reaction time. The products were separated using column chromatography and reacted with 2,4-dimethyl-3-ethylpyrrole to give the mono and bis-dipyrromethane derivatives. These were then converted to 1 and 3 by the one-pot sequential oxidation with p-chloranil followed by reaction with boron trifluoride etherate in the presence of a base (N,N-diisopropylethylamine) with reasonable yields (50% and 23%).
Dimethyl-4,4′-[(4-formylphenyl)amino]bis[benzoate] was prepared by performing a second Vilsmeier–Haack reaction on dimethyl-4,4′-(phenylamino)bis[benzoate] in 19% yield on account of the lower reactivity of the deactivated aniline derivative. 2 was then prepared in 23% yield from dimethyl-4,4′-[(4-formylphenyl)amino]bis[benzoate] using the same procedure as for 1 and 3. The presence of the ester groups was confirmed by a peak in the IR absorption spectrum in acetonitrile at 1740 cm−1 corresponding to the stretching frequency of the ester carbonyl. The chemical shift of the protons closest to the ester were shifted downfield from δ 7.35–7.30 for 1 to δ 7.98 for 2.
The distance between the amine and the carboxylic acid was extended for 4 and 5 by coupling 4,4′-[(4-bromophenyl)amino]bis-[benzaldehyde] to 4-carboxyphenylboronic acid using a palladium catalysed Suzuki coupling. This was then converted to the dye using the same procedure as for 1–3.
Dye-sensitized solar cells
p-DSCs were constructed for the dyes containing acid anchoring groups (4–7 and Bodipy-CO2H). The relevant parameters are shown in Table 1 and the photocurrent density–voltage curves are provided in Fig. 3. Dark current data is provided in Fig. S85 (ESI†).| Dye | V oc (mV) | J sc (mA cm−2) | FFa | η (%) | IPCEb (%) | APCEb (%) |
|---|---|---|---|---|---|---|
| a J sc is the short-circuit current density at the V = 0 intercept, Voc is the open-circuit voltage at the J = 0 intercept, FF is the device fill factor, η is the power conversion efficiency. b IPCE is the monochromatic incident photon-to-current conversion efficiency; APCE is calculated using APCE = IPCE/LHE where LHE = 1 × 10−A where A is the absorptivity of the dye-sensitized NiO at λmax. | ||||||
| Bodipy-CO2H | 95 ± 7.5 | 1.48 ± 0.03 | 0.36 ± 0.01 | 0.05 ± 0.005 | 20 ± 0.5 | 27 ± 1.0 |
| 4 | 97 ± 0.5 | 1.60 ± 0.07 | 0.38 ± 0.005 | 0.06 ± 0.005 | 27 ± 0.5 | 30 ± 0.5 |
| 5 | 109 ± 1.5 | 3.70 ± 0.17 | 0.35 ± 0.005 | 0.14 ± 0.005 | 44 ± 3.0 | 45 ± 3.0 |
| 6 | 95 ± 3.0 | 1.58 ± 0.22 | 0.35 ± 0.01 | 0.05 ± 0.005 | 23 ± 4.0 | 24 ± 4.0 |
| 7 | 106 | 5.87 | 0.31 | 0.20 | 53 | 53 |
 | ||
| Fig. 3 Current–voltage characteristics of the dye-sensitized NiO solar cells with 0.1 M I2, 1.0 M LiI electrolyte and 4 (red), 5 (purple), 6 (orange), 7 (blue) and Bodipy-CO2H (green) under AM 1.5G (100 mW cm−2) illumination. | ||
Modest results were obtained for devices made from the simple molecule Bodipy-CO2H which performed better than expected relative to the more synthetically challenging dye 4. In general, dyes containing bodipy groups which were less hindered around the β-positions (5 and 7) outperformed dyes with more restricted rotation (Bodipy-CO2H, 4 and 6). Substituting the more sterically hindered pyrroles of 4 for those of 5 leads to devices more than twice as efficient. We have attributed this increase in overall performance to the increased electronic communication between the donor and acceptor groups. A further increase in performance was achieved by the incorporation of a thiophene spacer (7). Comparing this result to the similarly unrestricted 5 highlights an increase in performance when the thiophene spacer is present. Dye 6 was previously reported22 with an electrolyte consisting of 1 M LiI and 0.5 M I2 (compared to 1 M LiI and 0.1 M I2 as used here) with results of Jsc = 3.15 mA cm−2, Voc = 79 mV, FF = 0.31, η = 0.08%, indicating an increase in the obtained photocurrent but a reduction of the photovoltage as the iodine concentration of the electrolyte increases.
The maximum IPCE for NiO solar cells with dyes 4 and 5 (Fig. 4) increases from 27 to 44% respectively. This is accompanied by a broadening of the spectral response, with dye 5 exhibiting the maximum photocurrent between ca. 480 and 600 nm compared to ca. 480 to 540 nm for dye 4. None of the dyes gave any reasonable photocurrent with the Co(II/III) tris(4,4′-ditert-butyl-2,2′-dipyridyl) perchlorate electrolyte.
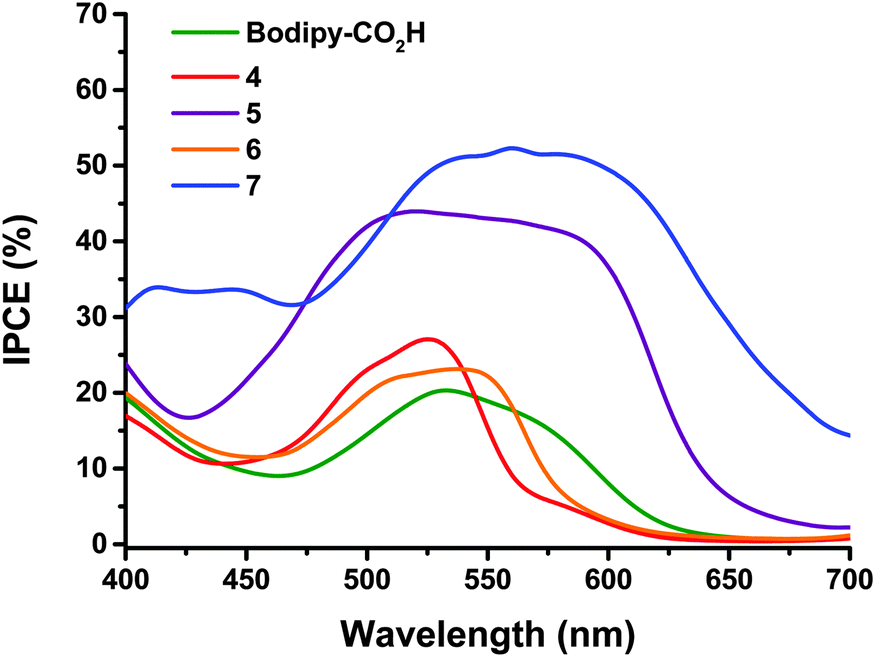 | ||
| Fig. 4 Comparison of IPCE spectra of the dye-sensitized NiO solar cells with 0.1 M I2, 1.0 M LiI electrolyte and 4 (red), 5 (purple), 6 (orange), 7 (blue) and Bodipy-CO2H (green). | ||
Steady state spectroscopic studies and excited state lifetimes
The UV-visible absorption and emission properties are listed in Table 2 and plotted in Fig. 5. For 1–4, the lowest energy absorption band (S0 to S1) has a maximum at 525–526 nm with a higher energy vibronic shoulder at ca. 30 nm from the main peak. The additional phenyl linker between the benzoic acid anchoring group and the triphenylamine in 4 did not affect the band at 525 nm but the absorption spectra contained an additional broad band at 360 nm. Adding a thiophene bridge between the amine and the bodipy to give 6 led to a 10 nm bathochromic shift in the absorption and emission spectra. Substituting the alkyl substituents around the pyrrole in 4 with a tertiary-butyl phenyl substituent in 5 lead to bathochromic shift of 14 nm in the absorption spectrum and 21 nm in the emission spectrum. Likewise the absorption and emission spectra of 7 were shifted by 25 nm and 48 nm respectively compared to 6. To summarise, the aniline auxochrome shifts the bodipy absorption maximum to the red when either a thiophene π-spacer is introduced or the methyl substituents at positions one and seven on the bodipy are absent.| Dye | λ abs/nm (ε/dm3 mol−1 cm−1) | λ em/nm | τ/ns | Φ | E 0–0 /eV | E g pred.c/eV |
|---|---|---|---|---|---|---|
| a Emission quantum yield (Φ) measurements were carried out in dichloromethane and compared to rhodamine 6G in ethanol (0.94). b Obtained from the intercept of the normalized absorption and emission spectra plotted in the ESI. c Obtained from TD-DFT calculations; HOMO values are with respect to vacuum. d Non-emissive in dichloromethane. | ||||||
| 1 | 525 (53800), 495 (sh, 11150) |
542 | 5.73 | — | 2.32 | 2.77 |
| 2 | 526 (53200), 497 (sh, 17900) |
542 | 6.65 | — | 2.32 | 2.77 |
| 3 | 525 (78800), 494 (sh, 25000) |
542 | 6.20 | — | 2.32 | 2.77 |
| 4 | 526 (81700), 360 (23900), 330 (19600) |
539 | 5.8 | 0.38 | 2.33 | 2.85 |
| 5 | 561 (65824), 351 (28237), 308 (41488) |
600 | 2.5 | 0.15 | 2.14 | 2.52 |
| 6 | 540 (112000), 507 (sh, 43000), 412 (sh, 16000), 365 (56000) |
560 | <1 | 0.03 | 2.27 | 2.84 |
| 7 | 565 (65700), 358 (32554) |
608 | <1 | —d | 2.11 | 2.26 |
| Bodipy-CO2H | 528 (52000) |
547 | 3.3 | — | 2.31 | 2.73 |
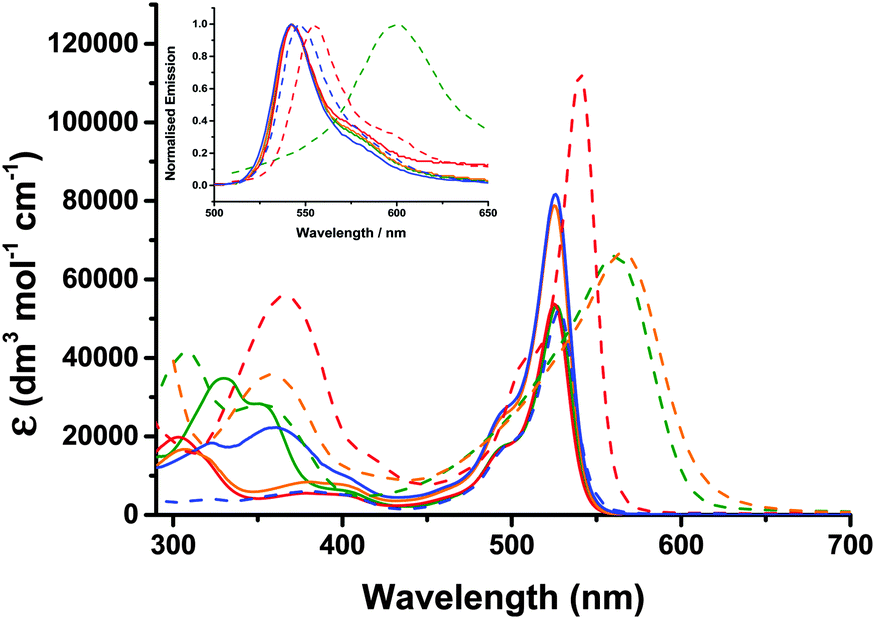 | ||
| Fig. 5 Plot of absorption and emission (inset) spectra for 1 (sold green line), 2 (solid red line), 3 (solid orange line), 4 (solid blue line), 5 (dashed green line), 6 (dashed red line), 7 (dashed orange line) and Bodipy-CO2H (dashed blue line) (λex = 500 nm) measured in dichloromethane. | ||
Appending a second dipyrromethene had no effect on the absorption maximum of 3 compared to 1 but did significantly increase the absorption coefficient of the dye from 53800 (1) to 78800 dm3 mol−1 cm−1 (3). We attribute this hyperchromic affect to the increased probability of the π–π* electronic transition when two bodipy moieties are present in one molecule. The absorption coefficient of 6 (112000 L mol−1 cm−1) is significantly higher than the molar absorption coefficient of 3 and 4 which both contain two bodipy chromophores. A bathochromic and hyperchromic shift is characteristic of increasing the conjugation of the chromophore. The shift between 4 and 6 may result from the smaller thiophene being less effected than phenyl by the steric hindrance of the 1,7 methyl substituents on the bodipy. By comparison, the absorption coefficient of 5 and 7 are similar, possibly because the rotation around the bodipy/thiophene (5) or bodipy/phenyl (7) is unhindered. The absorption spectrum of both 5 and 7 were significantly broadened compared to 4 and 6 but the absorption coefficients were much lower. We attribute the lowering of the absorption coefficient to a switch from a bodipy-localised π–π* transition (4 and 6) to a charge-transfer transition (5 and 7) from the amine to the bodipy due to the free rotation around the C–C bond between the bodipy and the thiophene or phenyl moiety.
The presence of the electron withdrawing ester groups on the triphenylamine in 3 had very little effect on the absorption wavelength or extinction coefficient, which is consistent with the DFT calculations (see below). For Bodipy-CO2H, the absorption and the emission maxima were red-shifted by 3 nm and 5 nm respectively relative to 1 and 2. The solvent dependence for Bodipy-CO2H was similar to the triphenyl-amine–bodipy conjugates. A bathochromic shift (7 nm for the absorption maximum and 14 nm for the emission maximum), rather than a slight hypsochromic shift, was observed when the solvent was switched from acetonitrile to dichloromethane.
Substantial fluorescence was observed in dichloromethane solution for 1–4 and 6 at room temperature (λex = 500 nm) and the spectra were mirror images of the S0 → S1 transition in the absorption spectra as expected (Fig. 5 (inset)). The wavelength at maximum emission was 542 nm for 1–3 and 539 nm for 4 in dichloromethane. The small stokes shift (17 nm) is indicative of little reorganisation in geometry between the ground and excited states. While 5 was more emissive in dichloromethane compared to acetonitrile and acetone (ϕ = 0.02 in acetonitrile, ϕ = 0.03 in acetone, ϕ = 0.15 in dichloromethane), the quantum yields were much lower than 4 (ϕ = 0.38 in dichloromethane). Excitation of the higher energy band of 5 (at 400 nm) led to very weak emission from the bodipy in acetone and tetrahydrofuran, no emission in acetonitrile and weak, dual emission bands centred at ca. 500 nm and 600 nm in dichloromethane. 7 was non-emissive in most solvents, displaying only weak emission in acetone and chloroform, as reported previously.
Kollmannsberger and coworkers observed quenching of a dimethylaniline-bodipy derivative in acetonitrile and restoration of the fluorescence on protonation.23 In their studies, emission from a charge-transfer band at ca. 600 nm was observed when diethyl ether was chosen as the solvent. We observed a very weak band at 700 nm (Fig. S47, ESI†) which was absent in the dichloromethane spectrum and could be due a small amount of charge transfer or intersystem crossing to the triplet state.24 Phosphorescence is rarely observed from bodipy chromophores because of the slow rate of intersystem crossing (106 s−1) in the absence of the heavy atom effect.16,25
The effect of solvent on the electronic spectra of 4–7 is consistent with the trend in increasing communication between the amine and the bodipy as (a) a thiophene spacer is introduced and (b) the methyl substituents at positions one and seven are removed. For 4 (and 1–3), only a slight shift in absorption maximum with solvent polarity (Fig. S36–S49, ESI†) was observed, which is not indicative of donor–acceptor interactions in the ground state. The emission quantum yield for 4 was Φ = 0.38 in dichloromethane, which is approximately half that reported elsewhere for 1,3,5,7-tetramethyl-2,6-diethyl-bodipy,17,26 and in acetonitrile the emission was quenched (Φ = 0.02). There was a 4 nm hypsochromic shift for 6 on switching the solvent from dichloromethane to acetonitrile but the molar absorption coefficient was the same in both solvents. In contrast, the emission was strongly solvent dependent (Fig. S48, ESI†). The Stokes-shift decreased with increasing polarity CH2Cl2 (16 nm) > THF (14 nm) > acetone (8 nm). Unlike the thiophene-free molecules, the emission spectrum of 6 was solvent dependent. When 6 was excited in the higher energy absorption band, the emission was similar in shape to 1–4 with the maximum at 560 nm. When 6 was excited in the higher energy absorption band (406 nm) it emitted at higher energy in CH3CN solution (475 nm) than when in CH2Cl2 (560 nm). According to DFT calculations (see below) the higher energy band is a triphenylamine-based transition and the lower energy band is a bodipy-based transition. TDDFT calculations (Fig. S28–S31, ESI†) are in agreement with the stabilisation of a charge transfer transition in acetonitrile compared to in dichloromethane where the bodipy π–π* transition is more favourable.
The excited state lifetimes for 1–4 were approximately 6 ns, which is characteristic of a bodipy singlet excited state.27 The excited state lifetimes measured using single photon counting (λex = 460 nm) decreased as the polarity of the solvent increased: dichloromethane (5.6 ns) > THF (4.6 ns) > acetone (3.7 ns) > acetonitrile (0.96 ns). The emission lifetime (λex = 400 nm) was slightly shorter for Bodipy-CO2H than 1 and 2 and was almost unaffected by the polarity of the solvent (a slight decrease was observed with decreasing polarity), whereas for 1 and 3 the emission lifetimes slightly increased. In dichloromethane solution, the emission lifetime of 5 was shorter than 4 and was shorter than the resolution of the instrument for the thiophene-containing dyes 6 and 7.
Density functional theory
To aid interpretation of the optical and electrochemical properties, DFT was used to predict the optimised geometry and the relative frontier orbital energies of Bodipy-CO2H and 1–7 (Fig. 6 and 7 and Fig. S23–S35, ESI†). The minimised structures are similar for each triphenylamine–bodipy conjugate: the triphenylamine geometry is trigonal planar; the planar motif contains a tetrahedral boron centre coordinated to two orthogonal fluorine atoms and each molecule has the aminophenyl spacer perpendicular to the plane of the dipyrromethene (Table S2, ESI†). Likewise, the calculated optimised geometry of 6 placed the bodipy at slightly smaller angles of 84°, 85° to the thiophene compared to 85°, 89° to the phenyl for 4. However, the dihedral angle between the aminophenyl spacer and the plane of the dipyrromethene is much smaller (52°, 54°) for 5 compared to 4. Likewise the dihedral angle between the thiophene spacer and the plane of the dipyrromethene is much smaller (44°, 48°) for 7 compared to 6. The increased ability to rotate around the bond between the bodipy and the spacer in 5 and 7 is consistent with the optical properties described above. These results suggest that the electronic coupling between the amine donor and bodipy chromophore increases when thiophene is introduced between the bodipy chromophore and the aniline donor and in the absence of methyl substituents at positions one and seven on the bodipy.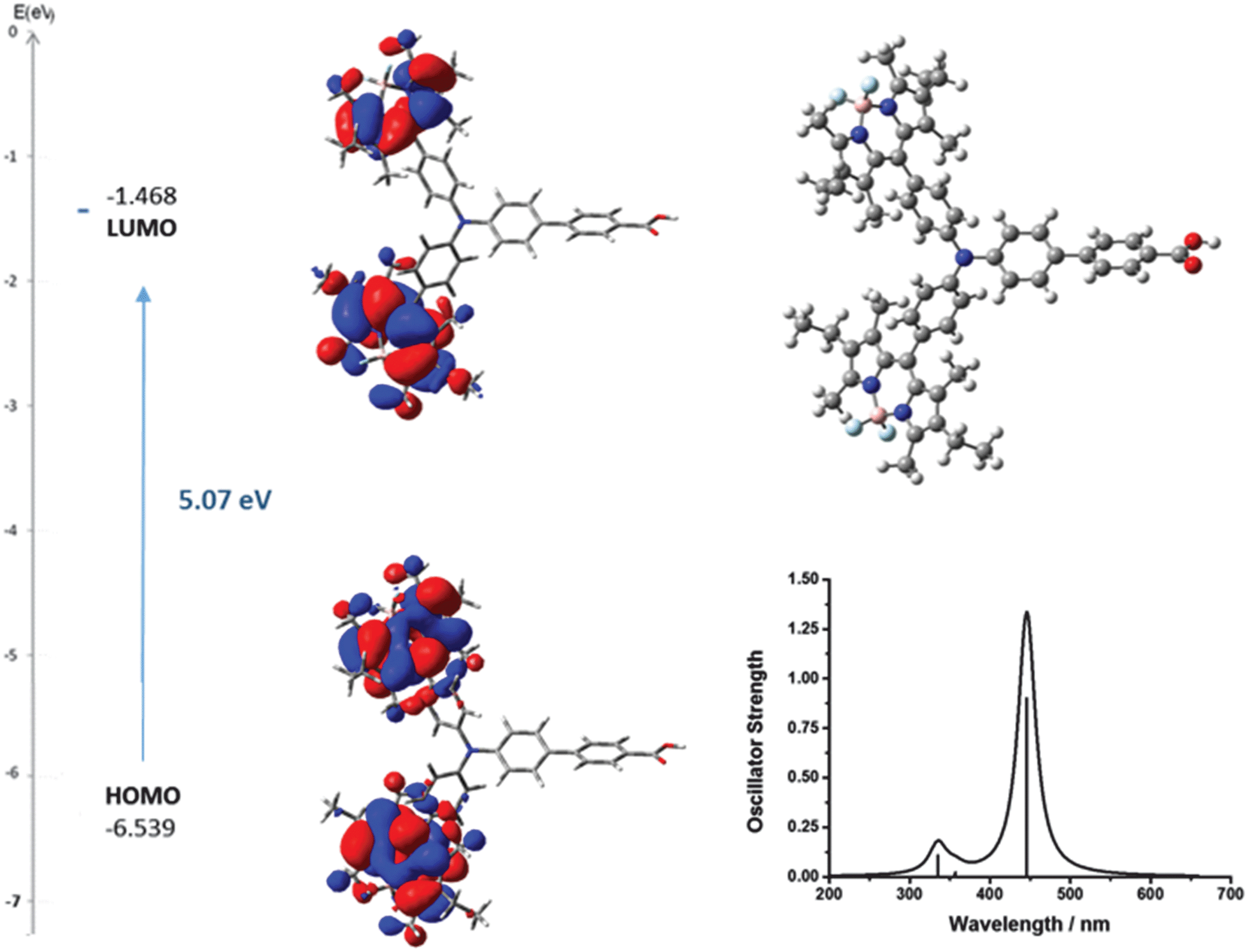 | ||
| Fig. 6 Optimised geometry and calculated principle energy transitions for 4 determined by DFT and TD-DFT (in dichloromethane solvent) using CAM-B3LYP/6-31G(d). | ||
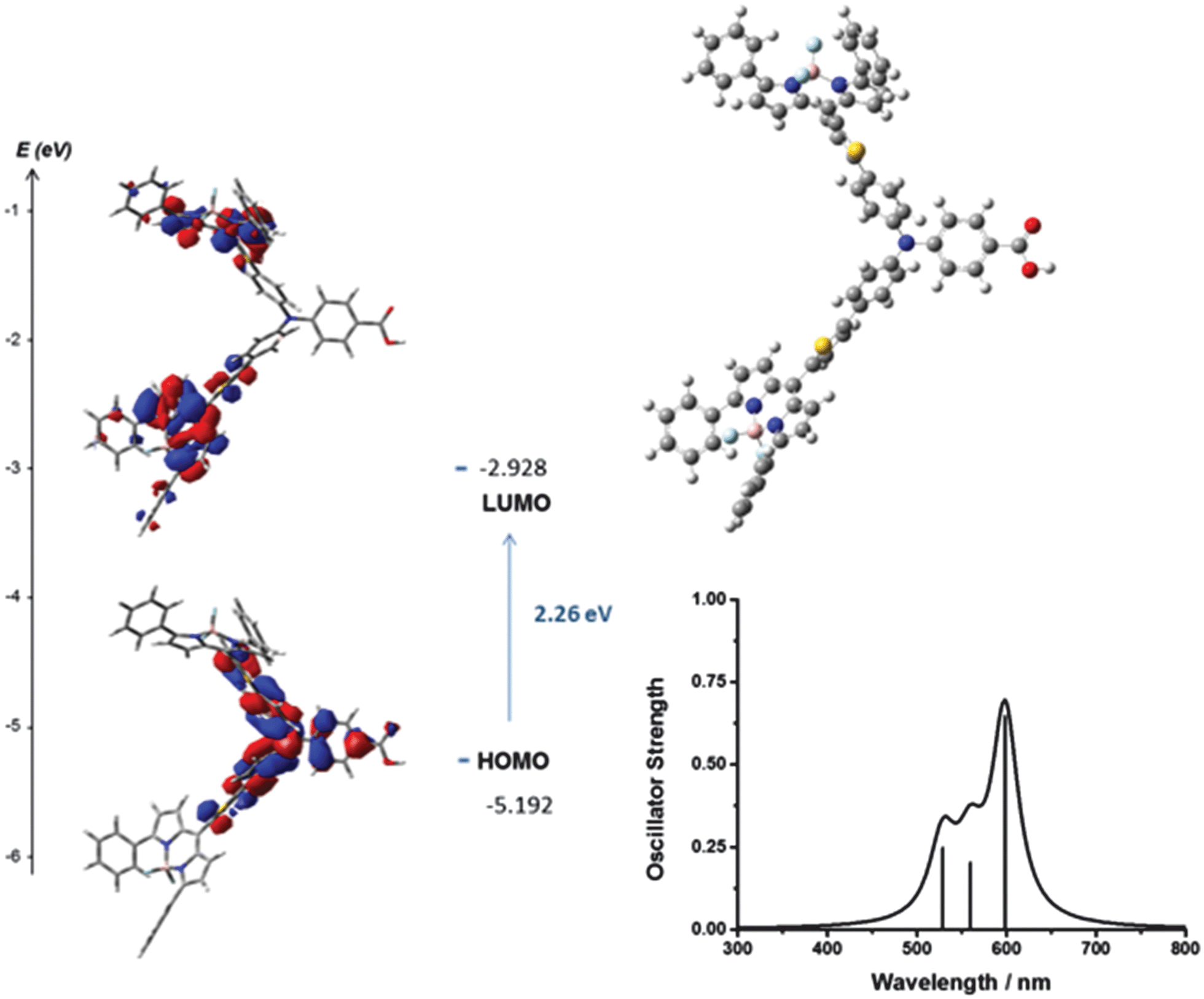 | ||
| Fig. 7 Optimised geometry and calculated principle energy transitions for 5 determined by DFT and TD-DFT (in dichloromethane solvent) using B3LYP/6-31G(d). | ||
Fig. S23–S35 (ESI†) show the electronic distribution in the frontier orbitals of 1–7. For 4 the HOMO/HOMO−1 and LUMO/LUMO+1 orbitals are localised on the dipyrromethene moieties while the HOMO−2 is localised on triphenylamine. In 5–7 the HOMO is localised on the triphenylamine donor-benzoic acid anchor (including the thiophene in 6 and 7). The HOMO−1 and HOMO−2 (degenerate in energy, shown in Fig. S24, ESI†) are localised on the dipyrromethene moieties. The LUMO is also delocalised across the dipyrromethene motifs, with 5 also having electron density on two out of three phenyl groups on the amine donor. The LUMO and LUMO+1 of 6 and 7 differ from 4 and 5 by the presence of electron density on the thiophene as well as the bodipy.
The selected major transitions, associated oscillator strengths and simulated electronic spectra are provided in Fig. 6 for 4, Fig. 7 for 5 and are presented in Table S3 and Fig. S23 and S24 (ESI†) for 1–3. The vertical excitation energies calculated from the optimised geometries using time-dependent DFT (TD-DFT) were in good agreement with the experimental values (above).
The lowest energy singlet excitations appear at 445 and 456 nm for 4 which correspond with a HOMO → LUMO bodipy localised π–π* transition (with a combined oscillator strength of 1.32) while for 5 these appear at 592 and 554 nm which correspond to HOMO → LUMO and HOMO → LUMO+1 transitions which involve charge-transfer from the triphenylamine to the bodipy. The higher energy transition for 5 corresponds with the HOMO−1 → LUMO transition, which is primarily localised on the bodipy and therefore has π–π* character.
The lowest energy singlet excitations calculated for 6 and 7 (calculated using B3LYP) were also charge-transfer in nature (from the triphenylamine-based HOMO – bodipy-based LUMO or LUMO+1) but for 6 the oscillator strength was very low (0.0066), with similar results observed for 4 calculated using B3LYP. As this was inconsistent with the high absorption coefficient of these dyes, we also carried out calculations using CAM-B3LYP for 6 and 7 (Fig. S30 and S32b, ESI†) which predicted the lowest energy transitions for 6 (458–459 nm) to be HOMO−1 → LUMO which is π–π* in character with a larger oscillator strength (total of 1.319), which fit more accurately with experimental observations. For 7 the lowest energy excitation (495 nm) was HOMO−1 → LUMO which is again bodipy based with high energy excitations (446 and 432 nm) corresponding to HOMO → LUMO transitions which have charge transfer character, however a closer agreement of spectral shape and excitation energy was seen for the B3LYP calculations. An overestimation of the excitation energies compared to the experimental values is observed for the TD-DFT calculations, which is consistent with calculations on similar molecules (ca. 0.3 eV for B3LYP and 0.5 eV for CAM-B3LYP).28
Electrochemical studies
The cyclic voltammograms for compounds 1–3 and 4–5 are shown in Fig. S77 (ESI†) and Fig. 8 respectively and Table 3 summarises the electrochemical data. In order to obtain a peak separation for the reversible waves of 59 mV, as expected for Nernstian behaviour, a strong concentration of supporting electrolyte (0.5 M) was required because of the low dielectric constant of the solvent. The voltammograms of the dyes contained reversible reduction and irreversible oxidation waves when scanning at 0.2 V s−1.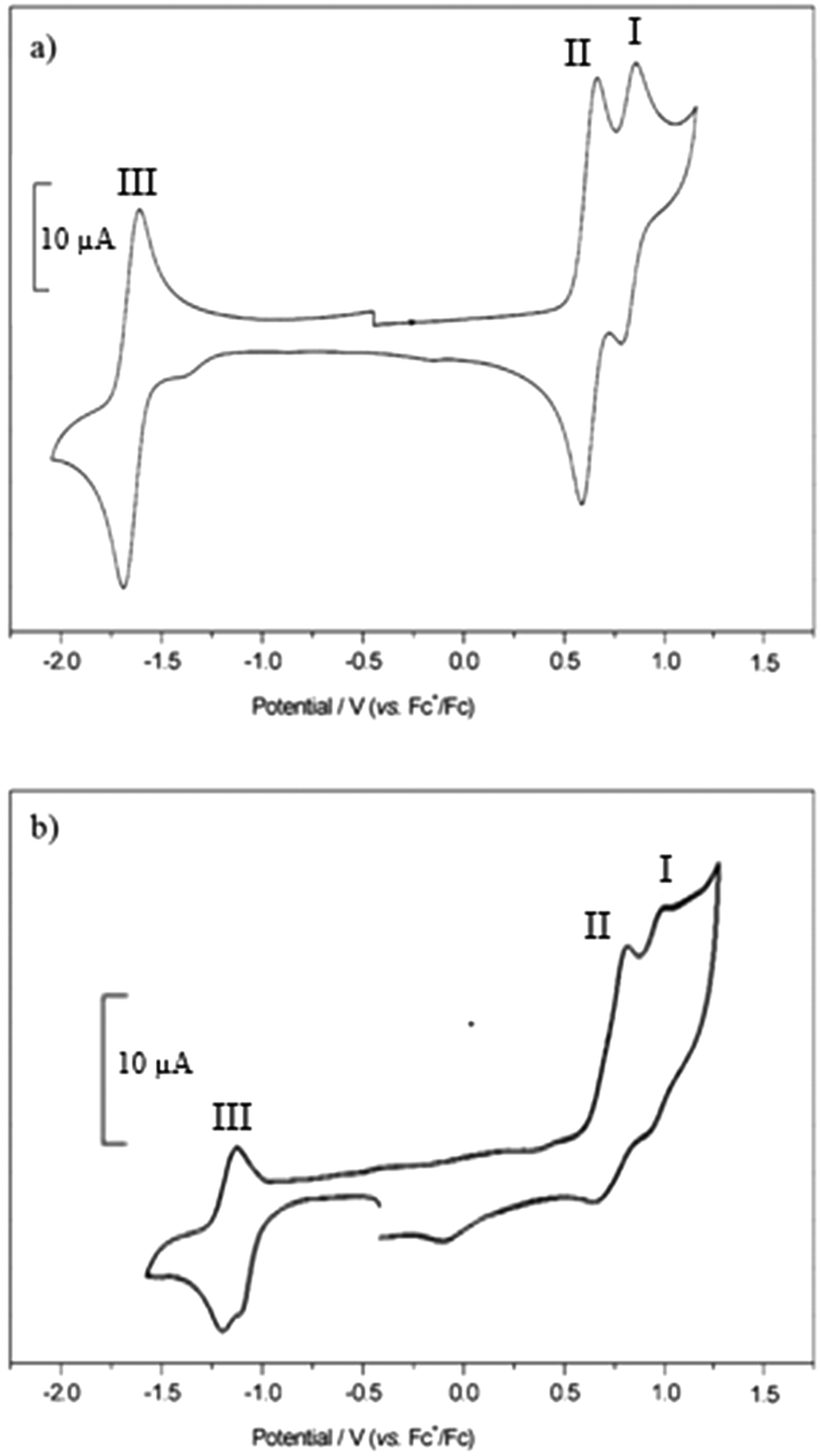 | ||
| Fig. 8 Cyclic voltammograms recorded for 4 (a) and 5 (b) in CH2Cl2 containing [Bu4N][ClO4] (0.5 M) at 0.1 V s−1. | ||
| 1st reduction (III) | 1st oxidation (II) | 2nd oxidation (I) | ΔE(Fc+/Fc) | |
|---|---|---|---|---|
| a For 1 mM solutions in CH2Cl2 containing 0.5 M [NBu4][ClO4] as supporting electrolyte. Potentials reported as E1/2 (= (Eap + Ecp)/2) in V vs. Fe(Cp)2+/Fe(Cp)2 at 0.1 V s−1 scan rate and quoted to the nearest 0.01 V. Values in parentheses are ΔE (= Eap − Ecp) for the couple at 0.01 V s−1. b Unresolved in cyclic voltammogram, peak maximum from square wave voltammetry. c E adp at a scan rate of 200 mV s−1. | ||||
| 1 | −1.68 (0.07) | +0.58b | +0.76c | (0.07) |
| 2 | −1.65 (0.07) | +0.63 (0.07) | +0.95 (0.10) | (0.07) |
| 3 | −1.69 (0.09) | +0.61 (0.09) | +0.89c | (0.07) |
| 4 | −1.66 (0.08) | +0.62 (0.08) | +0.81 (0.08) | (0.07) |
| 5 | −1.27 (0.08) | +0.74 (0.15) | +0.99 (0.10) | (0.15) |
| 6 | −1.48 (0.13) | +0.65b | +0.75b | (0.1) |
| 7 | −1.29 (0.14) | +0.63 (0.06) | +0.87 (0.08) | (0.15) |
| Bodipy-CO2H | −1.63 (0.25) | +0.64 (0.09) | +0.83b | (0.15) |
Scanning in the positive direction gave two redox couples (I and II) which overlapped for 1 and 3 in the cyclic voltammogram but were separated by 11 and 23 mV respectively in the square wave experiment (Fig. S22 and Table S1 in the ESI†). In the cyclic voltammetry experiment I and II are separated by 320 mV for 2, with I, assigned to the triphenylamine, shifted +190 mV relative to 1 because of the electron withdrawing esters. The electrode potential I, associated with the amine, is unaffected by the electron withdrawing carboxylic acid group when separated by the extra phenyl unit in 4 and 5.
Scanning in the negative direction gave one reversible redox couple (III). The peak currents were approximately equal, an indication of the good stability of the radical ions. The redox potential of III was similar for 1–4 which suggests that the electron withdrawing ester groups affect the oxidation potential of the dye but not the reduction potential, but shifted by around 400 mV to a more positive potential for 5. This is in agreement with the DFT calculations which indicate that the LUMO is located on the dipyrromethene at similar energies for each dye but shifted for 5. For 3 and 4, the potentials for II and III were similar to those for 1 but the peak currents were double those of 1 and 2, in agreement with two electron reduction and two electron oxidation of bodipy pair. Since there does not appear to be electronic communication between the two bodipy centres, these are considered to be two, one-electron processes and not one, two electron process. The peak current for the reversible waves in process I were approximately half the size of processes II and III, consistent with the hypothesis that this is associated with oxidation of the triphenylamine moiety.
Subtracting the E0–0 value (2.32 eV) from the reduction potential gives an estimate of the reduction potential of the excited state. These are 0.64 V for 1, 0.67 V for 2, 0.63 V for 3, 0.69 for 4 1.05 V for 5, 0.84 for 6, 1.03 for 7 and 0.69 for Bodipy-CO2H and are approximately equal to the first oxidation potentials measured by cyclic voltammetry. Only the redox potentials for process I in 4–7 are less positive than this indicating that charge-transfer from triphenylamine to the excited dipyrromethene is less likely to occur in 1–6 than in 4–7.
Spectroelectrochemistry
Spectroelectrochemistry was performed on 1, 2, 4, 5 and 7 in order to determine the absorption spectrum of the reduced and oxidised species (4/5 are shown in Fig. 9, 1, 2 and 7 are in the ESI,†6 has been reported previously22). Table S6 (ESI†) gives the peak maxima and extinction coefficients for the oxidised and reduced species for 1–4. This information can be used to assign the species in the cyclic voltammetry and transient absorption spectra (see below) as well as demonstrating the stability of the dyes on oxidation and reduction. | ||
| Fig. 9 UV-visible difference spectra recorded in CH2Cl2 containing [Bu4N][ClO4] (0.5 M) using spectroelectrochemical methods showing the inter-conversion of (a) 4 to 41−, (b) 5 to 51−, (c) 4 to 41+ and (d) 5 to 51+ at 248 K. Arrows indicate the progress of the oxidation or reduction. | ||
Reduction of the 1–6 was accompanied by a bleach of the bodipy absorption at 526 nm and the appearance of a peak at 582 nm. This is consistent with Hattori et al.'s results for a similar 1,3,5,7-tetramethyl-2,6-diethyl-bodipy.29 The absorption bands differed slightly in the UV region. Unlike 1 and 4, where the reduction progressed through a series of well-defined isosbestic points, the reduction of 2 showed the development, then depletion of bands, best observed in the region between 400–500 nm. This could be attributed to the electron withdrawing ester groups stabilising the bodipy radical anion. The reduction of dye 5 corresponded with a bleach of the bodipy absorption at 561 nm but unlike 1–4 no corresponding growth was observed at lower energies, only the appearance of a peak toward the UV region. For dye 6 a bleach of the bodipy at 540 nm and the formation of a peak at 595 nm was observed upon a potential sweep to −1.5 V.22 For dye 7 (Fig. S72, ESI†) the bleach of the bodipy at 565 nm was accompanied by the growth of a band at 630 nm.
Oxidation of 2 and 4 was accompanied by a bleach of the bodipy absorption at 526 nm and a growth in the peak at 407 nm attributed to oxidation of the bodipy as predicted above. For comparison, the spectroelectrochemistry of triphenylamine has been described previously. On oxidising triphenylamine, Chiu et al.30 reported a peak at 696 nm and Chung et al.31 reported a peak at 660 nm. Oxidation of 5 corresponded with the bleach of the absorption at 561 nm and the growth of a broad peak at 605 nm that extended toward the near-IR region alongside the growth of a band at 450 nm. Similarly, oxidation of 7 (Fig. S73, ESI†) was accompanied by the bleach at 565 nm and the growth of a broad absorption at 600–800 nm and a band at 450 nm.
The spectra recorded at the beginning and the end of the experiments show the extent of reversibility of the reduction and oxidation reactions and are provided in Fig. S74–S76 in the ESI.† We note that for 2, although the reduction appears to be chemically reversible from the cyclic voltammetry and there was almost complete restoration of the ground state absorption spectrum, a small shoulder developed at ca. 550 nm. Since this was not observed for the other dyes, we attribute this to irreversible changes associated with the ester. Subsequent reduction of 4+ returns the spectrum to the same spectral shape as 4 but there is a loss of intensity in the main band; oxidation of 4− restores the main band at 526 nm but the high energy region is changed. We attribute these changes to the presence of the carboxylic acid.
Transient absorption spectroscopy
Charge transfer dynamics of dyes Bodipy-CO2H, 4, 5 and 7 were investigated by femtosecond time-resolved transient absorption spectroscopy in order to evaluate the charge separation at the dye–NiO interface. Details are provided in the Experimental section. The dyes were studied when dissolved in CH2Cl2 to characterise the excited state and when adsorbed onto nanocrystalline NiO films to explore the formation and decay of the NiO+/dye− charge-separated state. The data was fitted to a single exponential function unless stated otherwise. The transient absorption spectra of 6 have been reported by us previously.22 The transient absorption spectra of Bodipy-CO2H, 4, 5, and 7 in CH2Cl2 and adsorbed on NiO are presented in Fig. 10. The time constants corresponding to the kinetic traces (Fig. 10 inset and Fig. S82–S84, ESI†) are presented in Table 4.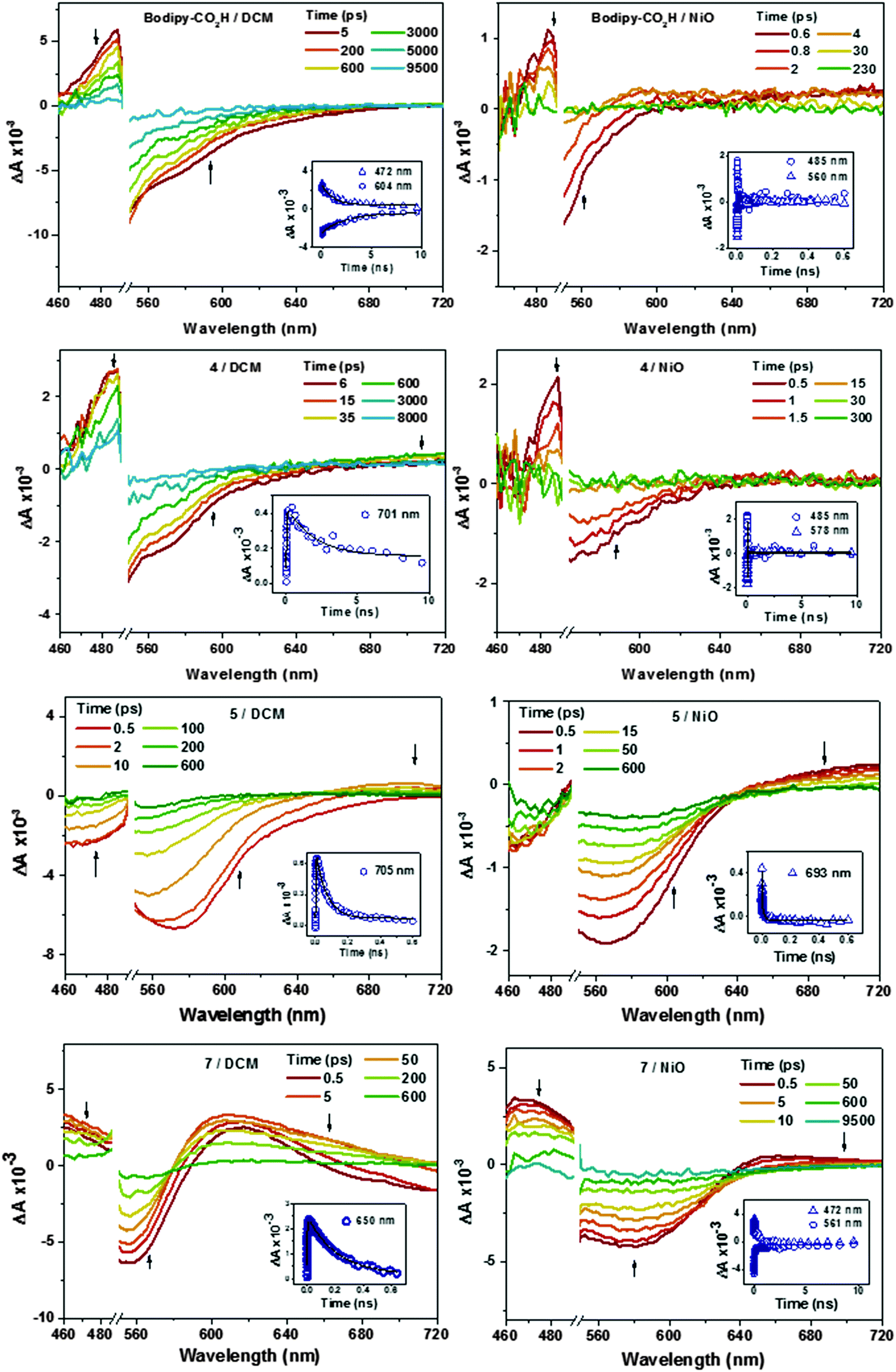 | ||
| Fig. 10 Time-resolved transient-absorption spectra of Bodipy-CO2H, 4, 5, and 7 in CH2Cl2 solution and absorbed onto NiO films at a number of delay times (ps) after excitation at 532 nm. (Inset) Corresponding kinetic traces at the given wavelengths. | ||
| Solution | NiO | |||||
|---|---|---|---|---|---|---|
| Dye | (λ/nm) | τ/ps | Transient (λ/nm) | τ/ps | ||
| a The growth and decay of the peak were fit separately to single exponential functions. | ||||||
| Bodipy-CO2H | 472 | Transient | 1.1 × 103 ± 0.1 × 103 | 488 | Transient | 4.1 ± 0.6 |
| 594 | Bleach | 2.3 × 103 ± 0. 3 × 103 | 560 | Bleach | 0.7 ± 0.1 | |
| 4 | 481 | Transient | 0.8 × 103 ± 0.1 × 103 | 485 | Transient | 5.0 ± 1.0 |
| 596 | Bleach | 18 ± 1 | 578 | Bleach | 2.6 ± 0.5 | |
| 2.7 × 103 ± 0.6 × 103 | ||||||
| 710 | Transient | 25 ± 3 (growth) | ||||
| 3.8 × 103 ± 0.9 × 103 (decay)a | ||||||
| 5 | 475 | Bleach | 5.8 ± 0.7 | |||
| 61 ± 4 | ||||||
| 611 | Bleach | 3.8 ± 0.1 | 606 | Bleach | 1.1 ± 0.1 | |
| 68 ± 4 | 470 ± 70 | |||||
| 705 | Transient | 1.2 ± 0.1 (growth) | 693 | Transient | 0.10 ± 0.01 | |
| 68 ± 2 (decay)a | 13 ± 1 | |||||
| 6a | 425 | Transient | 20.7 ± 2.9 | 425 | Transient | 12.5 ± 2.9 |
| 864 × 103 ± 28 × 103 | 49 × 103 ± 28 × 103 | |||||
| 470 | Transient | 9.52 ± 0.91 (growth) | 515 | Bleach | 9.76 ± 1.1 | |
| 396 × 103 ± 33 × 103 (decay) | 23 × 103 ± 10 × 103 | |||||
| 500 | Bleach | 14.1 ± 1.2 (growth) | 575 | Transient | 24.1 ± 3.0 | |
| 413 ± 50 and 896 × 103 ± 26 × 103 (decay) | 177 × 103 ± 59 × 103 | |||||
| 650 | Transient | 292 × 103 ± 31 × 103 | ||||
| 7 | 472 | Transient | 0.8 ± 0.04 (growth) | 472 | Transient | 4.9 ± 0.1 |
| 190 ± 10 (decay)a | 580 ± 40 | |||||
| 563 | Bleach | 0.6 ± 0.03 | 580 | Bleach | 2.8 ± 0.3 | |
| 190 ± 10 | 640 ± 80 | |||||
| 650 | Transient | 1.3 ± 0.1 (growth) | 700 | Transient | 780 ± 500 (rise) | |
| 210 ± 100 (decay)a | 6.5 ± 0.4 (decay) | |||||
The spectral shape of Bodipy-CO2H and 4 in CH2Cl2 excited at 532 nm are generally similar (Fig. 10). The bleach centered at ca. 530 nm corresponds to the ground state absorption of Bodipy-CO2H and 4. The ground state recovered with τ ≈ 2–3 ns.32 The difference in time constants for the decay of the excited state and the recovery of the ground state is probably due to the overlap of the ground state recovery and stimulated emission signals, which were observed at early time delays in the solution spectra of all four dyes. Consistent with the ground state absorption spectrum, excitation of 5 in solution generated a broader bleach, which extended across the visible region until 620 nm and decayed more rapidly with τ ≈ 65 ps.
A large positive absorption between 460–500 nm was observed for Bodipy-CO2H and 4, which was attributed to the singlet excited state. These transients formed within 1 ps and decayed with τ = 1.1 ± 0.3 ns. The lifetime of the singlet excited state of Bodipy-CO2H recorded in solution by transient absorption spectroscopy was slightly shorter than that measured by single photon counting (1.1 ns vs. 3 ns). This difference could be due the lifetime being close to the resolution of the instrument used for single photon counting. The transient spectra previously observed for 6 were similar in shape initially, but evolved over ca. 400 ps to be replaced by the triplet excited state spectrum (with absorption bands at 425 and 650 nm, τ ≈ 300 ns).21 In Fig. 10 there is no indication of triplet species in any of the spectra. However, in the transient spectra of 4 and 5 a second, small absorption at 710 nm grows in over τ = 25 ± 3 ps for 4 and τ = 1.4 ± 0.1 ps for 5. These subsequently decay on a similar timescale to the bleach (τ = 3.8 ± 0.9 ns for 4 and 68 ± 2 ps for 5). This transient resembles an oxidised triphenylamine (Seo et al. reported λmax [TPA+˙] = 640 nm in acetonitrile20). The appearance of this transient suggests that some charge-transfer from the amine group occurs but absorption corresponding to the bodipy-based radical anion (expected at λ = 582 nm for 4 from the spectroelectrochemistry data above) is either absent or is swamped by the stimulated emission. Excitation of 7 in CH2Cl2 solution generated a feature which absorbed broadly across the visible region up to 720 nm, which we have attributed to the first singlet excited state. This feature broadened, red-shifted slightly and increased in intensity over τ = 1.0 ± 0.4 ps. The broad absorption is consistent with the spectrum for a reduced bodipy and we infer that the excited dye transfers charge from the amine to the bodipy, forming an intramolecular charge-separated excited state. The peak at 700 nm which was observed for 4 and 5 was absent. The higher and lower wavelength regions of the transient were fitted to first-order decay and the time constants were consistent (τ472 = 0.19 ± ns and τ650 = 0.21 ± 0.1 ns) indicating that both are due to the same excited state. The bleach centered at 565 nm decayed with a fast (τ = 0.60 ± 0.03 ps) and slower (τ = 0.19 ± 0.01 ns) component. The faster component is likely to be due to overlap with stimulated emission which should be centred at 630 nm and causes a negative absorption at 700 nm at early time delays.
The spectral features between 460–500 nm in the transient spectra for the dyes adsorbed on NiO (Fig. 10) were similar to those of the dyes in solution and formed within the time resolution of the instrument. Stimulated emission was observed at early time delays for Bodipy-CO2H/NiO and 4/NiO but not in 5/NiO and 7/NiO. The excited state of the dyes on NiO decayed three orders of magnitude faster (τ485 = 4.1 ± 0.6 ps for Bodipy-CO2H/NiO, τ485 = 5 ± 1 ps for 4/NiO) than the dyes in solution, which suggests that in both cases the excited state is rapidly quenched by electron transfer from the NiO to the dye. Consistent with this, a broad, weak absorption persists at wavelengths longer than 600 nm in the transient spectra of Bodipy-CO2H/NiO, which is characteristic of holes introduced in NiO (τ700 = 24 ± 4 ps, see Fig. S81 in ESI†), and a weak feature at 580 nm, which is characteristic of the bodipy radical anion. Likewise, contrary to 5 in solution, a single isosbestic point at 620 nm was observed for 5/NiO (suggesting that a single transient species formed) and the transient at 700 nm decayed five times faster for 5/NiO (τ693 = 13 ± 1 ps), than for 5 in CH2Cl2. The presence of these features suggests that photoinduced electron transfer from NiO to the dye occurs with the subsequent formation of a charge-separated state, NiO+/bodipy−. The signals were too weak for 4/NiO to extract meaningful kinetics. In the transient absorption spectra for 7/NiO adsorbed onto NiO the negative absorption corresponding to the ground-state bleach was broader than for 7 in solution due to the electronic interactions between the dye and the NiO. A transient which absorbed broadly between 460 and 500 nm decayed on a longer timescale (τ472 = 0.58 ± 0.04 ns) than that for 7 in solution. The absorption at 650–700 nm is indicative of reduced dye, 7−. It was difficult to extract a time constant for the decay of this transient because of the overlap with the bleach in the same spectral region (τrise = 0.78 ± 0.5 ns is consistent with the kinetics of the ground state recovery at 560 nm). Despite the complexity of the spectra for 7, in general the lifetime for NiO+/7− was observed to be longer compared to the charge separated state lifetimes of 4/NiO and 5/NiO but shorter than 6/NiO.
Discussion
The performance of the p-DSCs, particularly the photocurrent, was strongly dependent on the dye structure. The IPCE of a solar cell is a product of the different charge transfer processes occurring within the operational device (IPCE = LHE·ηinj·ηcc·ηreg).33 We assume that the charge collection efficiency (ηcc) is similar for each of the dyes (1) because the charge transport times should be similar for each device as the semiconductor and redox mediator are the same; (2) the charge lifetimes were similar between the dyes, apart from a slight improvement for the larger dyes (5–7) (Fig. S91–S93, ESI†). The charge injection efficiency (ηinj) is estimated from the ratio of the excited state lifetime in the absence and the presence of NiO. The time resolved transient absorption experiments (see above) and time-resolved emission studies (Fig. S72–S74, ESI†) have indicated that electron transfer from the NiO to the dye occurs rapidly (<ps) for dyes 4–7 and Bodipy-CO2H. This is despite the differences in the rate of intramolecular charge separation in the dyes in solution, which were at least 10–20 times slower for 6 and 4 compared to 5 and 7. Also, this is despite shorter excited state lifetimes for 5 and 7 compared to 4 and 6 (although we note that the driving force for charge separation is ca. 100 meV larger for 5 compared to the other dyes). Thus, it appears that the dye regeneration efficiency (ηreg) is responsible for the more substantial differences in the IPCE. We expected that a long lived charge-separated state would favour photoinduced electron transfer between the dye radical anion and triiodide and increase the IPCE. The driving force for electron transfer from the reduced dye to triiodide is 4 > Bodipy-CO2H > 6 > 7 > 5. From the time resolved transient absorption spectroscopy, we have found that the charge-separated state lifetime follows the order of 6 > 7 > 5 > 4 > Bodipy-CO2H. The cell performance follows the similar trend of 7 > 5 > 4 > 6 > Bodipy-CO2H. Lefebvre et al. estimated a 50% charge separation efficiency for 6 by comparing the relative amplitudes of the signals at 425 nm (which contains both contribution from 11* and 1−) and 575 nm (which is NiO+/1− only) at early (2 ps) and late (2 ns) times. Therefore a possible explanation for the low IPCE of 6 compared to 7 could be that yield of the charge-separated state is lower for 6 compared to 7 because, in 6, the rotation around the bodipy/thiophene spacer is more restricted. The IPCE for dye 7 is high for a NiO solar cell despite τ < 1 ns for NiO+/7−. This implies that re-oxidation of the dye by the I3− electrolyte is very efficient. Further investigation is required to determine whether certain functional groups on the dye promote rapid electron transfer to I3−.Conclusions
We have successfully prepared and characterised a number of NiO photosensitizers based on triphenylamine–bodipy conjugates. The results have provided us with guidelines for designing molecular dyes for p-type solar cells. Adding the thiophene spacer between the amine and the bodipy has a more positive affect on the solar cell efficiency than adding phenyl between the anchor and the amine donor. This could be a result of the better electron donating properties of thiophene compared to phenyl. Bodipy-CO2H performed similarly to 4 suggesting that the amine donor is not required for charge separation. The less substituted bodipy dyes 5 and 7 had a broader spectral response and a higher IPCE which collectively led to a higher short-circuit photocurrent density. We attribute this to better electronic communication between the NiO and bodipy in 5 and 7 compared to the hindered dyes 4 and 6.Acknowledgements
EAG wishes to thank The Royal Society for a Dorothy Hodgkin Fellowship, The University of Nottingham for an Anne McLaren Research Fellowship and a Nottingham-Lund Joint Research Project, the EPSRC and Newcastle University for a studentship for Fiona A. Black (EPJ5002881), COST CM1202 and LaserLab Europe.References
- F. Odobel, Y. Pellegrin, E. A. Gibson, A. Hagfeldt, A. L. Smeigh and L. Hammarström, Coord. Chem. Rev., 2012, 256(21–22), 2414–2423 CrossRef CAS.
- D. Dini, Y. Halpin, J. G. Vos and E. A. Gibson, Coord. Chem. Rev., 2015, 304–305, 179–201 CrossRef CAS.
- L. Li, L. Duan, F. Wen, C. Li, M. Wang, A. Hagfeldt and L. Sun, Chem. Commun., 2012, 48(7), 988–990 RSC.
- U. Bach and T. Daeneke, Angew. Chem., Int. Ed., 2012, 51(42), 10451–10452 CrossRef CAS PubMed.
- J. He, H. Lindström, A. Hagfeldt and S.-E. Lindquist, Sol. Energy Mater. Sol. Cells, 2000, 62, 265–273 CrossRef CAS.
- S. P. Bremner, M. Y. Levy and C. B. Honsberg, Prog. Photovoltaics Res. Appl., 2008, 16, 225–233 CrossRef.
- H. J. Snaith, Adv. Funct. Mater., 2010, 20, 13–19 CrossRef CAS.
- S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, B. F. E. Curchod, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, M. K. Nazeeruddin and M. Grätzel, Nat. Chem., 2014, 6(3), 242–247 CrossRef CAS PubMed.
- C. J. Wood, G. H. Summers and E. A. Gibson, Chem. Commun., 2015, 51, 3915–3918 RSC.
- Q.-Q. Zhang, K.-J. Jiang, J.-H. Huang, C.-W. Zhao, L.-P. Zhang, X.-P. Cui, M.-J. Su, L.-M. Yang, Y.-L. Song and X.-Q. Zhou, J. Mater. Chem. A, 2015, 3, 7695–7698 CAS.
- E. A. Gibson, A. L. Smeigh, L. Le Pleux, L. Hammarström, F. Odobel, G. Boschloo and A. Hagfeldt, J. Phys. Chem. C, 2011, 115(19), 9772–9779 CAS.
- L. Le Pleux, A. L. Smeigh, E. A. Gibson, Y. Pellegrin, E. Blart, G. Boschloo, A. Hagfeldt, L. Hammarström and F. Odobel, Energy Environ. Sci., 2011, 4(6), 2075 CAS.
- S. Powar, D. Xiong, T. Daeneke, M. T. Ma, A. Gupta, G. Lee, S. Makuta, Y. Tachibana, W. Chen, L. Spiccia, Y. Cheng, G. Götz, P. Bäuerle and U. Bach, J. Phys. Chem. C, 2014, 118(30), 16375–16379 CAS.
- I. R. Perera, T. Daeneke, S. Makuta, Z. Yu, Y. Tachibana, A. Mishra, P. Bäuerle, C. A. Ohlin, U. Bach and L. Spiccia, Angew. Chem., 2015, 127(12), 3829–3833 CrossRef.
- N. Boens, V. Leen and W. Dehaen, Chem. Soc. Rev., 2012, 41(3), 1130–1172 RSC.
- A. C. Benniston and G. Copley, Phys. Chem. Chem. Phys., 2009, 11(21), 4124–4131 RSC.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107(11), 4891–4932 CrossRef CAS PubMed.
- D. P. Hagberg, T. Edvinsson, T. Marinado, G. Boschloo, A. Hagfeldt and L. Sun, Chem. Commun., 2006, 2245–2247 RSC.
- P. Qin, H. Zhu, T. Edvinsson, G. Boschloo, A. Hagfeldt and L. Sun, J. Am. Chem. Soc., 2008, 130(27), 8570–8571 CrossRef CAS PubMed.
- E. T. Seo, R. F. Nelson, J. M. Fritsch, L. S. Marcoux, D. W. Leedy and R. N. Adams, J. Am. Chem. Soc., 1966, 18(14), 3498–3503 CrossRef.
- K. A. Click, D. R. Beauchamp, B. R. Garrett, Z. Huang, C. M. Hadad and Y. Wu, Phys. Chem. Chem. Phys., 2014, 16(47), 26103–26111 RSC.
- J.-F. Lefebvre, X.-Z. Sun, J. A. Calladine, M. W. George and E. A. Gibson, Chem. Commun., 2014, 50(40), 5258–5260 RSC.
- M. Kollmannsberger, K. Rurack, U. Resch-genger and J. Daub, J. Phys. Chem. A, 1998, 102, 10211–10220 CrossRef CAS.
- R. P. Sabatini, T. M. McCormick, T. Lazarides, K. C. Wilson, R. Eisenberg and D. W. McCamant, J. Phys. Chem. Lett., 2011, 2(3), 223–227 CrossRef CAS.
- A. Harriman, J. P. Rostron, M. Cesario, G. Ulrich and R. Ziessel, J. Phys. Chem. A, 2006, 110(26), 7994–8002 CrossRef CAS PubMed.
- Y. Rio, W. Seitz, A. Gouloumis, P. Vázquez, J. L. Sessler, D. M. Guldi and T. Tomás, Chem. – Eur. J., 2010, 16(6), 1929–1940 CrossRef CAS PubMed.
- K. Krumova and G. Cosa, J. Am. Chem. Soc., 2010, 132(13), 17560–17569 CrossRef CAS PubMed.
- M. R. Momeni and A. Brown, J. Chem. Theory Comput., 2015, 11(6), 2619–2632 CrossRef CAS PubMed.
- S. Hattori, K. Ohkubo, Y. Urano, H. Sunahara, T. Nagano, Y. Wada, N. V. Tkachenko, H. Lemmetyinen and S. Fukuzumi, J. Phys. Chem. B, 2005, 109(32), 15368–15375 CrossRef CAS PubMed.
- K. Yuan Chiu, T. Xiang Su, J. Hong Li, T.-H. Lin, G.-S. Liou and S.-H. Cheng, J. Electroanal. Chem., 2005, 575(1), 95–101 CrossRef.
- Y. Chung and Y. O. Su, J. Chin. Chem. Soc., 2009, 56, 493–503 CrossRef CAS.
- A. C. Benniston, S. Clift, J. Hagon, H. Lemmetyinen, N. V. Tkachenko, W. Clegg and R. W. Harrington, ChemPhysChem, 2012, 13, 3672–3681 CrossRef CAS PubMed.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp05177k |
| ‡ These authors contributed equally to this work. |
| This journal is © the Owner Societies 2016 |