
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective partial hydrogenation of alkynes to (Z)-alkenes with ionic liquid-doped nickel nanocatalysts at near ambient conditions†
Hannelore
Konnerth
and
Martin H. G.
Prechtl
*
Department of Chemistry, Institute of Inorganic Chemistry, University of Cologne, Greinstraße 6, 50939 Cologne, Germany. E-mail: martin.prechtl@uni-koeln.de; Web: http://catalysis.uni-koeln.de Fax: +49 221 4701788
First published on 29th March 2016
Abstract
A selective hydrogenation method for forming (Z)-alkenes from alkynes has been developed using a catalyst system of cheap Ni-NPs in a nitrile functionalised imidazolium based ionic liquid (IL) operating under very mild reaction conditions of 30–50 °C and 1–4 bar H2 pressure.
The hydrogenation of alkynes to produce selectively (E)- or (Z)-alkenes is desirable as this reaction is a useful synthetic tool for the production of many bioactive natural products or pheromones.1–4 The synthesis of stilbene derivatives is especially interesting as these structural motifs are present in many relevant natural products currently under investigation in pharmacological studies.5–7 Many of these alkyne transformations to (Z)-alkenes make use of the famous Lindlar catalyst, consisting of Pd–CaCO3 partially poisoned with Pb(OAc)2 and quinoline.8 Various heterogeneous catalyst systems are also known which make use of noble metals like Pd, Pt or Au.9–14 An approach towards more abundant and low-cost metals, thus expanding the applicability of this synthetic strategy, would be beneficial. Efforts have been made to investigate homogeneous nickel catalysts which make use of hydrogen transfer agents like H3PO2 or HCO2H. These systems are either limited to internal alkynes to produce (E)-alkenes or require high reaction conditions.15,16 In 1981 a nickel on graphite catalyst was introduced which operates under mild reaction conditions; sensitivity towards oxidation prohibits reusability.17 Moreover, only a few nanoscale catalyst systems were reported using non-noble metals such as the binary GaNi18 or ternary CuFeNi19 system. Ni(0)-NPs, in combination with an alcohol as hydrogen source, have also been described, although stoichiometric amounts of an alkali metal and catalyst are needed.20 A nickel phosphide catalyst has also been reported, however the activity is low applied to diphenyl acetylene derivatives.21
In the field of catalysis, the use of metal catalysts in ionic liquids is of tremendous importance.22–24 Besides, the role as “greener” solvents, ILs also show remarkable properties as reducing agents to synthesis nanoparticles and they act as stabilising agents for these NPs.25,26 Special properties including low vapour pressure, and high thermal and chemical stability make ILs suitable for the use in liquid–liquid biphasic systems, enabling facile separation of the catalyst from the substrate phase. Moreover, the selectivity of a metal-catalysed reaction can be controlled by steric or electronic properties of either the cationic or anionic component of the ionic liquid, which interacts with the nanoparticle surface or the substrate. This multi-functionality promotes the use of ionic liquids which combines reducing agent, stabilising agent, solvent and ligand as selectivity controller in one single compound in the catalyst phase. As described in our previous works, the ionic liquid plays a crucial role for the chemoselectivity in the hydrogenation of alkynes to (Z)-alkenes.12,27 Nitrile-functionalised imidazolium based ionic liquids are especially suitable to tune the selectivity towards partial hydrogenation. Pd–NPs embedded in IL showed a tunable selectivity for the production of either the corresponding (Z)-alkenes or alkanes in the hydrogenation of both terminal and internal alkynes under very mild reaction conditions.12 Moreover, a system with Fe–NPs in the same ionic liquid also showed the conversion of a wide substrate scope of internal alkynes to the corresponding (Z)-alkenes; harsh reaction conditions were required however (80 °C, 60 bar H2). For the catalyst preparation, a strong reducing agent is necessary to form the active iron species in an organic solvent followed by extraction of the Fe–NPs into the IL; the procedure has to be carried out under strict absence of moisture and air to prevent oxidation of the sensitive catalyst.27
In order to recreate the advantages of noble metal catalysts whilst taking advantage of the benefits of abundant metals, we were interested in developing a catalyst system containing a cheap and abundant metal such as nickel, which is not as sensitive to oxidation as iron. We thus developed a Ni-NPs catalyst system, which operates under very mild reaction conditions, to hydrogenate internal and even terminal phenylalkynes to the corresponding (Z)-alkenes with high selectivity using molecular hydrogen (Scheme 1).
 | ||
| Scheme 1 Semihydrogenation of alkynes using Ni-NPs in nitrile functionalised IL. | ||
In a first step using diphenylacetylene as model substrate, an IL screening and optimisation of the reaction conditions towards a high selectivity for the (Z)-alkene was investigated. The NPs were synthesised using different ionic liquids, through modification to known literature protocols, for the formation of Ni-NPs in imidazolium based ILs.25,28–30 The reactivity of all samples was seen to be high using mild reaction conditions of 30 °C within 6–16 h reaction times (Table 1). In all reactions, an excellent stereoselectivity for the formation of the (Z)-isomer was apparent, in several cases however, hydrogenation to the corresponding alkane occurred (Table 1; entries 1–3). The quantitative Ni-NP catalysed hydrogenation of diphenylacetylene is already apparent after only 6 h when using the non-functionalised ILs [BMIM]NTf2 and [BMMIM]NTf2. Low selectivities for the production of the alkene relative to overreduction to the alkane are seen however (Table 1, entries 1 and 2). With the IL [C10MMIM]NTf2 (Table 1, entry 3) even a higher amount of alkane was produced with 83% yield. Compared to the imidazolium based ionic liquids bearing a butyl side chain, which result in Ni-NPs size of 7.8–8.2 nm, the NPs synthesised in the IL with the decyl side chain are much smaller with around 4.4 nm in diameter. Aside from the different size and shape of the formed NPs in different ILs, it is possible that the surrounding of the NPs by the IL itself influences the metal–surface interaction with the stabilizer and thus the outcome of this hydrogenation reaction. The longer hydrophobic alkylchain might increase the non-polar region around the nanoparticle surface and allows the less polar alkene, compared to alkyne, a better accessibility for close proximity to the surface. The solubility of the alkene in the catalyst phase could also be enhanced, this might lead to an increase of hydrogenation activity resulting in production of the alkane. In the alkyl sidechain functionalised ILs (Table 1, entries 4–9), longer reaction times of 16 h are needed for quantitative conversion of the alkyne, although in the dihydroxyl functionalised IL only 10% conversion was achieved after 16.5 h. Moreover, in all functionalised ILs the selectivity for the formation of the alkene is very high. These two results indicate a different coordination mode of the ILs on the Ni-NPs. As reported previously for nitrile-ILs, a coordination on the nanoparticle surface is suggested which involves the nitrile-group.12,26,27 A higher coordination tendency further impedes displacement with the substrate, and thus a longer reaction time is necessary. Steric factors might also shield the surface of the nanoparticle or electronic modifications by the stabilizer, which might change the absorption/desorption mode of the more electron-rich alkyne-moiety.31,32 In the case of the diol-functionalised IL drawing of conclusions is difficult due to the low conversion. A decomposition route of [Ni(COD)2] in [C1C1(EG)IM]NTf2-IL is proposed via the formation of transitory non-classical NHC-ligands, confirmed by the generation of 1,3-COD, rather than a decomposition by simple ligand exchange of the OH-groups on the surface of the Ni-NPs.28 Thus, the diol-functionality might simply create a very hydrophilic area around the nanoparticle surface in the hydrogenation reaction of the alkyne. In such an environment it would be difficult for the lipophilic substrates to get in close proximity to the nanoparticle surface. In the IL [CNC3MIM]NTf2 a decreased conversion was observed, which is in accordance with the previously reported Fe–NPs-catalysed reaction in the same ionic liquid.27 Here, a reduced activity, compared to the IL [CNC3MMIM]NTf2, was also observed for the hydrogenation of diphenylacetylene. In this case the generation of NHC-complexes might be involved, although the selectivity for the alkene is still very high with about 93% (Table 1, entry 9). In all investigated systems with different ILs the formed NPs have a similar shape and a size of about 7–8 nm, except the NPs which were synthesised in [C10MMIM]NTf2 and [C1C1(EG)IM]NTf2, which have a size of about 4 nm. Comparing only the systems with a similar size and shape, changes in activity and selectivity are obvious, which appear to be especially influenced by the respective ionic liquid.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | IL | Ni-NPs size [nm] | t [h] | Conv. [%] | Yield [%] | ||
| 2 | 3 | 4 | |||||
| The volume of cyclohexane was 1 ml in all reactions.a For the synthesis of the NPs 0.3 g IL was used, the other reactions were conducted with 0.15 g IL.b The synthesis of Ni-NPs was conducted accordingly to literature method.28c The hydrogenation reaction was conducted with a constant pressure of 1 bar H2. | |||||||
| 1a | [BMIM]NTf2 | 8.2 ± 1.3 | 6 | 99 | 34 | 3 | 62 |
| 2a | [BMMIM]NTf2 | 7.8 ± 1.4 | 6 | 100 | 59 | 1 | 38 |
| 3a | [C10MMIM]NTf2 | 4.4 ± 0.7 | 6 | 97 | 11 | 2 | 83 |
| 4b | [C1C1(EG)IM]NTf2 | 3.5 ± 0.528 | 22 | 9 | 7 | 0 | 2 |
| 5a | [CNC3MMIM]NTf2 | 7.0 ± 1.1 | 6 | 44 | 36 | 2 | 4 |
| 6a | [CNC3MMIM]NTf2 | 7.0 ± 1.1 | 16 | 97 | 88 | 1 | 8 |
| 7 | [CNC3MMIM]NTf2 | 7.8 ± 1.1 | 16 | 100 | 90 | 2 | 7 |
| 8c | [CNC3MMIM]NTf2 | 7.8 ± 1.1 | 22 | 63 | 57 | 2 | 4 |
| 9 | [CNC3MIM]NTf2 | 8.4 ± 1.6 | 16 | 65 | 58 | 2 | 4 |
Best results were achieved with [CNC3MMIM]NTf2 at 4 bar H2 at 30 °C as quantitative conversion was achieved after 16 h with high selectivity for (Z)-stilbene (Table 1, entry 7). Even a smaller amount of ionic liquid could be applied without loss of activity and selectivity; this makes its use in large scale even more attractive due to reduced costs (Table 1, entries 6 and 7). Interestingly, neither the synthesis of the Ni-NPs nor the hydrogenation conditions of diphenylacetylene (Table 1, entry 7) showed modification of the ionic liquid, which was confirmed by 1H, 13C or 19F-NMR spectroscopy (see ESI†). Only small amounts of cyclooctadiene were visible in the spectra after the decomposition of the [Ni(COD)2] precursor. This is in vast contrast to iron and ruthenium NPs in such ILs,26,27 and underlines the suitability of this IL with Ni-NPs under the applied reaction conditions. Test reactions without Ni-catalyst or without applied hydrogen pressure under an atmosphere of argon yielded in no conversion (see ESI†); this confirms the crucial role of nickel as the active catalyst. Furthermore, we tested the reaction in [BMMIM]NTf2 with acetonitrile as additive. The nitrile group might coordinate as a ligand on the NP surface, similar to the coordination mode suggested for the nitrile functionalised IL. Indeed, in the hydrogenation reaction of diphenylacetylene, quantitative conversion resulted in 90% (Z)-stilbene, which confirms the results of the IL screening that the functionalised side chain of the IL is responsible for the high chemoselectivity in this hydrogenation reaction.
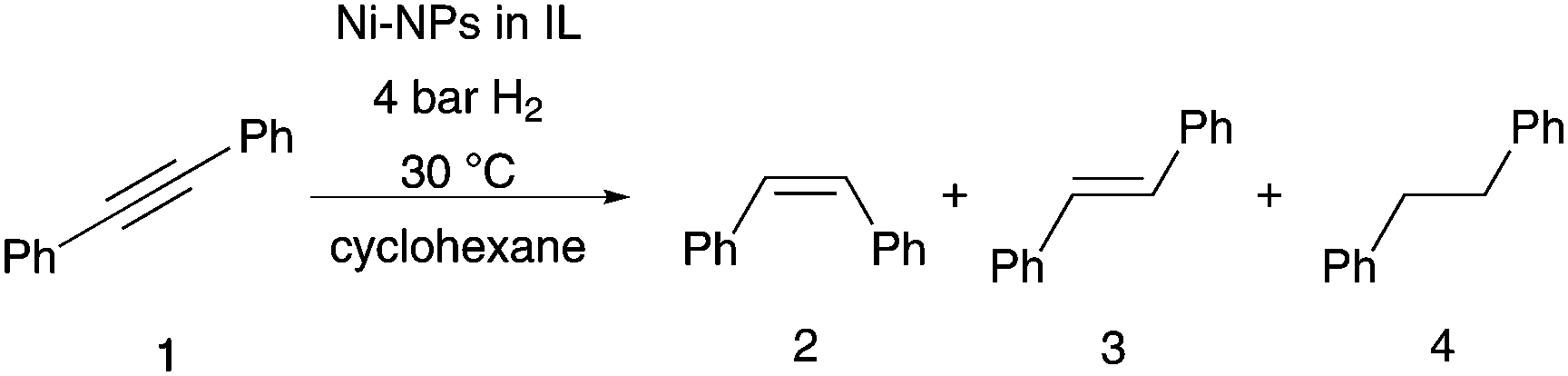
We concentrated our investigation to evaluate the optimized catalyst system in the hydrogenation of diphenylacetylene to (Z)-stilbene, using [Ni(COD)2] as precursor and the IL [CNC3MMIM]NTf2 (Table 1, entry 7). TEM analysis of the Ni-NPs embedded in IL shows well dispersed spherical shaped nanoparticles with a mean diameter of 7.8 ± 1.1 nm (Fig. 1). In previous works, we reported the formation of Ni(0)-NPs (<10 nm) and sponge-like agglomerates in [BMIM]NTf2.25 This underlines again the beneficial effect of the nitrile moiety for a more defined synthesis. Other works reported the preparation of Ni-NP systems in non-functionalised imidazolium based NTf2 ILs under hydrogen pressure with similar Ni(0)-NPs.29,30 It is well accepted that the IL acts as protective layer on the surface of the nanoparticles to inhibit further oxidation,23–25,29,31 this thin layer of IL around the NPs is also visible in the TEM images.
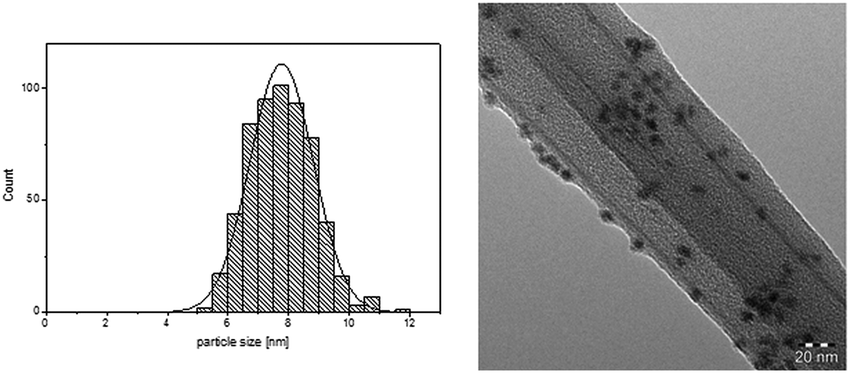 | ||
| Fig. 1 Particle size distribution and TEM image of Ni-NPs dispersed in [CNC3MMIM]NTf2 (38 μmol metal in 0.15 g IL, 70 °C; 20 h). | ||
A partially oxidation of the surface can never be excluded in nanoparticles owing to the exposure to air of the robust catalyst system.29,30 In addition, it is known that NiO can be reduced to Ni(0) under hydrogenative conditions, therefore our results show that this perspective does not play such a crucial role in contrast to highly air sensitive Fe–NPs.27
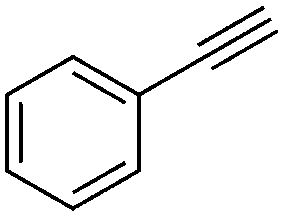
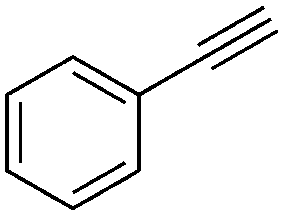
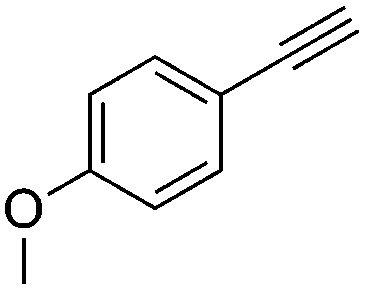
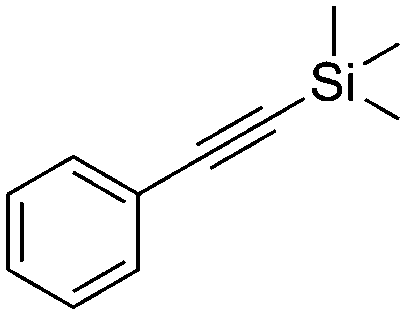
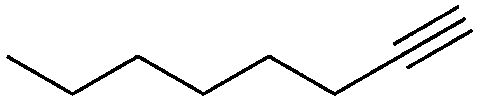
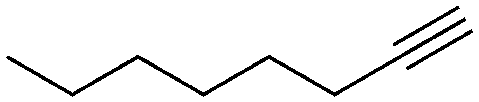
We further examined the application of the optimized catalyst system to a broader substrate scope of terminal phenyl alkynes, aliphatic internal and terminal alkynes as well as functionalised alkynes (Table 2). Interestingly, with precise control of the reaction conditions, including time and hydrogen pressure, even terminal alkynes bearing a phenyl-moiety such as phenylacetylene and the methoxy-substituted 1-ethynyl-4-methoxybenzene can be hydrogenated to the corresponding olefins with high selectivity of 79–81% (Table 2, entries 1 and 3). Interestingly, H2 pressure variation showed that even pressures below 5 bar H2 are sufficient and beneficial for the selectivity of the partial hydrogenation (Table 2, entries 1 and 2 and Fig. 1, ESI†). This makes this biphasic Ni-catalyst system superior to known heterogeneous literature procedures with non-noble metals for the hydrogenation of terminal alkynes to the alkenes with molecular hydrogen. The phenylic and aliphatic internal alkynes bearing a trimethylsilyl group reacted poorly under similar reaction conditions (Table 2, entries 4 and 9). Even with a higher reaction temperature of 50 °C, 22% conversion of 1-phenyl-2-trimethylsilylacetylene yielded only 12% of the alkene (Table 2, entry 4). The reaction might be hindered by the sterically bulky trialkylsilanes. However, aliphatic terminal alkynes like 1-octyne resulted in a mixture of alkene and alkane (Table 2, entries 5–7). Whilst the aliphatic internal alkyne 4-octyne required a higher reaction temperature of 40 °C for a high conversion of 92%, this alkyne was selectively converted to the corresponding (Z)-alkene with 82% yield (Table 2, entry 8). For the highly functional substrate dimethylacetylene dicarboxylate, the control of the chemoselectivity was difficult, as only 26% alkene were obtained after quantitative conversion of the starting material, the main product represents the overreduced alkane (Table 2, entry 11). The free electron pairs of the two methylester groups might additionally coordinate to the Ni-NPs and thus bring the alkyne/alkene moiety to close vicinity to the surface of the nanoparticles and the active sites. Additionally, better solubility of the polar substrate in the ionic liquid is plausible which would increase the reactivity. A change to a substrate with only one methylester group and an alkyl chain alters the reactivity, resulting in a higher selectivity of 45% for the alkene (Table 2, entry 12). For the terminal and internal aliphatic alkynes, no isomerisation products were detected by NMR-spectroscopy.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Alkyne | pH2 [bar] | t [h] | Conv. [%] | Yield [%] | S Z/E |
| The volume of the co-solvent cyclohexane was 0.5 ml.a Further experiments with the variation of the H2 pressure are shown in the ESI.b Reactions were conducted in a scale of 0.38 mmol substrate.c An external H2 reservoir was connected to the reactor.d A reaction temp. of 50 °C was used.e The reaction was conducted with a reduced catalyst loading with 1.8 mmol substrate.f A reaction temperature of 40 °C was used.g A volume of 1 ml of the co-solvent cyclohexane was used.h According to NMR analysis. | ||||||
| 1a |
|
4 | 3 | 100 | 79 | — |
| 2 |
|
1 | 16 | 100 | 73 | — |
| 3b,c |
|
4 | 3 | 100 | 81 | — |
| 4d |
|
4 | 17 | 22 | 12 | 95/5 |
| 5 |
|
1 | 16 | 100 | 54 | — |
| 6 |
|
1 | 7 | 72 | 47 | — |
| 7e |
|
1 | 16 | 100 | 53 | — |
| 8c,f |
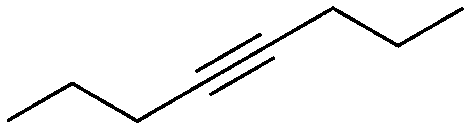
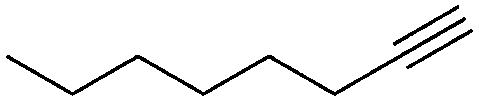
|
1 | 17 | 92 | 82 | 100h |
| 9e,f |
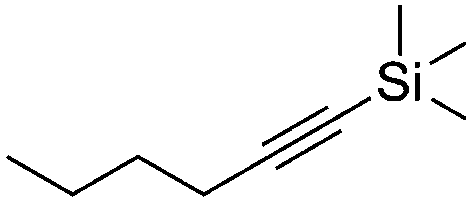
|
4 | 16 | 7 | 6 | 100h |
| 10b,c,d |

|
4 | 16 | 1 | 0 | — |
| 11g |
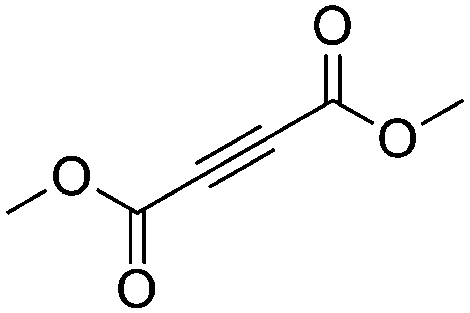
|
4 | 6 | 100 | 26 | 88/12 |
| 12b,c |
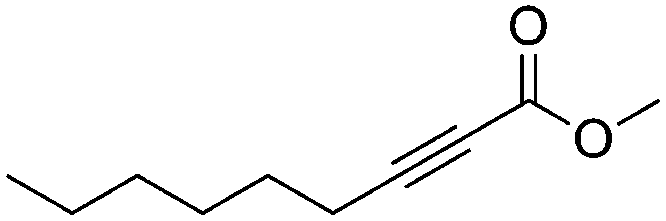
|
4 | 6 | 73 | 33 | 99/1 |
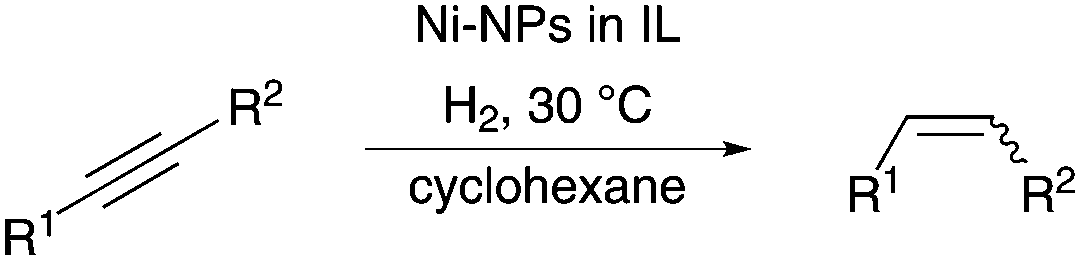
We tested the recycling of the NP-IL phase, using diphenylacetylene as the benchmark substrate, because a multiphase system allows for simple separation of product and catalyst phase, and enables easy handling and recycling of the NP-catalyst.27,33 After a reaction time of 8 h, a conversion of 88.1% was achieved with 92% selectivity for the alkene (Fig. 2). The catalyst is highly durable as it was recycled over 5 runs without significant loss of selectivity and activity. The total TON for these recycling experiments is about 518 for the formation of stilbene.
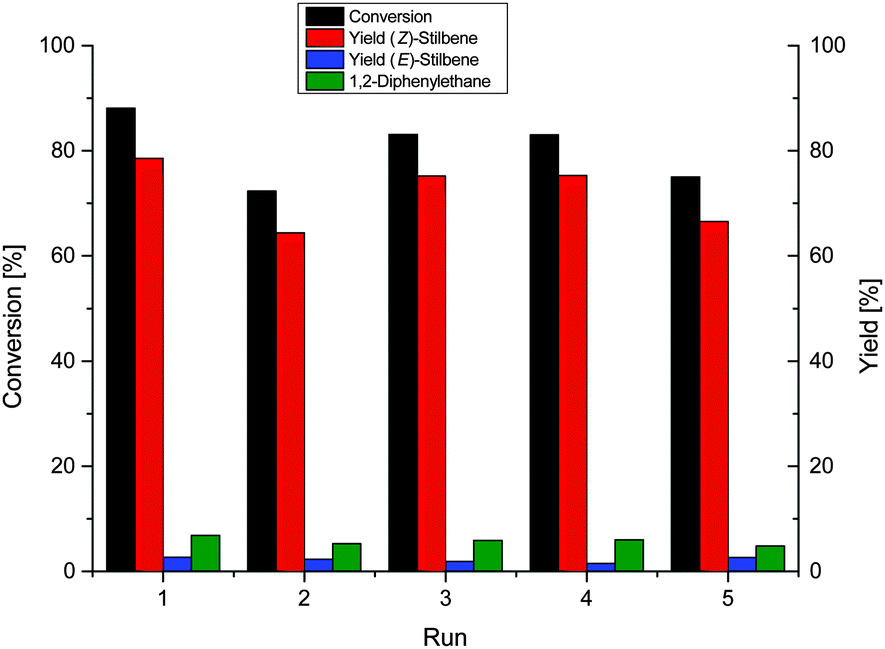 | ||
| Fig. 2 Recycling experiment of the hydrogenation of diphenylacetylene over Ni-NPs in [CNC3MMIM]NTf2 at 40 °C, 4 bar H2 for 8 h. | ||
To conclude, we developed a novel multiphase system with Ni-NPs as catalyst embedded in a nitrile-functionalised imidazolium based ionic liquid. The nitrile group is crucial for the selectivity of alkenes. This catalyst system shows high selectivity in the hydrogenation reaction of diphenylacetylene to the (Z)-alkene using very mild reaction conditions of 30 °C and 1–4 bar H2. Further studies examined that the method is also applicable to internal aliphatic alkynes as well as terminal phenylalkynes. Moreover, the catalyst is recyclable in multiphase systems with stable conversion rates and selectivity, and no precautions against moisture or air are required during the catalysis and the recycling of the active phase.
We gratefully acknowledge for financial support the Ministerium für Innovation, Wissenschaft und Forschung (NRW-returnee award 2009 for M. H. G. Prechtl), the Heisenberg-program (Deutsche Forschungsgemeinschaft), the Ernst-Haage Foundation (Max-Planck Institute for Chemical Energy Conversion) for the Ernst-Haage-Prize 2014, the Alexander-von-Humboldt Foundation (BRIGFOS Connect program) and H. Konnerth acknowledges the Jürgen Manchot Stiftung for a research scholarship. S. Roitsch is acknowledged for TEM analysis. And we are grateful to H. G. Schmalz for access to GC analysis and for some alkynes.
Notes and references
- M. Fouche, L. Rooney and A. G. M. Barrett, J. Org. Chem., 2012, 77, 3060–3070 CrossRef CAS PubMed.
- A. Furstner, O. Guth, A. Rumbo and G. Seidel, J. Am. Chem. Soc., 1999, 121, 11108–11113 CrossRef.
- V. N. Odinokov, Chem. Nat. Compd., 2000, 36, 11–39 CrossRef CAS.
- R. Rossi, Synthesis, 1977, 817–836 CrossRef CAS.
- N. Y. U. Anisimova, M. V. Kiselevsky, A. V. Sosnov, S. V. Sadovnikov, I. N. Stankov and A. A. Gakh, Chem. Cent. J., 2011, 5, 88 CrossRef CAS PubMed.
- J. Leiro, J. A. Arranz, N. Fraiz, M. L. Sanmartin, E. Quezada and F. Orallo, Int. Immunopharmacol., 2005, 5, 393–406 CrossRef CAS PubMed.
- K. A. Roupe, J. A. Yanez, X. W. Teng and N. M. Davies, J. Pharm. Pharmacol., 2006, 58, 1443–1450 CrossRef CAS PubMed.
- H. Lindlar, Helv. Chim. Acta, 1952, 35, 446–456 CrossRef CAS.
- M. J. Maccarrone, C. R. Lederhos, G. Torres, C. Betti, F. Coloma-Pascual, M. E. Quiroga and J. C. Yori, Appl. Catal., A, 2012, 441, 90–98 CrossRef.
- T. Mitsudome, Y. Takahashi, S. Ichikawa, T. Mizugaki, K. Jitsukawa and K. Kaneda, Angew. Chem., Int. Ed., 2013, 52, 1481–1485 CrossRef CAS PubMed.
- W. X. Niu, Y. J. Gao, W. Q. Zhang, N. Yan and X. M. Lu, Angew. Chem., Int. Ed., 2015, 54, 8271–8274 CrossRef CAS PubMed.
- R. Venkatesan, M. H. G. Prechtl, J. D. Scholten, R. P. Pezzi, G. Machado and J. Dupont, J. Mater. Chem., 2011, 21, 3030–3036 RSC.
- M. Jacobson, M. Beroza and W. A. Jones, J. Am. Chem. Soc., 1961, 83, 4819–4824 CrossRef CAS.
- M. Yan, T. Jin, Y. Ishikawa, T. Minato, T. Fujita, L. Y. Chen, M. Bao, N. Asao, M. W. Chen and Y. Yamamoto, J. Am. Chem. Soc., 2012, 134, 17536–17542 CrossRef CAS PubMed.
- T. Q. Chen, J. Xiao, Y. B. Zhou, S. F. Yin and L. B. Han, J. Organomet. Chem., 2014, 749, 51–54 CrossRef CAS.
- E. Richmond and J. Moran, J. Org. Chem., 2015, 80, 6922–6929 CrossRef CAS PubMed.
- D. Savoia, E. Tagliavini, C. Trombini and A. Umanironchi, J. Org. Chem., 1981, 46, 5340–5343 CrossRef CAS.
- K. Schutte, A. Doddi, C. Kroll, H. Meyer, C. Wiktor, C. Gemel, G. van Tendeloo, R. A. Fischer and C. Janiak, Nanoscale, 2014, 6, 5532–5544 RSC.
- B. Bridier, J. Perez-Ramirez, A. Knop-Gericke, R. Schlogl and D. Teschner, Chem. Sci., 2011, 2, 1379–1383 RSC.
- F. Alonso, M. Osante and M. Yus, Adv. Synth. Catal., 2006, 348, 305–308 CrossRef CAS.
- S. Carenco, A. Leyva-Perez, P. Concepcion, C. Boissiere, N. Mezailles, C. Sanchez and A. Corma, Nano Today, 2012, 7, 21–28 CrossRef CAS.
- J. D. Scholten, B. C. Leal and J. Dupont, ACS Catal., 2012, 2, 184–200 CrossRef CAS.
- P. S. Campbell, M. H. G. Prechtl, C. C. Santini and P. H. Haumesser, Curr. Org. Chem., 2013, 17, 414–429 CrossRef CAS.
- M. H. G. Prechtl and P. S. Campbell, Nanotechnol. Rev., 2013, 2, 577–595 CAS.
- M. H. G. Prechtl, P. S. Campbell, J. D. Scholten, G. B. Fraser, G. Machado, C. C. Santini, J. Dupont and Y. Chauvin, Nanoscale, 2010, 2, 2601–2606 RSC.
- M. H. G. Prechtl, J. D. Scholten and J. Dupont, J. Mol. Catal. A: Chem., 2009, 313, 74–78 CrossRef CAS.
- T. N. Gieshoff, A. Welther, M. T. Kessler, M. H. G. Prechtl and A. J. von Wangelin, Chem. Commun., 2014, 50, 2261–2264 RSC.
- W. Darwich, C. Gedig, H. Srour, C. C. Santini and M. H. G. Prechtl, RSC Adv., 2013, 3, 20324–20331 RSC.
- P. Migowski, G. Machado, S. R. Texeira, M. C. M. Alves, J. Morais, A. Traverse and J. Dupont, Phys. Chem. Chem. Phys., 2007, 9, 4814–4821 RSC.
- P. Migowski, S. R. Teixeira, G. Machado, M. C. M. Alves, J. Geshev and J. Dupont, J. Electron Spectrosc. Relat. Phenom., 2007, 156, 195–199 CrossRef.
- J. Dupont and J. D. Scholten, Chem. Soc. Rev., 2010, 39, 1780–1804 RSC.
- P. Migowski and J. Dupont, Chem. – Eur. J., 2007, 13, 32–39 CrossRef CAS PubMed.
- M. H. G. Prechtl, M. Scariot, J. D. Scholten, G. Machado, S. R. Teixeira and J. Dupont, Inorg. Chem., 2008, 47, 8995–9001 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cc00499g |
| This journal is © The Royal Society of Chemistry 2016 |