
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicrofluidics for the detection of minimal residual disease in acute myeloid leukemia patients using circulating leukemic cells selected from blood†
Joshua M.
Jackson
ab,
James B.
Taylor
ab,
Małgorzata A.
Witek
 bc,
Sally A.
Hunsucker
d,
Jennifer P.
Waugh
e,
Yuri
Fedoriw
de,
Thomas C.
Shea
e,
Steven A.
Soper
*abc and
Paul M.
Armistead
*de
bc,
Sally A.
Hunsucker
d,
Jennifer P.
Waugh
e,
Yuri
Fedoriw
de,
Thomas C.
Shea
e,
Steven A.
Soper
*abc and
Paul M.
Armistead
*de
aDepartment of Chemistry, UNC-Chapel Hill, USA. E-mail: ssoper@email.unc.edu
bCenter for Biomodular Multi-scale Systems for Precision Medicine, UNC-Chapel Hill, USA
cDepartment of Biomedical Engineering, UNC-Chapel Hill, USA
dUniversity of North Carolina Lineberger Comprehensive Cancer Center, UNC-Chapel Hill, USA. E-mail: paul_armistead@med.unc.edu
eDepartment of Medicine, UNC-Chapel Hill, USA
First published on 26th October 2015
Abstract
We report a highly sensitive microfluidic assay to detect minimal residual disease (MRD) in patients with acute myeloid leukemia (AML) that samples peripheral blood to search for circulating leukemic cells (CLCs). Antibodies immobilized within three separate microfluidic devices affinity-selected CLC subpopulations directly from peripheral blood without requiring pre-processing. The microfluidic devices targeted CD33, CD34, and CD117 cell surface antigens commonly expressed by AML leukemic cells so that each subpopulation's CLC numbers could be tracked to determine the onset of relapse. Staining against aberrant markers (e.g. CD7, CD56) identified low levels (11–2684 mL−1) of CLCs. The commonly used platforms for the detection of MRD for AML patients are multi-parameter flow cytometry (MFC), typically from highly invasive bone marrow biopsies, or PCR from blood samples, which is limited to <50% of AML patients. In contrast, the microfluidic assay is a highly sensitive blood test that permits frequent sampling for >90% of all AML patients using the markers selected for this study (selection markers CD33, CD34, CD117 and aberrant markers such as CD7 and CD56). We present data from AML patients after stem cell transplant (SCT) therapy using our assay. We observed high agreement of the microfluidic assay with therapeutic treatment and overall outcome. We could detect MRD at an earlier stage compared to both MFC and PCR directly from peripheral blood, obviating the need for a painful bone marrow biopsy. Using the microfluidic assay, we detected MRD 28 days following one patient‘s SCT and the onset of relapse at day 57, while PCR from a bone marrow biopsy did not detect MRD until day 85 for the same patient. Earlier detection of MRD in AML post-SCT enabled by peripheral blood sampling using the microfluidic assay we report herein can influence curative clinical decisions for AML patients.
Introduction
Leukemia is triggered by hematopoietic progenitor cells in the bone marrow that become mutated and clonally expand into leukemic blasts that do not fully differentiate into normally functioning blood cells.1 Leukemia can be divided into four major types by: (i) the rate of disease progression, acute (rapid, within weeks to months) or chronic (slow, within months to years); and (ii) the type of malignant cells, either originating from the lymphoid or myeloid lineage. Acute myeloid leukemia (AML) is the most common adult leukemia with ∼20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 new cases expected in 2015 with a 5-year survival rate of only 25%.2 The primary cause of death for AML patients is due to disease relapse.1
000 new cases expected in 2015 with a 5-year survival rate of only 25%.2 The primary cause of death for AML patients is due to disease relapse.1
Patients diagnosed with AML are treated with chemotherapy if they are considered fit enough for treatment with the goal of inducing complete remission, defined as a normal appearing bone marrow biopsy (<5% leukemic cells) and normal circulating blood counts. However, even when the patient is in complete remission, low levels of leukemic cells persist that are likely to have chemotherapy-resistance and stem cell properties. This minimal residual disease (MRD) can re-initiate AML within weeks to months.1,3 The consequences are significant: of 1108 patients in complete remission after therapy, 60% relapsed of which only 11% survived after 5 years.4 If clinicians can pinpoint when a patient's MRD begins progressing towards the rapid expansion to relapse, preemptive therapies can be taken with better patient outcome. Unfortunately, the classification of AML patients by risk according to age, white blood cell count, therapy response, and cytogenetic and genotypic abnormalities, if any,4–9 falls short of the ability to properly monitor MRD in individual patients. If MRD could be detected with high sensitivity at an early stage, the corresponding assay could assist in guiding therapy to enable precision medicine resulting in better patient outcome.5
A potentially curative therapy for AML is hematopoietic stem cell transplant (SCT), where a donor's hematopoietic stem cells, either in the peripheral blood or purified bone marrow, are introduced into the patient. The donor's graft transplanted into the recipient's bone marrow undergoes normal hematopoiesis and induces a donor-derived, T cell-mediated, anti-leukemia immunity, commonly called the graft-versus-leukemia effect. These transplants are typically reserved for patients at high risk of disease relapse, because while SCT lowers relapse risk, it is associated with a high treatment mortality (∼25%).10,11 Intense chemotherapy is needed to minimize AML relapse prior to grafting. In addition, T cell suppression is necessary to reduce graft rejection and graft-versus-host disease. These treatments are physically taxing and leave the patient susceptible to a host of foreign and dormant infections, leading to SCT's high rate of morbidity.
If relapse occurs after SCT, there are interventions that can be curative. A rapid withdrawal of immunosuppression and the infusion of donor lymphocytes can instigate an acute graft-versus-leukemia response that can result in sustained long term remission.9,12 However, the success of a donor lymphocyte infusion is intrinsically dependent upon the level of residual leukemia at the time of treatment. Donor lymphocyte infusion was only successful (overall survival >2 years) for 15% of patients with active AML, but the treatment was successful in 55% of patients when administered while in remission.9 Thus, the ability to detect low but rising levels of MRD that signal the start of relapse is not only prognostically important, but can enable clinicians to implement therapy earlier that can improve patient outcome.3
An ideal MRD assay would be sensitive to low MRD levels and suitable for frequent analysis. This goal has been hindered by two issues: (i) unlike other leukemias, AML's inter-patient heterogeneity is immense; there is no characteristic genetic mutation or aberrant protein expression pattern for all AML patients,3 thus complicating the broad applicability of PCR, fluorescence in situ hybridization (FISH) or multi-parameter flow cytometry (MFC) to test for MRD. (ii) AML relapse is rapid; it was calculated that long-term 42 day sampling intervals would be a minimum frequency to predict 75% of relapses.13
Assessing MRD from leukemic cells that circulate in a patient's peripheral blood (circulating leukemic cells – CLCs) is a viable option for achieving sensitive MRD detection that can be done on a frequent basis due to the minimally invasive nature of the test.3 While PCR-based MRD assays offer favorable analytical detection limits,14 ranging from 1 CLC in 104–106 normal blood cells, PCR assays are applicable to <50% of all AML patients due to AML's genetic heterogeneity.15,16
MFC is an approach that identifies aberrant expression of surface proteins (leukemia associated phenotypes), on mutated myeloid cells, which are present on almost all (>90%) AML cells.1,3,16 Two general leukemic associated phenotype patterns are: (i) immature, myeloid cells (common normal myeloid markers are CD33, CD34, and CD117) with lineage infidelity (abnormal co-expression of a myeloid and lymphoid marker, such as CD7); or (ii) asynchronous antigen expression (abnormal co-expression of an immature and mature myeloid marker, such as CD56).5 Two main limitations exist regarding MFC MRD analysis: (i) the assay requires flow cytometers with >5 colors and highly skilled operators to correctly identify a cluster of 20 CLCs amongst 200000 total bone marrow cells,5 although this detection limit varies between operators (bone marrow sensitivity ranges from 10−3–10−4).17 (ii) MFC is noise-limited for rare event analysis18,19 and is significantly affected by peripheral blood cells;5 hence bone marrow is generally required unless disease burden is very high. For example, MFC MRD analysis of peripheral blood yielded a log reduction in sensitivity compared to a bone marrow biopsy sample3 with a considerable number of false negatives below 1% MRD.20 Thus, MFC is only moderately sensitive and requires highly invasive bone marrow biopsies that limit test frequency due to the patient's physical burden.
Microfluidics has demonstrated success for the detection of epithelial solid cancers by isolating and interrogating circulating tumor cells (CTCs) that are extremely rare in blood (1–3000 CTCs per 109 normal blood cells).21 In particular, we have reported the use of a sinusoidal microfluidic device to isolate CTCs in pancreatic,22–24 ovarian, colorectal, breast, and prostate cancer.24 The sinusoidal microfluidic technology has demonstrated purities >90%,22–24 recoveries ∼97% for a model MCF-7 cell line25 and 80–100% clinical sensitivity for epithelial cancers when analyzing CTCs.24
The sinusoidal microfluidic device works on the principle of positive-affinity selection, where monoclonal antibodies (mAbs) are bound to a microfluidic device's surfaces and specifically select target antigen-bearing cells (Fig. 1B and C). In operation, these devices require no sample pre-processing. Peripheral blood is hydrodynamically infused into the device that contains ≥50 parallel microchannels,22,26 each possessing a 25 μm width and a sinusoidal architecture (Fig. 1A and B) that promote extensive cell interactions with mAbs covalently tethered to the microfluidic surfaces (Fig. 1B).23,27 Antigen-expressing cells are retained by the mAb-coated surfaces while all other blood components are removed from the device by high fluidic shear that disrupts weak, non-specific interactions (Fig. 1C).23,27,28 These fluidic shear forces, are an order of magnitude higher than in comparable microfluidic technologies22,23 and are not present in traditional magnetic bead isolation assays, which generally present low purities (0.01–0.1% for the CellSearch™ CTC selection platform)21 that complicate immunophenotyping and/or molecular analysis.27,28 The sinusoidal microfluidic technology has achieved the highest purities to date for rare cell isolation.22–24 Because of the high purity provided by the device, after isolation the cells can be immunostained and imaged22,23,28 or lysed to analyze mRNA expression,29 gDNA mutations,28 or membrane proteins,26 without the deleterious effects of high levels of contaminating cells into the assay.
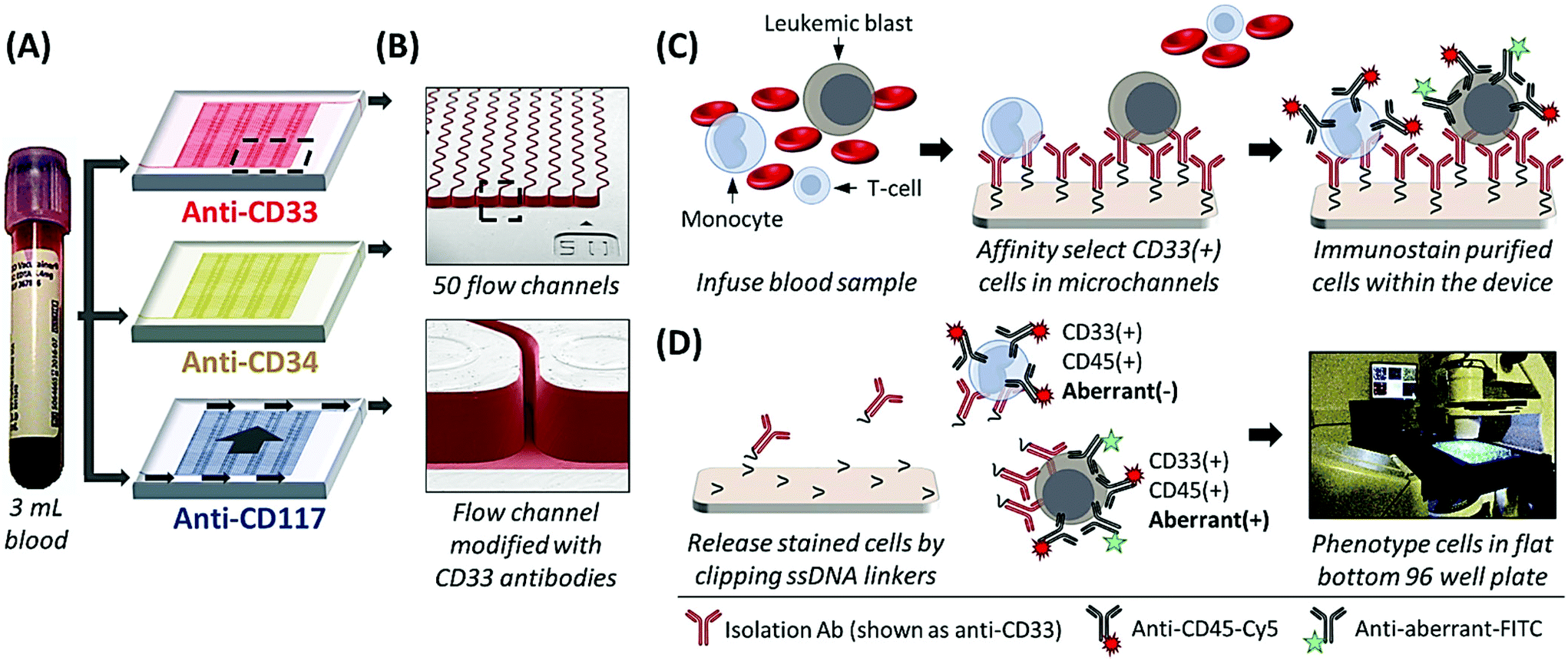 | ||
| Fig. 1 (A) Whole blood is processed through three microfluidic devices modified with mAbs specific for CD33 (red), CD34 (yellow), and CD117 (blue) expressing cells. Arrows indicate direction of blood flow through the device. (B) SEMs of the sinusoidal channel array (50 channels in the array) and the entrance of the single channel that addresses all sinusoidal channels. mAb-coated surfaces were false-colored red to represent an anti-CD33 mAb device. (C) Schematic of the affinity isolation assay. Antigen expressing cells (here CD33(+) cells used as an example) bind to surface-tethered mAbs and are retained in the device while other blood components are passed through the device. Selected cells are then immunostained against CD45 and the aberrant marker (e.g., with anti-CD7 or anti-CD56 fluorescent mAbs), followed by fixation and nuclear staining. (D) Selected cells are released from the capture surface and carried hydrodynamically into flat-bottomed wells, where the cells are imaged by semi-automated fluorescence microscopy. CLCs are identified by positive aberrant staining (aberrant(+)) and positive CD45 and DAPI staining, whereas other blood components only show CD45 and DAPI staining (aberrant(−)). | ||
Herein, we present a unique assay format and pilot clinical study where AML patients recovering from SCT were longitudinally tracked by isolating CD33, CD34, and CD117 expressing CLCs using three sinusoidal microfluidic devices arranged in a parallel configuration in which one blood sample was fluidically split into the three separate microfluidic devices (see Fig. 1A), co-staining against a patient-specific aberrant antigen, and immunophenotyping the cells by semi-automated fluorescence microscopy. As a minimally invasive blood sample (3 mL) was required, patients could be sampled frequently compared to a bone marrow biopsy to detect earlier the onset of relapse. We compared the results from the microfluidic assay to conventional MRD monitoring, which consisted of microscopy, MFC, PCR, and FISH analysis of bone marrow biopsy samples and, in cases where the disease burden was high, peripheral blood. MRD tracking by microfluidic CLC surveillance matched well with both therapeutic treatment and patient outcome but could detect the onset of relapse much earlier compared to PCR and MFC.
Experimental methods
Reagents and materials
Microfluidic devices were fabricated using 6013S-04 cyclic olefin copolymer (TOPAS Advanced Polymers), capillary tubing (365 μm OD, 150 μm ID, Polymicro Technologies), reagent-grade isopropyl alcohol, and Micro-90® (Sigma-Aldrich). Capillary connectors used Inner-Lok™ union capillary connectors (Polymicro Technologies) and barbed socket Luer Lock™ fittings (3/32′′ ID, McMaster-Carr). mAb immobilization reagents included: 1-ethyl-3-[3-dimethylaminopropyl] carbodiimide hydrochloride (EDC), N-hydroxysuccinimide (NHS), 2-(4-morpholino)-ethane sulfonic acid (MES) buffer (pH 4.8), phosphate buffered saline (PBS, pH 7.4), and bovine serum albumin (BSA, 7.5%) in PBS (Sigma-Aldrich); sodium carbonate anhydrous (EMD Millipore); sodium hydroxide (Fisher Scientific); nuclease-free water (BioExpress); DL-1,4-dithithreitol (DTT, molecular biology grade, Acros Organics); sulfosuccinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate (sulfo-SMCC), Zeba spin desalting columns (7 K MWCO), and a protein stabilizing cocktail (Thermo Scientific); and HPLC-purified, single-stranded, oligonucleotide linkers with 5′-amino and 3′-disulfide modifications with an internal dU residue (5′-NH2-C12-T8CCC TTC CTC ACT TCC CTT T-U-T9-C3-SS-C3OH, Integrated DNA Technologies). Other reagents included formaldehyde (Fisher Scientific), Triton-X100 (Sigma-Aldrich), 4′,6-diamidino-2-phenylindole (DAPI, eBioscience), and Uracil Specific Excision Reagent (USER™, New England Biosciences). Nuclease-free microfuge tubes (BioExpress) and centrifuge tubes (Corning) were used for preparation and storage of all samples and reagents.All mAbs used in this study were mouse anti-human. For cell isolation, anti-CD33 (clone WM53), anti-CD34 (clone 561, class III epitope), and anti-CD117 (c-kit, clone 104D2) mAbs (Biolegend) were used. Direct immunostaining mAbs were anti-CD45-AlexaFluor®647 (clone HI30), anti-CD7-FITC (clone CD7-6B7), anti-CD38-AlexaFluor®488 (clone HIT2), and anti-CD56-AlexaFluor®488 (NCAM, clone HCD56) from Biolegend. Indirect immunostaining mAbs were anti-CD7-biotin (clone MG34) and anti-CD33-biotin (clone HIM3-4) from Thermo Scientific and anti-CD34-biotin (clone 581, class III epitope), anti-CD56-biotin (NCAM, clone HCD56), and anti-CD117-biotin (c-kit, clone 104D2) from Biolegend. All biotinylated mAbs were counter-stained with streptavidin-DyLight®550 (Thermo Scientific). Fluorescent calibration beads, CELLQUANT Calibrator kit, were purchased from BioCytex and were prepared according to the manufacturer.
Cell selection device fabrication
Hot embossing was used to fabricate the microfluidic device in cyclic olefin copolymer as described previously.30,31 Briefly, mold masters were prepared using high precision-micromilling (KERN 44, KERN Micro- und Feinwerktechnik GmbH & Co. KG) and carbide bits (Performance Micro Tool).30 Hot embossing was performed using a HEX03 machine (Jenoptik Optical Systems GmbH) at 155 °C and 30 kN force for 120 s. Embossed devices were diced, cleaned with 10% Micro-90®, IPA, and DI water, assembled with capillary tubing and coverslip, clamped between glass plates, and thermal fusion bonded at 130 °C for 60 min. All protocols for activating the devices with cleavable oligonucleotide linkers and the cell selection mAbs were performed as previously described (ESI†).23,24,28Processing clinical samples
AML patients being prepared for allogeneic SCT were recruited according to a clinical protocol approved by the University of North Carolina's Institutional Review Board (IRB). All patient treatments and conventional MRD test results were kept blinded until conclusion of the study. Blood specimens were collected into BD Vacutainer® EDTA tubes and remained on a nutator until processing (<4 h). Assembled microfluidic devices were thoroughly washed with >1 mL 0.5% BSA/PBS buffer (50 μL min−1) to remove unbound mAb and passivate microchannel surfaces. Blood was transferred into disposable Luer Lock™ syringes (BD Biosciences) using a BD vacutainer female Luer transfer adapter. Filled syringes were connected to the devices via capillary connectors and processed through the microfluidic devices at 25 μL min−1 (2 mm s−1).23,25 Immediately thereafter, devices were rinsed with >1 mL 0.5% BSA/PBS (50 μL min−1, 4 mm s−1) to remove any nonspecifically bound cells.Immunostaining, cell release, and imaging
After blood processing and rinsing, isolated cells were incubated at 4 °C for 30 min with a cocktail of anti-CD45-Cy5 mAb and mAbs targeting the aberrant marker (CD7 or CD56) either conjugated to FITC or biotin. In some cases, the devices were stained with biotinylated mAbs targeting the isolation marker (i.e., anti-CD33-biotin on the CD33 cell isolation device). All biotinylated mAbs were indirectly stained via streptavidin-Cy3 incubation (4 °C, 30 min). After mAb incubation, cells were rinsed with 250 μL PBS, sequentially fixed, porated, and nuclear-stained via 10 min incubations with 2% formaldehyde, 0.1% Triton-X100, and 1 μg mL−1 DAPI. The devices were washed with 250 μL PBS and released by incubation with the USER™ enzyme (4 U per 10 μL PBS) for 45 min at 37 °C to cleave the oligonucleotide linker.28 Cells were then washed from the chip with PBS.The released cells were collected into separate wells of a flat-bottom 96 well plate (Argos Technologies) sealed with an optically clear Microseal® B adhesive film (Bio-Rad Laboratories), which was punctured just prior to release so that the device's capillary could be fed into the well. Before visualization, the plate was centrifuged for 7 min at 250 rcf and the wells were imaged using a Zeiss Axiovert 200 M microscope equipped with a 20× objective (0.4 NA, EC Plan NeoFluar®), an XBO 75 lamp, DAPI/FITC/Cy3/Cy5 filter sets (Omega Optical), a Cascade 1 K EMCCD (Photometrics) camera, and a MAC 5000 stage (Ludl Electronic Products), all of which were computer-controlled via Micro-Manager.32 DAPI, FITC, Cy3, and Cy5 exposure times were 50, 1000, 2500, and 3500 ms, respectively. Each well was imaged via Micro-Manager's grid collection software, and the image sets were stitched and analyzed via a custom ImageJ macro, which identified nuclei and displayed fluorescence panels for phenotyping. FITC-labeled calibration beads were imaged under the same conditions, and beads differing in antibody binding capacities were identified by surveying fluorescence intensities on the FITC color channel of the imaging microscope.
Results and discussion
Sample processing
The microfluidic assay used a minimally invasive peripheral blood sample that permitted frequent testing, which for this study consisted of biweekly or less MRD testing through the first 100 days of post-SCT and monthly thereafter. For most samples, ∼3 mL of peripheral blood was processed with 1 mL sent through one of three 50-channel, sinusoidal microfluidic devices that were modified with mAbs targeting CD33, CD34, and CD117 (Fig. 1). The blood sample, which was not fixed, fractionated, or diluted, was processed through each chip over the course of 40 min; although this time frame is flexible because the processing time can be reduced by scaling to >250 sinusoidal channels in each device.22,26 Isolated CLCs were then identified by staining against the aberrant marker (i.e., CD7 or CD56) and leukocyte specific antigen CD45. CD45 staining precluded non-hematopoietic cell types such as CD34(+) circulating endothelial cells.33 Isolated cells were then fixed and DAPI-stained, and each subpopulation (CD33, CD34 or CD117) was released for imaging into separate wells of a flat-bottom titer plate. Cell release was enabled by enzymatically cleaving DNA oligonucleotide bifunctional linkers containing a uracil residue that anchored mAbs to the activated microfluidic surfaces. We have recently optimized and validated the entirety of this assay for several cell lines and clinical samples,34 including the CD34(+) KG-1 AML cell line for which the assay achieved 65% recovery for the KG-1 cells and >80% release efficiency after fixation.28 While the recovery for the MCF7 cell line using EpCAM mAbs has been reported to be ∼97% for the sinusoidal chip, the smaller size of the KG-1 cell line account for the differences in the recovery.Fluorescence microscopy was chosen for immunophenotyping the selected cells because the cell abundance was low (<20 mL−1) in some cases, making it difficult to secure reliable results using MFC. The microscope imaging was semi-automated; the microscope stage was computer-controlled to automatically capture images of the wells for all fluorescence color channels. Custom image-processing macros were composed to stitch the images together, identify nuclei and display fluorescence panels. The user could call each cell's phenotype in a manner similar to a commercial CTC system.35 Cells that stained DAPI(+) and CD45(+) were identified as CLCs if they stained aberrant(+) (Fig. 2). CLC size (10–30 μm) and high nuclear-to-cytoplasm ratio were not regarded as absolute criteria. Conventional microscopic preparation flattens and enlarges cells when plating the coverslip.36,37 In general, CLCs were ∼10–15 μm in diameter, similar to KG1 cells.28
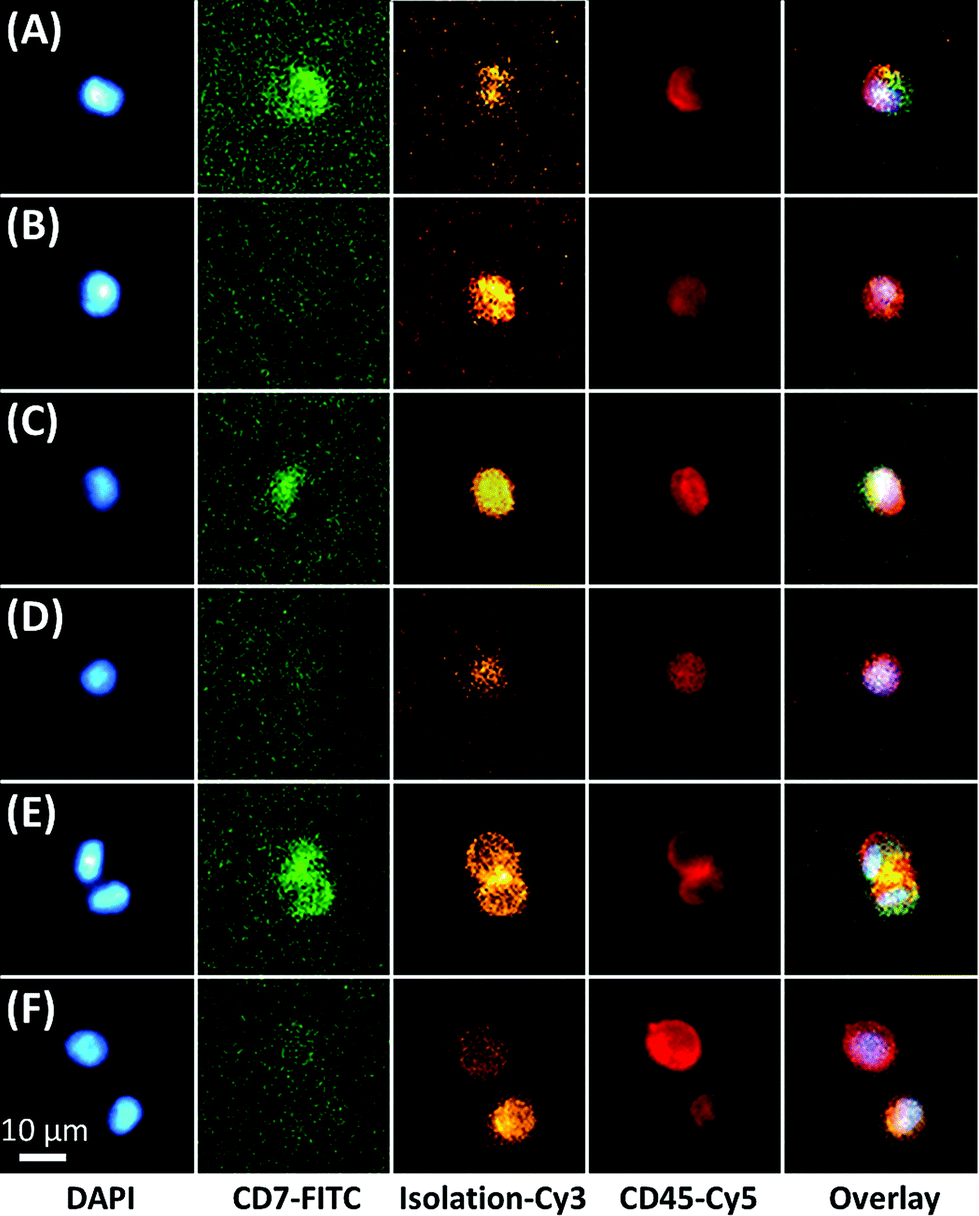 | ||
| Fig. 2 Immunophenotyping of aberrant(+) CLCs and aberrant(−) cells isolated by targeting (A,B) CD33, (C,D) CD34, and (E,F) CD117, respectively. All cells were DAPI(+)/CD45(+) and positive for at least one of the isolation markers (CD33, CD34, or CD117). All images were taken from Pt #1 (CD7 aberrant marker) 85 days post-SCT. The cells in this panel were stained using DAPI (nucleus), and mAbs directed against CD7 (FITC), CD45 (Cy5) and finally, the selection marker (CD33, CD34, CD117; Cy3). The images were acquired using an inverted microscope and the appropriate filter cube. | ||
The three devices were arranged in parallel rather than a serial configuration because co-expression of the isolation markers was observed for some AML cell lines (Fig. S1†) and was considered likely in CLCs isolated from clinical samples as well. Antigen co-expression could bias results secured from the first device in the series with a large number of cells while depleting target cells from devices positioned downstream. The parallel arrangement can also enable separate interrogation of the CLC subpopulations to determine drug resistance for each subpopulation resulting from chemotherapy and other factors.38,39 For example, circulating leukemic stem cells, which are the only leukemic cells capable of propagating AML,38 would be isolated in the anti-CD34 and/or anti-CD33 device. These stem cells could be phenotypically identified by CD38 and CLL-1 expression38,40 and further interrogated while simultaneously monitoring the leukemic blast progeny and normal blood cells.
Microfluidic affinity selection sensitivity and specificity
We assessed specificity of the microfluidic selection process by co-staining against the isolation marker, either CD33, CD34, or CD117 (Fig. 2). The observed specificity (count of cells staining positive for the isolation marker divided by the total cell count) was 88–99% with 2–33 cells per mL not showing discernable expression of the selection antigen. These results agree with our previous reports using this microfluidic in terms of the selection of CTCs in whole blood samples.41,42The efficiency of affinity selection depends on the cell's antigen expression and the density of surface-confined mAbs.24,27 Also, as the cell rolls along the microfluidic surfaces, the probability that a moving antigen will bind to the surface-confined mAb depends on the cell's forward velocity, the associated residence time of the mAb and antigen in the reaction radius, the Ab-antigen binding kinetics,43 and the steric likelihood that the mAb interacts with the targeted epitope (analogous to a steric factor in collision theory).
The overall probability of cell recovery can be improved by accumulating a large number of binding events for a single cell. In the sinusoidal microfluidic device, this accumulation is engineered into the device by using continuous sinusoidal channels that offer rolling distances >250 μm as opposed to discrete surfaces provided by micropillars.24 For example, assuming a monolayer of mAbs on the surface, a cell rolling for 250 μm would encounter in excess of 16000 mAb collisions. Because the number of binding events also scales with the number of antigens, recovery is dependent on the expression of the antigen and the target cell's antigen density. This has been empirically observed by several groups using cell lines with variable antigen expression.44
There is a fundamental lower limit to antigen expression for cell recovery, which is set by an insufficient number of mAb-antigen complexes to hold the cell to the surface against the blood's fluidic shear force. Bell45 provided a theoretical framework to assess the critical force at which a cell will detach from a surface (Fc) when it is bound by Nb bonds, each with a critical force of Fb and an equilibrium constant K (taken as 106 M−1) given by;
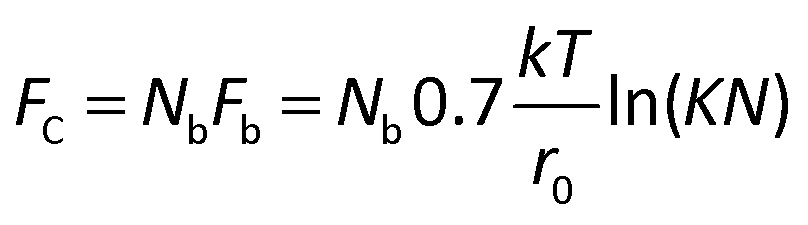 | (1) |
For a 12 μm diameter cell with a surface area of ∼450 μm2, the recovery limit for antigen expression is approximately 700 antigens per cell. In contrast, it is technically difficult to immunophenotype cells with only 700 antigens by fluorescence without a highly sensitive microscope. For example, we imaged ∼12 μm beads that were coated with different levels of anti-IgG antibodies and functionalized with IgG-FITC, ranging in antigen binding capacity from 940 to 259000 (Fig. 3). Beads with an antigen binding capacity of 7000 were detected but with weak signal. Beads with an antigen binding capacity of 940 could not reliably be detected from background.
 | ||
| Fig. 3 (A) FITC fluorescence, (B) brightfield, and (C) overlay images of calibration beads with different antigen binding capacity levels (see (C) annotations for the approximate load of the fluorescent antibodies per bead). Image contrast settings were selected to highlight low intensity fluorescence; brightly fluorescent beads were not saturating the CCD. FITC exposure times were identical to those used for CLC identification. | ||
Thus, it is possible to physically isolate a cell by positive-affinity selection but incorrectly classify the cell as negative for the marker by immunofluorescence (albeit, we did not consider the efficiency of isolation, which we discuss elsewhere24,27). Close inspection of the top left cell in Fig. 2F (FITC panel) shows extremely faint fluorescence signal similar to the 940 antigen binding capacity beads in Fig. 3, but this was not counted as a CLC. Also, the specificity reported, which is based on staining for the isolation marker with good results (88–99% with 2–33 non-target cells per mL), does not contradict previously reported purities for CTC isolation (3.2 ± 3.4 nonspecific leukocytes per mL blood, averaged from 66 samples) that identified nonspecific binding leukocytes using CD45 (∼200000 molecules per cell).22,23,28,46
Patient characteristics
Five AML patients (Pts #1–5) undergoing allogeneic SCT were recruited for post-SCT microfluidic AML MRD monitoring. Characteristics of the patients, such as cytogenetic/molecular risk, leukemic associated phenotype aberrant marker, and pre-SCT characteristics regarding disease burden are available in Table S1.† Full leukemic associated phenotype panels and aberrant markers that were identified by MFC are provided in Table S2.† An optimal aberrant marker that was expressed on a significant portion of the patient's leukemic blasts and not on normal blood cells was chosen to identify CLCs for each patient (further discussion in the ESI†). The assay was designed to accommodate any aberrant marker; however, the five patients enrolled in this study were a prior found to express either CD7 or CD56. It is possible that the leukemic associated phenotype could change during disease progression, which most often involves up-regulation of the isolation markers (CD33, CD34, CD117)1,47 that would improve assay recovery.24 However, CLCs may also lose or change the aberrant marker (i.e., CD7 or CD56).47 Thus, cells that stain aberrant(+) are referred to as CLCs while aberrant(−) cells are regarded as cells that include normal blood cells and potentially other leukemic cells even if they express the selection antigen (CD33, CD34 or CD117). In future studies, loss of the aberrant marker can be accommodated by employing wider staining panels that target all aberrant antigens (Table S2†) to provide more complete AML coverage.47 The rare presence of normal and immature CD34(+) cells that co-express CD7 has been noted during marrow regeneration and T-lymphopoiesis.48 These cells are unlikely to be present in peripheral blood after full engraftment (∼14 days post-SCT) and thus, unlikely to affect CLC identification by the microfluidic assay. The presence of either CD33(+) or CD117(+) cells that co-express CD7, however, should never occur in normal marrow or peripheral blood.CLCs in clinical samples and early signs of impending relapse by microfluidic MRD surveillance
All data for each patient sample including CLC and aberrant(−) cell counts for each device are provided in Table S3.† The five AML patients were sampled 39 times by the microfluidic assay; in comparison, only eight microscopy, PCR and/or MFC tests were administered over the same sampling interval because of the need for requiring the patient's bone marrow in most cases. Three healthy donors were also analyzed (Table S4†). An average of 151 ± 89, 19 ± 13, and 108 ± 103 aberrant(−) cells per mL blood and 2 ± 2, 0 ± 1, and 1 ± 1 cells nonspecifically stained aberrant(+) cells were isolated in the CD33, CD34, and CD117 devices, respectively, for these normal blood samples. Based on a 99% confidence level (3× the standard deviation), we established a threshold of 8, 3, and 5 aberrant(+) cells for MRD positivity in the CD33, CD34, and CD117 subpopulations, respectively.At 137, 254, and 178 days post-SCT, Pts #3, #4, and #5, respectively, were alive and showed no signs of relapse. Pts #1 and #2 relapsed and died 95 and 118 days post-SCT, respectively. For both patients that relapsed, the microfluidic assay detected MRD well before PCR, MFC, microscopic or FISH-based MRD testing performed on the same patient and detected patterns in MRD progression that may have indicated the onset of relapse (see Table S3† and the associated heat map). However, the MRD assessments made by the microfluidic assay agreed well to the result secured using less frequent PCR analysis when these tests were performed on the patients. Tracked MRD progressions are shown in Fig. 4 and 5 for Pt #1 and Pts #2–5, respectively, with annotations of MRD test results and antiviral treatments given in the figure.
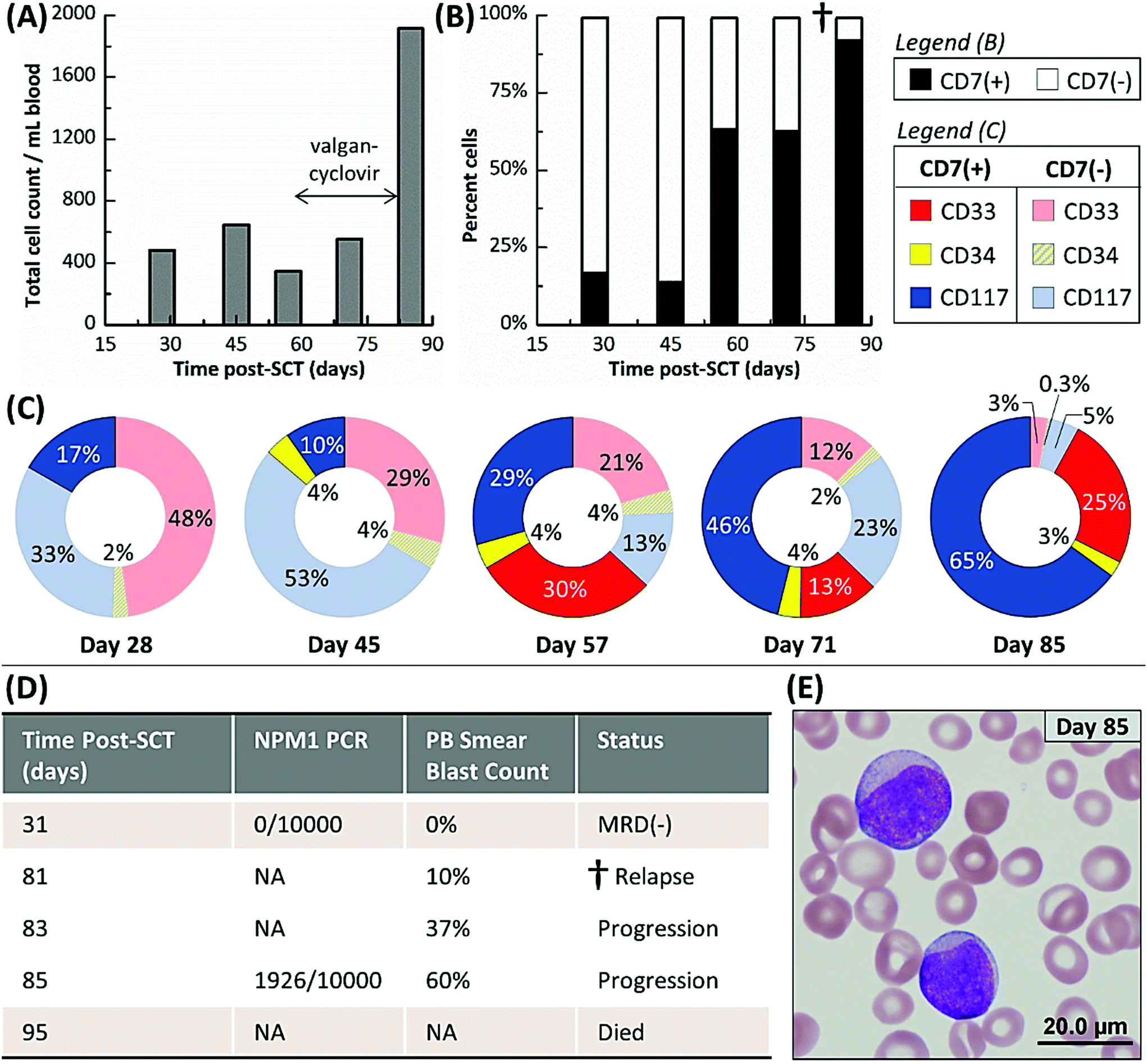 | ||
| Fig. 4 (A–C) Microfluidic monitoring of Pt #1 from 28 to 85 days post-SCT. (A) Total cell count, which represents the cumulative number of cells counted from all three subpopulations (CD33, CD34 and CD117) selected in the three separate microfluidic devices and all phenotypes (aberrant(+) and aberrant(−)). (B) Cell counts of aberrant(+) and aberrant(−) phenotypes but cumulative for all microfluidic devices used in the assay. (C) Cell counts of aberrant(+) and aberrant(−) cells discerned by each isolation antigen (CD33, CD34, or CD117). (D) Results for PCR (NPM1 gene) and peripheral blood smear MRD assays. Relapse was confirmed on day 81 by a peripheral blood smear test (dagger mark). This patient died 95 days post-SCT. (E) An image of the Wright-Geimsa stained peripheral blood smear from day 85, which showed two blasts with open chromatin and weak intensity from the cytoplasm (magnification was 100×). PB = Peripheral Blood; NA = not applicable. | ||
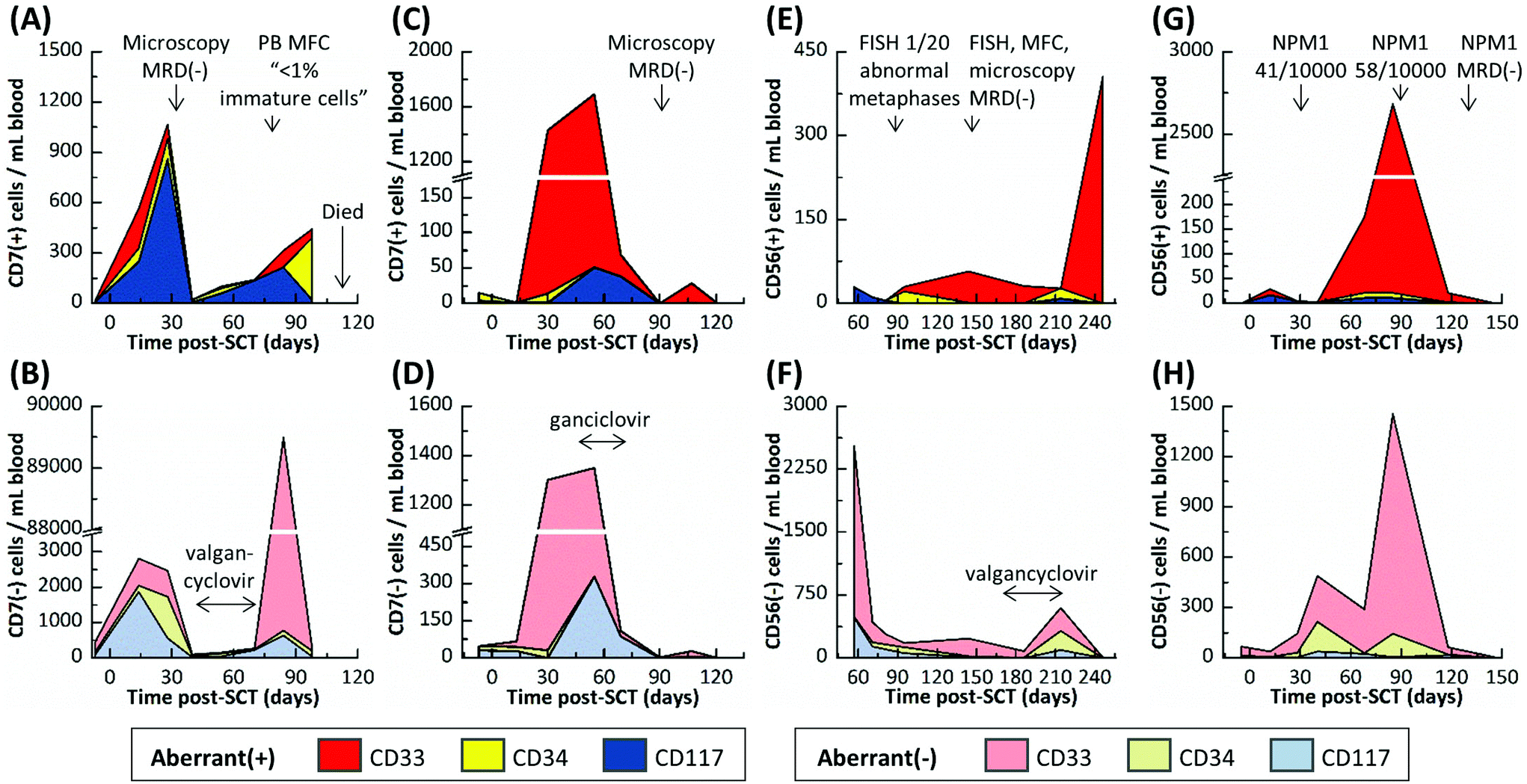 | ||
| Fig. 5 CLC counts and aberrant(−) cells for (A,B) Pt #2, (C,D) Pt #3, (E,F) Pt #4 and (G,H) Pt #5. Cell counts are color coded according to the targeted marker used for CLC selection. Results from FISH, PCR, blood smear and MFC MRD diagnostics, which used bone marrow biopsies unless noted otherwise, and the time frames for antiviral therapy are shown in the figures. Linear connections between events are for visualization purposes only. | ||
Active cytomegalovirus infections are common in SCT patients as the regulating lymphoid immune system remains suppressed to avoid graft-versus-host disease. In this study, Pts #1–4 experienced cytomegalovirus activation as detected by weekly PCR surveillance. These Pts were treated with antivirals (oral valgancyclovir or intravenous ganciclovir), which are known to cause myelosuppression, until cytomegalovirus was cleared. Cytomegalovirus replicates in myeloid cells and is in effect myelosuppressive, which is why it has been suggested that early cytomegalovirus infections can aid in long term remission. Cytomegalovirus may be cytotoxic to the MRD (virus-versus-leukemia effect) and/or cause myeloid cells to present antigens that induce a T-cell and/or natural killer cell attack (another graft-versus-leukemia mechanism).49
Fig. 4A shows Pt #1's total cell counts for CD33, CD34 and CD117 selection days 28 through 85 as determined by the microfluidic assay and Fig. 4B distinguishes these counts by aberrant staining. The microfluidic assay detected 17% CLCs for Pt #1 on day 28, whereas the NPM1 PCR assay of a bone marrow biopsy three days later was MRD(−) (Fig. 4D). The PCR assay's negative result indicated that the residual leukemic cells, at least those with the NPM1 mutation, were below the assay's detection limit (1 mutated gene in 10000 background copies). In the microfluidic MRD assay, aberrant(−) cells, which included normal donor cells, increased by ∼40% 17 days later while CLC levels remained approximately constant. In addition, the presence of aberrant marker (CD7) for the CD34 subpopulation was observed 45 days post-SCT. However, the total cell count decreased at day 57, perhaps due to cytomegalovirus activation (Fig. 4A), but CLC percentages rose to 63% with the observation of aberrant(+) cells in the CD33 subpopulation (Fig. 4B).
CLC counts were high after day 57 and spiked to 92% at day 85. Relapse was confirmed by a peripheral blood smear at day 81. Unfortunately, the disease burden was already high (10% of the bone marrow) and the AML burden approximately doubled every two days (Fig. 4D and E). Pt #1 died 95 days post-SCT. Considering myelosuppressive antiviral treatment from days 58 to 82, it may be suggested that the antiviral treatment delayed the rapid relapse progression that was observed after day 81 by microscopy (Fig. 4D). We retrospectively highlight the microfluidic assay's results at day 57 as a potential indicator of impending relapse for this patient characterized by increasing CLC levels and receding aberrant(−) cell numbers. Thus, the microfluidic assay was able to detect MRD 28 days and impending relapse 57 days following SCT, while microscopy detected relapse in a bone marrow biopsy at day 81 when disease burden was high.
Pt #2's MRD progression (Fig. 5A and B) from days 7 to 28 post-SCT was very similar to Pt #1's onset of relapse with CLC levels increasing and aberrant(−) cells receding at day 28. However, microscopic analysis was MRD(−) at day 30, and the microfluidic assay's cell counts dropped precipitously at day 40. Pt #2 was treated with antivirals for an active cytomegalovirus infection from days 41 to 69. During treatment, all cell counts recovered slowly and after lifting treatment, a surge of CD33(+)/aberrant(−) cells was observed at day 84 (Fig. S2†) that may be attributed to a “left shift” immune response to late-onset cytomegalovirus disease or severe physiological burden, where the marrow is stimulated to produce immature CD33(+)/aberrant(−) cells that spill into the peripheral blood (also supported by MFC analysis of peripheral blood on day 79 that indicated the presence of <1% immature cells). Regardless, CLC counts increased steadily, and the last sample for microfluidic analysis (day 98) was characterized by a low aberrant(−) cell count and high CLC count, most notably with CD34(+)/aberrant(+) CLCs comprising 54% of all selected cells. It is possible that the CD34(+)/CD7(+) subpopulation may have contained immature, non-leukemic blasts, which are sometimes observed in regenerating marrow;48 however, there were relatively few CD34(+)/CD7(+) cells at day 14, when the marrow may have been regenerating from initial engraftment and so we would expect the CD34(+)/CD7(+) subpopulation at day 98 to contain CLCs almost exclusively. Further, CD33(+) and CD117(+) cells aberrantly expressing CD7 (a T cell antigen) should never be observed in normal marrow, regenerating or otherwise, all of which suggested persistent leukemic MRD. Subsequent sampling of this patient was not possible as this patient died 118 days post-SCT.
Pt #3 had the most acute MRD progression with 1430 CLCs per mL (50% CLCs) developing at day 30 even though the patient was MRD(−) at day 13 as noted by blood smear assays using microscopy (Fig. 5C and D). This may reflect the patient's pre-SCT chemotherapy regimen, which was less intense than Pts #1 and #2 due to Pt #3's age (Table S1†). During antiviral treatment from days 41 to 69, the cell counts remained approximately the same as the day 30 results until day 69, when both CLC and aberrant(−) counts slowly declined. At 90 and 121 days post-SCT, Pt #3's samples were MRD(−) as was microscopic analysis at day 89.
Pt #4's MRD profile is rather unique, remaining MRD(+) but with low CLC counts (11–57 CLCs per mL) for 157 days except for one MRD(−) result at 81 days post-SCT (Fig. 5E and F). During this time, Pt #4's MRD was just detectable by FISH at day 88 but was not detected by several tests at day 145. Pt #4 incurred cytomegalovirus activation and was treated between days 174 and 214, and there was a notable increase in aberrant(−) cell counts at day 214. However, the next sample at day 246 showed a spike of CD33(+) CLCs and low aberrant(−) cell counts.
Pt #5 is the only patient that did not test positive for a cytomegalovirus infection during tracking for 146 days (Fig. 5H). Pt #5 had been consistently MRD(+) by NPM1-PCR, but with low disease burden. The microfluidic assay indicated consistently MRD(+) with CLC levels spiking at days 68 and 85 post-SCT but then sharply declining at day 118 (Fig. 5G). The microfluidic assay at day 146 was MRD(−), as was NPM1 PCR at day 132. The aberrant(−) cell counts correlated with the CLC counts (Fig. 5H); we speculate a graft-versus-leukemia response occurred.
In summary, Pts #1–3 had very high CLC counts and experienced the same viral activation within a few months post-SCT. These patients represent three divergent scenarios. Pt #1 rapidly progressed towards relapse; Pt #2 experienced a recession of all myeloid counts, presumably due to cytomegalovirus activation, but then relapsed; and Pt #3 CLC counts receded almost entirely and is currently in complete remission. While we sampled the patients at a sufficient frequency to observe the reported trends, we still have limited data for each sample to confidently determine the nature of these trends. However, we can speculate that virus-versus-leukemia effects played a role in the progression of Pts #2 and #3. Pt #5 was similar in progression to Pt #3 yet did not experience a viral infection. It is possible that the recession of Pt #5's MRD was due to graft-versus-leukemia effects that acted similarly to the proposed virus-versus-leukemia mechanism.49 Lastly, Pt #4 was anomalous to the other patients, progressing to high CLC counts and experiencing a viral infection far later (>six months post-SCT).
Shifts in CLC subpopulations through relapse
Frequent monitoring of CLC subpopulations can provide a real time insight into patient-specific MRD progression. It has been observed that both genetic38,39 and phenotypic1,47 evolution occurs as residual leukemic cells experience selective pressures, which range from chemotherapy to nutrient, oxygen, and space deprivation38 and likely from interplays between residual leukemia, the grafted immune system, infections, and clinical treatments. All of these variables contribute to heterogeneous clonal subpopulations that compete towards forming dominant AML clone(s) that are, in effect, relapse.38 Previous methods have been limited for retrospective comparisons of the primary and relapsed tumors. But, with the microfluidic assay and its high sensitivity even when sampling peripheral blood, it may be possible to monitor acute clonal responses to selective pressures.For this reason, the CLC and aberrant(−) subpopulations (differing by the target isolation antigen: CD33, CD34, or CD117) were independently analyzed by selecting them in different devices. While it is unlikely all CLC subpopulations are mutually exclusive due to co-expression of the targeted antigen, there may be CLC subpopulations that solely express one antigen or have very weak expression of the other antigens (Fig. S1†). One case in point is Pt #1 (Fig. 4C): from days 45 to 85, the CD34(+) CLCs remain fairly constant at 3–4%; CD117(+) CLCs steadily increased from 10% to 65%; and CD33(+) CLCs appeared at day 57 but their number density fluctuated thereafter. Similar patterns were not observed in the aberrant(−) subpopulations (Fig. S3†), which were isolated in the same devices as the CLC subpopulations. Thus, it is most likely that we have observed genetically distinct CLC clonal subpopulations or that the gene expression/translation of the CLCs is highly variable.
The CLC subpopulations in each patient's MRD presented a unique profile. We also found no clear pattern in the progression of any CLC or aberrant(−) subpopulation between patients (see Fig. S3†). While there were cases where the progression of the CLC subpopulations is mirrored by the aberrant(−) subpopulations, which may reflect physiological pressures on the bone marrow environment, there are many cases where progression of the CLC and aberrant(−) subpopulations are extremely dissimilar. These results may reflect AML's inter- and intra-patient heterogeneity.38 To explore the significance of these subpopulations as the AML evolves post-SCT, we plan future studies that will gather more information from each sample, gene expression and/or proteomic profiling and genome sequencing of the CLC subpopulations.
Conclusions
This study represents the first microfluidic endeavor for monitoring AML patients following stem cell transplantation. The microfluidic assay was able to isolate and phenotypically identify leukemic cells circulating in a patient's peripheral blood. In this pilot clinical study, we monitored five AML patients following SCT. Because the assay required peripheral blood and not a bone marrow biopsy, 39 microfluidic tests could be carried out compared to only eight PCR, MFC, FISH, and/or microscopy tests, because they required highly invasive bone marrow biopsies in most cases. Because we were able to frequently test and observe changes in MRD levels, we identified signs of impending relapse earlier than bone marrow-based tests, which could enable therapeutic interventions at low disease burden and result in better outcomes for the patients. We also observed a case where late PCR detection of a patient's MRD translated to rapid relapse with the tumor doubling every two days and patient death shortly thereafter. We also observed the heterogeneity in AML; the CLCs and non-aberrant cells progressed variably, unpredictably, and, as we suspect, in response to graft- and virus-versus-leukemia effects as the bone marrow replenished. We are now developing a multifaceted microfluidic system capable of quantitative and in-line microfluidic flow cytometry with integrated flow sorting that will be able to provide molecular information on various CLC subpopulations, such as gene expression and gene mutation analysis of the CLCs, as well as surveying the lymphoid system29 for its graft-versus-leukemia capability.The microfluidic assay demonstrated herein the ability to track response to therapy in a minimally invasive fashion. The assay could also be used to provide a venue for the detailed management of a particular patient's cancer, especially in monitoring a patient's response to initial chemotherapy regimens, as well as long term monitoring for disease recurrence. This microfluidic assay could also be adapted to manage chronic lymphocytic leukemia (CLL), B cell lymphomas,50 and Hodgkin's lymphoma by programming into the microfluidic chips the appropriate selection mAbs and aberrant markers.51 In addition, the presented microfluidic assay could also be envisaged as a companion diagnostic for the discovery of new therapies for various leukemic diseases. With these observations and the data presented in this manuscript, our reported microfluidic assay can assist in enabling precision medicine for leukemic-based diseases.
Acknowledgements
We are sincerely grateful to the patients participating in this study. The authors thank Dr Gregory Lizee at MD Anderson and Dr Douglas Graham at the University of Colorado, Denver for generously gifting AML cell lines. This work was supported through the NIH (NCI – IMAT; R21-CA173279 and NIBIB – P41-EB020594), the University Cancer Research Fund (UNC), and the Lineberger Comprehensive Cancer Center (UNC). JMJ thanks the ACS Division of Analytical Chemistry and the Society for Analytical Chemists of Pittsburgh for funding and JBT thanks the Royster Society of Fellows (UNC) for funding.References
- J. M. Jaso, S. A. Wang, J. L. Jorgensen and P. Lin, Bone Marrow Transplant., 2014, 49, 1129–1138 CrossRef CAS PubMed.
- A. C. Society, Cancer Facts and Figures, American Cancer Society, Atlanta, 2015 Search PubMed.
- G. J. Schuurhuis and G. Ossenkoppele, Expert Rev. Hematol., 2010, 3, 1–5 CrossRef PubMed.
- D. A. Breems, W. L. Van Putten, P. C. Huijgens, G. J. Ossenkoppele, G. E. Verhoef, L. F. Verdonck, E. Vellenga, G. E. De Greef, E. Jacky, J. Van der Lelie, M. A. Boogaerts and B. Lowenberg, J. Clin. Oncol., 2005, 23, 1969–1978 CrossRef PubMed.
- E. Paietta, Hematology/the Education Program of the American Society of Hematology. American Society of Hematology. Education Program, 2012, pp. 35–42 Search PubMed.
- A. K. Burnett, A. H. Goldstone, R. M. Stevens, I. M. Hann, J. K. Rees, R. G. Gray and K. Wheatley, Lancet, 1998, 351, 700–708 CrossRef CAS.
- D. Grimwade, R. K. Hills, A. V. Moorman, H. Walker, S. Chatters, A. H. Goldstone, K. Wheatley, C. J. Harrison, A. K. Burnett and G. National Cancer Research Institute Adult Leukaemia Working, Blood, 2010, 116, 354–365 CrossRef CAS PubMed.
- P. D. Kottaridis, R. E. Gale, M. E. Frew, G. Harrison, S. E. Langabeer, A. A. Belton, H. Walker, K. Wheatley, D. T. Bowen, A. K. Burnett, A. H. Goldstone and D. C. Linch, Blood, 2001, 98, 1752–1759 CrossRef CAS PubMed.
- C. Schmid, M. Labopin, A. Nagler, M. Bornhauser, J. Finke, A. Fassas, L. Volin, G. Gurman, J. Maertens, P. Bordigoni, E. Holler, G. Ehninger, E. Polge, N. C. Gorin, H. J. Kolb, V. Rocha and E. A. L. W. Party, J. Clin. Oncol., 2007, 25, 4938–4945 CrossRef CAS PubMed.
- J. J. Cornelissen, W. L. van Putten, L. F. Verdonck, M. Theobald, E. Jacky, S. M. Daenen, M. van Marwijk Kooy, P. Wijermans, H. Schouten, P. C. Huijgens, H. van der Lelie, M. Fey, A. Ferrant, J. Maertens, A. Gratwohl and B. Lowenberg, Blood, 2007, 109, 3658–3666 CrossRef CAS PubMed.
- M. L. Sorror, B. M. Sandmaier, B. E. Storer, M. B. Maris, F. Baron, D. G. Maloney, B. L. Scott, H. J. Deeg, F. R. Appelbaum and R. Storb, J. Clin. Oncol., 2007, 25, 4246–4254 CrossRef PubMed.
- R. Zeiser, A. Spyridonidis, R. Wasch, G. Ihorst, C. Grullich, H. Bertz and J. Finke, Leukemia, 2005, 19, 814–821 CrossRef CAS PubMed.
- S. Schnittger, W. Kern, C. Tschulik, T. Weiss, F. Dicker, B. Falini, C. Haferlach and T. Haferlach, Blood, 2009, 114, 2220–2231 CrossRef CAS PubMed.
- D. Steinbach and K. M. Debatin, Leukemia, 2008, 22, 1638–1639 CrossRef CAS PubMed.
- B. Oran and M. de Lima, Curr. Opin. Hematol., 2011, 18, 388–394 CrossRef CAS PubMed.
- A. Al-Mawali, D. Gillis and I. Lewis, Am. J. Clin. Pathol., 2009, 131, 16–26 CrossRef PubMed.
- D. Grimwade and S. D. Freeman, Blood, 2014, 124, 3345–3355 CrossRef CAS PubMed.
- J. J. van Dongen and D. H. Ryan, in Immunologic approaches to the classification and management of lymphomas and leukemias, ed. J. M. Bennett and K. A. Foon, Kluwer Academic, Dordrecht, The Netherlands, 1988, pp. 173–207 Search PubMed.
- A. D. Donnenberg and V. S. Donnenberg, Clin. Lab. Med., 2007, 27, 627–652 CrossRef PubMed.
- V. H. J. van der Velden, A. van der Sluijs-Geling, B. E. S. Gibson, J. G. te Marvelde, P. G. Hoogeveen, W. C. J. Hop, K. Wheatley, M. B. Bierings, G. J. Schuurhuis, S. S. N. de Graaf, E. R. van Wering and J. J. M. van Dongen, Leukemia, 2010, 24, 1599–1606 CrossRef CAS PubMed.
- W. J. Allard, J. Matera, M. C. Miller, M. Repollet, M. C. Connelly, C. Rao, A. G. J. Tibbe, J. W. Uhr and L. W. M. M. Terstappen, Clin. Cancer Res., 2004, 10, 6897–6904 CrossRef PubMed.
- J. W. Kamande, M. L. Hupert, M. A. Witek, H. Wang, R. J. Torphy, U. Dharmasiri, S. K. Njoroge, J. M. Jackson, R. D. Aufforth, A. Snavely, J. J. Yeh and S. A. Soper, Anal. Chem., 2013, 85, 9092–9100 CrossRef CAS PubMed.
- J. M. Jackson, M. A. Witek, M. L. Hupert, C. Brady, S. Pullagurla, J. Kamande, R. D. Aufforth, C. J. Tignanelli, R. J. Torphy, J. J. Yeh and S. A. Soper, Lab Chip, 2014, 14, 106–117 RSC.
- M. A. Witek, R. D. Aufforth, H. Wang, J. W. Kamande, J. M. Jackson, S. R. Pullagurla, M. L. Hupert, J. Usary, W. Z. Wysham, V. Bae-Jump, L. A. Carey, P. A. Gehrig, M. I. Milowsky, C. M. Perou, J. T. Soper, Y. E. Whang, J. J. Yeh, G. Martin and S. A. Soper, 2015, submitted for publication.
- A. A. Adams, P. I. Okagbare, J. Feng, M. L. Hupert, D. Patterson, J. Gottert, R. L. McCarley, D. Nikitopoulos, M. C. Murphy and S. A. Soper, J. Am. Chem. Soc., 2008, 130, 8633–8641 CrossRef CAS PubMed.
- K. N. Battle, J. M. Jackson, M. A. Witek, M. L. Hupert, S. A. Hunsucker, P. M. Armistead and S. A. Soper, Analyst, 2014, 139, 1355–1363 RSC.
- J. M. Jackson, M. A. Witek and S. A. Soper, Sinusoidal microchannels with high aspect ratios for CTC selection and analysis, in Circulating Tumor Cells: Isolation and Analysis, ed. H. Fan, Wiley Publishing, 2015, in press Search PubMed.
- S. V. Nair, M. A. Witek, J. M. Jackson, M. A. M. Lindell, S. A. Hunsucker, T. Sapp, E. Perry, M. L. Hupert, V. Bae-Jump, P. A. Gehrig, W. Z. Wysham, P. M. Armistead, P. Voorhees and S. A. Soper, Chem. Commun., 2015, 51, 3266–3269 RSC.
- S. R. Pullagurla, M. A. Witek, J. M. Jackson, M. A. M. Lindell, M. L. Hupert, I. V. Nesterova, A. E. Baird and S. A. Soper, Anal. Chem., 2014, 86, 4058–4065 CrossRef CAS PubMed.
- M. Hupert, W. J. Guy, S. Llopis, H. Shadpour, S. Rani, D. Nikitopoulos and S. Soper, Microfluid. Nanofluid., 2007, 3, 1–11 CrossRef CAS.
- M. L. Hupert, J. M. Jackson, H. Wang, M. A. Witek, J. Kamande, M. I. Milowsky, Y. E. Whang and S. A. Soper, Microsyst. Technol., 2014, 20, 1815–1825 CrossRef CAS PubMed.
- A. D. Edelstein, M. A. Tsuchida, N. Amodaj, H. Pinkard, R. D. Vale and N. Stuurman, J. Biol. Methods, 2014, 1 Search PubMed.
- P. K. Y. Goon, G. Y. H. Lip, C. J. Boos, P. S. Stonelake and A. D. Blann, Neoplasia, 2006, 8, 79–88 CrossRef CAS PubMed.
- A. A. Adams, P. I. Okagbare, J. Feng, M. L. Hupert, D. Patterson, J. Gottert, R. L. McCarley, D. Nikitopoulos, M. C. Murphy and S. A. Soper, J. Am. Chem. Soc., 2008, 130, 8633–8641 CrossRef CAS PubMed.
- A. G. Tibbe, M. C. Miller and L. W. Terstappen, Cytometry, Part A, 2007, 71, 154–162 CrossRef PubMed.
- G. W. Schmid-Schonbein, Y. Y. Shih and S. Chien, Blood, 1980, 56, 866–875 CAS.
- E. Sollier, D. E. Go, J. Che, D. R. Gossett, S. O'Byrne, W. M. Weaver, N. Kummer, M. Rettig, J. Goldman, N. Nickols, S. McCloskey, R. P. Kulkarni and D. Di Carlo, Lab Chip, 2014, 14, 63–77 RSC.
- S. J. Horton and B. J. Huntly, Haematologica, 2012, 97, 966–974 CrossRef CAS PubMed.
- L. Ding, T. J. Ley, D. E. Larson, C. A. Miller, D. C. Koboldt, J. S. Welch, J. K. Ritchey, M. A. Young, T. Lamprecht, M. D. McLellan, J. F. McMichael, J. W. Wallis, C. Lu, D. Shen, C. C. Harris, D. J. Dooling, R. S. Fulton, L. L. Fulton, K. Chen, H. Schmidt, J. Kalicki-Veizer, V. J. Magrini, L. Cook, S. D. McGrath, T. L. Vickery, M. C. Wendl, S. Heath, M. A. Watson, D. C. Link, M. H. Tomasson, W. D. Shannon, J. E. Payton, S. Kulkarni, P. Westervelt, M. J. Walter, T. A. Graubert, E. R. Mardis, R. K. Wilson and J. F. DiPersio, Nature, 2012, 481, 506–510 CrossRef CAS PubMed.
- D. Bonnet and J. E. Dick, Nat. Med., 1997, 3, 730–737 CrossRef CAS PubMed.
- J. W. Kamande, M. L. Hupert, M. A. Witek, H. Wang, R. J. Torphy, U. Dharmasiri, S. K. Njoroge, J. M. Jackson, R. D. Aufforth, A. Snavely, J. J. Yeh and S. A. Soper, Anal. Chem., 2013, 85, 9092–9100 CrossRef CAS PubMed.
- S. V. Nair, M. A. Witek, J. M. Jackson, M. A. M. Lindell, S. A. Hunsucker, T. Sapp, E. Perry, M. L. Hupert, V. Bae-Jump, P. A. Gehrig, W. Z. Wysham, P. M. Armistead, P. Voorhees and S. A. Soper, Chem. Commun., 2015, 51, 3266–3269 RSC.
- K. C. Chang and D. A. Hammer, Biophys. J., 1999, 76, 1280–1292 CrossRef CAS PubMed.
- J. Autebert, B. Coudert, J. Champ, L. Saias, E. T. Guneri, R. Lebofsky, F. C. Bidard, J. Y. Pierga, F. Farace, S. Descroix, L. Malaquin and J. L. Viovy, Lab Chip, 2015, 15, 2090–2101 RSC.
- G. I. Bell, Science, 1978, 200, 618–627 CAS.
- D. Sharma, M. R. Eichelberg, J. D. Haag, A. L. Meilahn, M. J. Muelbl, K. Schell, B. M. G. Smits and M. N. Gould, BioTechniques, 2012, 53, 57–60 CAS.
- M. R. Baer, G. C. Stewart, R. K. Dodge, G. Leget, N. Sule, K. Mrozek, C. A. Schiffer, B. L. Powel, J. E. Kolitz, J. O. Moore, R. M. Stone, F. R. Davey, A. J. Carrol, R. A. Larson and C. D. Bloomfield, Blood, 2001, 97, 3574–3580 CrossRef CAS PubMed.
- G. Awong, E. Herer, C. D. Surh, J. E. Dick, R. N. La Motte-Mohs and J. C. Zuniga-Pflucker, Blood, 2009, 114, 972–982 CrossRef CAS PubMed.
- A. H. Elmaagacli, N. K. Steckel, M. Koldehoff, Y. Hegerfeldt, R. Trenschel, M. Ditschkowski, S. Christoph, T. Gromke, L. Kordelas, H. D. Ottinger, R. S. Ross, P. A. Horn, S. Schnittger and D. W. Beelen, Blood, 2011, 118, 1402–1412 CrossRef CAS PubMed.
- H. Y. Dong, W. Gorczyca, Z. Liu, P. Tsang, C. D. Wu, P. Cohen and J. Weisberger, Am. J. Clin. Pathol., 2003, 119, 218–230 CrossRef PubMed.
- J. Irsch, S. Nitsch, M. L. Hansmann, K. Rajewsky, H. Tesch, V. Diehl, A. Jox, R. Kuppers and A. Radbruch, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 10117–10122 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5an01836f |
| This journal is © The Royal Society of Chemistry 2016 |