
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
pH-induced on/off-switchable graphene bioelectronics†
Onur
Parlak
a,
Anthony P. F.
Turner
a and
Ashutosh
Tiwari
*ab
aBiosensors and Bioelectronics Centre, IFM, Linköping University, S-58183 Linköping, Sweden. E-mail: ashutosh.tiwari@liu.se; Fax: +46 13 1375 68; Tel: +46 13 2823 95
bTekidag AB, Mjärdevi Science Park, Teknikringen 7, SE 583 30 Linköping, Sweden
First published on 17th August 2015
Abstract
Switchable interfaces can deliver functionally reversible reactivity with their corresponding analytes, which allows one to positively respond to the activity of biological elements, including enzymes and other biomolecules, through an encoded stimulus. We have realized this by the design of stimuli-responsive graphene interfaces for the pH-encoded operation of bioelectronics. Herein, we have demonstrated stimuli-responsive graphene interfaces for the pH-encoded operation of bioelectronics. The resulting switchable interfaces are capable of the highly specific, on-demand operation of biosensors, which has significant potential in a wide range of analytical applications.
Introduction
The stimuli-responsive bioelectronic interfaces, which are capable of properties-on-demand change upon communication with an external signal, are a challenging goal for both fundamental and applied studies in the area of biotechnology due to their potential applications in miniaturized devices.1–7 The integration of stimuli-responsive interfaces in these devices allows their operation as electrochemical gates, switching on and off or tuning the rate of the interfacial reaction to control and regulate mass transport, adhesive and, more importantly, electrochemical properties.8–13 Herein, we aim to address the design and development of pH-encoded bio-catalysis by employing pH-responsive poly(4-vinyl pyridine) (P4VP) graphene oxide bio-interfaces (pHGr) to control and regulate enzyme-based molecular interaction. Using electrochemical methods, we demonstrate that the interfacial electrochemical properties can be controlled by the relatively modest changes in the pH of the medium.Graphene oxide (GO), in the form of an atomic layer of carbon as a basic material for the preparation of individual graphene nanosheets, has attracted intense interest in recent years owing to its unique physical and chemical properties.14,15 In addition to high surface area, high electrical conductivity and high enzyme loading capacity,16,17 the copious oxygen-containing surface functionalities, such as epoxide, hydroxyl, and carboxylic groups, make GO an ideal material for various applications ranging from catalysis to energy storage.14,18–21 The biocompatibility, processability and availability of surface functional groups of GO provides feasibility for its use as an efficient chemical and biological carrier material for bioelectronics applications.22,23 The combination of graphene oxide with pH responsive polymer (P4VP) allows us to generate pH responsive interfaces to control and regulate biomolecular interactions. In principle, P4VP polymer has an intrinsic ability to respond to chemical change in the medium. The protonation of the pyridine units of the polymer backbone under the acidic conditions results in the swelling of the polymer structure on the surface, which allows the diffusion of an analyte through the electrode surface.24 However, when the pH of a medium reaches neutral conditions, the mobility of the polymer chains is restricted and the analyte cannot reach the electrode surface, resulting in a non-active state. The objective of our model system is to control a bioelectrocatalytic interaction on its interface by varying the pH of the reaction medium.25 At a relatively low pH (i.e., at pH 4), the pH encoded graphene (pHGr) and glucose oxidase (GOx, pI 4.2)26 possess opposite charges, which help them to assemble in an ordered structure due to the strong electrostatic interaction. Under these conditions, the bio-substrate (i.e., glucose) can easily access the enzyme, which facilitates electron transfer to the electrode surface. In contrast, at a relatively high pH range (i.e., at 6–10), the forces between GOx and P4VP-conjugated graphene oxide (pHGr) become repulsive due to the oppositely charged character of both components, which results in the drifting away of GOx from the graphene surface.26 As a result, even though a bio-substrate can freely access the enzyme, electron transport cannot occur efficiently. The responsiveness of such assemblies delivers tuneable bioelectrocatalytic interfaces as well as bioelectronics through ‘built-in’ programmable physical, structural or morphological properties upon receiving chemical change.27
Experimental
Materials
GOx (GOx, ≥100 units per g, from Aspergillus Niger species), glucose (≥99.5%), 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) diammonium (ABTS, ≥99%), hydrogen peroxide (H2O2, ACS reagent, 30 wt% in H2O), phenol (≥99.5%), potassium dihydrogen phosphate monobasic (KH2PO4, 99.99%), dipotassium hydrogen phosphate (K2HPO4, ≥99%), potassium chloride (KCl, ≥99%), potassium ferrocyanide (K3[Fe(CN)6], ≥98.5%), potassium ferricyanide (K4[Fe(CN)6]), poly(4-vinyl pyridine) (MW = ∼60 kDa), and sodium dodecylbenzenesulfonate (SDBS) were purchased from Sigma-Aldrich (St. Louis, MO, USA) and were used without further purification. Phosphate buffer saline (1× PBS) and ferri/ferro cyanide solutions were used as a supporting electrolyte for some amperometric measurements. Aqueous solutions were prepared with double-distilled water from a Millipore system (18.2 MΩ cm).Preparation of pH-encoded graphene oxide based hybrid structures
The pH encoded bio-interface was produced using a graphene oxide, which was obtained by the modified Hummers method (Graphene Supermarket, USA), glucose oxidase (GOx, from Aspergillus niger, ≥100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 units per g) and pH responsive poly(4-vinyl pyridine). To prevent aggregation and sustain chemical and physical properties, the surface of graphene oxide was modified using an anionic surfactant, sodium dodecyl benzene sulfonate (SDBS). The surfactant molecule contains an alkyl chain (dodecyl-C12H25), which gives it a hydrophobic nature, and a benzenesulfonate (C6H5SO3−) group, which is responsible for its hydrophilic character. First, 1.0 mg of graphene oxide was dispersed in 5 mL of 0.1 M SDBS aqueous solution. The reaction mixture was sonicated for 1 h, and mixed for 3 h at room temperature (RT). After 3 h incubation, the resulting suspension was subjected to three cycles of centrifugation to isolate the surface-capped graphene oxide nanosheets. The samples were then washed with distilled water to remove the excess SDBS and dried at room temperature prior to conjugation with glucose oxidase.
000 units per g) and pH responsive poly(4-vinyl pyridine). To prevent aggregation and sustain chemical and physical properties, the surface of graphene oxide was modified using an anionic surfactant, sodium dodecyl benzene sulfonate (SDBS). The surfactant molecule contains an alkyl chain (dodecyl-C12H25), which gives it a hydrophobic nature, and a benzenesulfonate (C6H5SO3−) group, which is responsible for its hydrophilic character. First, 1.0 mg of graphene oxide was dispersed in 5 mL of 0.1 M SDBS aqueous solution. The reaction mixture was sonicated for 1 h, and mixed for 3 h at room temperature (RT). After 3 h incubation, the resulting suspension was subjected to three cycles of centrifugation to isolate the surface-capped graphene oxide nanosheets. The samples were then washed with distilled water to remove the excess SDBS and dried at room temperature prior to conjugation with glucose oxidase.
For the conjugation of glucose oxidase with surface-modified graphene oxide, 1.0 mg of glucose oxidase was dissolved in 1 mL of pure water at pH 7.4, and the mixture was incubated for 3 h at 4 °C. The mixture was then centrifuged at 5000 rpm for 30 min and the supernatant of the solution was collected for the determination of enzyme-loading efficiency. The precipitate was washed with PBS and centrifuged successively three times to remove loosely attached enzymes from the graphene oxide surface. The same experiments were performed for different concentrations of graphene oxide and enzyme with different pH values to determine enzyme-loading efficiency. Subsequently, the graphene oxide/GOx hybrids were incorporated in a poly(4-vinyl pyridine) (P4VP) matrix to obtain the pH-responsive graphene oxide–enzyme–polymer hybrid structure. In a typical preparation, 1 mg of previously obtained graphene oxide–GOx conjugate was added to 1 mL of 1 mg mL−1 P4VP aqueous solution to form a homogenous solution under ultrasonication.16
Fabrication of bio-electrodes
Prior to the immobilisation of the graphene oxide based interfaces, glassy carbon electrodes (GCE) were carefully polished with 1, 0.3 and 0.5 μm α-alumina slurry, and then washed with deionised water. The solution containing graphene oxide–glucose oxidase and poly(4-vinyl pyridine) (10 μL) was sonicated for 5 min to obtain a homogenous dispersion. This suspension (10 μL) was drop-cast onto the GC electrode surface and dried for 8 h at 4 °C.Enzyme immobilization efficiency test
Enzyme immobilisation was carried out by adding the desired amount of pHGr to 1× PBS that contained the enzyme to be immobilised as described in detail. The mixture was incubated for 30 min on an ice-bath by shaking and then centrifuged. The supernatant was collected to determine the enzyme-loading efficiency. The immobilised enzymes were washed three times with the same buffer solution to remove the physically adsorbed enzymes. The enzyme activities of the resulting immobilised enzymes were evaluated using a colorimetric method.Apparatus and characterisation
The zeta potential of the graphene oxide dispersions was determined using a Nano ZS dynamic light-scattering (DLS)-zeta potential measuring instrument (Malvern Instruments, Worcestershire, UK). Transmission electron microscopy (TEM) was performed using a G2 Spirit/Biotwin (FEI-Technai, Hillsboro, OR, USA) with a working voltage of 120 kV. All the voltammetry and amperometric measurements were carried out with Ivium Stat.XR electrochemical analyser (Eindhoven, The Netherlands). Absorptions for enzymatic assay in PBS solution were measured with a Cary 50 UV-Vis Spectrometer (Varian, CA, USA). A three-electrode cell with a glassy carbon working electrode with the surface area of 0.07 cm2, a platinum auxiliary wire, and Ag/AgCl (3 M KCl) reference electrodes was used for the voltammetric and amperometric measurements.Results and discussion
Scheme 1 illustrates the reversible conformational change of pH-encoded graphene–GOx interaction upon change of reaction medium. To demonstrate the “on” and “off” performance of the bio-electrodes, pH 4 and 6 were chosen as representative states.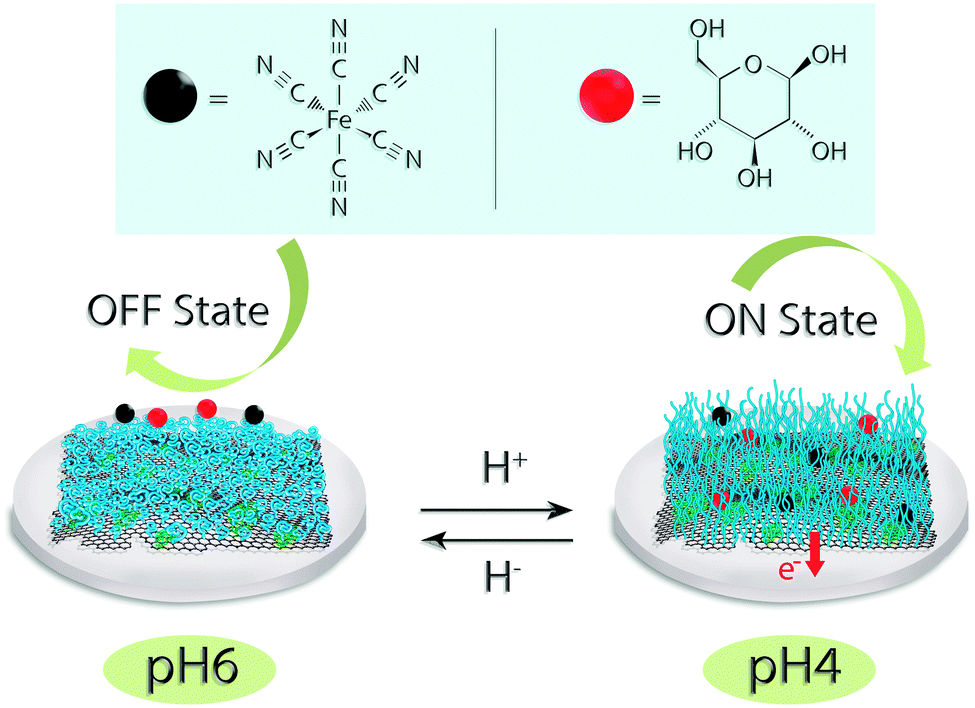 | ||
| Scheme 1 Schematic representations of a pH-encoded switchable graphene oxide interface at two different states. The tuneable character of the interface was tested using redox-active ferri/ferro cyanide probe (black circle) and glucose as a substrate (red circle). The diffusion of both molecules was controlled by varying the polymer states: at pH 6, (off state) polymer turns to a shrunken state, and at pH 4 (on state), polymer turns to a swelling state. | ||
Morphological characterisations
The structure and morphology of the electrodes prepared with the immobilised pHGr were characterised by transmission electron (TEM) and scanning electron (SEM) microscopy. The TEM samples were prepared by dropping 5 μL of a bare graphene oxide (1.0 mg mL−1) solution on a polymer-coated copper grid. Fig. 1(a and b) depicts the TEM images of graphene oxide with different magnifications. The TEM image of a sheet, as shown in Fig. 1a, demonstrated a wrinkled and folded sheet structure, presumably resulting from the available surface groups of graphene oxide. Owing to the hydrophilic character, it can be well-dispersed in an aqueous solution, and it generally does not form any agglomerates with a badly ordered architecture. The folded region in Fig. 1a was selected to observe the edges at high magnification (Fig. 1b). The edges of the GO sheets are consistent with their atomically-thin nature.28 The SEM images (Fig. 1c and d) of P4VP conjugated graphene oxide electrodes also show a paper-like wrinkled structure. The P4VP conjugated graphene oxide samples were prepared by mixing 1 mg of previously obtained graphene oxide–GOx conjugate with 1 mL of 1 mg mL−1 P4VP aqueous solution to obtain a homogenous solution at neutral pH. After 30 min of ultrasonication, 50 μL of graphene oxide–GOx–P4VP hybrids were drop-casted onto a SEM sample holder and dried overnight at 4 °C before the SEM observations. | ||
| Fig. 1 (a and b) Transmission electron microscopy images (TEM) of graphene oxide nanosheets and scanning electron microscopy (SEM) images of a pH-responsive polymer conjugated graphene oxide at low (c) and high (d) magnifications. | ||
Surface charge and enzyme immobilisation
The surface charge of a colloidal system also plays an important role in controlling and regulating the physicochemical interactions at the molecular level. In our design, pHGr and GOx immobilised pHGr were dispersed in an aqueous solution over a wide pH range to assess how does the surface charge changes before and after enzyme immobilisation. Fig. 2a shows that pHGr carries a negative charge ranging from −14 to −43 mV, depending on the pH of the medium. However, after the immobilisation of GOx onto pHGr, the total surface charge decreases depending on the charge of GOx. It is well-known that the isoelectric point (pI) of GOx is pH 4.2. Therefore, the enzyme structure carries a net positive charge at pH lower than 4.2, and a net negative charge at pH higher than that. The net surface charge of enzyme enables us to design a controllable bio-chemical interface over a range of pH conditions.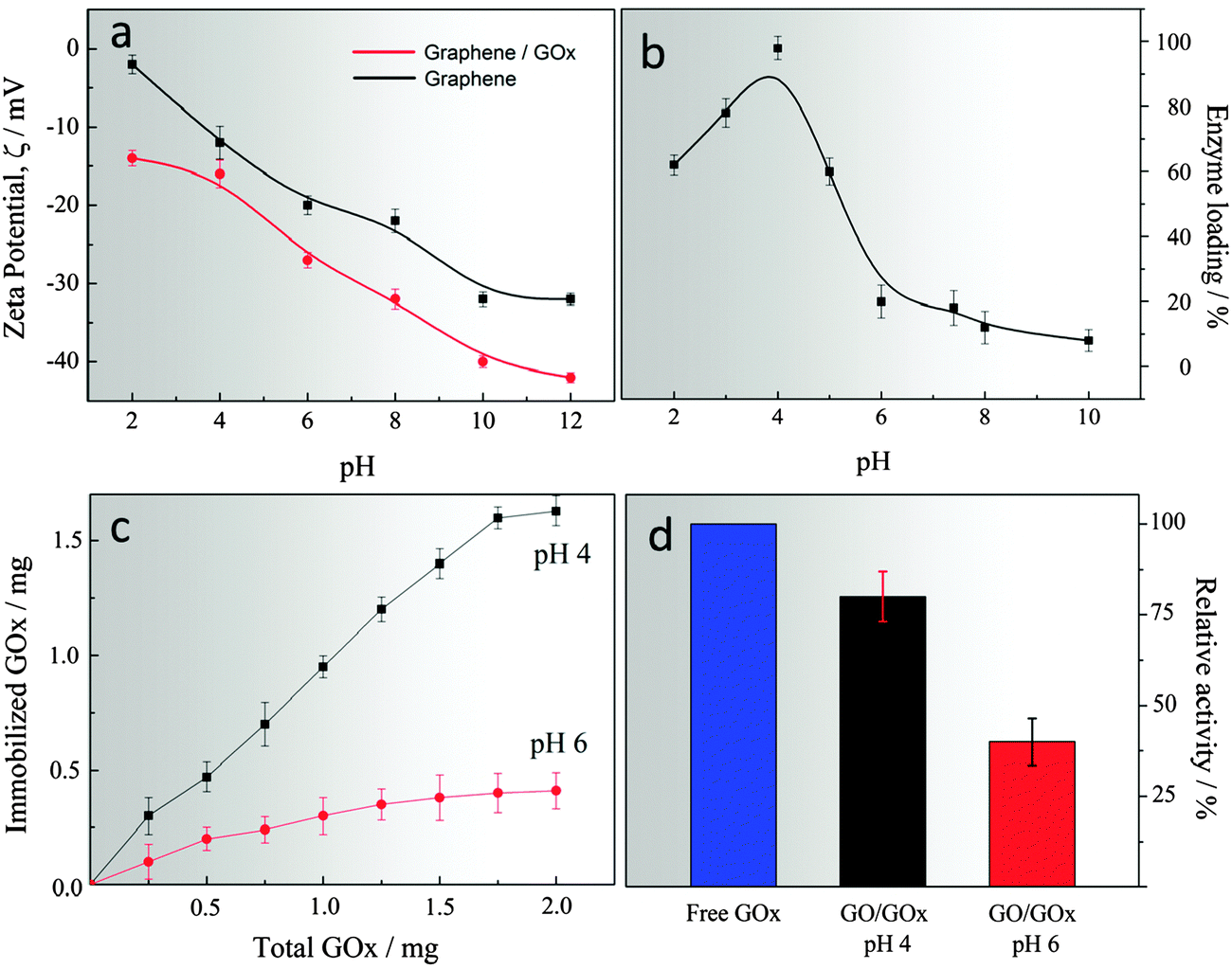 | ||
| Fig. 2 Zeta potential of pH-encoded graphene oxide before and after enzyme immobilisation at different pH (a), effect of pH on the enzyme loading on graphene oxide (b), enzyme loading on pH encoded graphene as a function of the total amount of enzyme (c) and relative enzyme activity under different pH conditions (d). | ||
The immobilisation efficiency of GOx on the pHGr sheets was assessed by incubating both pHGr and GOx in a solution at 4 °C between pH 2 and 12 (Fig. 2b). It was found that GOx can be spontaneously immobilised onto the pHGr surface within a relatively short incubation time (30 min). Presumably, the amine group of GOx forms amide bonds with the carboxyl group of pHGr. However, without any coupling reagent, this covalent interaction usually occurs very slowly. Therefore, the covalent bonding may not contribute to the interaction between pHGr and GOx. Another important outcome of an immobilisation efficiency test was the information regarding the effects of surface charge on bio-molecular loading. To elucidate this effect, pH 4 and pH 6 were chosen as representative pH-values. The zeta potential measurements (Fig. 2a) showed that pHGr and GOx carry an opposite charge at pH 4, which increases the enzyme loading on graphene oxide. The maximum loading efficiency was found to be 96% at pH 4. However, above pH 5, the enzyme loading efficiency decreased very rapidly from 96% to 18%. The reason for the sharp decrease in loading efficiency might be a change in the charge on the enzyme periphery. The presence of opposite charges of pHGr results in GOx drifting away from the surface at pH 6. The results show that the pH-dependent enzyme loading on GO is governed by the electrostatic interaction between pHGr and GOx. Moreover, to show that pHGr has different loading capacity at different pH, we incubated various amounts of GOx with fixed amount of pHGr at two different pH. It was found that GOx can be directly immobilised on pHGr and the maximum loading was about 1.65 mg mg−1 at pH 4 and 0.28 mg mg−1 at pH 6 (Fig. 2c). Fig. 2d shows the effects of pH on the activity of the immobilised enzyme at different pH relative to the free enzyme. The results showed that the strong electrostatic interactions between GOx and pHGr at pH 4 might help conserve the native structure of the enzyme rather than the loosely attached counterpart at pH 6. It shows that a lower pH might not only be useful for high bio-molecular loading, but also provide a flexible distance to each enzyme on the surface of graphene oxide to sustain its activity.
Water contact angle
To further understand the interactions between GOx and pHGr, the surface property of pHGr before and after the interaction with GOx at representative pH values (i.e., pH 4 and 6) was evaluated. As shown in Fig. S1 (ESI†), the water contact angle of graphene oxide was 54°. However, after the immobilisation of GOx at pH 4, the water contact angle decreased to 38° and then increased to 52° at pH 6. These results suggest that the hydrophobicity decreases on the surface of graphene oxide due to the strong interaction between the enzyme and pHGr. The surface of graphene oxide becomes more wettable due to the presence of hydrophilic enzyme biomolecules. However, when the pH increases to 6, interaction between the enzyme and pHGr becomes weaker and enzyme starts to move away from the surface due to high hydrophobicity and contact angle, which reached almost the same degree as bare pHGr.Electrochemical study
The GOx immobilised on pHGr was studied using cyclic voltammetry in 5 mM Fe[(CN)6]3−/4− and 0.1 M KCl(aq) at two different pH (pH 4 and pH 6) and a scan rate of 50 mV s−1. Cyclic voltammograms were recorded for GOx-immobilised pHGr electrodes and the results are shown in Fig. 3a and b. When the pHGr/GOx assembled structures were immobilised on the electrode surface at pH 4, a well-defined redox couple corresponding to the oxidation and reduction of ferri/ferro cyanide probe is obtained. However, when the same measurement was performed at pH 6, no well-defined redox couple was observed. This shows that the oxidation of the redox probe cannot occur at pH 6 due to the strong repulsive forces between GOx and pHGr. To define the reproducibility of this reversible enzymatic reaction for both pH values, the electrodes were cycled successively 8 times (Fig. 3b). The results were obtained based on each cyclic voltammetry measurement. The switching time between the two different pH values was no longer than 1 min for successive cycles. After 8 runs, no significant current difference was found between each cycle for both pH values.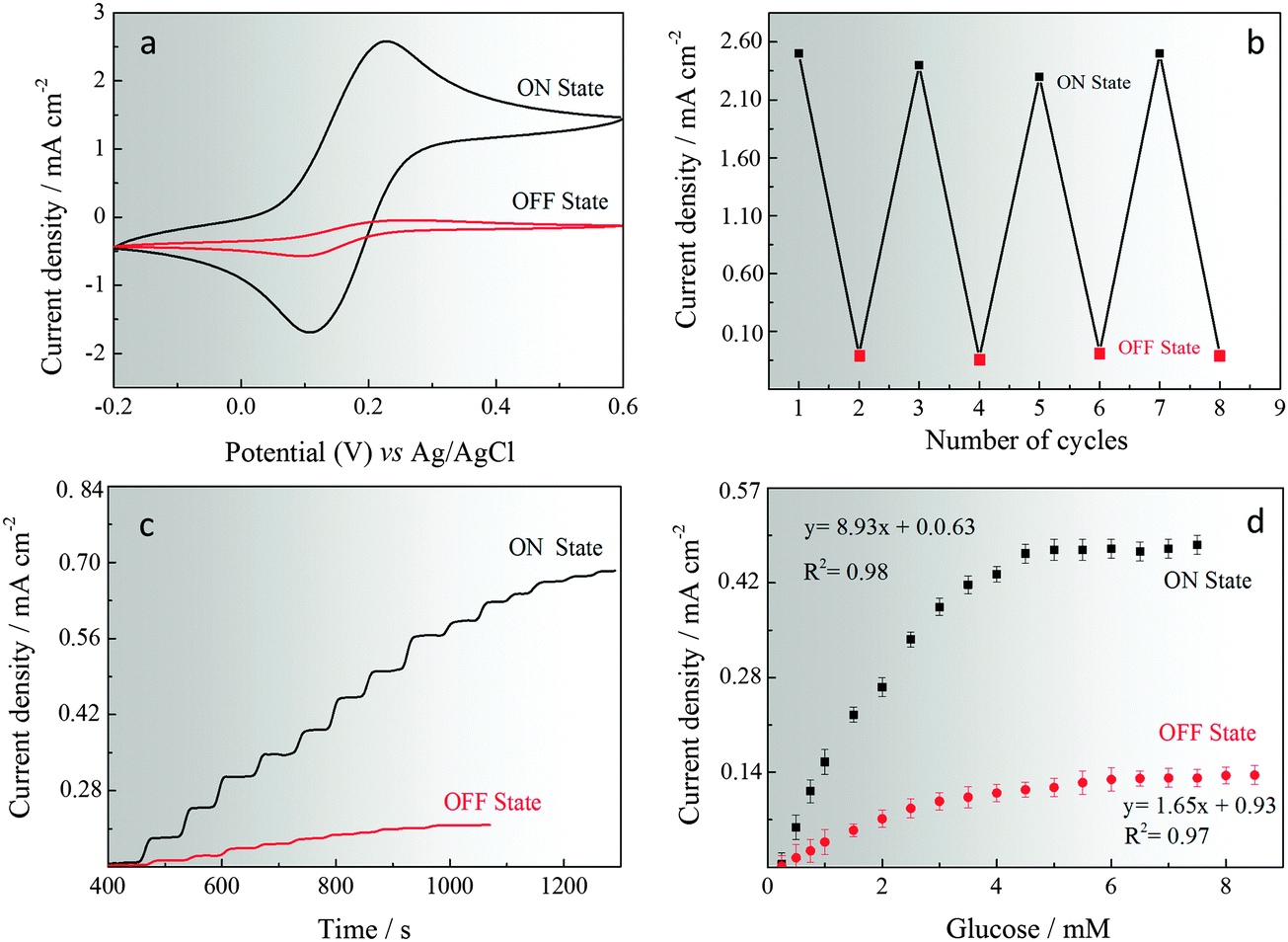 | ||
| Fig. 3 Cyclic voltammetry response (a) and repeatability (b) of P4VP-modified graphene electrodes in the “on-state” (pH 4) and “off-state” (pH 6). Choronoamperometric response (c) and calibration curve (d) of enzyme immobilised P4VP-modified graphene electrodes in the “on-state” (pH 4) and “off-state” (pH 6) at +0.3 V in 5 mM [Fe(CN)6]3−/4− and 0.1 M KCl(aq) at 50 mV s−1vs. Ag/AgCl reference electrode. | ||
Amperometric glucose sensing was also performed using a three-electrode set-up and a working potential (+0.60 V vs. Ag/AgCl) in 5 mM Fe[(CN)6]3−/4− and 0.1 M KCl(aq) at two different pH values (pH 4 and pH 6). The calibration curves of glucose sensing at two pH values were obtained by adding successive aliquots of increasing concentrations of glucose and measuring the steady-state signal response, which were achieved within 3 and 8 seconds. Fig. 3c and d show the amperometric responses and calibration curves obtained for the sensing of glucose at two different pH values. The dynamic range for the GOx immobilised pHGr electrode at pH 4 was between 0.01 and 6.0 mM and the sensitivity was 127.57 μA μM−1 cm−2. However, at pH 6, the dynamic range decreased to 0.025 from 3.0 mM and sensitivity decreased to 65.43 μA μM−1 cm−2. The detection limits were calculated to be 0.012 (at pH 4) and 0.043 (at pH 6) (S/N = 3).
Conclusions
The pH-switchable interfaces deliver the finest control and regulation functions to biodevices due to high-density bio-catalysis with sharp transitions upon stimuli. We demonstrated an alterable active-passive communication between pHGr and GOx as a model bio-system. In this approach, the programming of bioelectronic interfaces was evaluated by in situ change in the pH of the electrode. The electrochemical analysis supports the finding that interfacial properties and bio-catalytic interactions on the bio-electrodes can be tuned by encoded information. One another key goal of this study was to develop high surface areas to support greater enzyme loadings and thereby significantly increase the bio-catalytic activity within atomic-thick volumes.Acknowledgements
The authors wish to acknowledge the Swedish Research Council (VR-2011-6058357) for generous financial supports to carry out this research.Notes and references
- O. Parlak, A. P. F. Turner and A. Tiwari, Adv. Mater., 2014, 26, 482–486 CrossRef CAS PubMed.
- V. Bocharova and E. Katz, Chem. Rec., 2012, 12, 114–130 CrossRef CAS PubMed.
- O. Parlak and A. P. F. Turner, Biosens. Bioelectron., 2015 DOI:10.1016/j.bios.2015.06.023.
- P. M. Mendes, Chem. Soc. Rev., 2008, 37, 2512–2529 RSC.
- P. M. Mendes, Chem. Soc. Rev., 2013, 42, 9207–9218 RSC.
- E. Katz, M. LionDagan and I. Willner, J. Electroanal. Chem., 1996, 408, 107–112 CrossRef.
- M. Motornov, R. Sheparovych, E. Katz and S. Minko, ACS Nano, 2008, 2, 41–52 CrossRef CAS PubMed.
- I. Tokarev, V. Gopishetty, J. Zhou, M. Pita, M. Motornov, E. Katz and S. Minko, ACS Appl. Mater. Interfaces, 2009, 1, 532–536 CAS.
- A. P. F. Turner, Chem. Soc. Rev., 2013, 42, 3184–3196 RSC.
- R. Klajn, Chem. Soc. Rev., 2014, 43, 148–184 RSC.
- M. Gamella, N. Guz, S. Mailloux, J. M. Pingarron and E. Katz, ACS Appl. Mater. Interfaces, 2014, 6, 13349–13354 CAS.
- K. MacVittie and E. Katz, Chem. Commun., 2014, 50, 4816–4819 RSC.
- T. K. Tam, M. Pita, M. Motornov, I. Tokarev, S. Minko and E. Katz, Electroanalysis, 2010, 22, 35–40 CrossRef CAS PubMed.
- J. Kim, L. J. Cote, F. Kim, W. Yuan, K. R. Shull and J. X. Huang, J. Am. Chem. Soc., 2010, 132, 8180–8186 CrossRef CAS PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- O. Parlak, A. Tiwari, A. P. F. Turner and A. Tiwari, Biosens. Bioelectron., 2013, 49, 53–62 CrossRef CAS PubMed.
- J. L. Zhang, F. Zhang, H. J. Yang, X. L. Huang, H. Liu, J. Y. Zhang and S. W. Guo, Langmuir, 2010, 26, 6083–6085 CrossRef CAS PubMed.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- E. Morales-Narvaez and A. Merkoci, Adv. Mater., 2012, 24, 3298–3308 CrossRef CAS PubMed.
- Y. W. Zhu, S. Murali, M. D. Stoller, K. J. Ganesh, W. W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz, M. Thommes, D. Su, E. A. Stach and R. S. Ruoff, Science, 2011, 332, 1537–1541 CrossRef CAS PubMed.
- Y. Wang, Z. H. Li, J. Wang, J. H. Li and Y. H. Lin, Trends Biotechnol., 2011, 29, 205–212 CrossRef CAS PubMed.
- K. Yang, L. Z. Feng, X. Z. Shi and Z. Liu, Chem. Soc. Rev., 2013, 42, 530–547 RSC.
- Y. Wang, Z. H. Li, D. H. Hu, C. T. Lin, J. H. Li and Y. H. Lin, J. Am. Chem. Soc., 2010, 132, 9274–9276 CrossRef CAS PubMed.
- T. K. Tam, M. Ornatska, M. Pita, S. Minko and E. Katz, J. Phys. Chem. C, 2008, 112, 8438–8445 CAS.
- T. K. Tam, M. Pita, O. Trotsenko, M. Motornov, I. Tokarev, J. Halamek, S. Minko and E. Katz, Langmuir, 2010, 26, 4506–4513 CrossRef CAS PubMed.
- J. H. Pazar and K. Kleppe, J. Biochem., 1964, 64, 439–447 Search PubMed.
- O. Parlak, P. Seshadri, I. Lundstrom, A. P. F. Turner and A. Tiwari, Adv. Mater. Interfaces, 2014, 1, 1400136 Search PubMed.
- S. Eigler, C. Dotzer and A. Hirsch, Carbon, 2012, 50, 3666–3673 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5tb01355k |
| This journal is © The Royal Society of Chemistry 2015 |