
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePt/Ni(OH)2–NiOOH/Pd multi-walled hollow nanorod arrays as superior electrocatalysts for formic acid electrooxidation†
Han
Xu
a,
Liang-Xin
Ding
b,
Jin-Xian
Feng
a and
Gao-Ren
Li
*a
aMOE Laboratory of Bioinorganic and Synthetic Chemistry, KLGHEI of Environment and Energy Chemistry, School of Chemistry and Chemical Engineering, Sun Yat-sen University, Guangzhou 510275, China. E-mail: ligaoren@mail.sysu.edu.cn
bSchool of Chemistry & Chemical Engineering, South China University of Technology, Guangzhou 510640, China
First published on 28th August 2015
Abstract
The catalytic activity and durability are crucial for the development of high-performance electrocatalysts. To design electrocatalysts with excellent electroactivity and durability, the structure and composition are two important guiding principles. In this work, novel Pt/Ni(OH)2–NiOOH/Pd multi-walled hollow nanorod arrays (MHNRAs) are successfully synthesized. The unique MHNRAs provide fast transport and short diffusion paths for electroactive species and high utilization rate of catalysts. Because of the special surface and synergistic effects, the Pt/Ni(OH)2–NiOOH/Pd MHNRA electrocatalysts exhibit high catalytic activity, high durability and superior CO poisoning tolerance for the electrooxidation of formic acid in comparison with Pt@Pd MHNRAs, commercial Pt/C, Pd/C and PtRu/C catalysts.
1. Introduction
Direct formic acid fuel cells (DFAFCs) have attracted growing attention as promising energy converters with their high efficiency to supply energy and environmental friendliness for portable electronic devices,1–4 and they are expected to play a vital role in our future sustainable society. Formic acid as a fuel offers the following unique advantages in comparison to other fuels: formic acid is easier to store and transport compared with hydrogen, while compared with methanol, formic acid has a higher power density and lower crossover rate through a Nafion membrane.5,6 At present, Pt and Pd are considered efficient electrocatalysts for the oxidation of small organic molecules such as formic acid, methanol and ethanol.7–13 For formic acid oxidation, Pd electrocatalysts exhibit a high initial performance, but the high performance will be lost rapidly over time.14–16 However, Pt electrocatalysts are hindered by CO poisoning, which causes poor durability, and the scale of Pt use leads to a high cost of the electrocatalyst.17–20Transition metal oxides, such as CeO2, RuO2, TiO2, and MoO2, as co-catalysts have shown the potential to reduce the cost of catalysts, promote catalytic activity and improve the CO poisoning tolerance of Pt and Pd because of the low-cost and high cation-exchange capacity of metal oxides.21–26 Nevertheless, the wide-spread application of transition metal oxides in electrocatalysts was hampered by their low electrical conductivity. For now, transition metal hydroxides (such as Ni, Fe and Co hydroxides), which have been widely used for the oxygen evolution reaction (OER), organic photovoltaics and supercapacitors with significantly improved performances, have attracted much interest as electrocatalyst supports for the oxidation of small organic molecules. These transition metal hydroxides have better electrical conductivity and stability than transition metal oxides during the catalytic reaction process and can efficiently produce sufficient available OHads species to oxidize the majority of adsorbed CO on the surface of the catalyst by activation of water at lower potentials.27–31 In addition, tailoring the nanostructures of Pt-based electrocatalysts will provide an effective strategy to improve the performance of electrocatalysts.32–35 Noble metal nanomaterials with multi-walled hollow nanorod arrays have been highlighted in previous studies.22,36 In contrast to single-walled hollow nanorods, multi-walled hollow nanorods can obviously decrease noble metal utilization and have a strong synergistic effect on different materials, which will significantly improve the electrocatalytic activity and CO poisoning tolerance of the catalysts used for the electrooxidation of small organic molecules.37 However, to the best of our knowledge, almost no study has focused on Ni hydroxide-functionalized Pt–Pd electrocatalysts with multi-walled hollow nanostructures for the electrooxidation of formic acid for DFAFCs.
Based on the above considerations, novel Pt/Ni(OH)2–NiOOH/Pd multi-walled hollow nanorod array (MHNRA) electrocatalysts were designed and fabricated for formic acid electrooxidation for DFAFCs. A Ni hydroxide [Ni(OH)2–NiOOH] layer was employed as an interlayer and Pt and Pd layers were homogeneously coated on the outside and inside of the Ni(OH)2–NiOOH interlayer, respectively. Under these circumstances, Ni(OH)2–NiOOH provided fast electron transport and abundant OHads species to remove the adsorbed poisoning species (such as CO) on the surface of the Pt and Pd layers. As a result of the special surface and synergistic effects, the Pt/Ni(OH)2–NiOOH/Pd MHNRAs exhibited significantly improved electrocatalytic activity, CO poisoning tolerance and long-term cycling stability compared with Pt@Pd MHNRAs and commercial Pt/C, Pd/C and PtRu/C electrocatalysts. This paper represents a novel, prime strategy to design electrocatalysts with high catalytic activity and excellent long-term durability for formic acid oxidation.
2. Experimental section
Synthesis of Pt/Ni(OH)2–NiOOH/Pd multi-walled hollow nanorod arrays (MHNRAs)
In this study, all chemical reagents were of analytical (AR) grade. Electrodeposition was performed in a simple two-electrode cell via a galvanostatic method, and a graphite electrode was used as the counter electrode (spectral grade, 1.8 cm2). The Pt/Ni(OH)2–NiOOH/Pd multi-walled hollow nanorod arrays (MHNRAs) were fabricated via the following procedure:(1) ZnO nanorod arrays (ZnO NRAs) were electrodeposited in a solution of 0.005 M Zn(NO3)2·6H2O + 0.025 M NH4NO3 (10 ml) at a current density of 0.5 mA cm−2 at 70 °C for 90 min on Ti plates (99.99%, 2.0 cm × 2.5 cm), as shown in Fig. S1a.† The Ti plates were polished using SiC abrasive paper with 300 and 800 grits, then cleaned by sonication for 5 min in ethanol (50%) and distilled water, respectively, and finally dipped in anhydrous ethanol to clean.
(2) ZnO/Pt NRAs were synthesized by electrodeposition of Pt onto the surface of the ZnO nanorods in a solution of 0.001 M H2PtCl6·6H2O + 0.0005 M NaH2PO2·H2O + 0.0002 M C6H5Na3O7·2H2O (10 ml) (the pH was controlled to 3.5 ± 0.2 using a 1.0 M NaOH solution) at a current density of 0.25 mA cm−2 at 30 °C for 20 min, as shown in Fig. S1b.† Then, the Ni(OH)2–NiOOH layers were electrodeposited onto the surface of the ZnO/Pt NRAs in a solution of 0.01 M NiAc2 + 0.05 M NH4Cl + 0.05 M H3BO4 (10 ml) at a current density of 0.25 mA cm−2 at 30 °C for 20 min, and accordingly ZnO/Pt@Ni(OH)2–NiOOH NRAs were fabricated, as shown in Fig. S1c.† After that, the Pd layers were further electrodeposited onto the surface of the ZnO/Pt/Ni(OH)2–NiOOH NRAs to form ZnO/Pt/Ni(OH)2–NiOOH/Pd NRAs in a solution of 0.0008 M PdCl2 + 0.0005 M NaH2PO2·H2O (the pH was controlled to 4.0 ± 0.2 using a 1.0 M NaOH solution) at a current density of 0.25 mA cm−2 at 30 °C for 20 min, as shown in Fig. S1d.†
(3) ZnO was removed from the ZnO/Pt/Ni(OH)2–NiOOH/Pd NRAs by immersion in 3% NH3·H2O solution for 1 h and the Pt/Ni(OH)2–NiOOH/Pd MHNRAs were finally fabricated. For comparison, Ni(OH)2–NiOOH/Pt, Ni(OH)2–NiOOH/Pd and Pt/Pd MHNRAs were also fabricated by a similar process, and their SEM images are shown in Fig. S3a–c,† respectively.
Structural characterization
The surface morphology of the synthesized Pt/Ni(OH)2–NiOOH/Pd MHNRAs was characterized using thermal field emission environmental scanning electron microscopy (SEM, FEI, Quanta 400F) and Transmission Electron Microscopy (TEM, FEI Tecnai G2 F30). The noble metal loading of the synthesized catalysts was measured using an Inductively Coupled Plasma-Optical Emission Spectrometer (ICP-OES). Chemical-state analysis of the catalysts was carried out using X-ray photoelectron spectroscopy (XPS, ESCALab250). All XPS spectra were corrected using the C 1s line at 284.6 eV. Curve fitting and background subtraction were accomplished. The fabricated Pt@Ni(OH)2–NiOOH@Pd MHNRAs were also characterized using an X-ray diffractometer (XRD).Electrochemical characterization
Electrochemical measurements (CHI 760E Electrochemical Workstation) were performed in a standard three-electrode cell at room temperature (30 °C). A Pt foil and saturated calomel electrode (SCE) were used as the counter electrode and reference electrode, respectively. The electrocatalysts Pt/Ni(OH)2–NiOOH/Pd MHNRAs, Pt/Pd MHNRAs, Ni(OH)2–NiOOH/Pt MHNRAs and Ni(OH)2–NiOOH/Pd MHNRAs (the loading area of the catalysts is ∼2 cm2 on the Ti plate) acted directly as the working electrode. Commercial Pt/C catalysts (20% Pt on Vulcan XC-72, JM), Pd/C catalysts (20% Pd on Vulcan XC-72, JM), and PtRu/C catalysts (40% Pt and 20% Ru on carbon black, HISPEC 10000), loaded on a glassy carbon electrode (the apparent surface area is 0.1962 cm2), were also utilized as a comparison in this study. The noble metal (Pt, Pd) loading of the Pt/Ni(OH)2–NiOOH/Pd MHNRAs, Pt/Pd MHNRAs, Ni(OH)2–NiOOH/Pt MHNRAs, Ni(OH)2–NiOOH/Pd MHNRAs, Pt/C, Pd/C, and PtRu/C catalysts is 26.15, 31.91, 18.15, 13.26, 24.46, 24.46, and 24.46 μg cm−2, respectively. The electrochemically active surface area (ECSA) of the prepared electrocatalysts was measured in 0.5 M H2SO4 at a scan rate of 20 mV s−1 using cyclic voltammetry (CV). The ECSA (m2 gnoble metal−1) of the electrocatalysts was estimated according to the following equation.| ECSA = QH/(210 × Wnoble metal) | (1) |
Q H is the charge (μC) for hydrogen desorption, 210 represents the charge (μC cm−2) required to oxidize a monolayer of hydrogen on a bright noble metal surface, and Wnoble metal represents the noble metal loading (μg) in the electrode. Cyclic voltammograms (CVs) were recorded between −0.20 and 1.00 V vs. SCE at a scan rate of 100 mV s−1. Chronoamperometry curves were measured at 0.4 V. For the CVs and chronoamperometry measurements of the formic acid oxidation reaction, a solution of 0.5 M HCOOH + 0.5 M H2SO4 was utilized in this study. The CO anti-poisoning ability of the prepared electrocatalysts was measured in 0.5 M H2SO4 solution at a scan rate of 20 mV s−1. Prior to the measurements, high purity CO was bubbled into the H2SO4 solution for 15 min to achieve a maximum coverage of CO at the electrocatalyst surface while keeping the potential at 0 V. Then, the dissolved CO was purged out of the H2SO4 solution by bubbling high-purity N2 gas for 15 min.
3. Results and discussion
The Pt/Ni(OH)2–NiOOH/Pd MHNRAs were facilely synthesized by an electrodeposition method, as illustrated in Scheme 1. The details of the fabrication are described in the Experimental section. SEM images of the fabricated ZnO NRAs, ZnO/Pt NRAs, ZnO/Pt/Ni(OH)2–NiOOH NRAs and ZnO/Pt/Ni(OH)2–NiOOH/Pd NRAs are shown in Fig. S1a–d in the ESI,† respectively. It can be clearly seen that the diameters and lengths of the ZnO nanorods are ∼300 nm and 2 μm, respectively. Meanwhile, the Pt, Ni(OH)2–NiOOH and Pd wraps favorably share the surfaces of the ZnO nanorods and no deposit is seen among the interspaces of the nanorods. After dissolving ZnO, the Pt/Ni(OH)2–NiOOH/Pd MHNRAs were fabricated and a typical SEM image is shown in Fig. 1a, which clearly shows that the Pt/Ni(OH)2–NiOOH/Pd nanorods are separated from each other. The high void volume in the Pt/Ni(OH)2–NiOOH/Pd MHNRAs will provide an excellent three-dimensional space for the mass transfer of reactant and resultant molecules during the formic acid electrooxidation. An SEM image of a broken Pt/Ni(OH)2–NiOOH/Pd nanorod is shown in the inset of Fig. 1a. The hollow structure is clearly observed and the inner diameter and wall thickness are about 300 and 80 nm, respectively.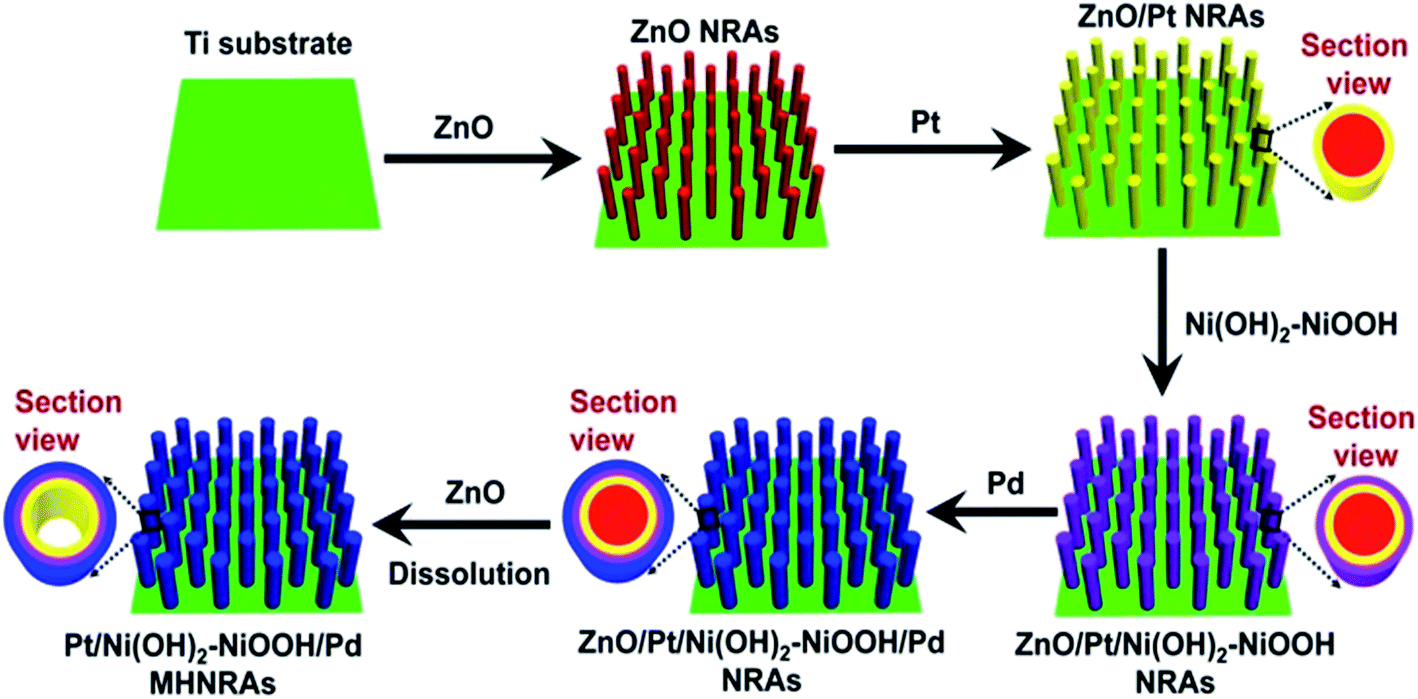 | ||
| Scheme 1 Fabrication of the Pt/Ni(OH)2–NiOOH/Pd MHNRAs. | ||
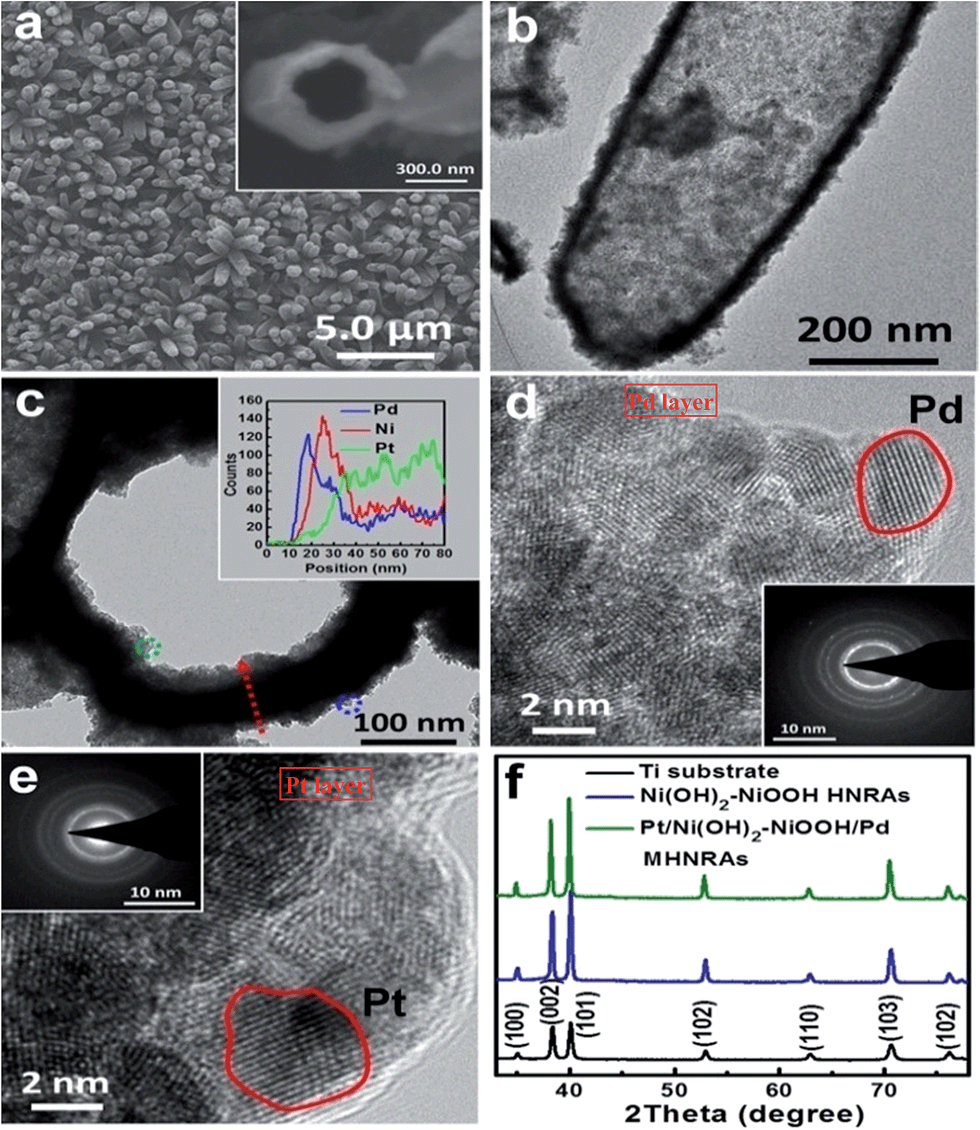 | ||
| Fig. 1 (a) SEM image of the Pt/Ni(OH)2–NiOOH/Pd MHNRAs and a broken hollow nanorod (inset); (b) TEM image of a Pt/Ni(OH)2–NiOOH/Pd hollow nanorod; (c) TEM image of a frontal Pt/Ni(OH)2–NiOOH/Pd hollow nanorod and EDX line scan along the red arrow (inset); (d) HRTEM image and SAED (inset) of the area marked in blue in (c); (e) HRTEM image and SAED (inset) of the area marked in green in (c); (f) XRD patterns of the Ti substrate, Ni(OH)2–NiOOH HNRAs and Pt/Ni(OH)2–NiOOH/Pd MHNRAs. | ||
To further confirm the hollow structure, a typical transmission electron microscopy (TEM) image of a Pt/Ni(OH)2–NiOOH/Pd hollow nanorod is shown in Fig. 1b, which shows the hollow structure, a homogeneous wall thickness of ∼80 nm and an inner diameter of ∼300 nm. Fig. 1c shows a TEM image of the frontal view of a Pt/Ni(OH)2–NiOOH/Pd hollow nanorod. To verify the multi-walled nanostructures in the Pt/Ni(OH)2–NiOOH/Pd MHNRAs, an EDX line scan of the wall marked with a red arrow was measured and is shown in the inset of Fig. 1c; it shows the multi-walled nanostructure, and there are some overlaps among the Pt, Pd and Ni layers, which will be very conducive to produce a strong synergistic effect between the Pt, Pd and Ni(OH)2–NiOOH layers. The thicknesses of the Pd, Ni(OH)2–NiOOH and Pt layers are ∼30, 30, and 50 nm, respectively. HRTEM images of the area marked with blue and green small circles in Fig. 1c are shown in Fig. 1d and e, respectively, and indicate that the Pd and Pt layers both consist of nanocrystals of ∼5 nm. The selected area electron diffraction (SAED) patterns of the Pd and Pt layers (inset of Fig. 1d and e, respectively) indicate that the Pd and Pt layers are polycrystalline and have small size characteristics. The HRTEM image and SAED pattern of the Ni(OH)2–NiOOH interlayer are shown in Fig. S2,† which show that the Ni(OH)2–NiOOH layer is amorphous. The XRD pattern of the Pt/Ni(OH)2–NiOOH/Pd MHNRAs is shown in Fig. 1f, which only shows the peaks of the Ti substrate. This can be attributed to the low content of Pt and Pd and the amorphous Ni(OH)2–NiOOH layers in the sample.30 The content of Pt in the Pt/Ni(OH)2–NiOOH/Pd MHNRAs is 9.94 at%, that of Pd is 9.26 at%, and that of Ni(OH)2–NiOOH is 80.80 at%. The mole ratio of Ni(OH)2/NiOOH is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Ni(OH)2 in acidic media can be converted to NiOOH, which will be a stable phase in the catalyst during the electrochemical measurements:38
:1. Ni(OH)2 in acidic media can be converted to NiOOH, which will be a stable phase in the catalyst during the electrochemical measurements:38
| 4Ni(OH)2 ↔ 4NiOOH + 4H+ + 4e− | (2) |
To investigate the effects of Ni(OH)2–NiOOH on the electron structures of Pt and Pd, XPS spectra of the Pt/Ni(OH)2–NiOOH/Pd MHNRAs, Pt HNRAs, Pd HNRAs and Ni(OH)2–NiOOH HNRAs in the Pt 4f, Pd 3d and Ni 2p regions were measured. A comparison of the relative areas of the integrated intensities of the noble metal (Pt0 and Pd0) and divalent metal (Pt2+ and Pd2+) peaks in Fig. S4 and S5† indicates that most of the Pt and Pd exist as Pt0 and Pd0 in the Pt/Ni(OH)2–NiOOH/Pd MHNRAs, whereas much more divalent Pt2+ and Pd2+ are seen in the Pt HNRAs and Pd HNRAs, as shown in Fig. S4 and S5,† respectively. Thus, the introduction of Ni(OH)2–NiOOH can significantly increase the content of Pt0 and Pd0 and decrease the relative content of divalent Pt2+ and Pd2+ in the Pt/Ni(OH)2–NiOOH/Pd MHNRAs. In addition, for the binding energy of Pt 4f and Pd 3d in the Pt/Ni(OH)2–NiOOH/Pd MHNRAs, we also observed a definite positive shift of ∼0.19 eV and 0.71 eV relative to the Pt HNRAs and Pd HNRAs, respectively, indicating the change of the electronic states of the Pt and Pd atoms. In addition, we also found a negative shift of ∼0.38 eV in the binding energy of Ni(OH)2 for the Pt/Ni(OH)2–NiOOH/Pd MHNRAs relative to the Ni(OH)2–NiOOH HNRAs, as shown in Fig. S6.† Hence, the above shifts in the binding energy of the Pt 4f, Pd 3d and Ni 2p peaks confirm the presence of electron interactions between Pt, Pd and Ni in the Pt/Ni(OH)2–NiOOH/Pd MHNRAs, which lead to a high content of metallic Pt and Pd and synergistic effects for catalytic reactions, and accordingly the catalytic performance of the Pt/Ni(OH)2–NiOOH/Pd MHNRAs for formic acid electrooxidation will obviously improve.
The electrochemical properties of the Pt/Ni(OH)2–NiOOH/Pd MHNRAs were firstly evaluated by the electrochemically active surface area (ECSA) that can be calculated from the hydrogen desorption via cyclic voltammetry measurements. Fig. 2a shows the cyclic voltammograms (CVs) of the Pt/Ni(OH)2–NiOOH/Pd MHNRAs, Pt/Pd MHNRAs and commercial Pt/C, Pd/C and PtRu/C catalysts in a deaerated 0.5 M H2SO4 solution at 20 mV s−1. Herein, the ECSA of the Pt/Ni(OH)2–NiOOH/Pd MHNRAs is calculated to be 94.84 m2 g−1, which is much larger than those of the Pt/Pd MHNRAs (68.22 m2 g−1) and commercial Pt/C (47.35 m2 g−1), Pd/C (43.28 m2 g−1) and PtRu/C (26.56 m2 g−1) catalysts. The ECSA enhancement of the Pt/Ni(OH)2–NiOOH/Pd MHNRAs can be attributed to the high content of metallic Pt and Pd, hollow nanorod structures, multi-walled Pt/Ni(OH)2–NiOOH/Pd structures, and the effect of Ni(OH)2–NiOOH on the electronic states of Pt and Pd. In addition, the Pt/Ni(OH)2–NiOOH/Pd MHNRAs exhibit a much larger ECSA than Ni(OH)2–NiOOH@Pt MHNRAs (5.95 m2 g−1) and Ni(OH)2–NiOOH@Pd MHNRAs (26.82 m2 g−1), as shown in Fig. S7a.†
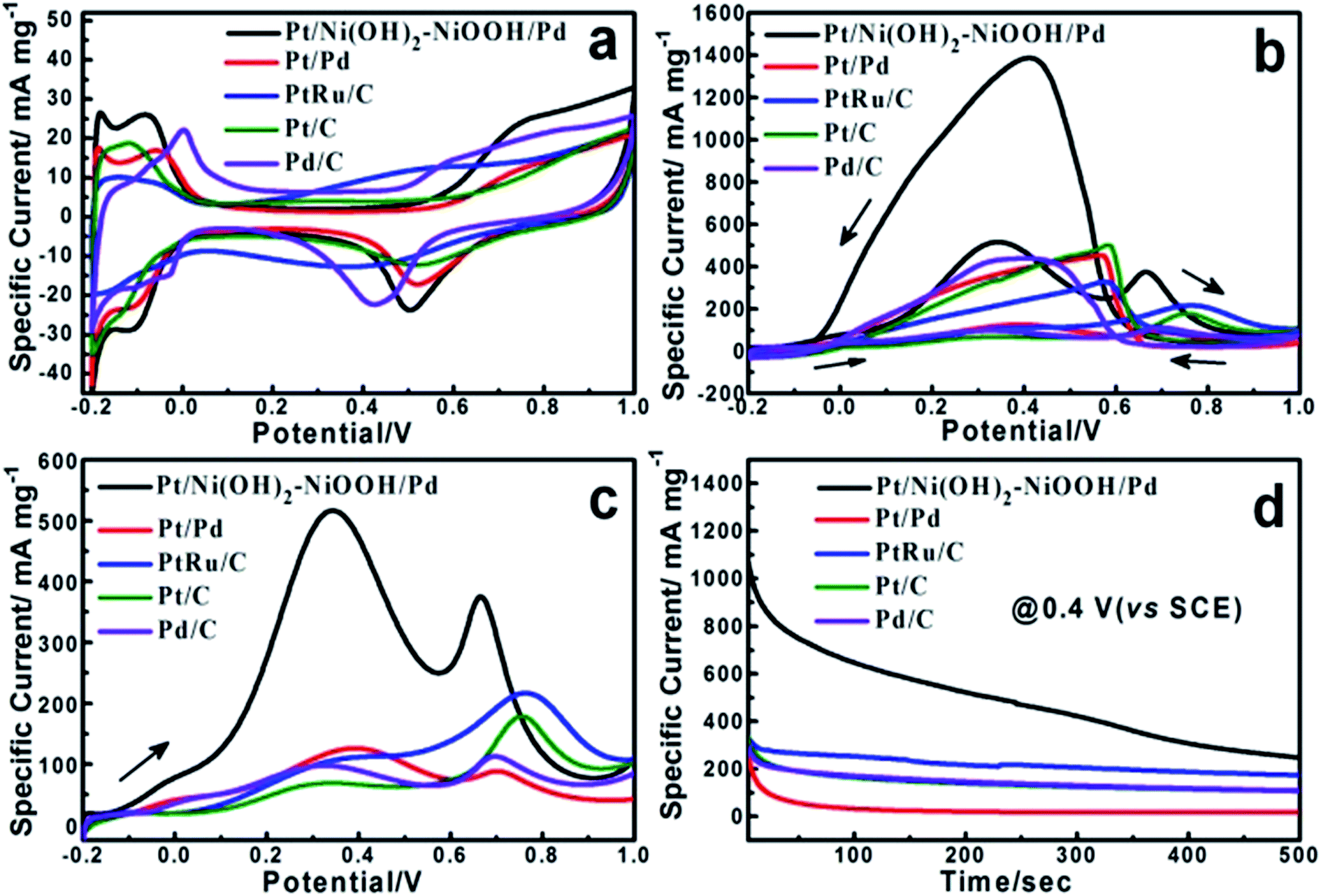 | ||
| Fig. 2 (a) CVs of the Pt/Ni(OH)2–NiOOH/Pd MHNRAs, Pt/Pd MHNRAs, and commercial Pt/C, Pd/C, PtRu/C catalysts in deaerated 0.5 M H2SO4 solution at 20 mV s−1; (b) CVs of the Pt/Ni(OH)2–NiOOH/Pd MHNRAs, Pt/Pd MHNRAs, and commercial Pt/C, Pd/C, PtRu/C catalysts in a solution of 0.5 M HCOOH + 0.5 M H2SO4 at 100 mV s−1; (c) the forward scan peaks of CVs between −0.2 and 1.0 V; (d) chronoamperometry curves of the Pt/Ni(OH)2–NiOOH/Pd MHNRAs, Pt/Pd MHNRAs, and commercial Pt/C, Pd/C, PtRu/C catalysts in 0.5 M HCOOH + 0.5 M H2SO4 at 0.4 V. | ||
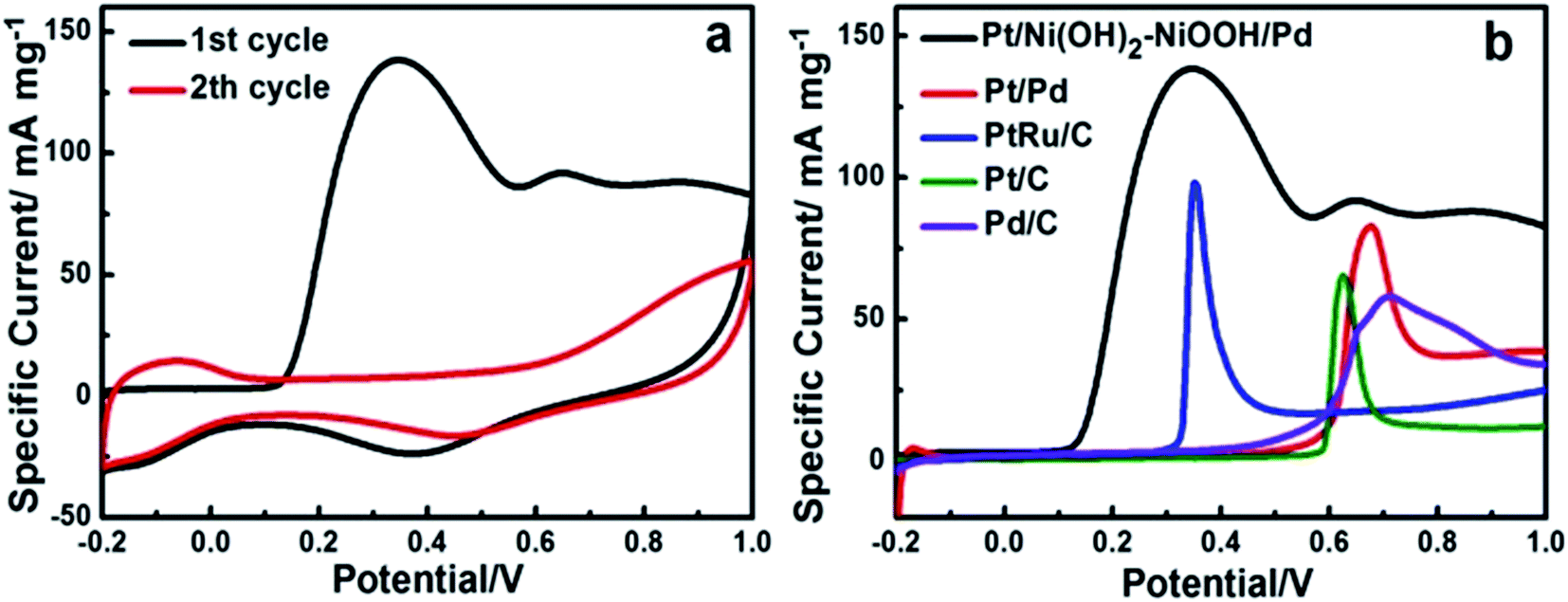
The electrocatalytic activity of the Pt/Ni(OH)2–NiOOH/Pd MHNRAs toward formic acid electrooxidation along with the Pt/Pd MHNRAs and commercial Pt/C, Pd/C, PtRu/C catalysts was investigated in a solution of 0.5 M HCOOH + 0.5 M H2SO4 at 100 mV s−1, and the CVs are shown in Fig. 2b. The Pt/Ni(OH)2–NiOOH/Pd MHNRAs exhibit a very high specific peak current density of ∼510 mA mg−1 at 0.4 V, which is almost 4.2, 7.5, 5.5 and 5.0 times higher than that of the Pt/Pd MHNRAs, commercial Pt/C, Pd/C, and PtRu/C catalysts (the current densities are all normalized to the mass of the noble metal in the catalyst), respectively, indicating the mass activity of the Pt/Ni(OH)2–NiOOH/Pd MHNRAs is much larger than that of the Pt/Pd MHNRAs, commercial Pt/C, Pd/C, and PtRu/C catalysts, as shown in Fig. 2c. Furthermore, the Pt/Ni(OH)2–NiOOH/Pd MHNRAs also exhibited an obviously lower onset potential than that of the Pt/Pd MHNRAs, commercial Pt/C, Pd/C and PtRu/C catalysts, indicating that the formic acid electrooxidation on the Pt/Ni(OH)2–NiOOH/Pd MHNRAs is much easier. Compared with Ni(OH)2–NiOOH@Pt and the Ni(OH)2–NiOOH@Pd MHNRAs, the Pt/Ni(OH)2–NiOOH/Pd MHNRAs also exhibit a significantly improved electrocatalytic activity toward formic acid electrooxidation, as shown in Fig. S7b.† In addition, we compared the specific activity of various catalysts based on the ECSA, as shown in Fig. S7c,† which shows that the Pt/Ni(OH)2–NiOOH/Pd MHNRAs also exhibit a significantly improved ECSA and electrocatalytic activity compared with the Pt/Pd MHNRAs, commercial Pt/C, Pd/C and PtRu/C catalysts.
Chronoamperometry (CA) was carried out to investigate the electrocatalytic activity and stability of the Pt/Ni(OH)2–NiOOH/Pd MHNRAs at an operating voltage of 0.4 V vs. SCE. The CA curves show that the Pt/Ni(OH)2–NiOOH/Pd MHNRAs have a slower current decay over time in comparison with Pt/Pd MHNRAs and commercial Pt/C, Pd/C and PtRu/C catalysts, as shown in Fig. 2d, indicating that the Pt/Ni(OH)2–NiOOH/Pd MHNRAs have a much higher tolerance to CO generated during formic acid oxidation. Fig. 2d also shows that the Pt/Ni(OH)2–NiOOH/Pd MHNRAs exhibit much higher specific current densities than the Pt/Pd MHNRAs and commercial Pt/C, Pd/C and PtRu/C catalysts at all times, further demonstrating that the Pt/Ni(OH)2–NiOOH/Pd MHNRAs have a significantly improved electrocatalytic activity for formic acid electrooxidation, which is in agreement with the CVs shown in Fig. 2b. The enhanced catalytic performance of the Pt/Ni(OH)2–NiOOH/Pd MHNRAs can be attributed to the synergistic effects between the Pt, Pd and Ni(OH)2–NiOOH and the special surface effects of the multi-walled hollow nanorods. Specifically, the introduction of Ni(OH)2–NiOOH could effectively increase the charge transfer ability of the Pt/Ni(OH)2–NiOOH/Pd MHNRAs, and this can be demonstrated using impedance measurements, as shown in Fig. S9.† Compared to the Pt/Pd MHNRAs, the Pt/Ni(OH)2–NiOOH/Pd MHNRAs show a well-defined small semicircle in the high frequency region and have a low charge transfer resistance. As we all know, a low charge transfer resistance corresponds to favorable charge transport kinetics in catalysts and will effectively enhance the catalytic activity of electrocatalysts.4,28
To evaluate the long-term cycling stability, Fig. 3a shows CVs of the Pt/Ni(OH)2–NiOOH/Pd MHNRAs from the 1st to 500th cycles and Fig. 3b shows the change in specific peak current density with increasing cycle number for formic acid electrooxidation in a solution of 0.5 M HCOOH + 0.5 M H2SO4 at 100 mV s−1. It is shown that the peak specific current density of Pt/Ni(OH)2–NiOOH/Pd MHNRAs drastically increases during the initial cycles, and the maximum specific peak current density appears at about the 100th cycle. After 100 cycles, the specific peak current density exhibits a slow attenuation with increasing cycle number. After 500 cycles, the conservation rate of the specific peak current density of the Pt/Ni(OH)2–NiOOH/Pd MHNRAs is ∼94.75% of the maximum value, which is much higher than those of the Pt/Pd MHNRAs (54.44%), commercial Pt/C (47.66%), Pd/C (17.04%) and PtRu/C (76.22%) catalysts, as shown in Fig. 3c, indicating that the Pt/Ni(OH)2–NiOOH/Pd MHNRAs have excellent cycling stability for formic acid oxidation and exhibit a significantly enhanced cycling stability compared with the Pt/Pd MHNRAs, commercial Pt/C, Pd/C and PtRu/C electrocatalysts. In addition, the Pt/Ni(OH)2–NiOOH/Pd MHNRAs exhibited a much higher cycling stability than the Ni(OH)2–NiOOH/Pt MHNRAs (44.75%) and Ni(OH)2–NiOOH/Pd MHNRAs (21.22%), as shown in Fig. S10.† After 500 cycles, the surface morphology of the Pt/Ni(OH)2–NiOOH/Pd MHNRAs has been maintained very well, as shown in Fig. S11,† indicating the high structural stability of the Pt/Ni(OH)2–NiOOH/Pd MHNRAs.
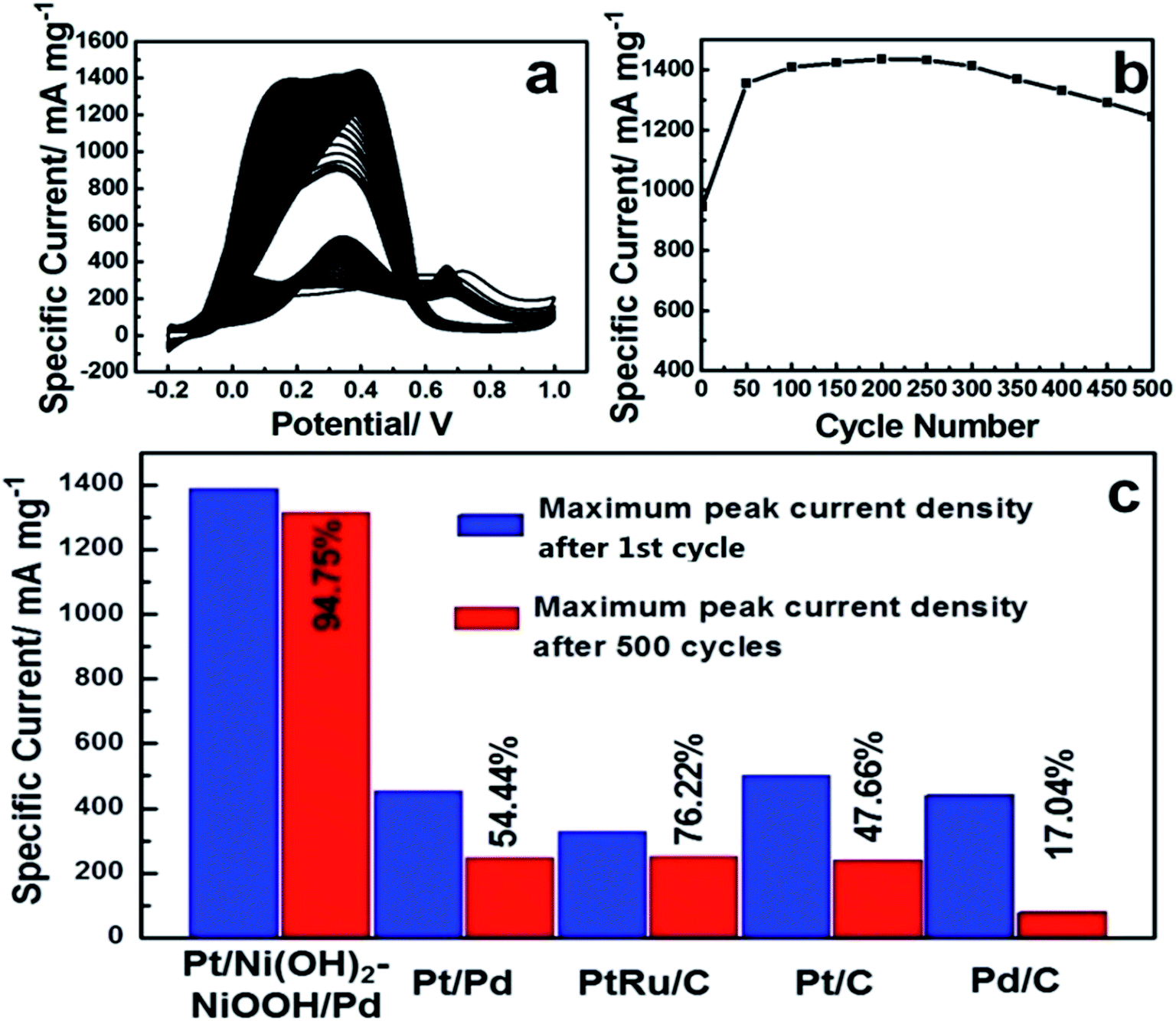 | ||
| Fig. 3 (a) CVs of the Pt/Ni(OH)2–NiOOH/Pd MHNRAs from the 1st to 500th cycles; (b) the change of the maximum specific peak current density with increasing cycle number for the Pt/Ni(OH)2–NiOOH/Pd MHNRAs; (c) the comparison of the maximum specific peak current density after the 1st cycle and the maximum specific peak current density after the 500th cycle for formic acid electrooxidation of the Pt/Ni(OH)2–NiOOH/Pd MHNRAs, Pt@Pd MHNRAs, and commercial Pt/C, Pd/C and PtRu/C catalysts. | ||
The superior CO anti-poisoning ability of the Pt/Ni(OH)2–NiOOH/Pd MHNRAs was demonstrated through CO stripping measurements, as shown in Fig. 4a. A remarkably larger CO oxidation peak can be clearly seen in the initial forward scan in the 1st cycle, indicating that the Pt/Ni(OH)2–NiOOH/Pd MHNRAs have a large ECSA and good oxidation activity for CO. During the 2nd cycle, the CV does not show any peak for CO oxidation, while the adsorption/desorption peak of hydrogen is clearly visible, indicating that the CO was oxidized completely during the 1st scan. In addition, in the initial forward scan of CO stripping, the onset potential (∼0.10 V) of CO oxidation on the Pt/Ni(OH)2–NiOOH/Pd MHNRAs is obviously more negative than those on the Pt@Pd MHNRAs (∼0.50 V) and commercial Pt/C (∼0.60 V), Pd/C (∼0.45 V) and PtRu/C (∼0.30 V) catalysts, as shown in Fig. 4b, indicating that the Pt/Ni(OH)2–NiOOH/Pd MHNRAs have a much higher CO oxidation ability than the Pt/Pd MHNRAs and commercial Pt/C, Pd/C and PtRu/C catalysts. The above result shows that the introduction of Ni(OH)2–NiOOH can facilitate the removal of CO from the catalyst surface as the Ni(OH)2–NiOOH can provide enough OHads species to oxidize CO and can realize the preferable electron delocalization among the hybrid Pt, Pd and Ni(OH)2–NiOOH layers. In addition, the Pt/Ni(OH)2–NiOOH/Pd MHNRAs exhibit a much larger CO oxidation peak than the Ni(OH)2–NiOOH/Pt MHNRAs and Ni(OH)2–NiOOH/Pd MHNRAs, as shown in Fig. S7d.†
 | ||
| Fig. 4 (a) CO stripping voltammograms on the Pt/Ni(OH)2–NiOOH/Pd MHNRAs performed in a solution of 0.5 M H2SO4 at 20 mV s−1; (b) the comparison of the CO stripping voltammograms of the Pt/Ni(OH)2–NiOOH/Pd MHNRAs, Pt/Pd MHNRAs, commercial Pt/C, Pd/C and PtRu/C catalysts. | ||
Based on the above results, the high performance of the Pt/Ni(OH)2–NiOOH/Pd MHNRAs is summarized as follows: (a) the merits of the surface morphology of the MHNRAs: (i) the MHNRAs can provide a large surface area, and fast electrolyte penetration/diffusion because of the hollow structures, (ii) the MHNRAs will be much less vulnerable to dissolution, Ostwald ripening, and aggregation, which is beneficial for the improvement of stability of the catalyst, (iii) the MHNRAs directly grown on the conductive substrate have an excellent electrical contact with the current collectors, and this would let each MHNRA effectively participate in the catalytic reactions with almost no “dead” volume; (b) the merits of the hybrid structure of Pt/Ni(OH)2–NiOOH/Pd: (i) the Ni hydroxide [Ni(OH)2–NiOOH] layers were employed as an interlayer and they will provide fast electron transport and abundant OHads species to remove the adsorbed poisoning species (such as CO) on the surfaces of the Pt and Pd layers, (ii) the Pt and Pd layers are homogeneously coated on the outside and inside surfaces of the Ni(OH)2–NiOOH interlayer, and accordingly the utilization rates of Pt and Pd will be obviously enhanced. In addition, the synergistic effects between Pt, Ni(OH)2–NiOOH and Pd can be well realized.
4. Conclusions
In summary, we have designed and fabricated a Ni hydroxide (Ni(OH)2–NiOOH) functionalized electrocatalyst by constructing Pt/Ni(OH)2–NiOOH/Pd multi-walled hollow nanorod arrays for formic acid electrooxidation. The introduction of Ni(OH)2–NiOOH as a co-catalyst can provide enough OHads species to remove CO adsorbed on the surfaces of Pt and Pd. Furthermore, the multi-walled nanostructure will lead to strong synergistic effects between the Pt, Pd and Ni(OH)2–NiOOH for the catalytic reactions. Compared with the Pt@Pd MHNRAs and commercial Pt/C, Pd/C and PtRu/C catalysts, the Pt/Ni(OH)2–NiOOH/Pd MHNRAs exhibit a significantly enhanced electrocatalytic activity, cycling stability and CO poisoning tolerance for formic acid electrooxidation. The hybridization method based on the Ni(OH)2–NiOOH and multi-walled hollow nanorod structure will provide a new route for the fabrication of electrocatalysts with low-cost and high-performance for the electrooxidation of small organic molecules.Acknowledgements
This work was supported by National Natural Science Foundation of China (51173212 and J1103305), National Basic Research Program of China (2015CB932304), Natural Science Foundation of Guangdong Province (S2013020012833), Fundamental Research Fund for the Central Universities (13lgpy51), SRF for ROCS, SEM ([2012]17071707), Project of High Level Talents in Higher School of Guangdong Province, Science and Technology Planning Project of Guangdong Province (2013B010403011) and Open-End Fund of Key Laboratory of Functional Inorganic Material Chemistry (Heilongjiang University), Ministry of Education.References
- (a) Y. Wang, D. Wilkinson and J. Zhang, Chem. Rev., 2011, 111, 7625–7651 CrossRef CAS PubMed; (b) V. Mazumder and S. Sun, J. Am. Chem. Soc., 2009, 131, 4588–4589 CrossRef CAS.
- X. Ji, T. L. Kyu, C. Martin, F. N. Linda, H. Reanne, L. Zhang, J. Zhang, A. B. Gianluigi and B. Albert, Nat. Chem., 2010, 2, 286–293 CrossRef CAS PubMed.
- (a) J. Chang, L. Feng, C. Liu, W. Xing and X. Hu, Angew. Chem., Int. Ed., 2014, 53, 122–126 CrossRef CAS PubMed; (b) D. Zhao, Y.-H. Wang and B.-Q. Xu, J. Phys. Chem. C, 2009, 113, 20903–20911 CrossRef CAS.
- (a) C. Guo, L. Zhang, J. Miao, J. Zhang and C. M. Li, Adv. Energy Mater., 2013, 3, 167–171 CrossRef CAS; (b) R. Lyyamperu-mal, L. Zhang, G. Henkelman and R. M. Crooks, J. Am. Chem. Soc., 2013, 135, 5521–5524 CrossRef.
- (a) J. Joo, T. Uchida, A. Cuesta, M. Koper and M. Osawa, J. Am. Chem. Soc., 2013, 135, 9991 CrossRef CAS; (b) F. Vidal-Iglesias, A. López-Cudero, J. Solla-Gullón and J. M. Feliu, Angew. Chem., Int. Ed., 2013, 52, 964–967 CrossRef CAS PubMed.
- (a) X. Xia, S. Choi, J. A. Herron, N. Lu, J. Scaranto, H.-C. Peng, J. Wang, M. Mavrikakis, M. J. Kim and Y. Xia, J. Am. Chem. Soc., 2013, 135, 15706–15709 CrossRef CAS PubMed; (b) J. V. Perales-Rondón, A. Ferre-Vilaplana, J. Feliu and E. Herrero, J. Am. Chem. Soc., 2014, 136, 13110–13113 CrossRef PubMed.
- A. Chen and P. Holt-Hindle, Chem. Rev., 2010, 110, 3767–3804 CrossRef CAS.
- (a) S. Zhang, Y. Shao, G. Yin and Y. Lin, Angew. Chem., Int. Ed., 2010, 49, 2211–2214 CrossRef CAS PubMed; (b) F. J. Vidal-Iglesias, J. Solla-Gullón, E. Herrero, A. Aldaz and J. M. Feliu, Angew. Chem., Int. Ed., 2010, 49, 6998–7001 CrossRef CAS PubMed.
- J. Xiao, S. Liu, N. Tian, Z. Y. Zhou, H. X. Liu, B. B. Xu and S. G. Sun, J. Am. Chem. Soc., 2013, 135, 18754–18757 CrossRef CAS PubMed.
- (a) N. Tian, Z. Zhou, S. G. Sun, Y. Ding and Z. L. Wang, Science, 2007, 316, 732 CrossRef CAS; (b) M. Osawa, K. Komatsu, G. Samjeské, T. Uchida, T. Ikeshoji, A. Cuesta and C. Gutiérrez, Angew. Chem., Int. Ed., 2011, 50, 1159–1163 CrossRef CAS PubMed.
- (a) N. Tian, Z. Y. Zhou, N. F. Yu, L. Y. Wang and S. G. Sun, J. Am. Chem. Soc., 2010, 132, 7580–7581 CrossRef CAS PubMed; (b) X. Wang, J. Yang, H. Yin, R. Song and Z. Tang, Adv. Mater., 2013, 25, 2728–2732 CrossRef CAS PubMed.
- (a) B. Y. Xia, H. B. Wu, Y. Yan, X. W. Lou and X. Wang, J. Am. Chem. Soc., 2013, 135, 9480–9485 CrossRef CAS PubMed; (b) B. Y. Xia, H. B. Wu, N. Li, Y. Yan, X. W. Lou and X. Wang, Angew. Chem., Int. Ed., 2015, 54, 3797–3801 CrossRef CAS PubMed.
- V. Mazumder and S. Sun, J. Am. Chem. Soc., 2009, 131, 4588–4589 CrossRef CAS PubMed.
- D. Xu, S. Bliznakov, Z. Liu, J. Fang and N. Dimitrov, Angew. Chem., Int. Ed., 2010, 49, 1282–1285 CrossRef CAS PubMed.
- (a) L. Hyunjoo, E. Susan, A. Gabor and P. Yang, J. Am. Chem. Soc., 2008, 130, 5406–5540 CrossRef; (b) X. Huang, S. Tang, H. Zhang, Z. Zhou and N. Zheng, J. Am. Chem. Soc., 2009, 131, 13916–13917 CrossRef CAS.
- H. Ataee-Esfahani, M. Imura and Y. Yamauchi, Angew. Chem., Int. Ed., 2013, 52, 13611–13615 CrossRef CAS.
- (a) L. Zhang, N. Li, F. Gao, L. Hou and Z. Xu, J. Am. Chem. Soc., 2012, 134, 11326–11329 CrossRef CAS PubMed; (b) L. Wang and Y. Yamauchi, J. Am. Chem. Soc., 2013, 135, 16762–16765 CrossRef CAS.
- J. Suntivich, Z. Xu, C. E. Carlton, J. Kim, B. Han, S. W. Lee, N. Bonnet, N. Marzari, L. Allard, H. A. Gasteiger, K. Hamad-Schifferli and Y. Shao-Horn, J. Am. Chem. Soc., 2013, 135, 7985–7991 CrossRef CAS.
- S. Chen, Z. Wei, X. Qi, L. Dong, Y.-G. Guo, L. Wan, Z. Shao and L. Li, J. Am. Chem. Soc., 2012, 134, 13252–13255 CrossRef CAS PubMed.
- G. H. Wang, J. Hilgert, F. H. Richter, F. Wang, H. J. Bongard, B. Splie-thoff, C. Weidenthaler and F. Schuth, Nat. Mater., 2014, 13, 293 CrossRef CAS.
- R. Erik, S. Anthony, G. Bogdan, V. Rameshkrishnan, S. Sarangapani, S. S. Eugene and E. M. Thomas, Science, 1998, 280, 1735–1737 CrossRef.
- K. Yoon, Y. Yang, P. Lu, D. Wan, H. Peng, K. Stamm Masias, P. Fanson, C. Campbell and Y. Xia, Angew. Chem., Int. Ed., 2012, 51, 9543 CrossRef CAS.
- Y. Y. Tong, H. S. Kim, P. K. Babu, P. Waszczuk, A. Wieckowski and E. Oldfield, J. Am. Chem. Soc., 2002, 124, 468–473 CrossRef CAS PubMed.
- H. P. Zhou, J. Shen, A. X. Yin, L. D. Sun and C. H. Yan, J. Am. Chem. Soc., 2010, 132, 4998–4999 CrossRef CAS PubMed.
- X. Zhao, L. Ma, L. Liang, C. Liu, J. Liao, T. Lu and X. Wei, Energy Environ. Sci., 2011, 4, 2736–2753 CAS.
- V. B. Dmitry and C. W. Frank, Adv. Mater., 2006, 18, 2807–2824 CrossRef PubMed.
- L. Trotochaud, S. Young, J. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS PubMed.
- Z. Zhao, H. Wu, H. He, X. Xu and Y. Jin, Adv. Funct. Mater., 2014, 24, 4698–4705 CrossRef CAS PubMed.
- L. R. Erin, M. Jens, K. Xerxes Steirer, G. Andres, J. Joseph, S. G. David, C. O. Dana, K. Antoine and R. A. Neal, Chem. Mater., 2011, 23, 4988 CrossRef.
- H. B. Li, M. H. Yu, F. X. Wang, P. Liu, Y. Liang, J. Xiao, C. X. Wang, Y. X. Tong and G. W. Yang, Nat. Commun., 2013, 4, 1–7 CAS.
- D. Bediako, B. Lassalle-Kaiser, Y. Surendranath, J. Yano, V. Yachandra and D. G. Nocera, J. Am. Chem. Soc., 2012, 134, 6801–6809 CrossRef CAS.
- H. H. Li, S. Zhao, M. Gong, C. H. Cui, D. He, H. W. Liang, L. Wu and S. H. Yu, Angew. Chem., Int. Ed., 2013, 52, 1–6 CrossRef CAS.
- S. J. Jongmin, Y. Ye, J. Hwang, S.-K. Kim, T.-H. Lim, U. Wiesner and J. Lee, ACS Nano, 2012, 6, 6870–6881 CrossRef PubMed.
- (a) S. Zhang, Y. Shao, G. Yin and Y. Lin, Angew. Chem., Int. Ed., 2010, 49, 2211–2214 CrossRef CAS PubMed; (b) L. Wang, Y. Nemoto and Y. Yamauchi, J. Am. Chem. Soc., 2011, 133, 9674–9677 CrossRef CAS PubMed.
- X. Ge, L. Chen, J. Kang, T. Fujita, A. Hirata, W. Zhang, J. Jiang and M. A. Chen, Adv. Funct. Mater., 2013, 23, 4156–4162 CrossRef CAS PubMed.
- (a) L. Ding, G. Li, Z. Wang, Z. Liu, H. Liu and Y. Tong, Chem.–Eur. J., 2012, 18, 8386–8391 CrossRef CAS PubMed; (b) Z. Wen, S. Ci, F. Zhang, X. Feng, S. Cui, S. Mao, S. Luo, Z. He and J. Chen, Adv. Mater., 2012, 24, 1399–1404 CrossRef CAS PubMed.
- (a) A. L. Wang, X. J. He, X. F. Lu, H. Xu, Y. X. Tong and G. R. Li, Angew. Chem., Int. Ed., 2015, 54, 3669–3673 CrossRef CAS PubMed; (b) A. L. Wang, H. Xu, J. X. Feng, L. X. Ding, Y. X. Tong and G. R. Li, J. Am. Chem. Soc., 2013, 135, 10703–10709 CrossRef CAS PubMed.
- (a) R. Manoharan and J. B. Goodenough, J. Mater. Chem., 1992, 2, 875–887 RSC; (b) K.-W. Park, J. Choi, B.-K. Kwon, S.-A. Lee, Y.-E. Sung, H.-Y. Ha, S.-A. Hong, H. Kim and A. Wieckowski, J. Phys. Chem. B, 2002, 106, 1869–1877 CrossRef CAS; (c) A. Arun, M. Gowdhamamoorthi, K. Ponmani, S. Kiruthika and B. Muthukumaran, RSC Adv., 2015, 5, 49643–49650 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc02544c |
| This journal is © The Royal Society of Chemistry 2015 |