
Synthesis of monolateral and bilateral sulfur-heterocycle fused naphthalene diimides (NDIs) from monobromo and dibromo NDIs†
Bing
Leng
*a,
Dong
Lu
ab,
Xueshun
Jia
b,
Xiaodi
Yang
c and
Xike
Gao
*a
aKey Laboratory of Synthetic and Self-Assembly Chemistry for Organic Functional Molecules, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: gaoxk@mail.sioc.ac.cn; lengbing@sioc.ac.cn
bDepartment of Chemistry, Shanghai University, 99 Shangda Road, Shanghai 200444, China
cLaboratory of Advanced Materials, Fudan University, Shanghai 200433, China
First published on 11th February 2015
Abstract
Monolateral and bilateral sulfur-heterocycle fused naphthalene diimides (NDIs) are successfully synthesized from monobromo and dibromo NDIs via nucleophilic aromatic substitution reactions (SNAr) and the subsequent oxidative aromatization processes. The bromo-substituted monolateral fused NDI can be further functionalized with other aromatic or heteroaromatic rings to construct new electron-deficient π-functional materials with fine-tuned molecular orbital energy levels. The low-lying LUMO levels of these materials afford them great potential as n-type organic semiconductors.
The unique delocalized π-electron structure of organic π-conjugated molecules endows them with unique optoelectronic properties, like strong absorption, light emission and charge carrier transport. Such π-conjugated molecules have been widely investigated for their academic values and potential applications in the field of organic electronics.1 For example, they can act as hole and/or electron transport semiconductors in organic thin film transistors (OTFTs),2 light-harvesting and charge conductive materials in organic photovoltaics (OPVs),3 and electronic conduction and light emission agents in organic light emitting diodes (OLEDs).4
Among organic semiconductors (OSCs) studied, most are electron-rich π-systems and serve as electron donors with hole transport (p-type) behaviors in organic electronic devices. However, the development of electron-deficient π-functional materials that act as electron acceptors with electron conductive properties (n-type) lag far behind their p-type counterparts, which limit their real applications in complementary logic circuits and OPVs (as electron acceptors).5 Therefore, it is highly required to develop electron-deficient π-materials with novel designed structures and/or new synthetic methods.
Naphthalene diimide (NDI) is a widely studied prototypical π-core that possesses a planar aromatic scaffold. The structure of a parent NDI comprises a naphthalene core which bears two electron-withdrawing imide groups at its four α-positions. Diverse substituents at the imide position can alter the solubility and self-assembly of NDI derivatives, but have a little effect on their optical and electrochemical properties. Tuning of the absorption, emission and electronic properties which correlate to structures of π-scaffolds are achievable by substitution at the 2, 3, 6 and 7 core positions. Great progress has been made on the synthesis and applications of NDIs.6 Due to their electron-deficient nature, NDIs have been used extensively as electron acceptors, n-type OSCs and intercalators in aromatic donor–acceptor complexes.6 As a result of the two annulated electron-withdrawing imide groups, NDIs have low-lying LUMO levels, which facilitate electron transport and stable device performance.6a,j,7 As shown in Fig. 1, further modifications of the NDI-cores could fine tune the electronic structures and molecular orbital energy levels according to the methodology investigated, including electron-withdrawing moieties substitution,8 core-expanded by heterocycles fusion9,10 and incorporation of electron-donor units to form donor–acceptor (D–A) structures.11 Gao and co-workers have developed a series of NDI-based OSCs, through NDI core-expansion with sulfur-containing heterocycles bearing terminal electron-withdrawing groups.9 The expanded π-conjugation promotes intermolecular interaction, and the low-lying LUMO is profitable for facile electron injection and stable electron transport. Therein, OTFTs based on core-expanded NDIs fused with two 2-(1,3-dithiol-2-ylidene)malononitrile (DTYM) moieties exhibited good device stability and high electron mobility (maximum electron mobility can reach 3.5 cm2 V−1 s−1),12 which is among the best performances of solution-processed n-channel OTFTs.
 | ||
| Fig. 1 The strategies for core-modifications of NDIs. | ||
The annulation of heterocyclic rings on one side (monolateral fusion)10,13 or both sides (bilateral fusion)9,10,11d of the NDI core have been reported. The bilaterally extended NDIs were usually synthesized from dibromo or tetrabromo NDIs, having centrosymmetric or axisymmetric π-conjugation structures. In contrast, monolateral fused NDIs bear an asymmetric π-core along the naphthalene axis. The symmetric structures were often used to design OSCs for their more efficient and facile synthesis processes, while some of the asymmetric OSCs were proved to have comparable or even better performance in comparison with their symmetric counterparts.14 In addition, asymmetric materials may have unique characteristics; the investigation of their molecular packing in thin films and their device performance could afford us more helpful insights into molecular structure–film morphology–device performance relationship.15
Being inspired by the excellent device performance of NDI-DTYM2 derivatives,9,12 we naturally speculate about the characteristics and potential application of monolateral substituted NDI analogues with the same sulfur-heterocycles as n-type OSCs. In this paper, monobromo and dibromo NDIs were used as the initial materials to synthesize the one-side annulated derivatives 1 and 2 (Scheme 1). The optical and electrochemical properties of both objective compounds were investigated. Besides, the reactive site of bromo on the NDI core of 2 enabled substitution by aromatic or heteroaromatic rings to construct new π-conjugation materials with fine-tuned molecular orbital energy levels.
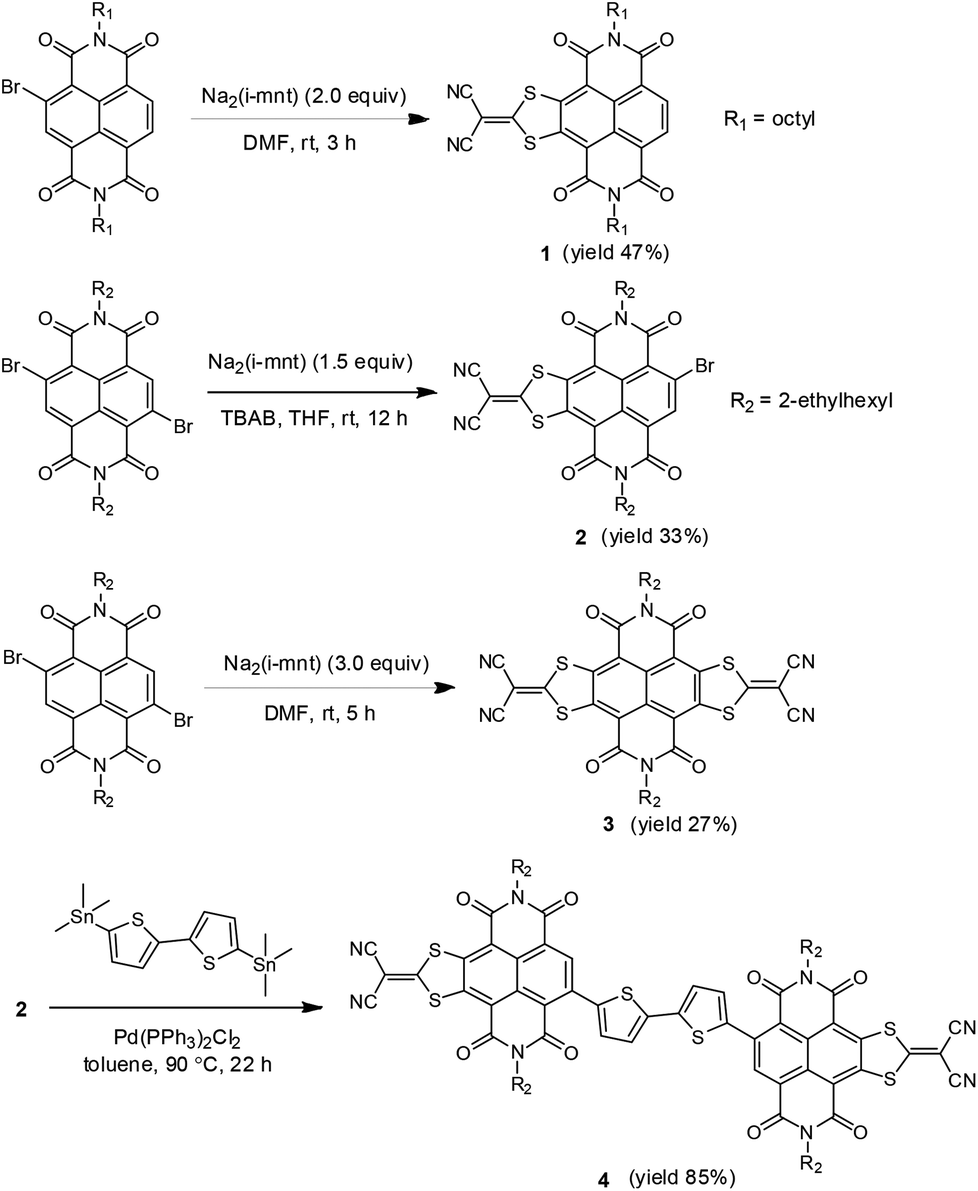 | ||
| Scheme 1 Structures and synthesis of monolateral/bilateral sulfur-heterocycle fused NDIs (1, 2 and 3) and A–D–A type NDI derivative 4. | ||
According to the synthesis of bilateral analogue NDI-DTYM2,9a,b we initially used the nucleophilic aromatic substitution reaction (SNAr) of the corresponding 2,3,6,7-tetrabromonaphthalene diimide (TBNDI) and sodium 1,1-dicyanoethylene-2,2-dithiolate (Na2(i-mnt)) to produce the monolateral derivative, but unfortunately, we could not obtain the desired product in spite of several attempts under different reaction conditions. As per our previous findings on the one-pot synthesis of NDI-DTYM2,9c we proposed that the monolateral fused intermediate 5 (Scheme 2) was produced at first via one side SNAr reaction, which might be more electron deficient than its precursor TBNDI and hence more prone to perform a subsequent nucleophilic substitution with another Na2(i-mnt), affording both sides substituted compound 3 as the only isolated product. To obtain the desired monolateral product, less reactive reagents would be favorable for avoiding the two side substitution reaction. Through trial and error, we found that monobromo and dibromo NDIs are adequate to synthesize one side fused 1 and 2 that are stable enough to avoid further reaction and can be isolated.
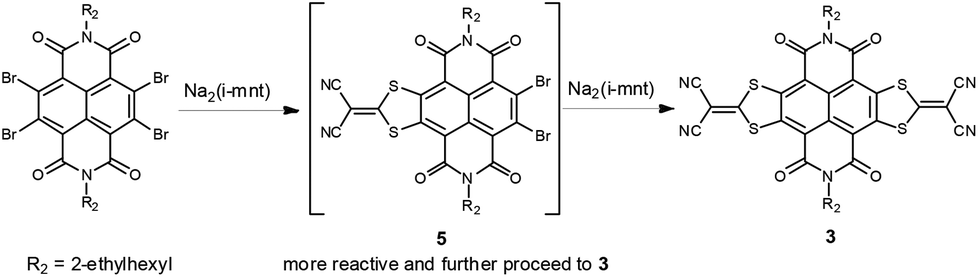 | ||
| Scheme 2 Proposed reaction process of Na2(i-mnt) with TBNDI. | ||
Compound 1 was synthesized by addition of 2 equiv. of Na2(i-mnt) to a solution of monobromo NDI in DMF at room temperature under ambient conditions, giving the desired product in 47% yield. Changing solvents, temperatures, or appending more nucleophilic sodium salts did not achieve higher yields. Batch-wise addition of 1.5 equiv. of Na2(i-mnt) to a solution of dibromo NDI in THF in the presence of tetrabutylammonium bromide (TBAB) as the phase-transfer catalyst affords product 2 in 33% yield. It should be noted that the solvents polarity and water miscibility are considerable factors in producing 2 in view of an appropriate reactivity between hydrophilic salts and hydrophobic molecules. For example, bilateral derivative 3 instead of monolateral fused 2 was synthesized as the only product when carrying out the reaction in DMF, while in the case of dichloromethane, reaction did not proceed even in the presence of TBAB. For comparison, bilateral counterpart 3 was synthesized using dibromo NDI as the starting material with a yield of 27%, which is lower than that achieved from TBNDI (over 50%),9a indicating a relatively lower reactivity of dibromo NDIs than that of tetrabromo ones. The acceptor–donor–acceptor (A–D–A) type derivative 4 was prepared in excellent yield of 85% via palladium-catalyzed Stille coupling between distannyl bithiophene and 2. The reaction was performed in a Schlenk flask under a nitrogen atmosphere by using 5 mol% Pd(PPh3)2Cl2 as the catalyst.
The reaction processes of Na2(i-mnt) with monobromo and tetrabromo NDIs are obviously diverse. According to previous reports, we propose that the reaction mechanism should involve an intermolecular nucleophilic attack at the C-Br position initially, followed by an intramolecular Michael addition process (Scheme 3).10e,11d,13c,16 The resulting intermediate may undergo an oxidative aromatization process with the elimination of hydride anions to form the desired products. Note that oxidants involved in the last step could probably be O2, H2O (from air) or NDI itself.10e,11d In order to gain further insights, comparative experiments were performed to clarify the effects of O2 and H2O by conducting the reactions with/without N2 protection and using super dry DMF (moisture content <50 ppm, J&K Corporation). As a result, a 51% yield of 1 was obtained under an N2 atmosphere and by using super dry DMF, which approximates to that (1, 47%) performed under ambient conditions, indicating that O2 and H2O may not be essential elements for the oxidation process. In addition, when a trace of water was added to the reaction mixture, the yield of 1 decreased significantly to 32%, demonstrating a somewhat negative effect of H2O. Although the participation of H2O could not be fully excluded even use super dry DMF, NDI itself more probably serves as an electrophile and an oxidant to accept the hydrogen anion. Zhang's and Takimiya's groups also proposed similar oxidative processes in synthesizing thiazole-fused NDIs10e and thiophene-fused NDIs11d respectively. Since separation of the NDI reductive byproduct is unsuccessful because of the complex impurities, the reaction mechanism needs to be further investigated.
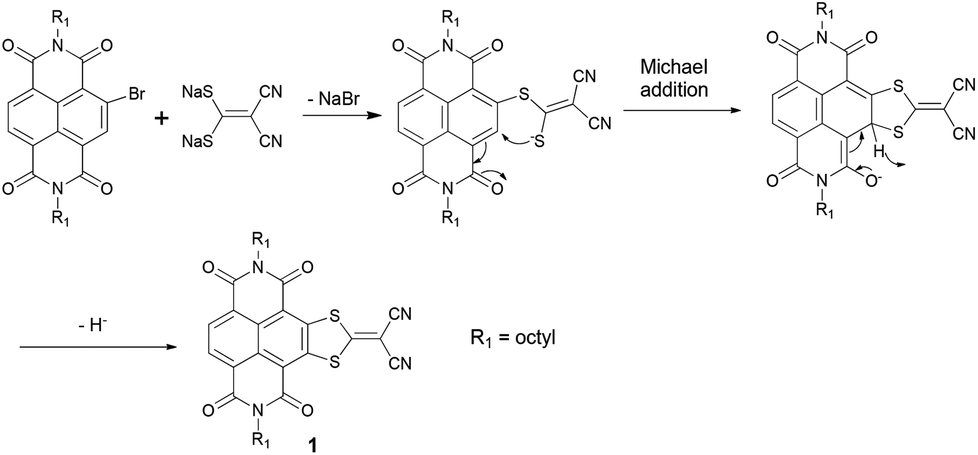 | ||
| Scheme 3 Proposed reaction mechanism of Na2(i-mnt) with monobromo NDI. | ||
Thermogravimetric analyses (TGA) and differential scanning calorimetry (DSC) analyses of 1, 2, 3 and 4 were carried out under a nitrogen atmosphere. TGA analyses (Fig. S1†) of 1, 2, 3 and 4 exhibit thermolysis onset temperatures of 401, 313, 396, 410 °C, respectively. The similar onset decomposition temperatures (Td) of 1 and 3 indicate high thermal stability of both monolateral and bilateral DTYM-fused NDIs. DSC measurements (Fig. S2–5†) displayed the melting points of 273 and 211 °C for 1 and 2, respectively, while the melting points of 3 and 4 were undetectable in our measurement range up to 300 °C. A lower Td and melting points of 2 bearing a bromo-substituent demonstrate a relatively low thermal stability and weak intermolecular interaction.
To estimate the position and energies of frontier molecular orbitals of 1, 2, 3 and 4, density functional theory (DFT) calculations were performed using Gaussian 09 at the B3LYP/6-31G(d,p) level. All alkyl chains were replaced by methyl groups to reduce the time required for calculations. HOMO and LUMO wave function distributions are depicted in Fig. 2. The largest coefficients in the HOMO orbitals of 1, 2 and 3 possess similar distributive characteristics and are delocalized along the entire lateral axis of the conjugation skeleton. The HOMO wave functions of 4 are localized on the central electron-rich bithiophene unit, demonstrating that the HOMO energy distribution is dominated by the donor moiety.11c,17 The electron density of the LUMO orbitals of all four compounds are mainly positioned on the NDI units, of which the LUMO wave functions of 4 are almost entirely localized on the two NDI segments. The result indicates that the LUMO energy distribution is primarily determined by the acceptor moiety.11c,17 HOMO and LUMO energies of 1–4 are also estimated by DFT calculations (Table 1). In comparison with 1, a slightly reduced band gap of 2 is ascribed to the bromo substituent. The lower LUMO level of 3 is derived from the strong electron-withdrawing ability of another DTYM unit, while HOMO energy of 4 is effectively increased after incorporation of the bithiophene donor moiety, giving rise to narrower band gaps.
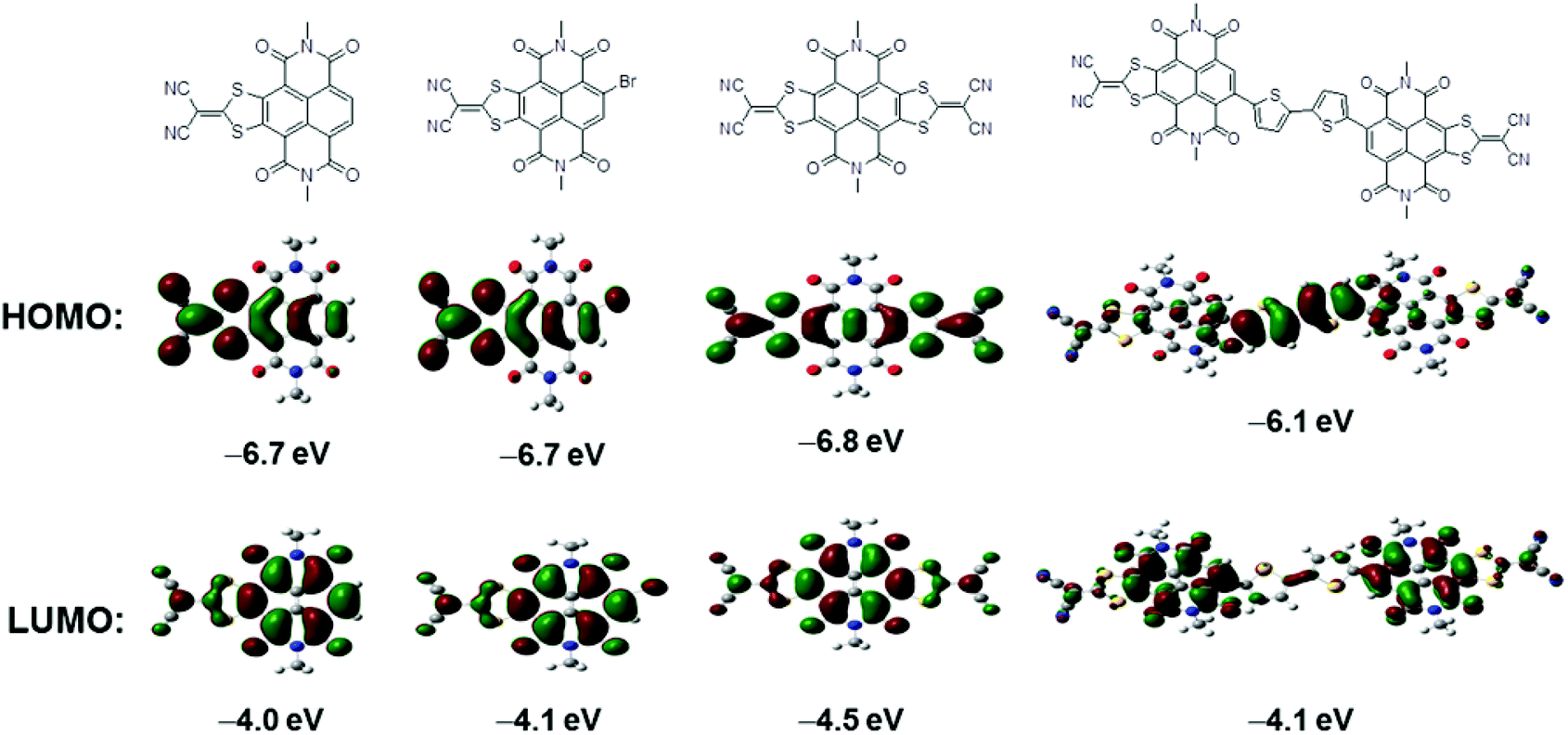 | ||
| Fig. 2 Frontier molecular orbitals and HOMO/LUMO levels of N,N′-bis(methyl)-substituted model molecules of 1, 2, 3 and 4 estimated by DFT calculations. | ||
| Compound |
T
d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (°C) a (°C) |
λ absmax (nm)/ε × 10−3 (L mol−1 cm−1) | λ emmax (nm)/ΦF | HOMOc (eV) | LUMOb (eV) | E optg (eV) |
|---|---|---|---|---|---|---|
| a Onset decomposition temperature measured by TGA under a nitrogen flow with a heating rate of 10 °C min−1. b Estimated from the equation: LUMO = −(Ered11/2 − E(Fc/Fc+)1/2 + 4.8) eV. c Estimated from the equation: HOMO = LUMO − Eoptg. d The HOMO or LUMO energy values that were optimized by simulation with DFT calculation, Eg = LUMO – HOMO. | ||||||
| 1 | 401 | 492/32.8, 409/32.8, 386/20.6 | 529/0.032 | −6.4 (−6.7)d | −4.0 (−4.0)d | 2.37 (2.74)d |
| 2 | 313 | 503/37.4, 401/23.6, 388/15.6 | 539/0.014 | −6.3 (−6.7)d | −4.0 (−4.1)d | 2.30 (2.67)d |
| 3 | 396 | 573/101.6, 531/50.6, 433/25, 408/13.8 | 593/0.019 | −6.4 (−6.8)d | −4.3 (−4.5)d | 2.07 (2.33)d |
| 4 | 410 | 598/38, 488/48.6, 411/51.8, 388/51 | −5.8 (−6.1)d | −4.1 (−4.1)d | 1.69 (1.95)d | |
The UV-Vis spectrum of 1 shows a maximum absorption at 492 nm, and the two sides annulated 3 exhibits a bathochromic-shifted absorption band at 573 nm resulting from a more expanded π-conjugated system (Fig. 3). Compound 2 displays an 11 nm red shift relative to 1 due to the bromo-substitution, in line with a previous finding that the four bromo substituted NDIs showed a more red-shifted absorption than that of unsubstituted NDIs.18 For compound 4, in addition to a typical rigid-core absorption peak at 488 nm, a broad band centered at 598 nm is ascribed to the intramolecular charge transfer (ICT) between bithiophene and NDI moieties.11c Optical energy gaps (Eoptg) for 1, 2, 3 and 4, estimated from the onset of absorption in the solution, are 2.37, 2.3, 2.07 and 1.69 eV, respectively. The sequential long wavelength shifts of absorption and degressive band gaps are in good agreement with molecular simulation through theoretical calculations (Fig. S6† and Table 1). Fluorescence spectra were investigated and demonstrated in Fig. S7.† Compound 1 displayed a green-yellow emission centered at 529 nm with a rather low quantum yield of 0.032, an even depressed emission yield was obtained in the case of 2 due to the heavy atom effect19 with a fluorescence band at 539 nm. A more red-shifted emission spectrum of 3 centered at 593 nm is attributed to the extended conjugated backbone. The fluorescence of 4 is totally quenched through the incorporation of a bithiophene donor unit.
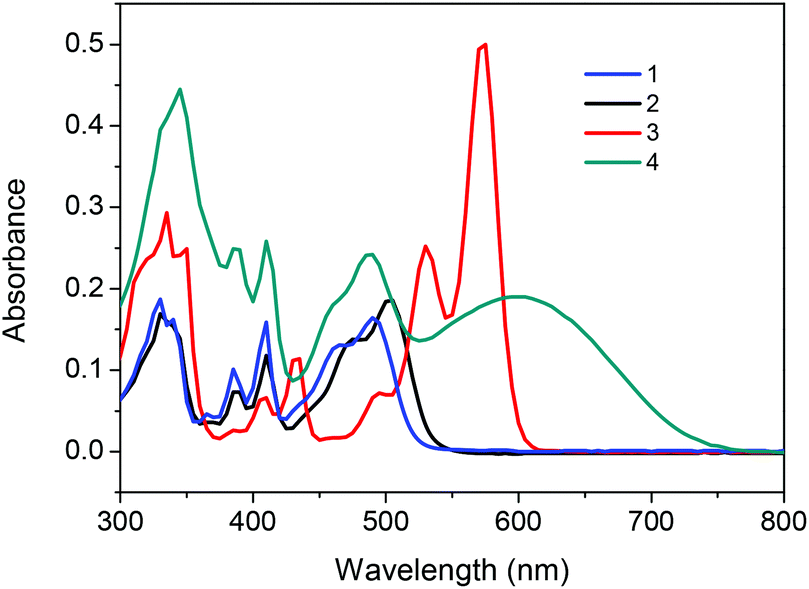 | ||
| Fig. 3 UV-Vis spectra of compounds 1, 2, 3 and 4 in dichloromethane solution at 5.0 × 10−6 M. | ||
Cyclic voltammetry (CV) measurements were performed with 0.1 M Bu4NPF6 in dichloromethane solution to investigate the electrochemical properties of NDI derivatives (Fig. 4). In the case of 1, 2 and 3, two reversible reduction processes are observed. The first half-wave reductive potentials (E1/2red1) for each compound are about −0.35, −0.3 and −0.06 V, respectively. In comparison with 1, a slightly positive shift of 2 (0.05 V) is assigned to weak electron withdrawing ability of the bromine atom. By fusion with another heterocyclic ring, the enlarged electronic orbital delocalization and strong electron withdrawing ability of the DTYM unit lead to a larger positive shift of 3 relative to 1 (about 0.3 V). The bithiophene-bridged derivative 4 shows three unexpected reversible reduction waves with a first half-wave reductive potential of approximately −0.25 V, which is anodically shifted by 0.1 V compared to that of 1, indicating a moderate influence of the bithiophene bridge on the molecular LUMO energy level. Based on the respective currents associated with the three waves, the first and second waves are rationally assigned to successive reduction of each NDI moiety to their radical anions to give a molecular dianion and the final feature is assigned to the second reductions of both NDIs to give a tetraanion.11c,20 The LUMO energies of 1, 2, 3 and 4 estimated from CV are −4.0, −4.0, −4.3 and −4.1 eV, respectively. The CV results are in accordance with DFT calculations and indicate that all the compounds are typical electron acceptors, making them potential n-type semiconducting materials. It is worth noting that the incorporation of the electron-donor bithiophene unit did not lead to any oxidative process during CV measurements of 4. However, according to the literature,11c donor-induced reversible oxidative waves were observed for similar A–D–A type NDI-based derivatives and shifted to a positive direction with increasing the electron-richness of the bridging groups. These results illustrate that our monolateral heterocycle fused NDI is more electron-deficient and easier to be reduced than the unsubstituted NDI and therefore is more favorable to exhibit pure n-channel behavior.11c,20
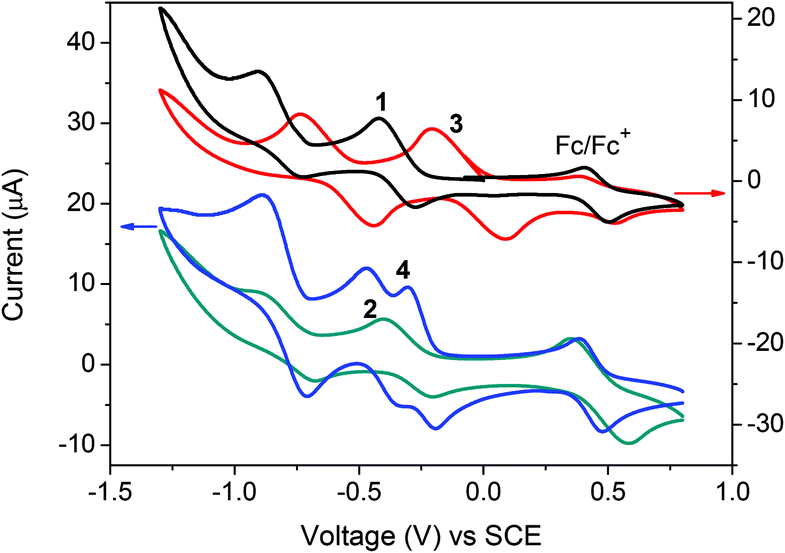 | ||
| Fig. 4 Cyclic voltammograms of compounds 1, 2, 3 and 4 with 0.1 M Bu4NPF6 in dichloromethane solution under a scan rate of 50 mV s−1. | ||
Conclusions
In summary, monolateral and bilateral sulfur-heterocycle fused NDIs have been successfully synthesized from the monobromo and dibromo NDIs via SNAr reactions and subsequent oxidative aromatization processes. Their thermal, optical and electrochemical properties have been investigated and compared with each other. The results reveal that the monolateral derivative has comparable thermal stability with the bilateral counterpart and low-lying LUMO energy (about −4.0 eV), which make it a favourable candidate for n-type OSCs. In addition, through incorporation of different aromatic or heteroaromatic rings with the monolateral fused NDI, diversatile π-conjugation materials could be constructed with fine-tuned molecular orbital energy levels that favor carrier conduction. Ongoing studies focus on their molecular stacking patterns in thin films, their device performances, and the molecular structure–device performance relationship.Acknowledgements
We thank the National Natural Science Foundation of China (21302212, 51173200 and 201103023) and the “Strategic Priority Research Program” (XDB12010100) for funding this work.Notes and references
- J. Zaumseil and H. Sirringhaus, Chem. Rev., 2007, 107, 1296 CrossRef CAS PubMed.
- (a) J. E. Anthony, A. Facchetti, M. Heeney, S. R. Marder and X. Zhan, Adv. Mater., 2010, 22, 3876 CrossRef CAS PubMed; (b) H. Usta, A. Facchetti and T. J. Marks, Acc. Chem. Res., 2011, 44, 501 CrossRef CAS PubMed; (c) C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208 CrossRef CAS PubMed; (d) Y. Zhao, Y. Guo and Y. Liu, Adv. Mater., 2013, 25, 5372 CrossRef CAS PubMed; (e) J. Mei, Y. Diao, A. Appleton, L. Fang and Z. Bao, J. Am. Chem. Soc., 2013, 135, 6724 CrossRef CAS PubMed; (f) C. B. Nielsen, M. Turbiez and I. McCulloch, Adv. Mater., 2013, 25, 1859 CrossRef CAS PubMed; (g) H. Sirringhaus, Adv. Mater., 2014, 26, 1319 CrossRef CAS PubMed; (h) Z. Liu, G. Zhang, Z. Cai, X. Chen, H. Luo, Y. Li, J. Wang and D. Zhang, Adv. Mater., 2014, 26, 6965 CrossRef CAS PubMed.
- (a) Y. Li, Acc. Chem. Res., 2012, 45, 723 CrossRef CAS PubMed; (b) Y. Lin, Y. Li and X. Zhan, Chem. Soc. Rev., 2012, 41, 4245 RSC.
- C. Ego, D. Marsitzky, S. Becker, J. Zhang, A. Grimsdale, K. Müllen, J. MacKenzie, C. Silva and R. Friend, J. Am. Chem. Soc., 2003, 125, 437 CrossRef CAS PubMed.
- B. A. Jones, A. Facchetti, M. R. Wasielewski and T. J. Marks, J. Am. Chem. Soc., 2007, 129, 15259 CrossRef CAS PubMed.
- (a) H. E. Katz, A. J. Lovinger, J. Johnson, C. Kloc, T. Siegrist, W. Li, Y.-Y. Lin and A. Dodabalapur, Nature, 2000, 404, 478 CrossRef CAS PubMed; (b) N. Sakai, J. Mareda, E. Vauthey and S. Matile, Chem. Commun., 2010, 46, 4225 RSC; (c) X. Zhan, A. Facchetti, S. Barlow, T. J. Marks, M. A. Ratner, M. R. Wasielewski and S. R. Marder, Adv. Mater., 2011, 23, 268 CrossRef CAS PubMed; (d) F. Würthner and M. Stolte, Chem. Commun., 2011, 47, 5109 RSC; (e) Q. Meng and W. Hu, Phys. Chem. Chem. Phys., 2012, 14, 14152 RSC; (f) S. V. Bhosale, S. V. Bhosale and S. K. Bhargava, Org. Biomol. Chem., 2012, 10, 6455 RSC; (g) F. Doria, M. Nadai, G. Sattin, L. Pasotti, S. N. Richter and M. Freccero, Org. Biomol. Chem., 2012, 10, 3830 RSC; (h) F. Doria, C. M. Gallati and M. Freccero, Org. Biomol. Chem., 2013, 11, 7838 RSC; (i) F. Doria, I. Manet, V. Grande, S. Monti and M. Freccero, J. Org. Chem., 2013, 78, 8065 CrossRef CAS PubMed; (j) X. Gao and Y. Hu, J. Mater. Chem. C, 2014, 2, 3099 RSC; (k) S.-L. Suraru and F. Würthner, Angew. Chem., Int. Ed., 2014, 53, 7428 CrossRef CAS PubMed; (l) A. Das and S. Ghosh, Angew. Chem., Int. Ed., 2014, 53, 2038 CrossRef CAS PubMed.
- (a) B. J. Jung, N. J. Tremblay, M.-L. Yeh and H. E. Katz, Chem. Mater., 2011, 23, 568 CrossRef CAS; (b) M. Sommer, J. Mater. Chem. C, 2014, 2, 3088 RSC.
- (a) J. Chang, Q. Ye, K.-W. Huang, J. Zhang, Z.-K. Chen, J. Wu and C. Chi, Org. Lett., 2012, 14, 2964 CrossRef CAS PubMed; (b) T. He, M. Stolte and F. Würthner, Adv. Mater., 2013, 25, 6951 CrossRef CAS PubMed.
- (a) X. Gao, C.-an Di, Y. Hu, X. Yang, H. Fan, F. Zhang, Y. Liu, H. Li and D. Zhu, J. Am. Chem. Soc., 2010, 132, 3697 CrossRef CAS PubMed; (b) Y. Hu, X. Gao, C.-an Di, X. Yang, F. Zhang, Y. Liu, H. Li and D. Zhu, Chem. Mater., 2011, 23, 1204 CrossRef CAS; (c) Y. Hu, Y. Qin, X. Gao, F. Zhang, C.-an Di, Z. Zhao, H. Li and D. Zhu, Org. Lett., 2012, 14, 292 CrossRef CAS PubMed.
- (a) S.-L. Suraru, U. Zschieschang, H. Klauk and F. Würthner, Chem. Commun., 2011, 47, 11504 RSC; (b) C. Li, C. Xiao, Y. Li and Z. Wang, Org. Lett., 2013, 15, 682 CrossRef CAS PubMed; (c) J. Gao, Y. Li and Z. Wang, Org. Lett., 2013, 15, 1366 CrossRef CAS PubMed; (d) L. Tan, Y. Guo, Y. Yang, G. Zhang, D. Zhang, G. Yu, W. Xu and Y. Liu, Chem. Sci., 2012, 3, 2530 RSC; (e) X. Chen, Y. Guo, L. Tan, G. Yang, Y. Li, G. Zhang, Z. Liu, W. Xu and D. Zhang, J. Mater. Chem. C, 2013, 1, 1087 RSC.
- (a) H. Yan, Z. Chen, Y. Zheng, C. Newman, J. R. Quinn, F. Dötz, M. Kastler and A. Facchetti, Nature, 2009, 457, 679 CrossRef CAS PubMed; (b) Z. Chen, Y. Zheng, H. Yan and A. Facchetti, J. Am. Chem. Soc., 2009, 131, 8 CrossRef CAS PubMed; (c) L. E. Polander, S. P. Tiwari, L. Pandey, B. M. Seifried, Q. Zhang, S. Barlow, C. Risko, J.-L. Brédas, B. Kippelen and S. R. Marder, Chem. Mater., 2011, 23, 3408 CrossRef CAS; (d) Y. Fukutomi, M. Nakano, J.-Y. Hu, I. Osaka and K. Takimiya, J. Am. Chem. Soc., 2013, 135, 11445 CrossRef CAS PubMed; (e) R. Steyrleuthner, R. D. Pietro, B. A. Collins, F. Polzer, S. Himmelberger, M. Schubert, Z. Chen, S. Zhang, A. Salleo, H. Ade, A. Facchetti and D. Neher, J. Am. Chem. Soc., 2014, 136, 4245 CrossRef CAS PubMed.
- F. Zhang, Y. Hu, T. Schuettfort, C.-an Di, X. Gao, C. R. McNeill, L. Thomsen, S. C. B. Mannsfeld, W. Yuan, H. Sirringhaus and D. Zhu, J. Am. Chem. Soc., 2013, 135, 2338 CrossRef CAS PubMed.
- (a) F. Doria, M. di Antonio, M. Benotti, D. Verga and M. Freccero, J. Org. Chem., 2009, 74, 8616 CrossRef CAS PubMed; (b) H. Langhals and S. Kinzel, J. Org. Chem., 2010, 75, 7781 CrossRef CAS PubMed; (c) C. Zhou, Y. Li, Y. Zhao, J. Zhang, W. Yang and Y. Li, Org. Lett., 2011, 13, 292 CrossRef CAS PubMed.
- (a) M. L. Tang, T. Okamoto and Z. Bao, J. Am. Chem. Soc., 2006, 128, 16002 CrossRef CAS PubMed; (b) M. L. Tang, M. E. Roberts, J. J. Locklin, M. M. Ling, H. Meng and Z. Bao, Chem. Mater., 2006, 18, 6250 CrossRef CAS.
- C. Du, Y. Guo, Y. Liu, W. Qiu, H. Zhang, X. Gao, Y. Liu, T. Qi, K. Lu and G. Yu, Chem. Mater., 2008, 20, 4188 CrossRef CAS.
- D. S. Baranov, B. Gold, S. F. Vasilevsky and I. V. Alabugin, J. Org. Chem., 2013, 78, 2074 CrossRef CAS PubMed.
- X. Guo, F. S. Kim, M. J. Seger, S. A. Jenekhe and M. D. Watson, Chem. Mater., 2012, 24, 1434 CrossRef CAS.
- X. Gao, W. Qiu, X. Yang, Y. Liu, Y. Wang, H. Zhang, T. Qi, Y. Liu, K. Lu, C. Du, Z. Shuai, G. Yu and D. Zhu, Org. Lett., 2007, 9, 3917 CrossRef CAS PubMed.
- T. D. Hudnall and F. P. Gabbai, Chem. Commun., 2008, 4596 RSC.
- D. K. Hwang, R. R. Dasari, M. Fenoll, V. Alain-Rizzo, A. Dindar, J. W. Shim, N. Deb, C. Fuentes-Hernandez, S. Barlow, D. G. Bucknall, P. Audebert, S. R. Marder and B. Kippelen, Adv. Mater., 2012, 24, 4445 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4qo00252k |
| This journal is © the Partner Organisations 2015 |