DOI:
10.1039/C5QI00169B
(Highlight)
Inorg. Chem. Front., 2015,
2, 1070-1079
CO2 and ambient air in metal–oxygen batteries: steps towards reality
Received
2nd September 2015
, Accepted 22nd September 2015
First published on 5th October 2015
Abstract
Metal–air batteries, especially lithium and sodium air technologies, have attracted significant research attention in the past decade. The high theoretical specific energy (3500 Wh kg−1 for Li–O2 and 1600 Wh kg−1 for Na–O2) and moderate equilibrium potential (2.96 V for Li–O2 and 2.3 V for Na–O2) make these chemistries attractive energy storage platforms for transportation, autonomous aircraft, and emergent robotics technologies. The term metal–air battery, however, hardly describes the cell design under most active investigation by researchers; in most studies, O2 is used in place of air as the active material in the battery cathode. This change, designed to eliminate the formation of electrochemically stable metal hydroxide and metal carbonate discharge products when CO2 and moisture present in ambient air react with metal ions in the cathode, introduces significant new complications for practical metal–air battery design and operation that largely defeat the competitive advantages of this storage technology. Recent work has shown that when a mixture of O2 and CO2 is used as the active material in the cathode, it is possible to recharge a metal–O2/CO2 cell provided steps are taken to prevent electrolyte decomposition during recharge. In this highlight, we critically review the literature on metal–O2/CO2 cells, focusing on how the presence of CO2 in the active cathode material changes electrochemistry at the cathode and rechargeability of the cells. We also assess the progress and future prospects for metal–air battery technologies involving ambient air as the cathode gas.
 Shaomao Xu | Shaomao Xu received his M. Eng. (chemical engineering) from Cornell University and B. S. (chemical engineering) from China University of Petroleum. He is currently a Ph.D. candidate in Professor Lynden Archer's group in the School of Chemical and Biomolecular Engineering at Cornell University. His research focuses on metal–O2/CO2 batteries as dual platforms for carbon capture and electrical energy storage. His most recent efforts include fundamental studies of the reaction mechanisms in such batteries to improve their cyclability. |
 Sampson Lau | Sampson Lau received his bachelor's degree in chemical engineering from The Cooper Union in 2011 and is currently pursuing a Ph.D. in the School of Chemical and Biomolecular Engineering at Cornell University. He is doing research with Professor Lynden Archer on lithium–oxygen batteries, including theoretical studies on capacity prediction and experimental strategies for extending the capacity and cycle life. |
 Lynden A. Archer | Lynden A. Archer is the William C. Hooey Director and Professor of Chemical and Biomolecular Engineering at Cornell University and co-Director of the KAUST-Cornell Center for Energy and Sustainability. Archer's research focuses on transport properties of polymers and organic–inorganic hybrid materials relevant for applications in energy storage technologies. He received his Ph.D. in chemical engineering from Stanford University in 1994 and was a postdoctoral member of the Technical Staff at AT&T Bell Laboratories. Archer is a fellow of the American Physical Society, Associate editor of Science Advances, and a member of the joint editorial board for Macromolecules and ACS Macro Letters. |
1. Introduction
With a theoretical energy density of 3505 Wh kg−1,13 including the weight of oxygen during discharge, the rechargeable Li–air battery is considered among the most promising electrical energy storage platforms for electrified transportation.1–3,6–12 While it is understood that in actual practice only a fraction of this high theoretical energy density can be achieved in a reversible energy4,5 storage device, the much higher efficiency of electric motors over internal combustion engines means that an electric vehicle powered by a Li–air cell can still attain a usable energy density competitive with that of conventional fossil fuels, such as gasoline (1750 Wh kg−1).9 Unlike the Li-ion battery, however, which is based on highly reversible ion intercalation reactions at both the anode and cathode, the Li–air cell involves an interfacial electrochemical conversion reaction between lithium ions in an electrolyte and oxygen, dissolved in the electrolyte from ambient air, at a porous carbon cathode. At the anode, electrochemical stripping and plating of metallic lithium is the source and sink of lithium ions consumed during the discharge reaction and produced during Li–air cell recharge.
These fundamental differences between the electrochemical processes at work in Li-ion and Li–air cells are largely responsible for the contrasting states of commercial readiness of the two technologies. On the anode side, it is known that reversible stripping and plating of metallic lithium in liquid electrolytes is frustrated by multiple issues, including side reactions with the electrolyte, which deplete the lithium and electrolyte over time, and growth of dendrites, which may lead to internal short circuiting and is therefore a serious safety concern.14,15 Electrolyte decomposition may also be exacerbated by the formation of reactive O2˙− species in the electrolyte.16,17 Additionally, the vast majority of the “Li–air” batteries that have been studied in the literature are in reality lithium–oxygen cells because these cells utilize pure oxygen, as opposed to ambient air, in the cathode. While this practice is important for advancing fundamental knowledge, the presence of significant amounts of moisture and CO2 in ambient air, produces fundamental changes in the chemistry of an actual Li–air cell, with new insulating species such as LiOH and Li2CO3 forming at the cathode, making the battery less rechargeable.18,19 It is possible to remove these complications in a practical Li–air cell by adding to the cell design a gas pretreatment/purification system to remove H2O and CO2 from ambient air or by including an on-board storage tank suitable for delivering pure oxygen as desired during battery operation.20 Unsurprisingly, the introduction of these additional components leads to substantial increases in the weight and capital cost of a Li–air system, which cancel out the most important advantages of a Li–air battery, compared to the Li-ion system, for applications in transportation.
As a first step towards advancing knowledge of the effect of CO2 in ambient air on the operation of the Li–air cell, several recent studies have focused on Li–O2/CO2 batteries, which utilize a mixture of oxygen and carbon dioxide at the cathode. An added impetus for such work comes from the prospect of using such metal–O2/CO2 cells in a hybrid EV design where the battery plays the dual role of a carbon capture–conversion system able to reduce emissions and enhance the range of a conventional IC-based vehicle powered by fossil fuels. Indeed, while a variety of chemical and physical methodologies are under active investigation to capture and sequester thousands of tons of CO2 emitted every year in transportation,21–23 the vast amount of energy consumed to regenerate the capture fluid or sorbent, is a common shortcoming of all approaches. Thus, without some form of government intervention to alter the economics of CO2 capture from mobile sources, it is unlikely that any of the carbon capture technologies under development will find widespread use in transportation. A metal–air battery that utilizes a mixed fuel of O2 and CO2 therefore provides a model for studying the effects of CO2 in ambient air on the Li–O2 cell operation and at the same time, provides a novel hybrid platform for electrical energy generation and carbon capture. This review focuses on recent developments in both areas and highlights progress in metal–O2/CO2 and metal–ambient air batteries that are enabled by these developments.
2. Metal–O2/CO2 batteries
2.1 Li–O2/CO2 battery
The first lithium–air battery system involving a mixture of O2 and CO2 gas in the cathode was reported by Takechi et al.24 It is known that O2˙− can be captured by CO2 and this reaction has been widely used in CO2 sensors, as well as in molten-carbonate fuel cells (MCFCs).25,26 Based on this reaction, the electrochemical processes in the cathode of a Li–O2/CO2 battery have been argued to involve the following five-step reaction sequence:| | | C2O6˙− + O2˙− → C2O62− + O2 | (4) |
| | | C2O62− + 2O2˙− + 4Li+ → 2Li2CO3 + 2O2 | (5) |
The discharge product is therefore thought to be Li2CO3. One interesting fact about this Li–O2/CO2 battery is that the specific discharge capacity is two times higher than that of the Li–O2 battery, as shown in Fig. 1. It has also been observed that the void space in the cathode is fully filled by the discharge products of the cell, which is not the case for a Li–O2 battery. The reason for this difference is believed to revolve around differences in the reaction rate. The first reaction in the proposed mechanism is common to both Li–O2 and Li–O2/CO2 batteries. Following this reaction, in a Li–O2 battery, the O2˙− radical reacts with Li+ ions to form 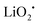 , and this reaction is believed to be slower than the reactions (2)–(4) in the analogous Li–O2/CO2 battery. Furthermore, although the intermediate products were not detected, it is believed that the intermediate C2O6− compound can stably diffuse in the electrolyte, thus slowing down the precipitation of Li2CO3, allowing it to completely fill the void space in the cathode. Unfortunately, these studies are all based on carbonate electrolytes, which are now known to undergo additional electrochemical decomposition reactions during recharge. This finding leads to the widespread, but incorrect, view that in contrast to the Li–O2 battery, a Li–O2/CO2 battery is not rechargeable.
, and this reaction is believed to be slower than the reactions (2)–(4) in the analogous Li–O2/CO2 battery. Furthermore, although the intermediate products were not detected, it is believed that the intermediate C2O6− compound can stably diffuse in the electrolyte, thus slowing down the precipitation of Li2CO3, allowing it to completely fill the void space in the cathode. Unfortunately, these studies are all based on carbonate electrolytes, which are now known to undergo additional electrochemical decomposition reactions during recharge. This finding leads to the widespread, but incorrect, view that in contrast to the Li–O2 battery, a Li–O2/CO2 battery is not rechargeable.
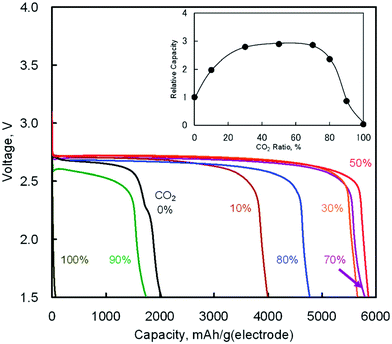 |
| | Fig. 1 Discharge curves of the Li–O2/CO2 batteries with various ratios of CO2 in O2/CO2 mixed gas at 25 °C (current density: 0.2 mA cm−2). The inset shows CO2 ratio dependence on relative capacities as compared with the Li–O2 battery (CO2 0%). Adapted from ref. 24. | |
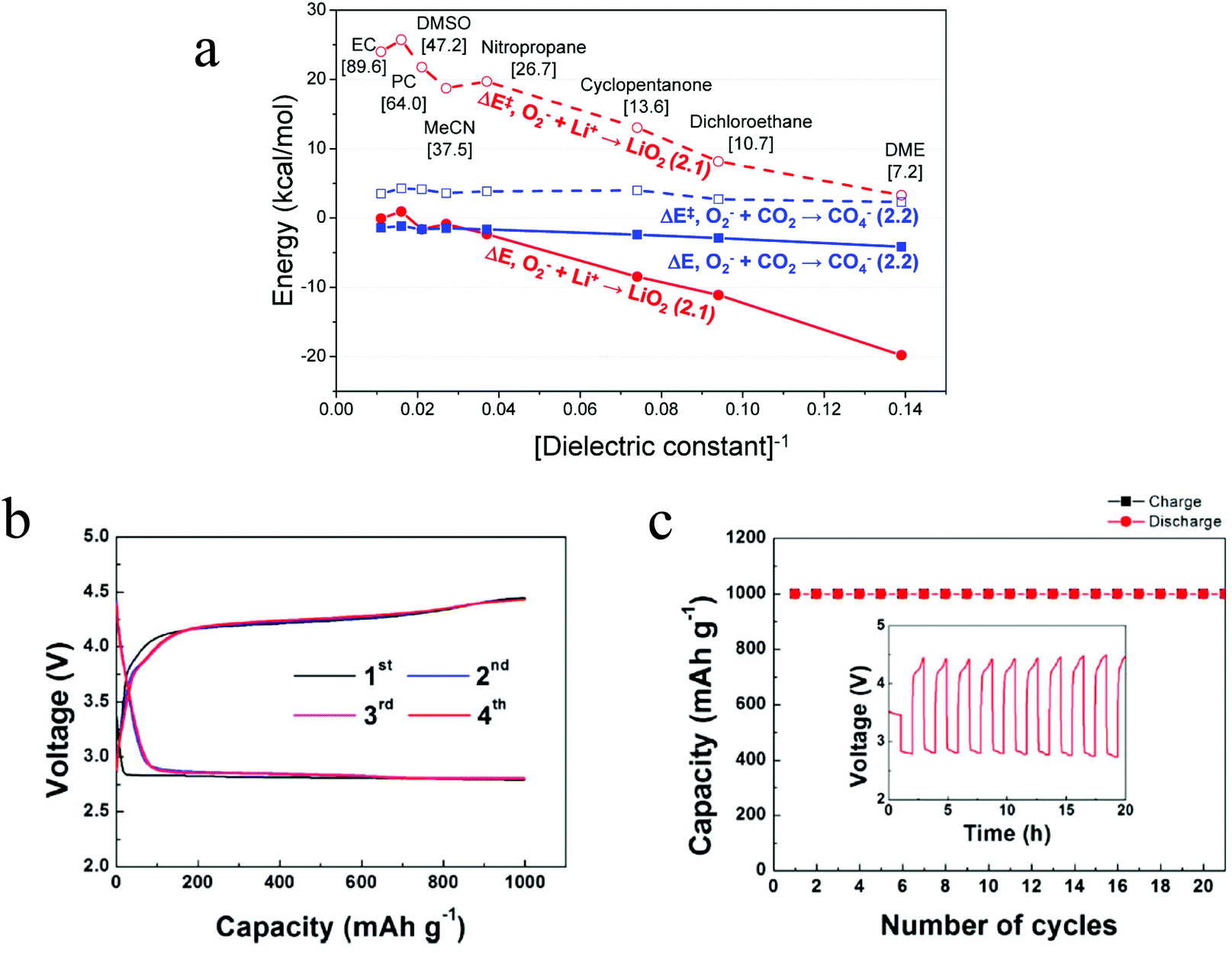
The reaction mechanism of the Li–O2/CO2 battery has been reported to depend on the type of electrolyte used. Kim and Kang et al. studied Li–O2/CO2 cells with DME and DMSO electrolytes.27 Using density functional theoretical (DFT) analysis, the authors contended that in low dielectric constant (ε) electrolytes such as DME (ε = 7.2) the O2˙− radical is favored to react with Li+ and form 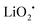 , so the reaction mechanism in the Li–O2/CO2 battery is thought to be the same as in the Li–O2 battery, with Li2O2 as the discharge product. However, in high ε electrolytes like carbonates and DMSO (ε = 47.2), the reaction between the O2˙− radical and CO2 is more favored, so in this case CO2 does take part in the reaction, and the discharge product is Li2CO3 (Fig. 2a). These predictions are consistent with the limited experimental data available. In the high ε electrolyte (DMSO) case, it is worth noting that even though the discharge product is Li2CO3, which is thermodynamically stable and thought to be undegradable during recharge, the cells exhibit some amount of rechargeability and are stably cycled for over 20 cycles, as shown in Fig. 2b and c. The formation of the discharge product Li2CO3 was detected after discharge and the disappearance of the product after recharge was also observed. These findings therefore demonstrate that the Li–O2/CO2 battery can be recharged, which opens the possibility for Li–O2/CO2 rechargeable cells to be used in more or less the same applications targeted for Li–O2 and Li–air.
, so the reaction mechanism in the Li–O2/CO2 battery is thought to be the same as in the Li–O2 battery, with Li2O2 as the discharge product. However, in high ε electrolytes like carbonates and DMSO (ε = 47.2), the reaction between the O2˙− radical and CO2 is more favored, so in this case CO2 does take part in the reaction, and the discharge product is Li2CO3 (Fig. 2a). These predictions are consistent with the limited experimental data available. In the high ε electrolyte (DMSO) case, it is worth noting that even though the discharge product is Li2CO3, which is thermodynamically stable and thought to be undegradable during recharge, the cells exhibit some amount of rechargeability and are stably cycled for over 20 cycles, as shown in Fig. 2b and c. The formation of the discharge product Li2CO3 was detected after discharge and the disappearance of the product after recharge was also observed. These findings therefore demonstrate that the Li–O2/CO2 battery can be recharged, which opens the possibility for Li–O2/CO2 rechargeable cells to be used in more or less the same applications targeted for Li–O2 and Li–air.
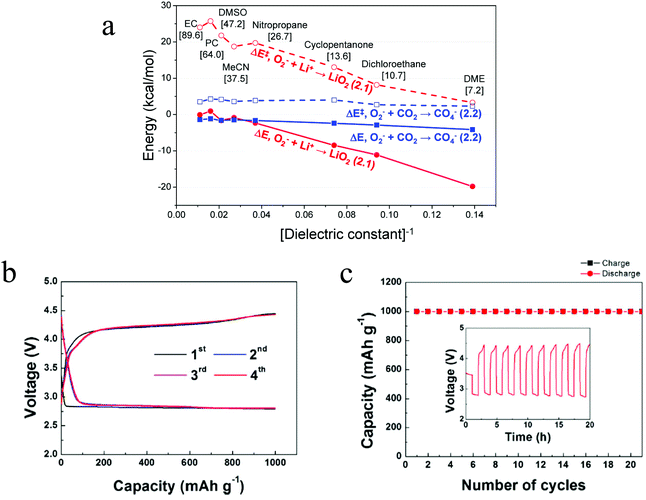 |
| | Fig. 2 (a) DFT-predicted trend of the activation barrier (ΔE‡, dashed) and binding reaction energy (ΔE, solid) plotted against [dielectric constant]−1 for the initial complex formation (ICF) steps of reactions, forming LiO2 (red, circle) and CO4− (blue, square), respectively. The values enclosed in brackets are the dielectric constants of various solvents. (b) The voltage profiles of the first four cycles and (c) the related cyclability of the Li–O2/CO2 battery with the DMSO electrolyte. Current density = 0.4 mA cm−2. Adapted from ref. 27. | |
For electrolytes such as DME, in which Li2O2 is the main discharge product, the amount of CO2 in the O2/CO2 mixture has been reported to markedly influence cell operation. Mekonnen et al. found that a 1% CO2 atmosphere (99% O2) increased the capacity of a Li–O2 cell to ∼120% of the pure O2 case, but a 50% CO2 atmosphere reduced the capacity to ∼4% of the pure O2 case, effectively deactivating the battery.42 The complementary DFT study found that CO2 adsorption is most favorable at the step valley sites of the (1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 00) Li2O2 surface, with an adsorption energy of −0.73 eV. The authors conclude that low CO2 concentrations block surface-active nucleation sites, which increase the capacity of the battery but at the expense of an increased overpotential. A follow-up DFT study of the Li2CO3@Li2O2 interface found that electron polaron hopping can occur within the peroxide side of the interface with a low energy barrier of less than 0.5 eV.43 These interfaces can increase the discharge product conductivity versus pure Li2O2, offering another possible explanation for the extended capacity in the presence of CO2.
00) Li2O2 surface, with an adsorption energy of −0.73 eV. The authors conclude that low CO2 concentrations block surface-active nucleation sites, which increase the capacity of the battery but at the expense of an increased overpotential. A follow-up DFT study of the Li2CO3@Li2O2 interface found that electron polaron hopping can occur within the peroxide side of the interface with a low energy barrier of less than 0.5 eV.43 These interfaces can increase the discharge product conductivity versus pure Li2O2, offering another possible explanation for the extended capacity in the presence of CO2.
2.2 Na–O2/CO2 battery
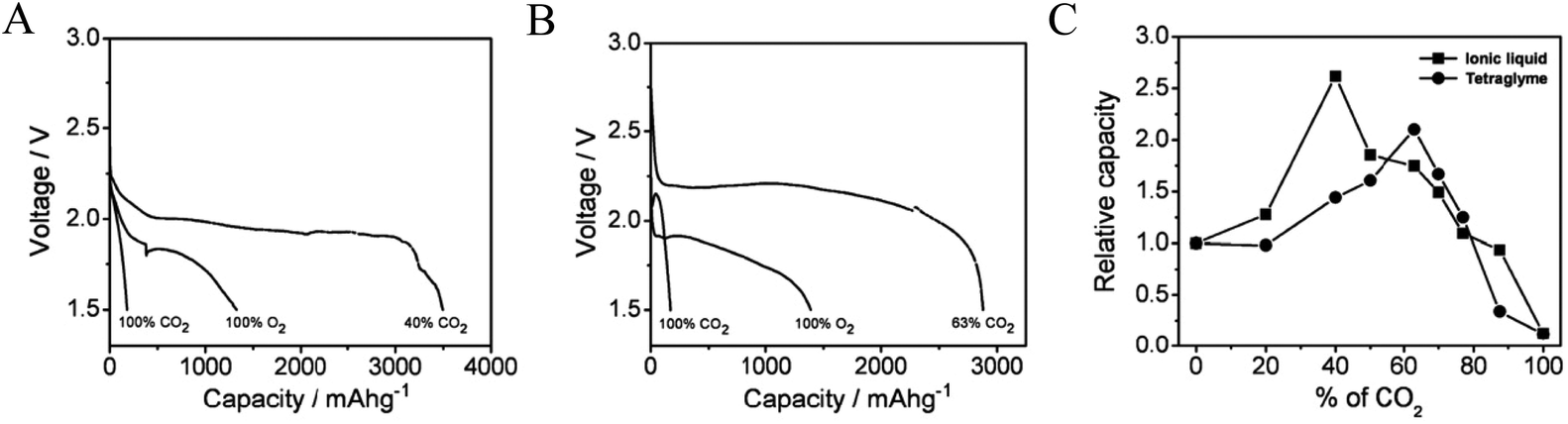
The gradual depletion of lithium resources and anticipated rise in the cost of lithium has increased interest in alternatives to metallic lithium as the anode in metal–air batteries.28–32 Sodium has a high earth-abundance (sixth most abundant element), is relatively easy to extract, and is available at a fraction of the cost of lithium. It is therefore unsurprising that sodium-based battery systems are attracting significant attention from research groups worldwide.33–35 With this increased interest, rising attention is being given to the benefits of Na–air and Na–O2 cells as energy storage platforms that can compete with Li–O2 technology. Das et al. reported the first example of the Na–O2/CO2 battery and pointed out how the lower cost and high natural abundance of Na would enable potential use of such cells for simultaneously capturing CO2 and generating electrical energy.36 Consistent with what has been found for the Li–O2/CO2 battery, these authors reported that by adding CO2 between 40% and 63% in the cathode gas streams, Na–O2/CO2 batteries with discharge capacity 2.1 and 2.6 times (Fig. 3A and B) higher than the corresponding Na–O2 cell are possible in the ionic liquid 1-ethyl-3-methylimidazolium trifluoromethanesulfonate and tetraglyme-based electrolytes, respectively. Postmortem analysis of the cathode using XRD showed that the specific choice of electrolyte chemistry profoundly influences the chemistry of the discharge product. In the IL-based electrolyte, the discharge product was found to be Na2CO3, while in a tetraglyme-based electrolyte, a mixture of Na2CO3 and Na2C2O4 was argued to be the discharge product. The authors reported the following reaction mechanisms in the two electrolyte systems: (i) in the ionic liquid based electrolyte, the reaction is analogous to the reaction in a Li–O2/CO2 battery:| | | C2O6˙− + O2˙− → C2O62− + O2 | (9) |
| | | C2O62− + 2O2˙− + 4Na+ → 2Na2CO3 + 2O2 | (10) |
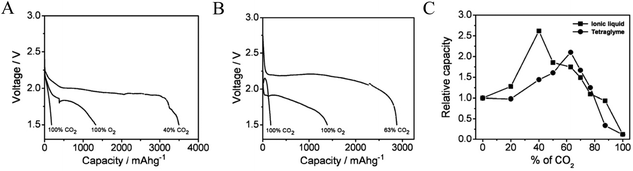 |
| | Fig. 3 Galvanostatic discharge profiles for Na–CO2/O2 cells operated with a mixed O2/CO2 feed. (A) Measurements performed in an electrolyte based on the ionic liquid (IL) 1-ethyl-3-methylimidazolium trifluoromethanesulfonate and (B) measurements performed in tetraglyme-based electrolytes; (C) variation of capacity with the CO2 concentration. Adapted from ref. 36. | |
(ii) In the tetraglyme-based electrolyte, the above reactions occur in tandem with the reaction summarized below, during discharge:
| | | CO42− + CO2 + 2Na+ → Na2C2O4 + O2 | (13) |
In this case, the discharge product is a mixture of Na
2CO
3 and Na
2C
2O
4.
Due to the stability of sodium oxides, the charge potential of the Na–O2/CO2 battery is above 4.5 V (vs. Na/Na+), which is beyond the stability window of most electrolytes. As a consequence, it is difficult to develop a rechargeable Na–O2/CO2 battery based on conventional electrolyte systems. The Archer group has reported that addition of ionic liquid tethered silica nanoparticles as additives in propylene carbonate-based electrolytes can enhance high-voltage stability of the electrolyte.50,51Fig. 4a illustrates how such SiO2-tethered ILs can be used as electrolyte additives in Na–O2/CO2 batteries and Fig. 4(b) and (c) show how the addition of additives leads to rechargeability of the Na–O2/CO2 battery.37 In particular, with the addition of 10% of the SiO2-IL additive to a propylene carbonate based electrolyte, the cathodic stability of the electrolyte was increased by almost 1 V. Therefore, even though the charge potential is above 4.5 V, no electrolyte decomposition is observed and the Na–O2/CO2 battery can be stably recharged for over 20 cycles. Besides, during recharge of the battery, the evolution of both O2 and CO2 is detected, which means the gases that took part in the discharge get released during recharge. This again proves the rechargeability of the battery. Postmortem analyses of the cathode using XRD and vibration spectroscopy indicate that the discharge product is NaHCO3 and that it decomposes upon cell recharge. Trace amounts of H2O introduced during the preparation of electrolyte is thought to be the source of NaHCO3 as the principal discharge product. The relatively instability of NaHCO3, compared to Na2CO3, may be a reason for the rechargeability of the Na–O2/CO2 battery. However, considering the fact that in a Li–O2/CO2 battery Li2CO3 can be decomposed during recharge, it is possible that even if the cell discharge product is the more stable Na2CO3, a rechargeable Na–O2/CO2 battery is perhaps possible.
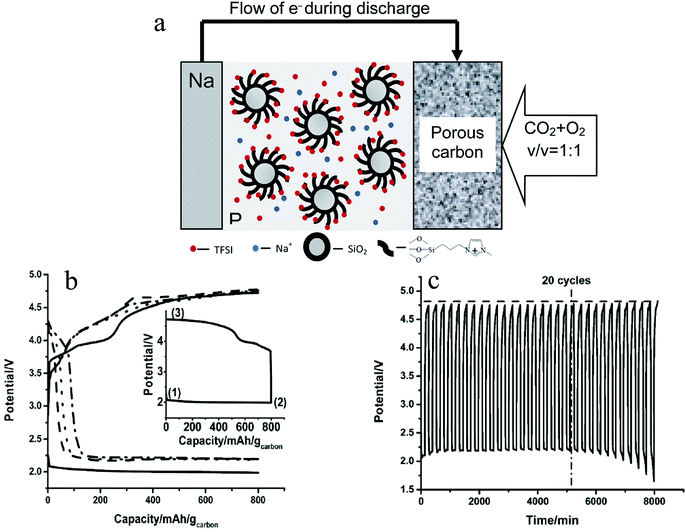 |
| | Fig. 4 (a) Schematic diagram of the Na–O2/CO2 rechargeable battery; (b) discharge–charge profiles of the Na–O2/CO2 battery in the first cycle (solid line), 5th cycle (dash line), 10th cycle (dot line) and 20th cycle (dash dot line); (c) cycling profiles for the Na–CO2/O2 battery with the SiO2-IL-TFSI/PC electrolyte. The dash line shows the trend of change of the charge potential. The dash-dot line indicates the 20th cycle where the battery shows a stable cycling. The current density employed is 200 mA g−1 and the capacity cutoff is 800 mA h gcarbon−1. Adapted from ref. 37. | |
2.3. Li–CO2 battery
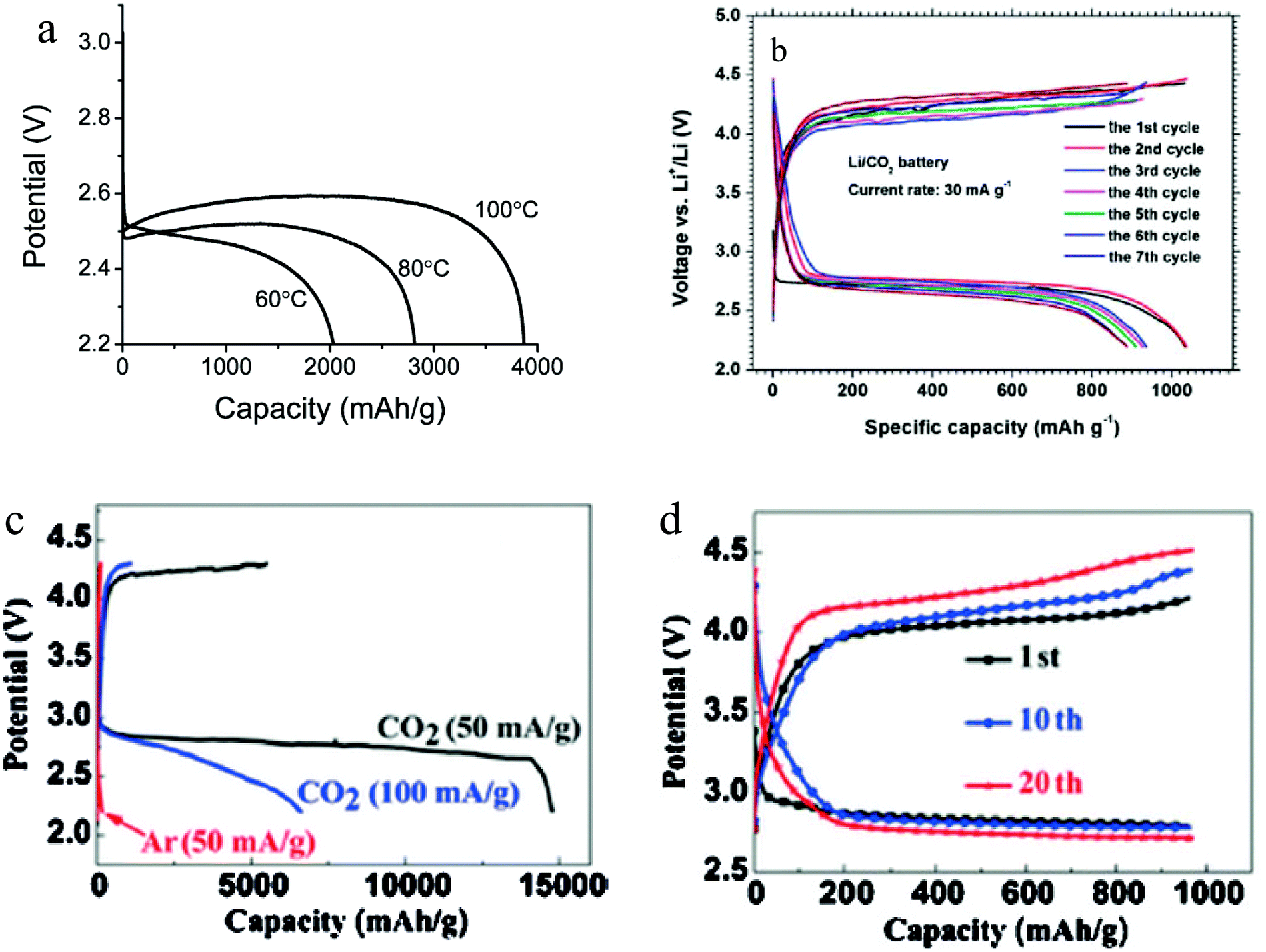
In the early studies of Li–O2/CO2 batteries, it was shown that with pure CO2 as the cathode gas, the battery has very low capacity.24 A report from Xu et al. is to our knowledge among the first to disclose a high-temperature Li–CO2 battery system that exhibits attractive electrochemical energy storage capacity.38 High temperature operation was possible using a 1-butyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide IL-based electrolyte. It is found that whereas the Li–CO2 battery has a very low storage capacity at room temperature, by increasing the operating temperature to 60 °C–100 °C, the discharge capacity of the battery increases dramatically (Fig. 5a). It is believed that operating the Li–CO2 battery at high temperature brings about two beneficial effects: Firstly, the solubility of the discharge product is thought to increase with temperature, thus limiting the thickness of the insulating deposition on the cathode surface. Additionally, a high temperature is thought to improve electrode kinetics by lowering transport barriers at the electrolyte–cathode interface. Based on thermodynamic analysis, it is believed that the discharge products are Li2CO3 and carbon:| | | 4Li + 3CO2 → 2Li2CO3 + C | (14) |
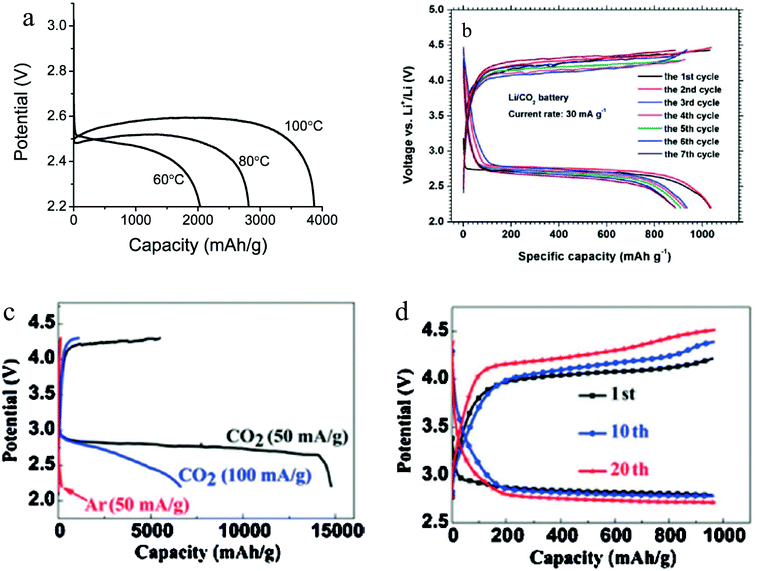 |
| | Fig. 5 (a) Galvanostatic discharge curves of Li–CO2 cells operated at various temperatures in the range 60–100 °C at a current density of 0.05 mA cm−2 to the potential of 2.2 V. (b) Charge–discharge voltage profiles of the Li–CO2 battery with the cut-off voltage of 2.2 V. (c) The initial discharge curves of the batteries with graphene cathodes at a current density of 50 mA g−1 and 100 mA g−1 under a CO2 atmosphere, as well as at 50 mA g−1 under an Ar atmosphere. (d) Voltage profiles of the 1st, 10th and 20th cycles with a curtailing capacity of 1000 mA h g−1 at a current density of 50 mA g−1. Adapted from ref. 38–40. | |
Li2CO3 was found to deposit on the surface of cathode, meaning that it is one of the major discharge products. However, since the carbon cathode was utilized in this study, the formation of carbon is hard to detect.
Li et al. studied the Li–CO2 battery using a porous gold cathode.39 After discharge, the formation of amorphous carbon was detected on the cathode, consistent with expectations based on the mechanism in reaction (14). Apart from this, the authors found that with a conventional tetraglyme based electrolyte, the Li–CO2 battery still shows a decent capacity of around 1000 mA h g−1, as shown in Fig. 5b. Even though the discharge product forms a thick layer of fibers on the surface of the cathode, it can be decomposed during recharge and the battery can be stably cycled. In order to increase the capacity of the Li–CO2 battery, Zhou et al. introduced graphene as the cathode material.40 Due to the exceptional catalytic activity of graphene, the Li–CO2 battery shows an incredibly high capacity of over 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mA h g−1. With a cutoff capacity of 1000 mA h g−1, the battery can be stably cycled for over 20 cycles (Fig. 5c and d). Even though the Li–CO2 cells used in the study were investigated only at low current density (J = 50–100 mA h g−1) and with a high overpotential, a growing consensus is that with additional research the Li–CO2 battery has the potential to become a prominent member among the new generation of energy storage systems.
000 mA h g−1. With a cutoff capacity of 1000 mA h g−1, the battery can be stably cycled for over 20 cycles (Fig. 5c and d). Even though the Li–CO2 cells used in the study were investigated only at low current density (J = 50–100 mA h g−1) and with a high overpotential, a growing consensus is that with additional research the Li–CO2 battery has the potential to become a prominent member among the new generation of energy storage systems.
2.4. Li–air battery with ambient air
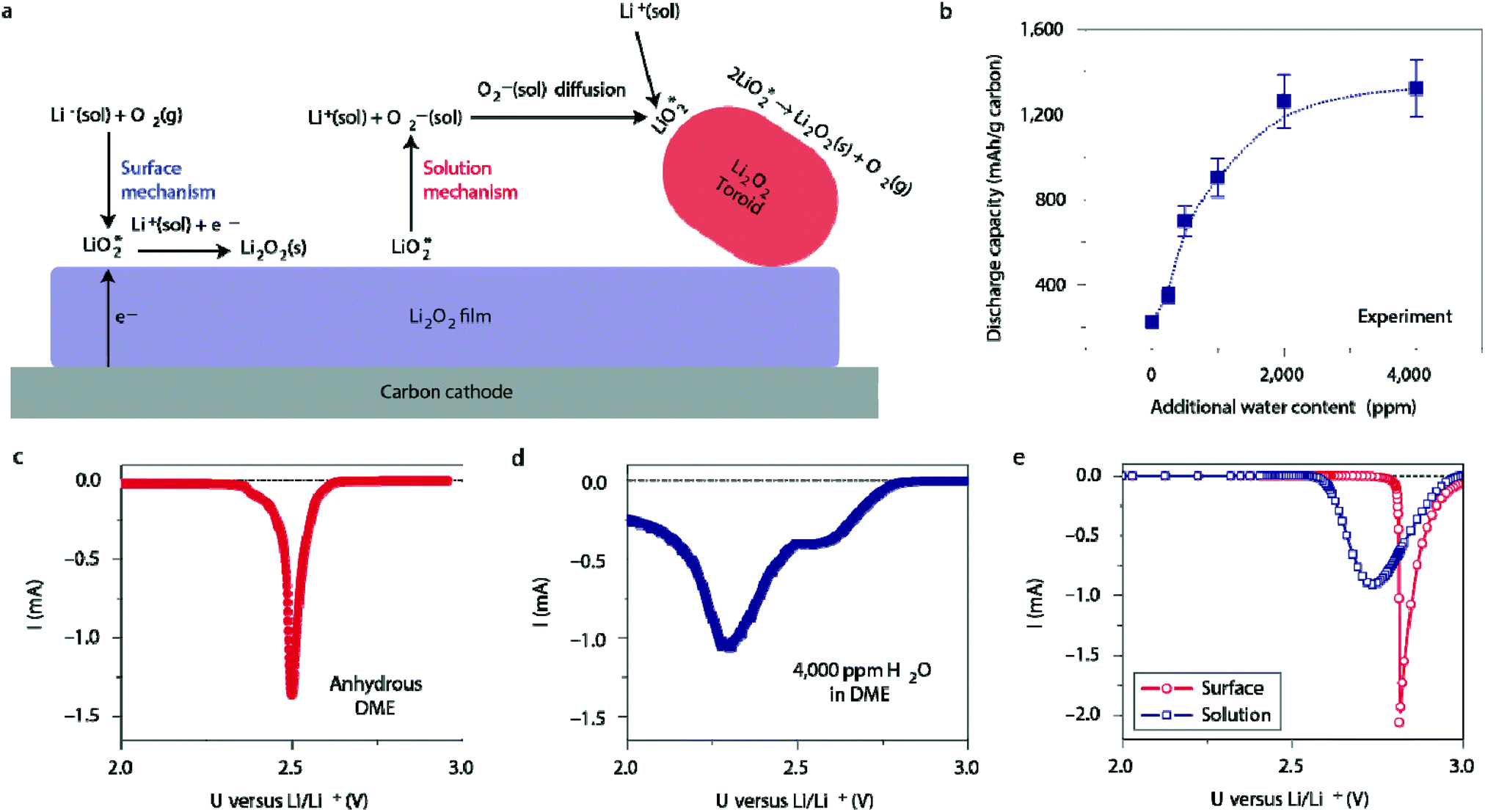
Ambient air introduces yet another impurity to the Li–O2 battery: water. The amount of water in the air can reach up to 4% depending on humidity, and is generally much greater than the amount of CO2 (400 ppm or 0.00040%) in ambient air.44 Thus, it is important that the effects of water on Li–O2 cell electrochemistry are studied at the fundamental level. Meini et al. performed one of the earliest of these studies by comparing the discharge performance of Li–O2 cells in a dry environment to those exposed to water vapor.45 They observed that the presence of water vapor increased the first cycle capacity of the cell by a factor of ten. The authors proposed that the superoxide radical, O2˙−, reacts preferentially with the trace water in the electrolyte to form soluble reaction products. A detailed study by Aetukuri et al. also showed a clear and consistent increase in the discharge capacity as a function of water content, shown in Fig. 6b.46 XRD analysis of the discharge product confirmed that Li2O2 is the dominant species and SEM imaging of the cathode revealed that Li2O2 forms as toroid-shaped particles, which increase in the size as the water content in the electrolyte rises. This phenomenon was ascribed to the ability of water to solvate O2−, which promotes a solution-phase mechanism in which LiO2 can disproportionate to produce Li2O2 without the need for a direct electron conduction pathway, as illustrated in Fig. 6a. Linear sweep voltammetry (LSV) appears to confirm this hypothesis. The anhydrous cell produces a sharp peak at a higher potential (Fig. 6c), agreeing with the theoretically predicted curve for the surface mechanism, while the cell with 4000 ppm of water produces a broader peak at a reduced potential (Fig. 6d), agreeing with the theoretically predicted curve for the solution mechanism.
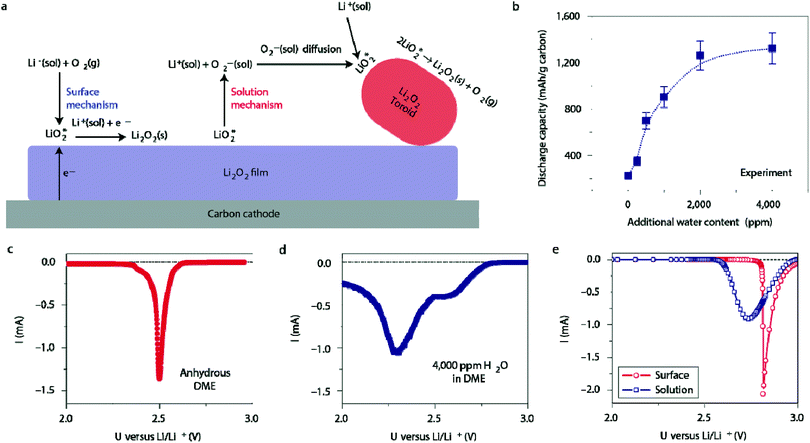 |
| | Fig. 6 (a) Illustration of surface-mediated and solution-mediated Li2O2 formation mechanisms, the latter of which is promoted by the presence of water. (b) The dependence of discharge capacity on the water content. Discharge linear sweep voltammetries (LSVs) of cells using (c) the anhydrous DME electrolyte and (d) electrolyte with 4000 ppm water added. (e) Discharge LSVs predicted by a model for surface- and solution-mediated Li2O2 formation. Adapted from ref. 46. | |
The trace amounts of water were also found to catalyze the oxygen evolution (charging) reaction. Li et al. constructed cells with a Super P carbon cathode containing ruthenium and manganese dioxide nanoparticles, an electrolyte containing 120 ppm of H2O, and a LiFePO4 anode.47 Although LiFePO4 could not be used in a practical cell, it was used in place of Li in this study to eliminate anodic side reactions. The presence of water was found to significantly decrease the charge overpotential from 0.70 V to 0.21 V. This was attributed to water acting as a redox mediator during charge. Specifically, Li2O2 reacts with water to form LiOH and H2O2, which are then oxidized on the Ru and MnO2 nanoparticles, respectively. The result is a cathode and electrolyte system that could cycle 200 times with only a discharge/charge potential gap of 0.32 V.
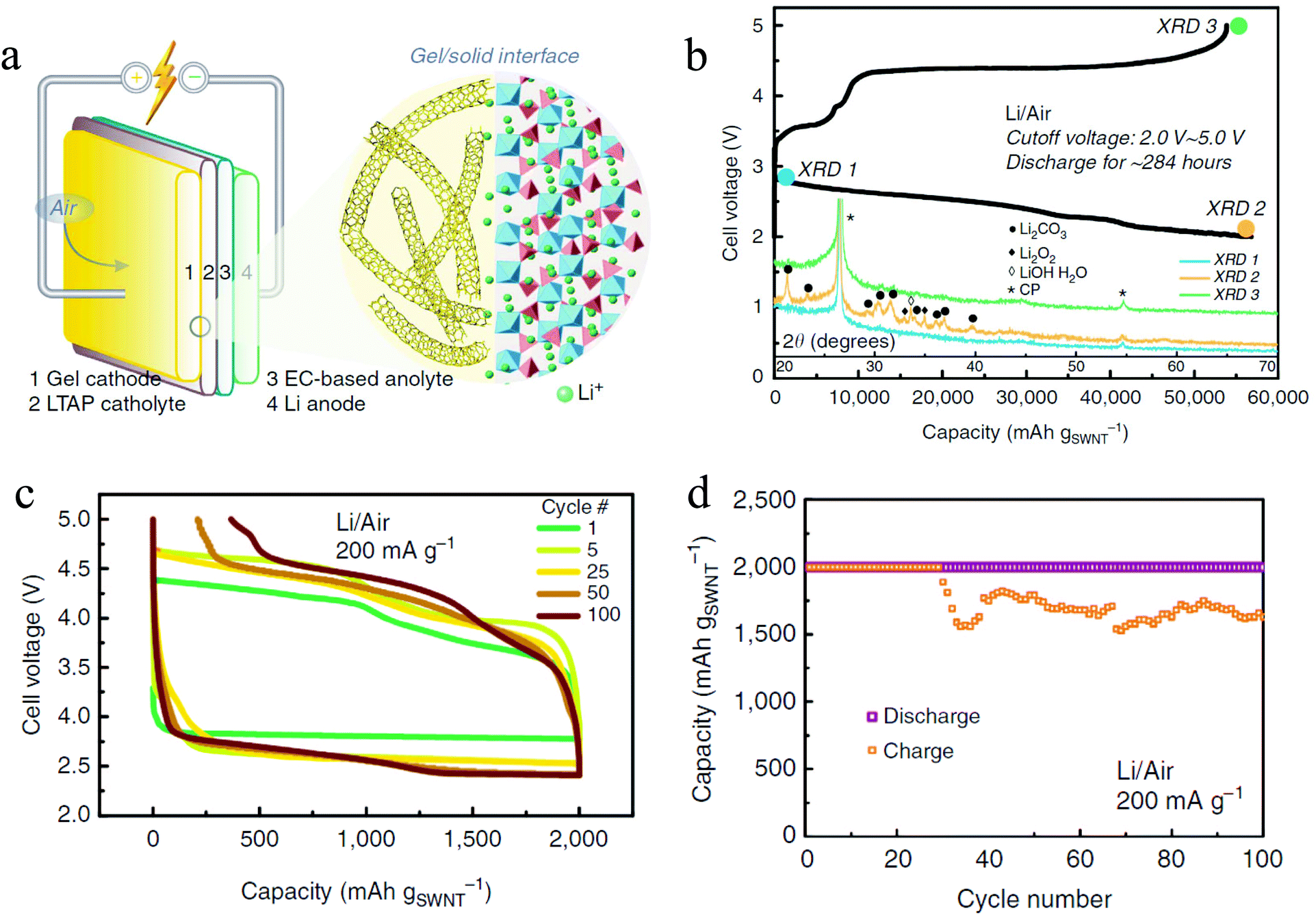
With the increase in the understanding of the effect of impurities in a Li–O2 battery, it is therefore becoming increasingly possible to develop a Li–air battery with ambient air as the cathode gas. By utilizing a hybrid electrolyte system with a lithium-ion conducting film separating the organic phase and the aqueous phase, a rechargeable Li–air battery with ambient air has been realized.48,49 The presence of an aqueous electrolyte in the cells, however, introduces extra complexity to the battery system, especially during the charge cycles.49 Zhou et al. used a different approach to develop a rechargeable Li–air battery with ambient air.41 The battery has a unique structure (Fig. 7a) with a hybrid electrolyte: the catholyte is a solid conductor Li1.35Ti1.75Al0.25P2.7Si0.3O12 while the analyte is a conventional EC based liquid electrolyte. With this electrolyte, the authors contended that it is possible to prevent contact between the liquid electrolyte and the O2− radical, so the problem of electrolyte decomposition is diminished. Also, instead of a conventional carbon cathode, a Li-salt modified crosslinked network gel air cathode was used, which consists of single-walled carbon nanotubes and a ionic liquid of 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide is utilized in order to form a stable electrode–electrolyte interface. The battery exhibits exceptional performance, showing a discharge capacity of 56800 mA h g−1 (Fig. 7b) and stable rechargeability of over 100 cycles with a capacity cutoff of 1000 mA h g−1 (Fig. 7c and d). The discharge product is a mixture of Li2O2, LiOH, and Li2CO3 which in the end is converted to Li2CO3 due to CO2 in the ambient air. Even though the existence of Li2CO3 leads to instability of charge in later cycles, the decomposition of the product during recharge is observed, the mechanism of which is not clear. This shows that if problems such as electrolyte decomposition and a Li parasitic reaction with air can be prevented, the Li–air battery can be operated with high performance for many stable cycles.
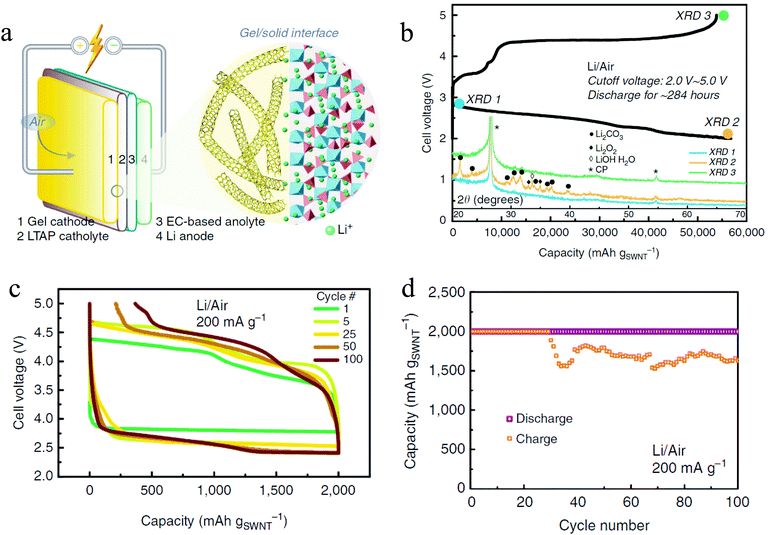 |
| | Fig. 7 (a) Schematic illustration of the Li–air battery with ambient air; (b) discharge–charge profiles and XRD analysis of the Li–air battery in ambient air; (c) and (d) cycling performance of the Li–air battery in ambient air: (c) voltage profiles and (d) cycling profiles under a capacity limitation of 2000 mA h gSWNT−1. Adapted from ref. 41. | |
3. Conclusions & perspectives
The emergence of rechargeable metal–air batteries in which a mixture of O2 & CO2 gas or ambient air is the active material in the cathode, is fueling renewed interest in the development of this storage technology for commercial applications. It also provides opportunities for designing novel electrochemical platforms for simultaneously storing electrical energy and for capturing CO2 emissions from exhaust streams from mobile and stationary sources. Using the results from the literature investigations of Li–O2/CO2 and Na–O2/CO2 electrochemical cells, we show that these systems exhibit two important traits that will drive significant future interest. First, at an optimum CO2/O2 composition close to 1/1, Li–O2/CO2 and Na–O2/CO2 electrochemical cells display higher energy storage capacities, compared to Li–O2 or Na–O2 technology. The source of the increase is shown to originate from new reactions enabled by CO2 in the cathode which, depending on the electrolyte solvent utilized, produces metal carbonates and oxalates—instead of metal peroxides and oxides, as the principal discharge products. Second, even in the absence of redox mediators, the batteries are rechargeable and the discharge curves exhibit stable plateaus at voltages comparable to those in Li–O2 and Na–O2 batteries. A particularly important observation that deserves deeper studies is that rechargeability of Li–O2/CO2 and Na–O2/CO2 electrochemical cells hinges on the ability to stabilize the electrolyte from degradation during the recharge cycle. Additional research is also needed to more fully understand the electrode reaction mechanisms in these cells. For example, although metal carbonates are easily identified as the dominant discharge products in Li–O2/CO2 and Na–O2/CO2 cells, some studies have reported that in Na–O2/CO2 cells more soluble bicarbonates may also predominate in the discharge product, if there is a source of hydrogen (e.g. trace amounts of water) in the electrolyte. This finding means that concerns about the negative effects on metal–air battery performance of CO2 and H2O contaminants in ambient air may be misplaced and, with appropriate steps taken to protect the metallic anode from side reactions with these species in the electrolyte, they may ultimately prove beneficial for stable cycling. Finally, the electrically insulating nature and poor solubility of metal carbonates in commonly used electrolytes create high overpotential during recharge, which means that new electrolytes with intrinsically high voltage stability windows, or cell designs where the voltage stability can be enhanced by electrolyte additives or electrode surface modification, are required to achieve the thousands of trouble-free charge and discharge cycles requried for transportation applications.
Acknowledgements
This work was supported by the National Science Foundation, Award no. DMR-1006323 and by Award no. KUS-C1-018-02, made by King Abdullah University of Science and Technology (KAUST).
References
- L. Grande, E. Paillard, J. Hassoun, J. Park, Y. Lee, Y. Sun, S. Passerini and B. Scrosati, Adv. Mater., 2015, 27, 784 CrossRef CAS PubMed.
- N. Choi, N. Chen, S. Freunberger, X. Ji, Y. Sun, K. Amine, G. Yushin, L. F. Nazar, J. Cho and P. G. Bruce, Angew. Chem., Int. Ed., 2012, 51, 9994 CrossRef CAS PubMed.
- A. Rahman, X. Wang and C. Wen, J. Appl. Electrochem., 2014, 44, 5 CrossRef.
-
M. Anderman, F. R. Kalhammer and D. MacArthur, Advanced batteries for electric vehicles: an assessment of performance, cost, and availability. Prepared for the State of California Air Resources Board, Sacramento, 2000.
- A. Taniguchi, N. Fujioka, M. Ikoma and A. Ohta, J. Power Sources, 2001, 100, 117 CrossRef CAS.
- M. Park, H. Sun, H. Lee, J. Lee and J. Cho, Adv. Energy Mater., 2012, 2, 780 CrossRef CAS PubMed.
- J. Wang, Y. Li and X. Sun, Nano Energy, 2013, 2, 443 CrossRef CAS PubMed.
- J. Lee, S. Kim, R. Cao, N. Choi, M. Liu, K. Lee and J. Cho, Adv. Energy Mater., 2011, 1, 34 CrossRef CAS PubMed.
- G. Girishkumar, B. McCloskey, A. C. Luntz, S. Swanson and W. Wilcke, J. Phys. Chem. Lett., 2010, 1, 2193 CrossRef CAS.
- R. Padbury and X. Zhang, J. Power Sources, 2011, 196, 4436 CrossRef CAS PubMed.
- R. Black, B. Adams and L. F. Nazar, Adv. Energy Mater., 2012, 2, 801 CrossRef CAS PubMed.
- Y. Lu, B. M. Gallant, D. G. Kwabi, J. R. Harding, R. R. Mitchell, M. S. Whittingham and S. Yang, Energy Environ. Sci., 2013, 6, 750 CAS.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J.-M. Tarascon, Nat. Mater., 2012, 11(1), 19 CrossRef CAS PubMed.
- D. Aurbach, E. Zinigrad, H. Teller and P. Dan, J. Electrochem. Soc., 2000, 147, 1274 CrossRef CAS PubMed.
- J.-M. Tarascon and M. Armand, Nature, 2001, 414, 359 CrossRef CAS PubMed.
- S. Freunberger, Y. Chen, Z. Peng, J. Griffin, L. Hardwick, F. Barde, P. Novak and P. G. Bruce, J. Am. Chem. Soc., 2011, 133, 8040 CrossRef CAS PubMed.
- S. Freunberger, Y. Chen, N. Drewett, L. Hardwick, F. Barde and P. G. Bruce, Angew. Chem., Int. Ed., 2011, 50, 8609 CrossRef CAS PubMed.
- D. Aurbach, Y. Gofer, M. Ben-Zion and P. Aped, J. Electroanal. Chem., 1992, 339, 451 CrossRef CAS.
- D. Aurbach, I. Weissman, A. Zaban and O. Chusid, Electrochim. Acta, 1994, 39, 51 CrossRef CAS.
- K. Gallagher, S. Goebel, T. Greszler, M. Mathias, W. Oelerich, D. Eroglu and V. Srinivasan, Energy Environ. Sci., 2014, 7, 1555 CAS.
- D. P. Scharg, Science, 2007, 315, 812 CrossRef PubMed.
- C. Gough, Int. J. Greenhouse Gas Control, 2008, 2, 155 CrossRef CAS.
- S. M. Benn and F. M. Orr, MRS Bull., 2008, 33, 297 CrossRef.
- K. Takechi, T. Shiga and T. Asaoka, Chem. Commun., 2011, 47, 3463 RSC.
- J. L. Roberts, T. S. Calderwood and D. T. Sawyer, J. Am. Chem. Soc., 1984, 106, 4667 CrossRef CAS.
- J. D. Wadhawan, P. J. Welford, H. B. McPeak, C. E. W. Hahn and R. G. Compton, Sens. Actuators, B, 2003, 88, 40 CrossRef CAS.
- H. Lim, H. Lim, K. Park, D. Seo, H. Gwon, J. Hong, W. Goddard, H. Kim and K. Kang, J. Am. Chem. Soc., 2013, 135, 9733–9742 CrossRef CAS PubMed.
- V. Palomares, P. Serras, I. Villaluenga, K. B. Hueso, J. C. Gonzalez and T. Rojo, Energy Environ. Sci., 2012, 5, 5884 CAS.
- H. Pan, Y.-S. Hu and L. Chen, Energy Environ. Sci., 2013, 6, 2338 CAS.
- B. L. Ellis and L. F. Nazar, Curr. Opin. Solid State Mater. Sci., 2012, 16, 168 CrossRef CAS PubMed.
- Y.-U. Park, D.-H. Seo, H.-S. Kwon, B. Kim, J. Kim, H. Kim, I. Kim, H.-I. Yoo and K. Kang, J. Am. Chem. Soc., 2013, 135, 13870 CrossRef CAS PubMed.
- H. Xiong, M. D. Slater, M. Balasubramanian, C. S. Johnson and T. Rajh, J. Phys. Chem. Lett., 2011, 2, 2560 CrossRef CAS.
- S. K. Das, S. Lau and L. A. Archer, J. Mater. Chem. A, 2014, 2, 12623 CAS.
- P. Hartmann, C. Bender, M. Vracar, A. K. Durr, A. Garsuch, J. Janek and P. Adelhelm, Nat. Mater., 2013, 12, 228 CrossRef CAS PubMed.
- E. Peled, D. Golodnitsky, H. Mazor, M. Goor and S. Avshalovmov, J. Power Sources, 2011, 196, 6835 CrossRef CAS PubMed.
- S. K. Das, S. Xu and L. A. Archer, Electrochem. Commun., 2013, 27, 59 CrossRef CAS PubMed.
- S. Xu, Y. Lu, H. Wang, H. D. Abruna and L. A. Archer, J. Mater. Chem. A, 2014, 2, 17723 CAS.
- S. Xu, S. K. Das and L. A. Archer, RSC Adv., 2013, 3, 6656 RSC.
- Y. Liu, R. Wang, Y. Lyu, H. Li and L. Chen, Energy Environ. Sci., 2014, 7, 677 CAS.
- Z. Zhang, Q. Zhang, Y. Chen, J. Bao, X. Zhou, Z. Xie, J. Wei and Z. Zhou, Angew. Chem., Int. Ed., 2015, 54, 6550 CrossRef CAS PubMed.
- T. Zhang and H. Zhou, Nat. Commun., 2013, 4, 1817 CrossRef PubMed.
- Y. S. Mekonnen, K. B. Knudsen, J. S. G. Mýrdal, R. Younesi, J. Højberg, J. Hjelm, P. Norby and T. Vegge, J. Chem. Phys., 2014, 140(12), 121101 CrossRef PubMed.
- Y. S. Mekonnen, J. M. Garcia-Lastra, J. S. Hummelshøj, C. Jin and T. Vegge, J. Phys. Chem. C, 2015, 119, 18066 CAS.
-
E. Dlugokencky and P. Tans, NOAA/ESRL, http://www.esrl.noaa.gov/gmd/ccgg/trends/.
- S. Meini, M. Piana, N. Tsiouvaras, A. Garsuch and H. A. Gasteiger, Electrochem. Solid-State Lett., 2012, 15(4), A45 CrossRef CAS PubMed.
- N. B. Aetukuri, B. D. McCloskey, J. M. García, L. E. Krupp, V. Viswanathan and A. C. Luntz, Nat. Chem., 2014, 7(1), 50 CrossRef PubMed.
- F. Li, S. Wu, D. Li, T. Zhang, P. He, A. Yamada and H. Zhou, Nat. Commun., 2015, 6, 7843 CrossRef CAS PubMed.
- Y. Wang, P. He and H. Zhou, Energy Environ. Sci., 2011, 4, 4994 CAS.
- Y. Wang and H. Zhou, J. Power Sources, 2010, 195, 358 CrossRef CAS PubMed.
- Y. Lu, S. K. Das, S. S. Moganty and L. A. Archer, Adv. Mater., 2012, 24, 4430 CrossRef CAS PubMed.
- Y. Lu, K. Korf, Y. Kambe, Z. Tu and L. A. Archer, Angew. Chem., Int. Ed., 2014, 53, 488 CrossRef CAS PubMed.
|
| This journal is © the Partner Organisations 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 


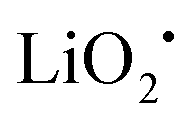 , and this reaction is believed to be slower than the reactions (2)–(4) in the analogous Li–O2/CO2 battery. Furthermore, although the intermediate products were not detected, it is believed that the intermediate C2O6− compound can stably diffuse in the electrolyte, thus slowing down the precipitation of Li2CO3, allowing it to completely fill the void space in the cathode. Unfortunately, these studies are all based on carbonate electrolytes, which are now known to undergo additional electrochemical decomposition reactions during recharge. This finding leads to the widespread, but incorrect, view that in contrast to the Li–O2 battery, a Li–O2/CO2 battery is not rechargeable.
, and this reaction is believed to be slower than the reactions (2)–(4) in the analogous Li–O2/CO2 battery. Furthermore, although the intermediate products were not detected, it is believed that the intermediate C2O6− compound can stably diffuse in the electrolyte, thus slowing down the precipitation of Li2CO3, allowing it to completely fill the void space in the cathode. Unfortunately, these studies are all based on carbonate electrolytes, which are now known to undergo additional electrochemical decomposition reactions during recharge. This finding leads to the widespread, but incorrect, view that in contrast to the Li–O2 battery, a Li–O2/CO2 battery is not rechargeable.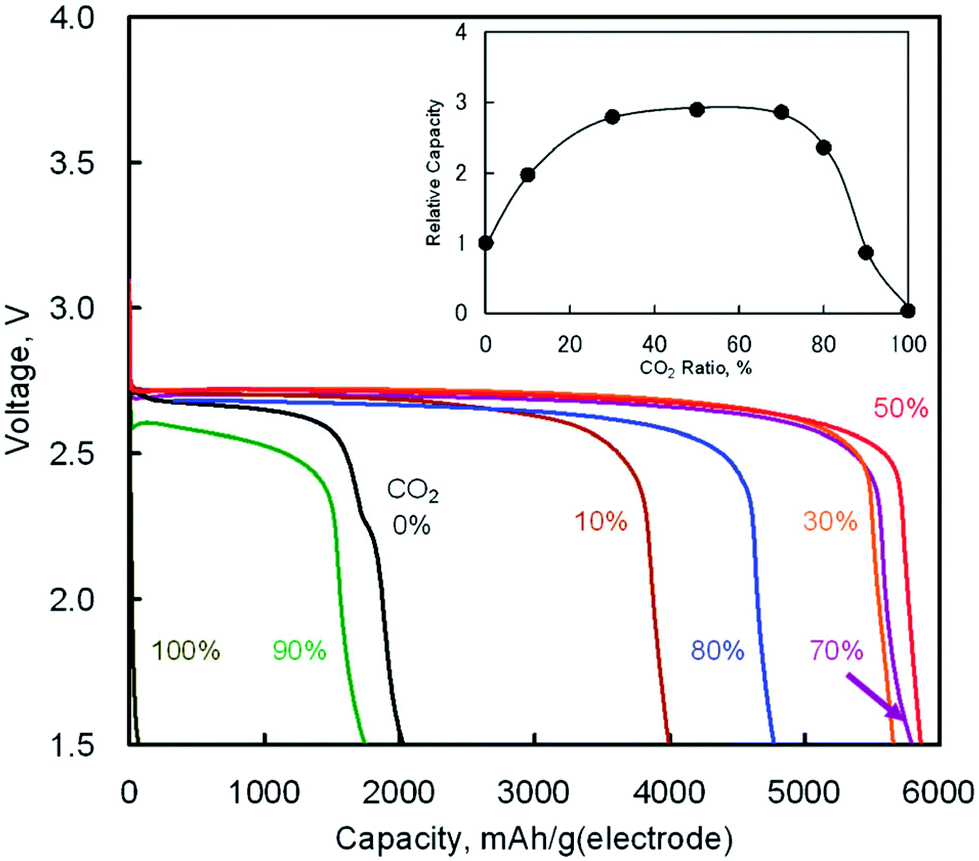
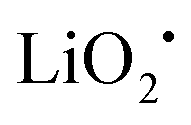 , so the reaction mechanism in the Li–O2/CO2 battery is thought to be the same as in the Li–O2 battery, with Li2O2 as the discharge product. However, in high ε electrolytes like carbonates and DMSO (ε = 47.2), the reaction between the O2˙− radical and CO2 is more favored, so in this case CO2 does take part in the reaction, and the discharge product is Li2CO3 (Fig. 2a). These predictions are consistent with the limited experimental data available. In the high ε electrolyte (DMSO) case, it is worth noting that even though the discharge product is Li2CO3, which is thermodynamically stable and thought to be undegradable during recharge, the cells exhibit some amount of rechargeability and are stably cycled for over 20 cycles, as shown in Fig. 2b and c. The formation of the discharge product Li2CO3 was detected after discharge and the disappearance of the product after recharge was also observed. These findings therefore demonstrate that the Li–O2/CO2 battery can be recharged, which opens the possibility for Li–O2/CO2 rechargeable cells to be used in more or less the same applications targeted for Li–O2 and Li–air.
, so the reaction mechanism in the Li–O2/CO2 battery is thought to be the same as in the Li–O2 battery, with Li2O2 as the discharge product. However, in high ε electrolytes like carbonates and DMSO (ε = 47.2), the reaction between the O2˙− radical and CO2 is more favored, so in this case CO2 does take part in the reaction, and the discharge product is Li2CO3 (Fig. 2a). These predictions are consistent with the limited experimental data available. In the high ε electrolyte (DMSO) case, it is worth noting that even though the discharge product is Li2CO3, which is thermodynamically stable and thought to be undegradable during recharge, the cells exhibit some amount of rechargeability and are stably cycled for over 20 cycles, as shown in Fig. 2b and c. The formation of the discharge product Li2CO3 was detected after discharge and the disappearance of the product after recharge was also observed. These findings therefore demonstrate that the Li–O2/CO2 battery can be recharged, which opens the possibility for Li–O2/CO2 rechargeable cells to be used in more or less the same applications targeted for Li–O2 and Li–air.