DOI:
10.1039/C4QI00244J
(Research Article)
Inorg. Chem. Front., 2015,
2, 467-472
Synthesis, structure, and alkyne insertion of a mixed-sandwich zirconacarborane alkyl†
Received
31st December 2014
, Accepted 10th March 2015
First published on 12th March 2015
Abstract
A neutral mixed-sandwich zirconacarborane alkyl [η1:σ:η5-{MeN(CH2)CH2CH2}C2B9H10]Zr(η5-Cp′′) (Cp′′ = 1,3-(Me3Si)2C5H3) was prepared via methane elimination reaction of 7-Me2N(H)CH2CH2-7,8-C2B9H11 with (η5-Cp′′)ZrMe3, followed by an intramolecular C–H activation. It reacted with internal alkynes and trimethylsilylacetylene to give the Zr–C σ bond mono-insertion products; however, the terminal alkyne 3,3-dimethyl-1-butyne underwent an acid–base reaction to produce a zirconium alkynyl complex. Both electronic and steric factors affected the regioselectivity of the insertion process. The results showed that the coordination of the sidearm nitrogen atom could enhance the thermal stability of the resulting zirconacarborane alkyl, and on the other hand, lowered the Lewis acidity of the Zr atom, leading to relatively poor reactivity. All complexes were fully characterized by NMR spectroscopy and elemental analyses. Most of them were further confirmed by single-crystal X-ray analyses.
Introduction
Cationic group 4 metallocene alkyls of the general type (C5R5)2M(R)+ have received great interest as they exhibit rich insertion, olefin polymerization, and C–H bond activation chemistry, which are highly sensitive to the structural and electronic properties of (C5R5)2M fragments.1 Replacement of a uninegative C5R5− in (C5R5)2M(R)+ by a dinegative isolobal C2B9H112− ligand results in the formation of a class of neutral mixed-sandwich group 4 metallacarborane alkyls (C5Me5)(C2B9H11)M(R) that have similar properties to those of cationic (C5R5)2M(R)+.2 Unfortunately, these neutral metal alkyls such as (C5Me5)(C2B9H11)Zr(Me) are not stable even at 45 °C, which limits their applications.2a We attempted to increase their thermal stability by introducing carbon-bridged hybrid ligands [Me2C(C5H4)(C2B9H10)]3−/[Me2C(C9H6)(C2B9H10)]3−/[H2C(C5Me4)(C2B9H10)]3−, leading to the isolation of ansa-metallocene dialkyl anionic complexes rather than the neutral metallocene alkyls.3 This can be ascribed to the much increased “bite angles”3,4 of the resulting ansa-metallocenes compared with the corresponding unbridged ones,2 which results in a more open coordination sphere around the metal center, thus facilitating the binding of additional anionic ligands. It seems that ansa-ligands are not a good solution to solve the thermal stability problem of (C5Me5)(C2B9H11)M(R). We thought an alternative way to address this issue is to introduce a functional sidearm to the dicarbollyl ligand in the hope that coordination of the sidearm to the central metal could stabilize the resulting metal alkyls while keeping the metal geometry unchanged.5 Herein, we report the synthesis, characterization and reactivity of a mixed-sandwich zirconacarborane alkyl [η1:σ:η5-{MeN(CH2)CH2CH2}C2B9H10]Zr(η5-Cp′′) (Cp′′ = 1,3-(Me3Si)2C5H3).
Results and discussion
Synthesis
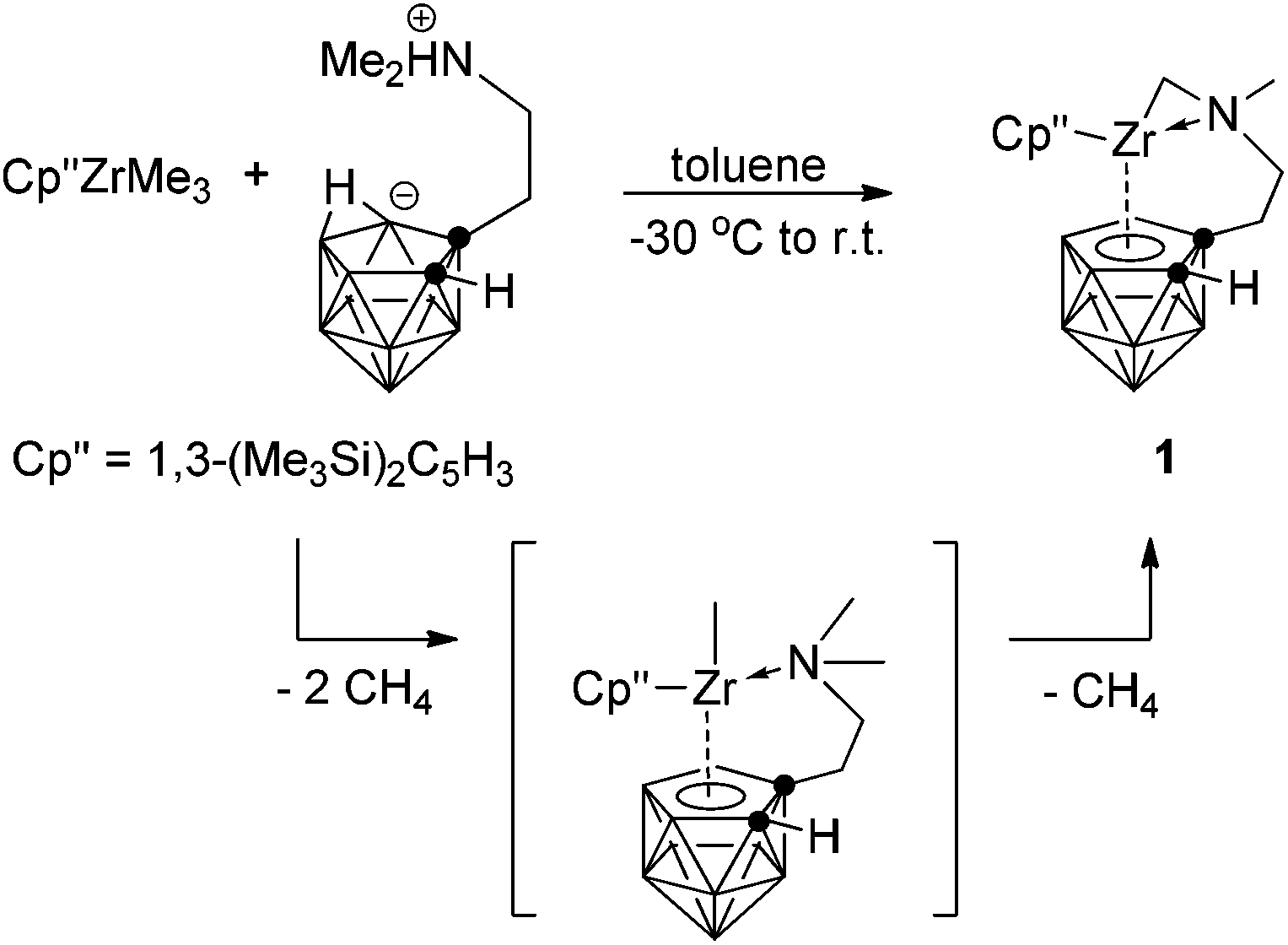
Alkane elimination is often used for the clean formation of zirconacarborane alkyl complexes.6 Treatment of (η5-Cp′′)ZrMe3 with 1 equiv. of the zwitterionic salt 7-Me2N(H)CH2CH2-7,8-C2B9H11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 5g,7 in toluene at temperatures between −30 °C and 28 °C overnight afforded a mixed-sandwich zirconacarborane alkyl [η1:σ:η5-{MeN(CH2)CH2CH2}C2B9H10]Zr(η5-Cp′′) (1) in 80% isolated yield (Scheme 1). The expected zirconacarborane methyl complex (η5-Cp′′)[η1:η5-(Me2NCH2CH2)C2B9H10]Zr(Me) was not obtained probably due to its thermal instability. However, it should serve as an intermediate for alkane elimination reaction and subsequently eliminates one CH4 molecule via the rupture of a C–H bond at one of the N-methyl groups to give the final product 1, as the Zr-methyl is more basic than the N-methyl.5g Such an intramolecular C–H activation is normally observed in methylzirconocenes8 and group 4 metallacarborane alkyls.9
5g,7 in toluene at temperatures between −30 °C and 28 °C overnight afforded a mixed-sandwich zirconacarborane alkyl [η1:σ:η5-{MeN(CH2)CH2CH2}C2B9H10]Zr(η5-Cp′′) (1) in 80% isolated yield (Scheme 1). The expected zirconacarborane methyl complex (η5-Cp′′)[η1:η5-(Me2NCH2CH2)C2B9H10]Zr(Me) was not obtained probably due to its thermal instability. However, it should serve as an intermediate for alkane elimination reaction and subsequently eliminates one CH4 molecule via the rupture of a C–H bond at one of the N-methyl groups to give the final product 1, as the Zr-methyl is more basic than the N-methyl.5g Such an intramolecular C–H activation is normally observed in methylzirconocenes8 and group 4 metallacarborane alkyls.9
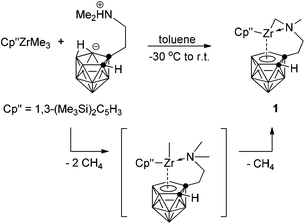 |
| | Scheme 1 Synthesis of mixed-sandwich zirconacarborane alkyl. | |
Complex 1 was fully characterized by various spectroscopic techniques and elemental analyses. The 1H NMR spectrum showed one broad singlet at 3.15 ppm attributable to the cage CH, two doublets at 2.21 and 2.19 ppm with J = 6.0 Hz assignable to the Zr–CH2 unit in addition to the resonances corresponding to the protons of Cp′′ and sidearm. Its 11B NMR spectrum exhibited a 1:1:2:1:2:1:1 pattern.
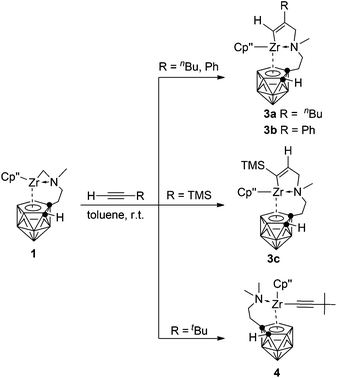
Reaction with internal alkynes
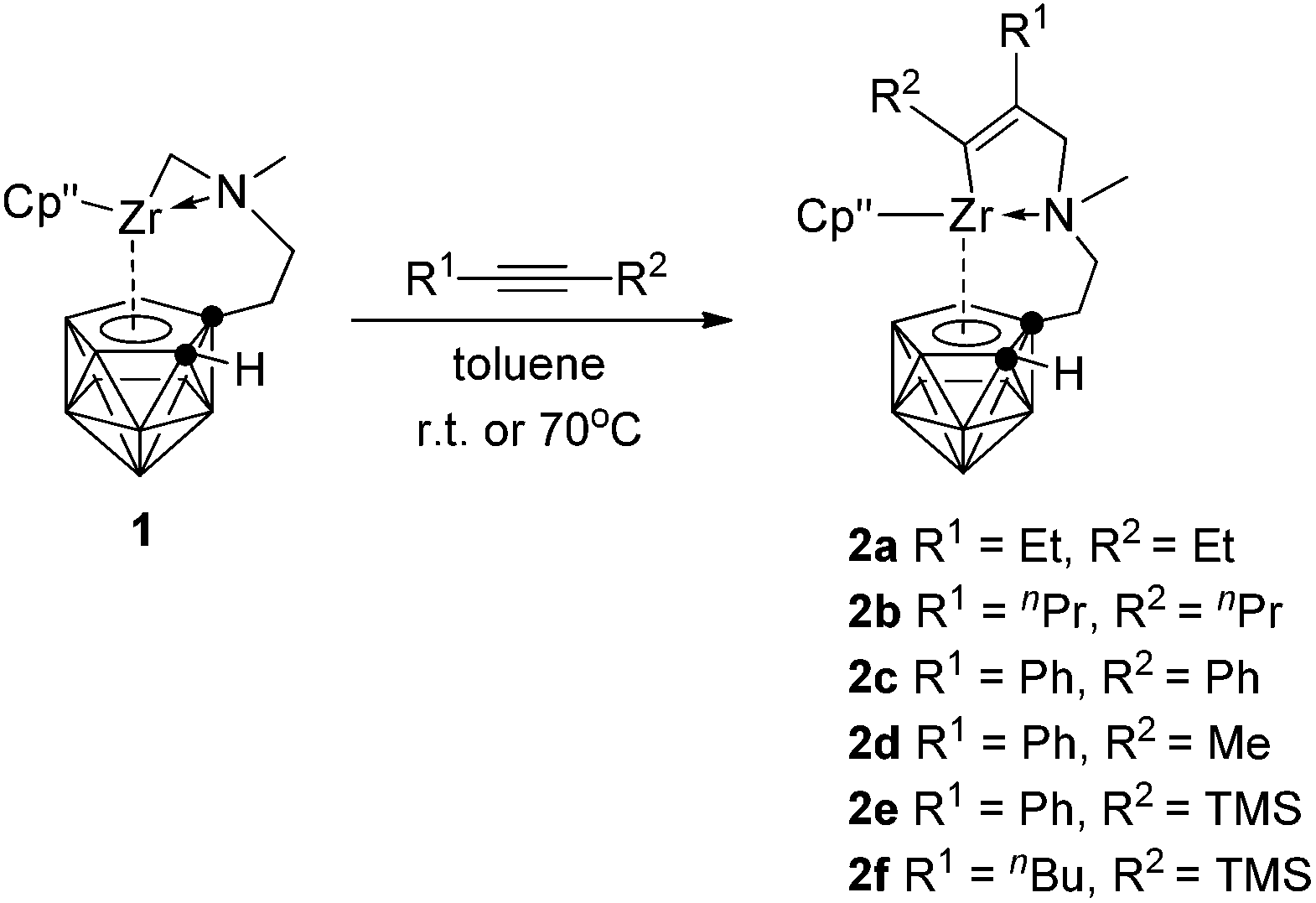
The migratory insertion reaction is a key step in transition metal catalyzed olefin polymerization.10 It has been documented that [Cp*(C2B9H11)ZrCH3]n can catalyze ethylene polymerization in the absence of any co-catalyst.2a,b However, zirconacarborane alkyl 1 showed no reactivity toward alkenes, such as ethylene, 1-hexene and styrene, even under the harsh conditions due probably to the intramolecular coordination of the N atom, leading to decreased Lewis acidity of the Zr center. On the other hand, alkynes can insert into the Zr–C σ bond in 1 to form mono-insertion products. Treatment of symmetrical internal alkynes with 1 in a 1:1 molar ratio in toluene afforded [η1:σ:η5-{MeN[CH2(R)C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(R)]CH2CH2}C2B9H10]Zr(η5-Cp′′) [R = Et (2a), nPr (2b), Ph (2c)] in ∼70% isolated yields (Scheme 2). Only mono-insertion products were formed even in the presence of an excess amount of alkynes in refluxing toluene. It was noted that the steric factors played an important role in the reactions. Sterically less demanding linear dialkylalkynes worked at room temperature; however, heating at 70 °C was required for the insertion of diphenylacetylene. No reaction proceeded for sterically demanding TMSC
C(R)]CH2CH2}C2B9H10]Zr(η5-Cp′′) [R = Et (2a), nPr (2b), Ph (2c)] in ∼70% isolated yields (Scheme 2). Only mono-insertion products were formed even in the presence of an excess amount of alkynes in refluxing toluene. It was noted that the steric factors played an important role in the reactions. Sterically less demanding linear dialkylalkynes worked at room temperature; however, heating at 70 °C was required for the insertion of diphenylacetylene. No reaction proceeded for sterically demanding TMSC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CTMS.
CTMS.
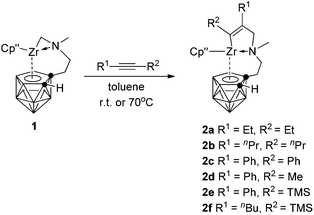 |
| | Scheme 2 Reaction of 1 with internal alkynes. | |
When unsymmetrical alkynes were used, regioselectivity would be an issue. The reaction of 1 with 1 equiv. of unsymmetrical internal alkynes (PhCCMe, PhCCTMS and nBuCCTMS) in toluene at 70 °C afforded the mono-insertion products [η1:σ:η5-{MeN[CH2(R1)CC(R2)]CH2CH2}C2B9H10]Zr(η5-Cp′′) [R1 = Ph, R2 = Me (2d); R1 = Ph, R2 = TMS (2e); R1 = nBu, R2 = TMS (2f)] in ∼80% isolated yields (Scheme 2). The regioselectivity observed in 2d may result from the more steric hindrance of Ph over the Me group since phenyl is often considered as an electron-withdrawing group.11 Complexes 2e and 2f show a totally different regioselectivity from that of 2d, in which the more bulky TMS group is closer to the Zr center. This result could be rationalized on the basis of electronic effects12 originating from the electron withdrawing SiMe3 group which can override the normal steric preference for 1,2-insertion of alkynes into the Zr–C bonds. These results suggest that both electronic and steric factors affect the regioselectivity of the insertion reaction, depending on the nature of substituents of alkynes.
Complexes 2a–f were characterized by various spectroscopic techniques and elemental analyses. The unique features of their NMR spectra are two doublets at 3.1–4.6 and 2.1–3.2 ppm attributable to the methylene protons of NCH2C(sp2) in the 1H NMR spectra and two characteristic resonances at 191–202 and 138–153 ppm corresponding to the two vinyl carbons of the Zr(CαCβ) unit in the 13C NMR spectra. The latter are very much comparable to the 13C chemical shifts of ∼194 and ∼142 ppm observed in Cp2Zr[C(R)C(R)]2.13
Reaction with terminal alkynes
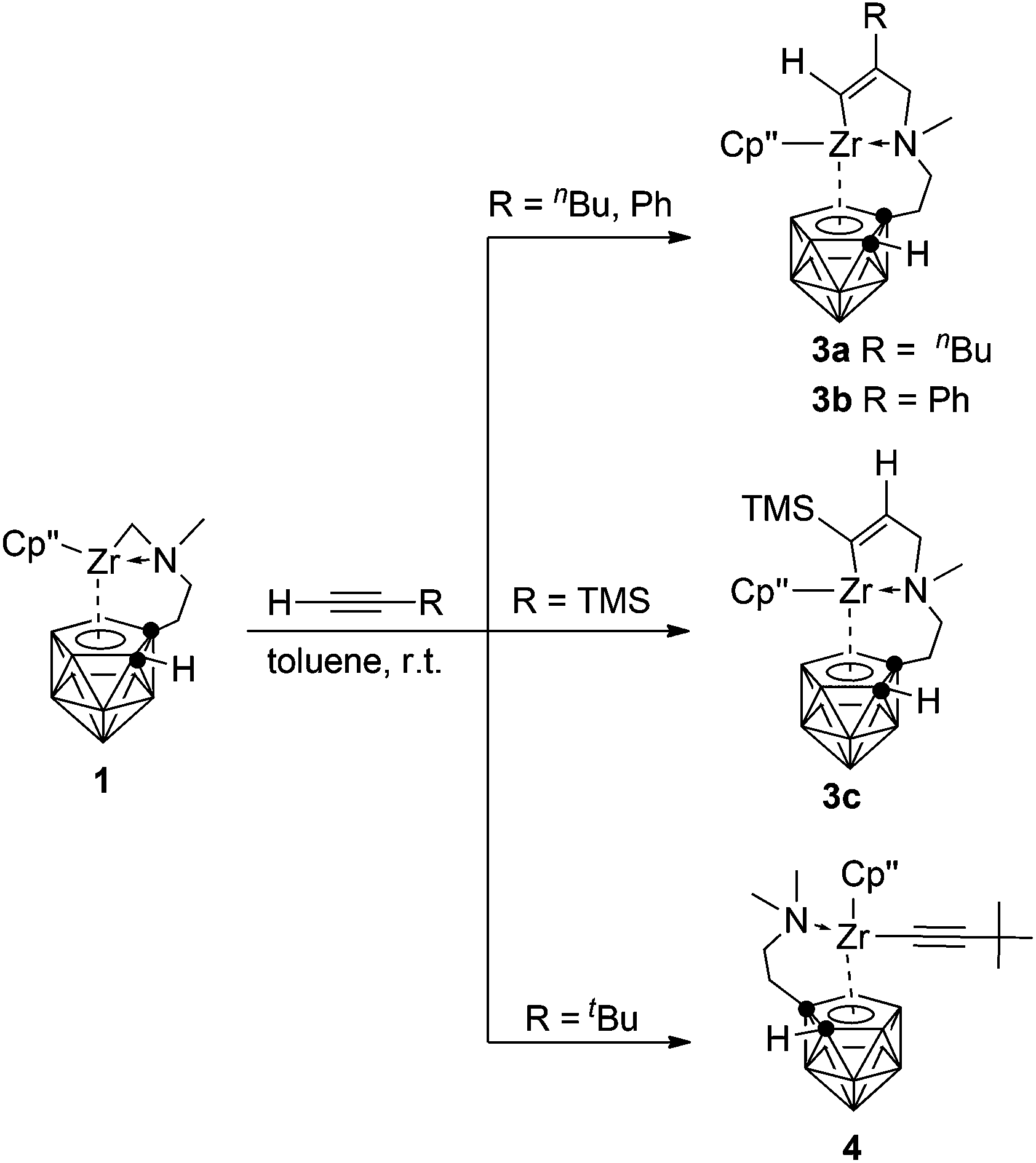
As the sp-CH protons in terminal alkynes are acidic, they may undergo either acid–base or insertion reaction with 1. Treatment of 1 with 1 equiv. of phenylacetylene, 1-hexyne or trimethylsilylacetylene in toluene at room temperature afforded mono-insertion zirconacyclic products [η1:σ:η5-{MeN[CH2(R1)CC(H)]CH2CH2}C2B9H10]Zr(η5-Cp′′) [R1 = nBu (3a); Ph (3b)] or [η1:σ:η5-{MeN[CH2(H)CC(TMS)]CH2CH2}C2B9H10]Zr(η5-Cp′′) (3c), respectively, in ∼80% isolated yields (Scheme 3). The results show that the regioselectivity observed in the above reactions is the same as that found in internal alkynes. In view of the different electronic properties of Ph and n-butyl groups, the regioselectivity in 3a and 3b is dominated by steric and electronic factors, respectively. On the other hand, the bulky TMS is closer to the Zr center in 3c, suggesting that it is an electronically-controlled 1,2-insertion. Unexpectedly, the reaction of 1 with 1 equiv. of 3,3-dimethyl-1-butyne (tBuCCH) in toluene at room temperature produced a zirconium alkynyl complex (η5-Cp′′)[η1:η5-(Me2NCH2CH2)C2B9H10]Zr(CCtBu) (4) in 75% isolated yield (Scheme 3). Only acid–base reaction product 4 was isolated, and no insertion product was observed. The reasons are not clear at this stage. Theoretically, both protonation and insertion reaction could proceed with terminal alkynes, in which both electronic and steric factors play a role. It was noted that terminal alkynes 4-NO2-C6H4CCH, 2-NO2-C6H4CCH and 4-CF3-C6H4CCH were also examined. These reactions were complicated as indicated by NMR spectra, from which no pure products were isolated.
 |
| | Scheme 3 Reaction of 1 with terminal alkynes. | |
Complexes 3 and 4 were characterized by using various spectroscopic data and elemental analyses. In the 1H NMR spectra of 3, one singlet at 4.85 (3a)/7.07 (3b) ppm attributable to the vinylic Zr[CαHCβ(R)] proton and one doublet of doublets at 6.43 ppm (3c) with J = 1.8 and 3.0 Hz corresponding to the vinylic Zr[Cα(R)CβH–CH2] were observed, respectively, for two different types of regioselective insertion products. The ZrNCH2C(sp2) protons showed two doublets at 3.07 and 2.12 ppm with J = 13.5 Hz for 3a, 3.45 and 2.85 ppm with J = 15.0 Hz for 3b, and two doublets of doublets at 3.05 (J = 1.8 and 15.0 Hz) and 2.15 (J = 3.0 and 15.0 Hz) ppm for 3c. Accordingly, the characteristic 13C resonances at 181.9/143.4 ppm for 3a, 187.5/141.8 ppm for 3b, and 211.7/140.3 ppm for 3c assignable to the Zr[CαCβ] were observed in their 13C NMR spectra. For 4, two singlets at 2.10 and 2.05 ppm corresponding to the NMe2 protons were observed in its 1H NMR spectrum, indicating the protonation of the zirconium alkyl moiety. In addition, its 13C NMR spectrum showed a signal of the Zr–CC carbon at 90.2 ppm. The 11B NMR spectra displayed a pattern of 2:2:1:1:1:2 for 3a, 2:2:2:2:1 for 3b, 1:3:1:1:1:1:1 for 3c and 1:1:2:1:1:1:1:1 for 4, respectively.
Molecular structure
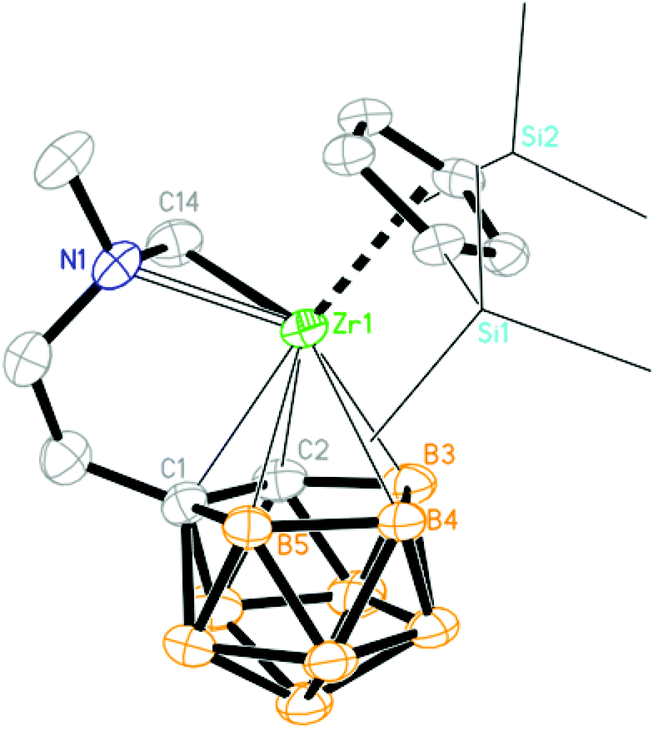
The molecular structures of 1, 2a, 2c–f, 3b, 3c and 4 were further confirmed by single-crystal X-ray analyses. Their key structural parameters are compiled in Table 1 for comparison. The Zr atom in 1 is η5-bound to both the Cp′′ ring and dicarbollyl ligand, σ-bound to one carbon atom from the NCH2 group and coordinated to the N atom of the amino group in a distorted-tetrahedral geometry (Fig. 1). The average Zr–C5 ring distance of 2.509(5) Å is comparable to that of 2.517(5)/2.522(5) Å in (η5-Cp′′)2ZrMe(μ-Me)B(C6F5)3,14 2.523(3) Å in (η5-Cp′′)(C4H4BC6F5)Zr(C6F5)(CNtBu),15 and 2.530(5) Å in (η5-Cp′′)(η5-C13H9)ZrCl2.16 The average Zr–cage atom distance of 2.522(5) Å is comparable to that of 2.540(8) Å in [η5:η5-H2C(C5Me4)(C2B9H10)]Zr[σ:σ-CH2(NMe2)-o-C6H4],3c 2.499 Å in (Cp*)(C2B9H11)Zr[C(Me)CMe2],17 2.535/2.533 Å in [(Cp*)(C2B9H11)Zr]2(μ-CH2),17 and 2.544(6) Å in [η1:σ:η5-{MeN(CH2)CH2CH2}C2B9H10]Zr(CH2SiMe3)(THF).5g The Zr–C σ bond distance of 2.270(4) Å is close to that of 2.282(2) Å in (η5-Cp′′)(η5-C5Me5)ZrH(Me),18 and 2.250(12) Å [Zr–C(sp3)] in Cp2Zr[(Ph)CC(Ph)CH(C6H4)CH],19 but is longer than that of 2.233(6)/2.242(6) Å (Zr–C(N)/Zr–C(TMS)) in [η1:σ:η5-{MeN(CH2)CH2CH2}C2B9H10]Zr(CH2SiMe3)(THF),5g 2.198(4) Å in (Cp*)(C2B9H11)Zr[C(Me)CMe2],17 and 2.187(6)/2.176(7) Å in [(Cp*)(C2B9H11)Zr]2(μ-CH2).17 The Cent(C5)–Zr–Cent(C2B3) angle of 140.0° is comparable to that of 141.3° in (Cp*)(C2B9H11)Zr[C(Me)CMe2],17 is larger than that of 134.9° in [(Cp*)(C2B9H11)Zr]2(μ-CH2),17 but is significantly larger than those observed in the corresponding ansa-metallocenes, such as [η5:η5-Me2C(C5Me4)(C2B9H10)]Zr(NHC6H3Pri2)(THF) (119.2°),3a [η5:η5-Me2C(C9H6)(C2B9H10)]Zr(NMe2)(NHMe2) (115.9°)3b and [η5:η5-Me2C(C5Me4)(C2B9H10)]Zr[σ:σ-CH2(NMe2)-o-C6H4] (120.2°).3c
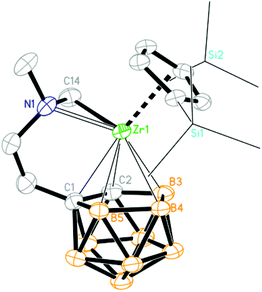 |
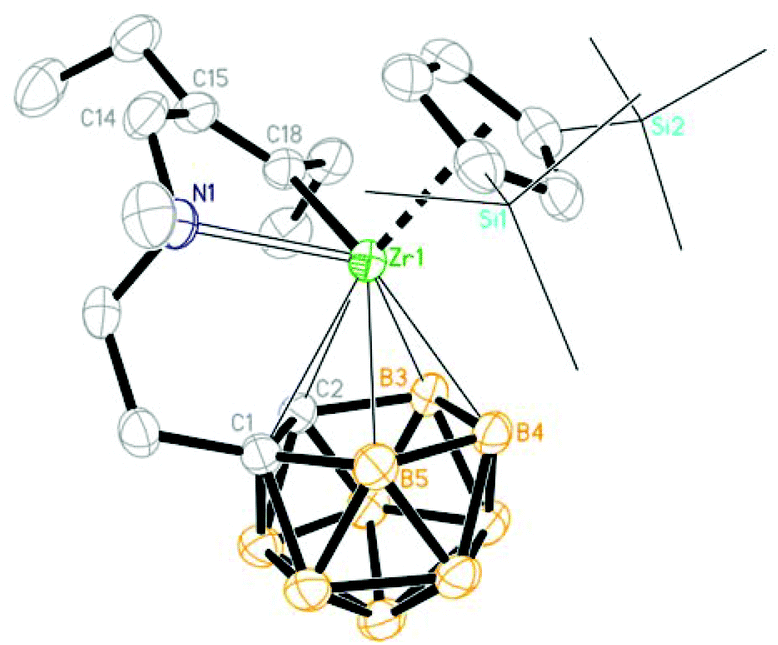
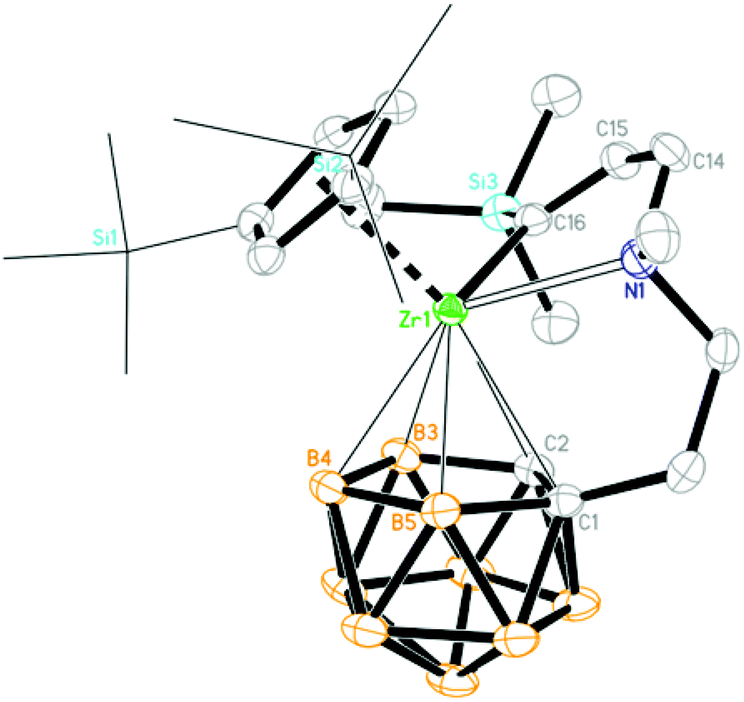
| | Fig. 1 Molecular structure of [η1:σ:η5-{MeN(CH2)CH2CH2}C2B9H10]Zr(Cp′′) (1) (two TMS groups are represented by lines for clarity). | |
Table 1 Selected bond lengths (Å) and angles (°) for 1, 2a, 2c–f, 3b, 3c and 4
| |
1
|
2a
|
2c
|
2d
|
2e
|
2f
|
3b
|
3c
|
4
|
Cent(C5), Cent(C2B3): the centroid of the cyclopentadienyl ring and the C2B3 bonding face, respectively.
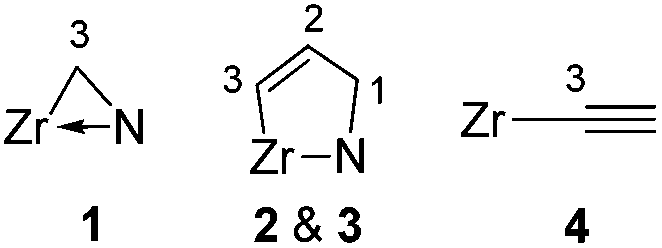
For numbering system, see:  . .
|
| Av. Zr–Cring |
2.509(5) |
2.559(3) |
2.533(8) |
2.544(3) |
2.567(4) |
2.566(5) |
2.537(7) |
2.553(3) |
2.539(4) |
| Av. Zr–cage atom |
2.522(5) |
2.554(2) |
2.537(7) |
2.551(4) |
2.554(6) |
2.561(5) |
2.550(7) |
2.551(3) |
2.560(6) |
| Zr–N |
2.267(3) |
2.368(2) |
2.355(3) |
2.353(3) |
2.349(4) |
2.351(4) |
2.376(5) |
2.379(3) |
2.415(4) |
| Zr–C(3)b |
2.270(4) |
2.295(2) |
2.329(4) |
2.284(2) |
2.338(4) |
2.346(4) |
2.279(6) |
2.304(3) |
2.190(4) |
| C(2)–C(3)b |
|
1.339(3) |
1.345(6) |
1.336(5) |
1.340(5) |
1.354(6) |
1.331(8) |
1.342(5) |
|
| C(1)–C(2)b |
|
1.506(3) |
1.498(7) |
1.513(4) |
1.523(7) |
1.507(5) |
1.500(8) |
1.503(4) |
|
| N–C(1)b |
|
1.493(3) |
1.496(7) |
1.499(3) |
1.480(7) |
1.492(5) |
1.498(8) |
1.501(4) |
|
| Zr–Cent(C5)a |
2.204 |
2.258 |
2.232 |
2.241 |
2.267 |
2.266 |
2.234 |
2.249 |
2.236 |
| Zr–Cent(C2B3)a |
2.075 |
2.115 |
2.100 |
2.111 |
2.114 |
2.121 |
2.107 |
2.106 |
2.120 |
| Cent(C5)–M–Cent(C2B3) |
140.0 |
137.6 |
136.5 |
138.2 |
135.4 |
135.3 |
138.6 |
135.9 |
136.8 |
In the molecular structures of 2a, c–f and 3b, c, the Zr atom is η5-bound to both the Cp′′ ring and dicarbollyl ligand, σ-bound to the sp2-C atom of the vinyl unit and coordinated to the N atom in a distorted-tetrahedral geometry. Their representative structures are shown in Fig. 2–6, respectively. The average Zr–C(C5 ring) and Zr–cage atom distances in these complexes are close to each other and comparable to those in 1. Their Cent(C5 ring)–Zr–Cent(C2B3) angles fall in the range 135.3°–138.6°, which is slightly smaller than that of 140.0° in 1. The Zr–C(sp2) σ bond distances (2.28–2.35 Å) are comparable to that of 2.298(10) Å in Cp2Zr[(Ph)CC(Ph)CH(C6H4)CH]19 and 2.27–2.31 Å observed in Cp2Zr[C(R)C(R)–C2B10H10],20 but longer than that of 2.198(4) Å in (Cp*)(C2B9H11)Zr[C(Me)CMe2].17 The above X-ray structures unambiguously confirm the regioselective insertion products of alkynes into the Zr–C σ bond in 1.
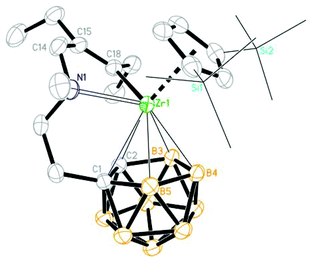 |
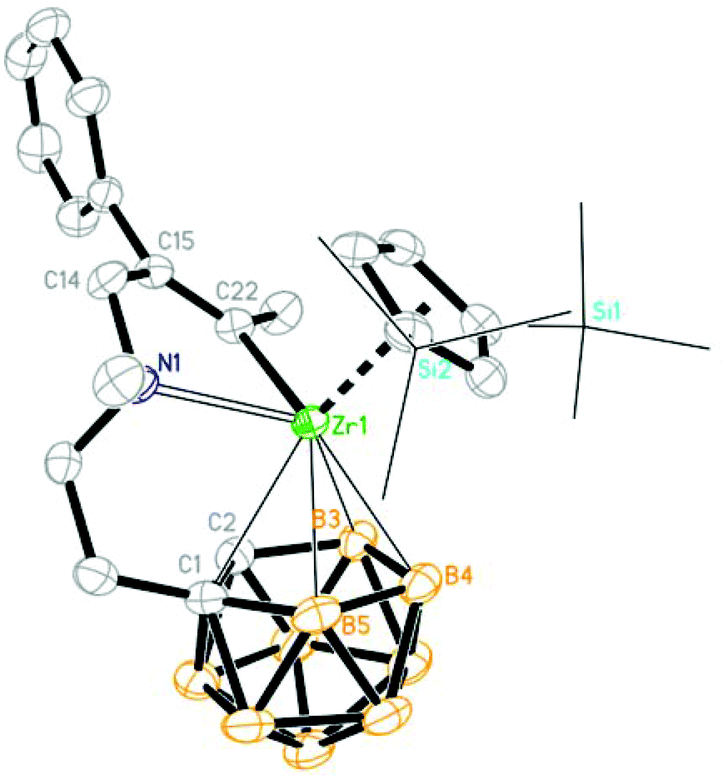
| | Fig. 2 Molecular structure of [η1:σ:η5-{MeN[CH2(Et)CC(Et)]CH2CH2}C2B9H10]Zr(η5-Cp′′) (2a) (two TMS groups are represented by lines for clarity). | |
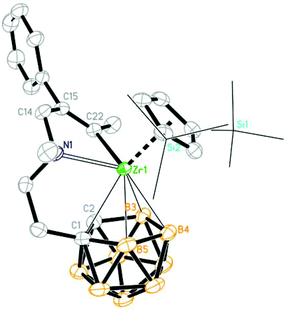 |
| | Fig. 3 Molecular structure of [η1:σ:η5-{MeN[CH2(Ph)CC(Me)]CH2CH2}C2B9H10]Zr(η5-Cp′′) (2d) (two TMS groups are represented by lines for clarity). | |
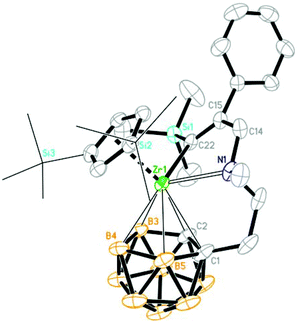 |
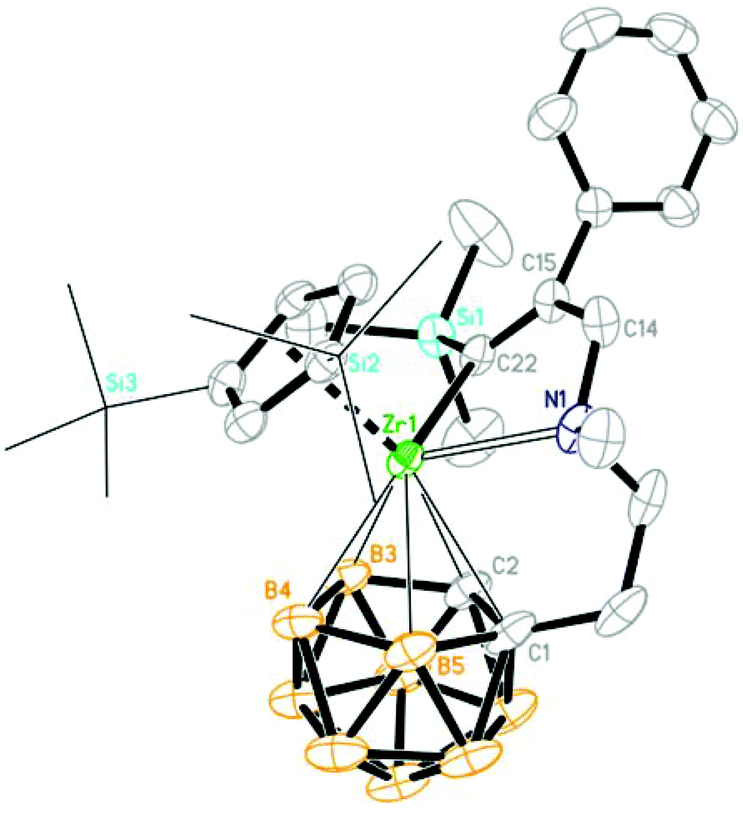
| | Fig. 4 Molecular structure of [η1:σ:η5-{MeN[CH2(Ph)CC(TMS)]CH2CH2}C2B9H10]Zr(η5-Cp′′) (2e) (two TMS groups of Cp′′ are represented by lines for clarity). | |
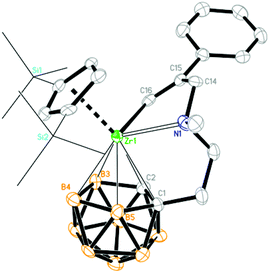 |
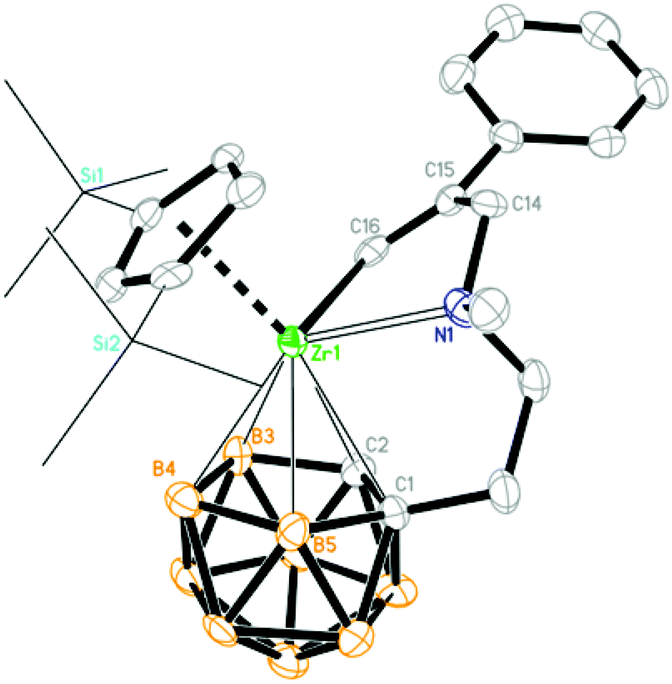
| | Fig. 5 Molecular structure of [η1:σ:η5-{MeN[CH2(Ph)CCH]CH2CH2}C2B9H10]Zr(η5-Cp′′) (3b) (two TMS groups are represented by lines for clarity). | |
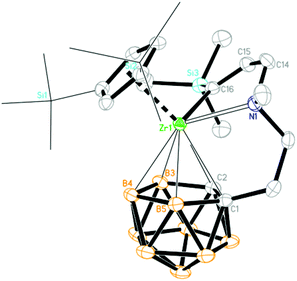 |
| | Fig. 6 Molecular structure of [η1:σ:η5-{MeN[CH2CHC(TMS)]CH2CH2}C2B9H10]Zr(η5-Cp′′) (3c) (two TMS groups of Cp′′ are represented by lines for clarity). | |
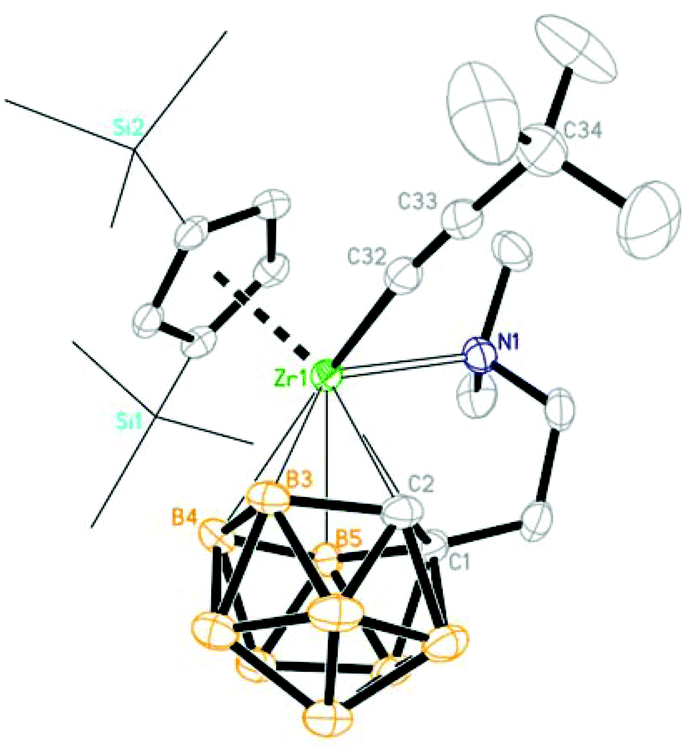
In the solid-state structure of 4, the Zr atom is η5-bound to both the Cp′′ ring and dicarbollyl ligand, σ-bound to the sp-C atom and coordinated to the sidearm nitrogen atom in a distorted tetrahedral geometry. The average Zr–C(C5 ring)/Zr–cage atom distances and the Cent(C5 ring)–Zr–Cent(C2B3) angle of 2.539(4) Å/2.560(5) Å/136.8° are similar to those of 2.537(7) Å/2.550(7) Å/138.6° in 3b. The Zr–C(sp) σ bond distance of 2.190(4) Å is similar to that of 2.215 Å in Cp2Zr[η1-CC(CHCH2CH2)]2,21 and 2.227(4)/2.223(5) Å in (η5-C5Me4H)2Zr(η1-CCSiMe3)2.22 The Zr–C(32)–C(33) angle of 172.1(4)° and the C(32)–C(33) distance of 1.240(7) Å clearly suggest that the alkynyl unit is σ-bonded to the Zr atom (Fig. 7).
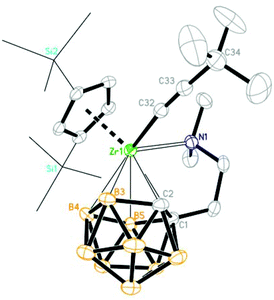 |
| | Fig. 7 Molecular structure of (Cp′′)[η1:η5-(Me2NCH2CH2)C2B9H10]Zr(CCtBu) (4) (two TMS groups are represented by lines for clarity). | |
Conclusions
Treatment of Cp′′ZrMe3 with 7-Me2N(H)CH2CH2-7,8-C2B9H11 afforded a neutral mixed-sandwich zirconacarborane alkyl complex (η5-Cp′′)[η1:σ:η5-{MeN(CH2)CH2CH2}C2B9H10]Zr (1) via methane elimination reaction followed by intramolecular C–H activation. It reacted with internal alkynes RCCR′ and terminal alkynes, such as PhCCH, nBuCCH and TMSCCH, to give the Zr–C σ bond mono-insertion products. The results showed that both electronic and steric factors played a role in controlling the regioselectivity of the insertion process. On the other hand, in sharp contrast to [(C5Me5)(C2B9H11)]ZrCH3 that can catalyze ethylene polymerization in the absence of any co-catalyst,2a,b1 does not react with alkenes although both complexes have a comparable geometry. Such differences may be ascribed to the relatively low Lewis acidity of the Zr atom in 1 because of the nitrogen coordination. It is reasonable to suggest that a soft heteroatom should be used to replace the N atom, thus enhancing Lewis acidity of the metal center. This work should shed some light on the design of new ligands for active yet thermally stable metallacarborane alkyls.
Acknowledgements
This work was supported by grants from the Research Grants Council of Hong Kong (project no. 14306114), National Natural Sciences Foundation of China (no. 21372245 to Z.Q.), and CAS-Croucher Funding Scheme.
Notes and references
- For reviews, see:
(a) U. Rosenthal, P.-M. Pellny, F. G. Kirchbauer and V. V. Burlakov, Acc. Chem. Res., 2000, 33, 119 CrossRef CAS PubMed;
(b) S. Lin and R. M. Waymouth, Acc. Chem. Res., 2002, 35, 765 CrossRef CAS PubMed;
(c) G. Erker, Acc. Chem. Res., 2001, 34, 309 CrossRef CAS PubMed;
(d) Z. Xie, Coord. Chem. Rev., 2006, 250, 259 CrossRef CAS PubMed;
(e)
E. Negishi and J.-L. Montchamp, in Metallocenes – Synthesis, Reactivity, Applications, ed. A. Togni and R. L. Halterman, Wiley-VCH, Weinheim, 1998, vol. 1, pp. 241 Search PubMed;
(f)
A. H. Hoveyda and J. P. Morken, in Metallocenes – Synthesis, Reactivity, Applications, ed. A. Togni and R. L. Halterman, Wiley-VCH, Weinheim, 1998, vol. 2, pp 625 Search PubMed;
(g) K. Angermund, G. Fink, V. R. Jensen and R. Kleinschmidt, Chem. Rev., 2000, 100, 1457 CrossRef CAS PubMed;
(h) H. G. Alt and A. Köppl, Chem. Rev., 2000, 100, 1205 CrossRef CAS PubMed;
(i) R. H. Grubbs and G. W. Coates, Acc. Chem. Res., 1996, 29, 85 CrossRef CAS;
(j) G. W. Coates, Chem. Rev., 2000, 100, 1223 CrossRef CAS PubMed;
(k) B. Wang, Coord. Chem. Rev., 2006, 250, 242 CrossRef CAS PubMed;
(l) L. Resconi, L. Cavallo, A. Fait and F. Piemontesi, Chem. Rev., 2000, 100, 1253 CrossRef CAS PubMed;
(m) R. L. Halterman, Chem. Rev., 1992, 92, 965 CrossRef CAS.
-
(a) D. J. Crowther, N. C. Baenziger and R. F. Jordan, J. Am. Chem. Soc., 1991, 113, 1455 CrossRef CAS;
(b) C. Kreuder and R. F. Jordan, Organometallics, 1995, 14, 2993 CrossRef CAS;
(c) D. J. Crowther, D. C. Swenson and R. F. Jordan, J. Am. Chem. Soc., 1995, 117, 10403 CrossRef CAS;
(d) M. Yoshida, D. J. Crowther and R. F. Jordan, Organometallics, 1997, 16, 1349 CrossRef CAS;
(e) M. Yoshida and R. F. Jordan, Organometallics, 1997, 16, 4508 CrossRef CAS;
(f) X. Bei, C. Kreuder, D. C. Swenson and R. F. Jordan, Organometallics, 1998, 17, 1085 CrossRef CAS;
(g) X. Bei Jr., V. G. Young and R. F. Jordan, Organometallics, 2001, 20, 355 CrossRef.
-
(a) Y. Wang, D. Liu, H.-S. Chan and Z. Xie, Organometallics, 2008, 27, 2825 CrossRef CAS;
(b) D. Liu, Y. Wang, H.-S. Chan, Y. Tang and Z. Xie, Organometallics, 2008, 27, 5295 CrossRef CAS;
(c) D. Liu, H. Shen, Y. Wang, Y. Cai and Z. Xie, Chem. – Asian J., 2011, 6, 628 CrossRef CAS PubMed.
-
(a) P. W. N. M. van Leeuwen, P. C. J. Kamer, J. N. H. Reek and P. Dierkes, Chem. Rev., 2000, 100, 274 CrossRef PubMed;
(b) M.-N. Birkholz, Z. Freixa and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2009, 38, 1099 RSC.
-
(a) D.-H. Kim, J. H. Won, S.-J. Kim, J. Ko, S.-H. Kim, S. Cho and S. O. Kang, Organometallics, 2001, 20, 4298 CrossRef CAS;
(b) Y.-J. Lee, J.-D. Lee, J. Ko, S.-H. Kim and S. O. Kang, Chem. Commun., 2003, 1364 RSC;
(c) J.-D. Lee, Y.-J. Lee, K.-C. Son, W.-S. Han, M. Cheong, J. Ko and S. O. Kang, J. Organomet. Chem., 2007, 692, 5403 CrossRef CAS PubMed;
(d) Y. Zhu, Y. Zhong, K. Carpenter, J. A. Maguire and N. S. Hosmane, J. Organomet. Chem., 2005, 690, 2802 CrossRef CAS PubMed;
(e) Y.-J. Lee, J.-D. Lee, H.-J. Jeong, K.-C. Son, J. Ko, M. Cheong and S. O. Kang, Organometallics, 2005, 24, 3008 CrossRef CAS;
(f) J.-D. Lee, Y.-J. Lee, K.-C. Son, M. Cheong, J. Ko and S. O. Kang, Organometallics, 2007, 26, 3374 CrossRef CAS;
(g) M.-S. Cheung, H.-S. Chan and Z. Xie, Organometallics, 2005, 24, 5217 CrossRef CAS;
(h) M. Gao, Y. Tang, M. Xie, C. Qian and Z. Xie, Organometallics, 2006, 25, 2578 CrossRef CAS;
(i) X. Li, K. Mashima and Z. Xie, Chem. Commun., 2013, 49, 9039 RSC.
- For examples:
(a) J. Pflug, A. Bertuleit, G. Kehr, R. Fröhlich and G. Erker, Organometallics, 1999, 18, 3818 CrossRef CAS;
(b) J. Pflug, G. Erker, G. Kehr and R. Fröhlich, Eur. J. Inorg. Chem., 2000, 1795 CrossRef CAS;
(c) N. Marquet, E. Kirillov, T. Roisnel, A. Razavi and J.-F. Carpentier, Organometallics, 2009, 28, 606 CrossRef CAS;
(d) R. F. Munhá, M. A. Antunes, L. G. Alves, L. F. Veiros, M. D. Fryzuk and A. M. Martins, Organometallics, 2010, 29, 3753 CrossRef;
(e) F. G. Schröder, C. Lichtenberg, M. Elfferding and J. Sundermeyer, Organometallics, 2013, 32, 5082 CrossRef.
- M.-S. Cheung, H.-S. Chan and Z. Xie, Dalton Trans., 2005, 2375 RSC.
-
(a) S. L. Buchwald, B. T. Watson, M. W. Wannamaker and J. C. Dewadd, J. Am. Chem. Soc., 1989, 111, 4486 CrossRef CAS;
(b) J. Pflug, A. Bertuleit, G. Kehr, R. Fröhlich and G. Erker, Organometallics, 1999, 18, 3818 CrossRef CAS;
(c) J. Pflug, G. Erker, G. Kehr and R. Fröhlich, Eur. J. Inorg. Chem., 2000, 1795 CrossRef CAS;
(d) L. Rocchigiani, G. Bellachioma, G. Ciancaleoni, A. Macchioni, D. Zuccaccia and C. Zuccaccia, Organometallics, 2011, 30, 100 CrossRef CAS.
-
(a) C. Kreuder, R. F. Jordan and H. Zhang, Organometallics, 1995, 14, 2993 CrossRef CAS;
(b) X. Bei, V. G. Young and R. F. Jordan, Organometallics, 2001, 20, 355 CrossRef CAS.
-
Y. V. Kissin, Alkene Polymerization Reactions with Transition Metal Catalysts, Elsevier, Amsterdam, 2008, ch. 4, p. 207 Search PubMed.
-
(a) M. A. Bennett, S. A. Macgregor and E. Wenger, Helv. Chim. Acta, 2001, 84, 3084 CrossRef CAS;
(b) L. Deng, H.-S. Chan and Z. Xie, J. Am. Chem. Soc., 2006, 128, 7728 CrossRef CAS PubMed;
(c) Z. Qiu and Z. Xie, J. Am. Chem. Soc., 2009, 131, 2084 CrossRef CAS PubMed;
(d) Z. Qiu and Z. Xie, J. Am. Chem. Soc., 2010, 132, 16085 CrossRef CAS PubMed.
-
(a)
E. W. Colvin, Silicon Reagents in Organic synthesis, Butterworths, London, 1981 Search PubMed;
(b) T. G. Traylor, W. Hanstein, H. J. Berwin, N. A. Clinton and R. S. Brown, J. Am. Chem. Soc., 1971, 93, 5715 CrossRef CAS;
(c) J. B. Lambert, G. Wang, R. B. Finzel and D. H. Teramura, J. Am. Chem. Soc., 1987, 109, 7838 CrossRef CAS;
(d) M. R. Ibrahim and W. L. Jorgensen, J. Am. Chem. Soc., 1989, 111, 819 CrossRef CAS;
(e) S. G. Wierschke, J. Chandrasekhar and W. L. Jorgensen, J. Am. Chem. Soc., 1985, 107, 1496 CrossRef CAS;
(f) N. Koga and K. Morokuma, J. Am. Chem. Soc., 1988, 110, 108 CrossRef CAS;
(g) A. S. Guram and R. F. Jordan, Organometallics, 1990, 9, 2190 CrossRef CAS;
(h) A. S. Guram, R. F. Jordan and D. F. Taylor, J. Am. Chem. Soc., 1991, 113, 1833 CrossRef CAS;
(i) Y. Quan, J. Zhang and Z. Xie, J. Am. Chem. Soc., 2013, 135, 18742 CrossRef CAS PubMed;
(j) Y. Quan, Z. Qiu and Z. Xie, J. Am. Chem. Soc., 2014, 136, 7599 CrossRef CAS PubMed.
-
(a) S. B. Jones and J. L. Peterson, Organometallics, 1985, 4, 966 CrossRef CAS;
(b) G. Erker, R. Zwettler, C. Krueger, I. Hyla-Kryspin and R. Gleiter, Organometallics, 1990, 9, 524 CrossRef CAS;
(c) S. S. H. Mao and T. D. Tilley, J. Am. Chem. Soc., 1995, 117, 7031 CrossRef CAS;
(d) C. L. Hilton and B. T. King, Organometallics, 2006, 25, 4058 CrossRef CAS;
(e) A. D. Miller, J. F. Tannaci, S. A. Johnson, H. Lee, J. L. McBee and T. D. Tilley, J. Am. Chem. Soc., 2009, 131, 4917 CrossRef CAS PubMed.
- M. Bochmann and S. J. Lancaster, Organometallics, 1994, 13, 2235 CrossRef CAS.
- T. J. Woodman, M. Thornton-Pett, D. L. Hughes and M. Bochmann, Organometallics, 2001, 20, 4080 CrossRef CAS.
- P. Bazinet and T. D. Tilley, Organometallics, 2008, 27, 1267 CrossRef CAS.
- D. J. Crowther, N. C. Baenziger and R. F. Jordan, J. Am. Chem. Soc., 1991, 113, 1455 CrossRef CAS.
- J. A. Pool, E. Lobkovsky and P. J. Chirik, Organometallics, 2003, 22, 2797 CrossRef CAS.
- T. V. V. Ramakrishna, S. Lushnikova and P. R. Sharp, Organometallics, 2002, 21, 5685 CrossRef CAS.
-
(a) S. Ren, H.-S. Chan and Z. Xie, Organometallics, 2009, 28, 4106 CrossRef CAS;
(b) S. Ren, H.-S. Chan and Z. Xie, J. Am. Chem. Soc., 2009, 131, 3862 CrossRef CAS PubMed.
- G. Dierker, G. Kehr, R. Fröhlich, G. Erker and S. Grimme, Chem. Commun., 2006, 3912 RSC.
- V. Varga, J. Hiller, U. Thewalt, M. Poláške and K. Mach, J. Organomet. Chem., 1998, 553, 15 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, complete characterization data of 1–4, and crystallographic data in the CIF format for 1, 2a, 2c–f, 3b, 3c and 4. CCDC 1041216–1041224. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qi00244j |
|
| This journal is © the Partner Organisations 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 .
.