
Guest dependent reversible single-crystal to single-crystal structural transformation in a flexible Gd(III)-coordination polymer†
Tapan K.
Pal
,
Dinesh
De
,
Subhadip
Neogi
and
Parimal K.
Bharadwaj
*
Department of Chemistry, Indian Institute of Technology, Kanpur 208016, India. E-mail: pkb@iitk.ac.in; Fax: (+91) 512-259-7637; Tel: (+91) 512-259-7034
First published on 12th February 2015
Abstract
The ligand 2,6,2′,6′-tetranitro-biphenyl-4,4′-dicarboxylic acid (H2L) reacts solvothermally with [Gd(NO3)3]·6H2O to produce a flexible and porous metal–organic framework, {[Gd2(L)3(DMF)4]·(4DMF)·(3H2O)}n (1) (DMF = N,N′-dimethylformamide). An X-ray crystallographic study reveals that compound 1 contains a 3D framework structure with two different 1D channels (A and B) that are occupied by solvent DMF and water molecules. Crystals of 1 when kept in a dichloromethane solution of 4-chlorobenzaldedhyde (4-ClPhCHO) afford the daughter product {[Gd2(L)3(DMF)4]·(4-ClPhCHO)·(4DMF)}n (1a), via single-crystal to single-crystal (SC–SC) transformation, where lattice water molecules of channel B are replaced by guest aldehyde molecules. Likewise, exposure of 4-fluorobenzaldehyde (4-FPhCHO) and 4-methylbenzaldehyde (4-MePhCHO) vapors to fresh crystals of 1 afforded two isostructural daughter frameworks, {[Gd(L)1.5(DMF)(H2O)3]·(4-FPhCHO)·(DMF)·(3H2O)}n (1b) and {[Gd(L)1.5(DMF)(H2O)3]·(4-MePhCHO)·(2DMF)·(H2O)}n (1c), respectively. Here, the guest aldehyde molecules occupy both the channels of the framework. Interestingly, the latter transformations exhibit a drastic rearrangement of the framework channels followed by several ‘carboxylate-shift’ processes, and concomitant movement of the water molecules from the cavity to the metal center. Importantly, all the host–guest complexes revert back to the as-synthesized crystal when kept in fresh DMF, rendering the mother framework a flexible and dynamic container for the aromatic aldehydes. All these transformations transpire through an SC–SC fashion under ambient conditions, pointing to the high flexibility of the framework and “guest-responsive fitting” of the channels. All the compounds are characterized by X-ray crystallography, thermogravimetry, elemental analysis, powder X-ray diffraction measurements and infrared spectroscopy.
Introduction
As typical porous materials, most of the studies on metal–organic frameworks (MOFs) have concentrated on the creation of robust frameworks, where particular emphasis is placed on their ability to retain open networks after solvent removal. Nevertheless, high framework stability is essential for many practical applications, including gas storage and separation; flexible MOFs can undergo structural changeovers during the removal or uptake of guest molecules, resulting in highly selective guest inclusion, stepwise guest uptake, gate-opening type adsorption behavior and so on.1 Particularly interesting are single-crystal to single-crystal (SC–SC) transformations2 involving movement of atoms, which provide crystallographic snapshots3 during such dynamic structural changes. Thus, SC–SC transformations should allow a deeper insight into the relationship between the host and the guest molecules, offering ways to fabricate systems with enhanced functionalities and novel applications.4Although a few SC–SC transformations (structural dynamism)5 on metal–organic single crystals, caused by solvent exchange, temperature change or framework distortion, have been reported,6 studies focused on the adsorption and separation of organic molecules, especially using lanthanide frameworks, are relatively rare.7 For most of the frameworks, the major obstacle is associated with the catastrophic failure of single crystallinity during these transformation processes.
Using the preferences of different coordination specifics of metal ions in combination with topology of organic linkers, crystal engineering can be very fruitful8 in the design and construction of porous flexible frameworks. To our perception, lanthanide (Ln) metal ions with their high and variable coordination numbers and different coordination geometries should be ideal for the fabrication of flexible MOFs. On the other hand, our interest in the ligand H2L (see Scheme 1) is mainly based on the fact that the presence of four nitro groups on the benzene ring creates steric hindrance, allowing the formation of a non-interpenetrating 3D structure with sufficient pore size to host small organic molecule(s). Besides, it may afford observation of the carboxylate coordination mode and its changes, termed as a “carboxylate shift”, which is a low-energy process and allows structural flexibility to influence the overall structure.9 Earlier, we used this ligand to construct flexible frameworks, which showed direct crystallographic observation of catalytic reactions inside the pores.10 Herein, we report the SC–SC encapsulation of various aromatic aldehydes inside the pores of the flexible Gd(III) framework (1), having two different types of channels (channels A and B). While 4-chlorobenzaldedhyde as the guest preferentially occupies channel B without substantial changes in the overall structure, encapsulation of 4-fluorobenzaldehyde and/or 4-methylbenzaldehyde as guests in 1 involves drastic changes in the framework channels, along with concomitant movement of the lattice water molecules to the metal center. Given the importance of particularly desirable SC–SC transformations in dynamic frameworks, the understanding of such non-coordinating guest-driven structural transitions followed by molecular motion can provide better insight to fabricate advanced molecular devices with the possibility of discriminating explosive or toxic organics. Furthermore, all the guest aldehydes can be recovered by soaking 1 in fresh DMF, whereupon the daughter frameworks revert back to the mother framework, again in an SC–SC manner, rendering the host framework a dynamic and recoverable container11 for the aromatic aldehydes.
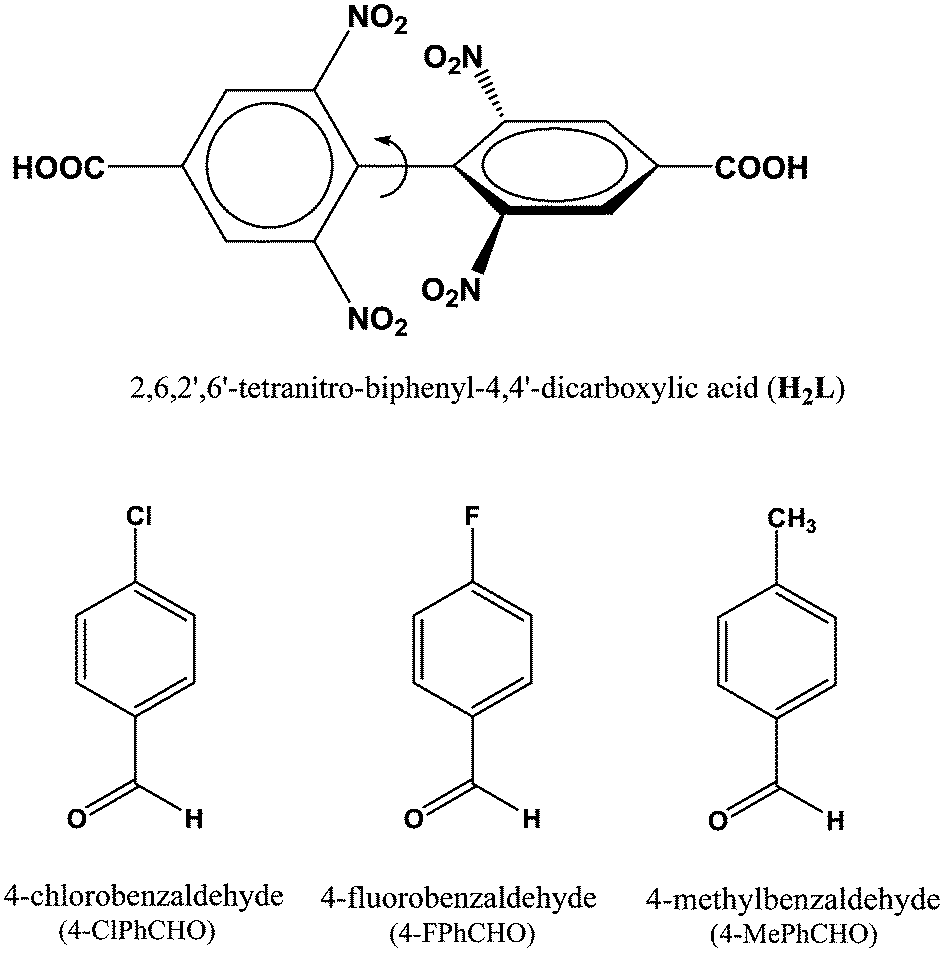 | ||
| Scheme 1 Schematic diagram of H2L18 and guest aldehyde molecules used in the study. | ||
Experimental section
Materials
Reagent-grade 4-chloro-3,5-dinitrobenzoic acid (97%), Gd(NO3)3·6H2O (99.99%), copper powder (electrolytic grade), 4-fluorobenzaldehyde, 4-chlorobenzaldehyde and 4-methylbenzaldehyde were acquired from Sigma-Aldrich and used as received. All solvents, such as DMF, benzene, dichloromethane and ethanol, were procured from S. D. Fine Chemicals, India. These solvents were purified following a standard method prior to use.Physical measurements
Infrared (IR) spectra were recorded (KBr disk, 400–4000 cm−1) on a Perkin-Elmer model 1320 spectrometer. Powder X-ray diffraction spectra (CuKα radiation, scan rate 3° min−1, 293 K) were obtained on a Bruker D8 Advance Series 2 powder X-ray diffractometer. Thermogravimetric analyses (TGA) were acquired on a Mettler Toledo Star system (heating rate, 5 °C min−1). Microanalyses of all the compounds were carried out using a CE-440 elemental analyzer (Exeter Analytical Inc.). 1H NMR and 13C NMR spectra were recorded on a JEOL-ECX 500 FT (500 and 125 MHz respectively) instrument in CDCl3 or in DMSO-d6 with Me4Si as the internal standard. ESI mass spectra were recorded on a Waters Q-TOF Premier mass spectrometer.Single-crystal X-ray studies
Single-crystal X-ray data of compound 1 and the daughter compounds, 1a–1c, were collected at 100 K on a Bruker Smart Apex CCD diffractometer using graphite monochromated MoKα radiation (λ = 0.71073 Å). The linear absorption coefficients, scattering factors for the atoms, and the anomalous dispersion corrections were taken from the International Tables for X-ray Crystallography.12 The data integration and reduction were carried out with SAINT13 software. Empirical absorption correction was applied to the collected reflections with SADABS14 and the space group was determined using XPREP.15 The structure was solved by direct methods using SHELXL-9716 and refined on F2 by full-matrix least-squares using the SHELXL-9717 program package. In compound 1, all the non-hydrogen atoms were refined anisotropically. In compound 1a, the atom C(74) was refined isotropically and all other non-hydrogen atoms were refined anisotropically. In compound 1b, atoms C(26), C(32) and F(1) were refined isotropically and all other non-hydrogen atoms were refined anisotropically. In compound 1c, atoms C(34), C(35), C(37), C(40), C(41), N(8), N(9) and OW4 were refined isotropically and all other non-hydrogen atoms were refined anisotropically. The hydrogen atoms of the coordinated water molecules and cavity water molecules in compound 1b and compound 1c could not be located by difference Fourier synthesis. The hydrogen atoms attached to carbon atoms were positioned geometrically and treated as riding atoms using SHELXL default parameters. Several DFIX commands were used to fix the bond distances in compounds 1a–1c. The amounts of guest molecules present in compounds 1a, 1b and 1c were also confirmed by thermogravimetric analysis (TGA) calculations. In compound 1a, two large residual electron density peaks (2.14 and 2.12 e Å−3) are present that are 0.835 and 0.914 Å away from the Gd(1) and Gd(2) respectively. In compound 1b, two large residual electron density peaks (2.68 and 2.38 e Å−3) are present that are 1.01 and 1.09 Å away from the Gd(1) respectively. These are likely due to inefficient absorption correction for Gd(III). Selected bond distances and bond angles are given in Table S1 (ESI†). Details of crystal parameters of all the compounds, data collection and refinement for the compounds are summarized in Table S2 (ESI†).Results and discussion
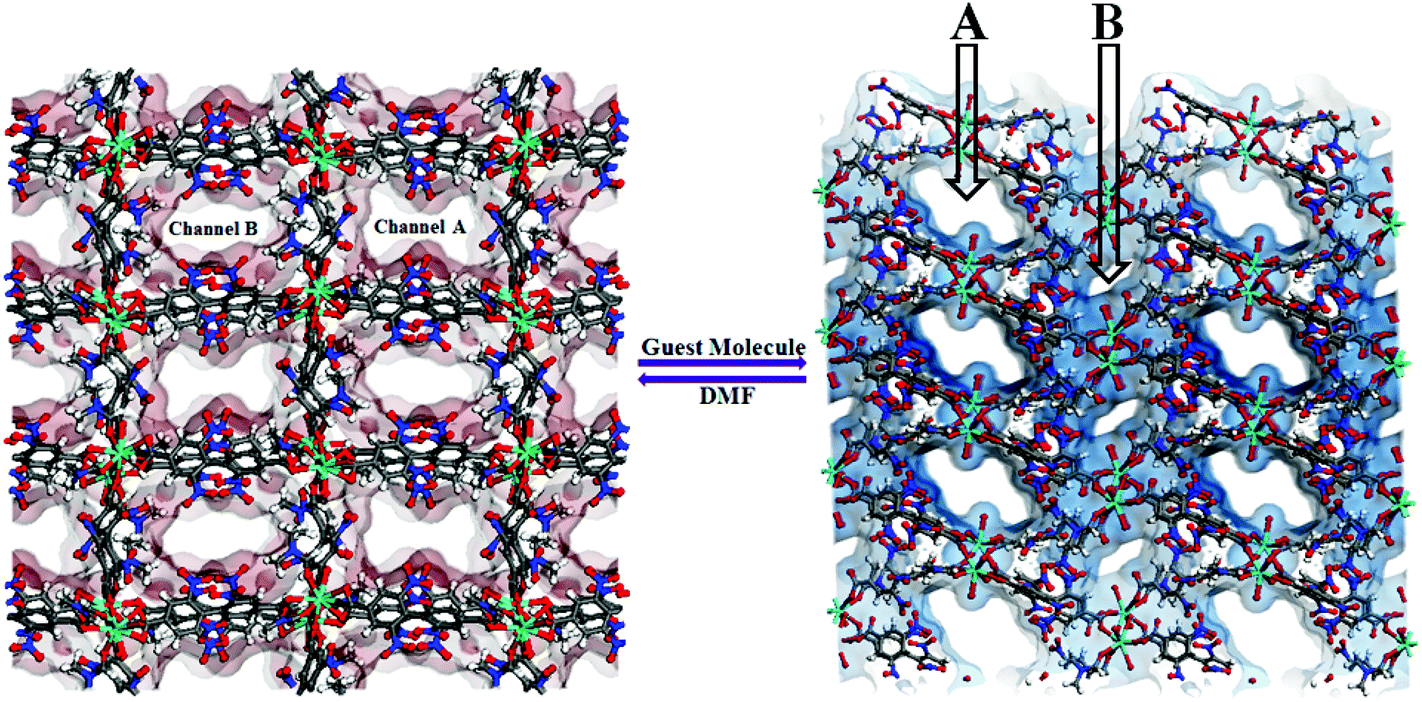
The framework {[Gd2(L)3·(DMF)4]·(4DMF)·(3H2O)}n (1) is solvothermally synthesized in high yield by treating Gd(NO3)3·6H2O and the ligand H2L (Scheme 1), as reported earlier.10 Once isolated, the compound is insoluble in common organic solvents as well as in water. The IR stretching frequency of compound 1 divulges (Fig. S1, ESI†) a broad band at 3379 cm−1 besides strong peaks at 1652 and 1351–1537 cm−1, indicating the presence of water, DMF and coordinated carboxylates, respectively. A single crystal X-ray study reveals that 1 crystallizes in the monoclinic space group P21/n. The structure of 1 consists of a dimeric Gd2 (M⋯M = 4.0481(11) Å) secondary building unit (SBU), constructed from two syn–syn bridging carboxylates, two terminal chelating carboxylates and two chelating as well as bridging carboxylates from six different L2− units (Fig. 1a). Each Gd(III) ion is further coordinated to two DMF molecules, providing a total of 9-coordination at the metal centre with a distorted tri-capped trigonal prismatic geometry (Fig. 1b). Two such binuclear Gd2 clusters are connected by ligand L2− to form a 3D framework, where distances between dimeric units are 15.84 and 9.94 Å along the crystallographic a-axis (Fig. 2).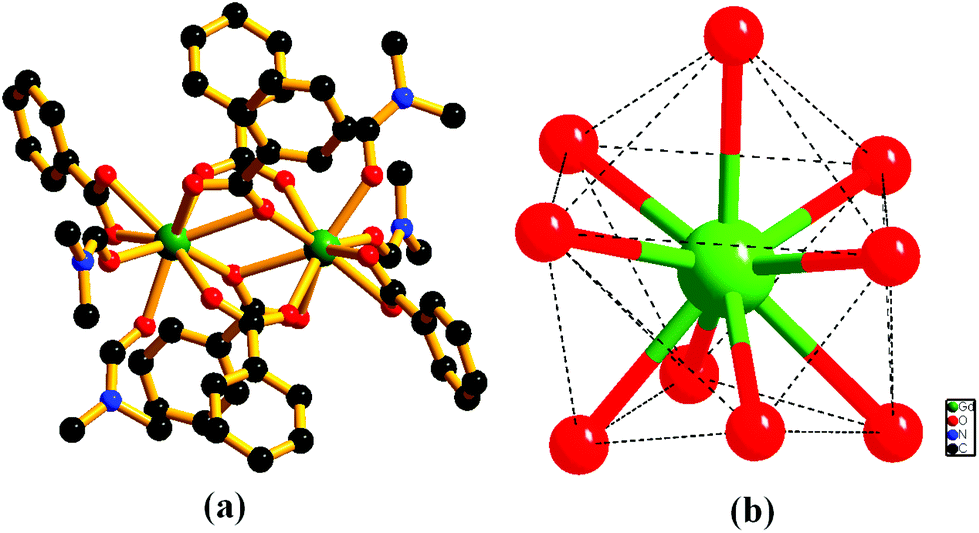 | ||
| Fig. 1 Perspective view of (a) a coordination environment around the Gd2 dimer in compound 1, (b) geometry around the metal center (hydrogen atoms are omitted for clarity). | ||
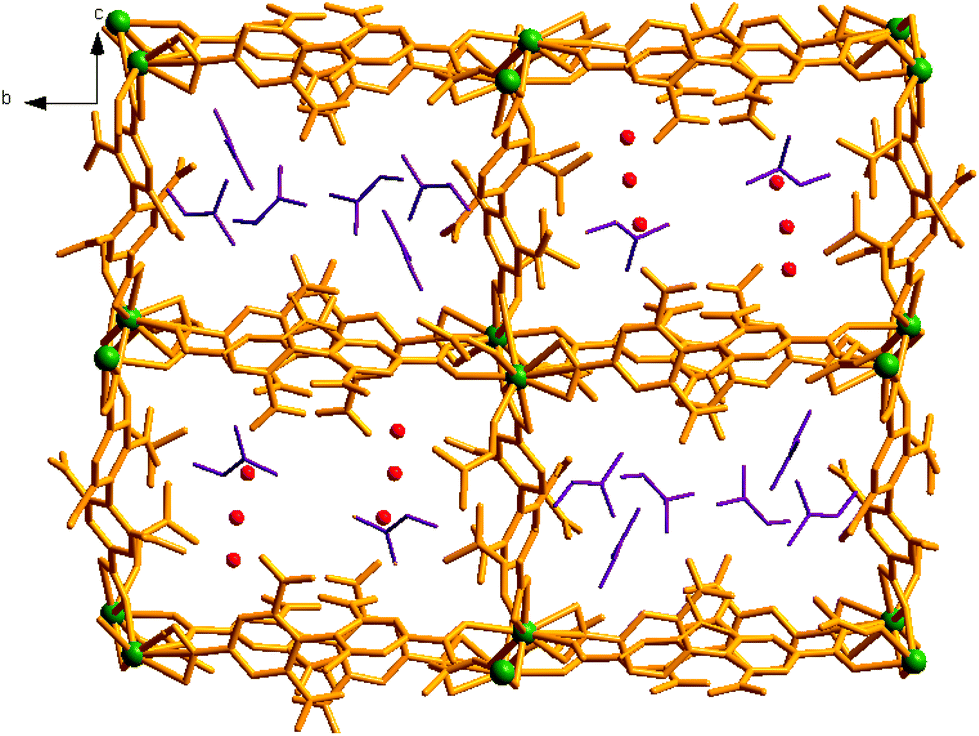 | ||
| Fig. 2 Perspective view of two dissimilar channels A and B in compound 1, hosting different solvent guests. | ||
View along the a-axis reveals (Fig. 2) that the 3D structure contains two different channels with dimensions of 3.33 × 8.65 Å2 (channel A) and 2.40 × 8.24 Å2 (channel B, channel sizes are measured by taking atom-to-atom distances considering the van der Waals radii of constituting atoms). Owing to the different disposition of nitro groups, attached to the L2− ligand unit, these two channels possess different electronic environments, which also allow guest molecules to rest inside the channels in diverse ways. Thus, channel A is occupied by six DMF molecules, while channel B includes two DMF and six water molecules (Fig. 2). The –NO2 groups are directed toward the centre of each channel and are involved in H-bonding interactions (3.037–3.63 Å) with the solvent molecules. It should be noted that the guest DMF and aldehyde molecules residing inside the metal–organic framework cavity are disordered in nature, while inherent disorder of the nitro groups in the L2− ligand unit has been realized earlier.10
Hydrogen bonding interactions (2.66–2.82 Å) also exist between the methyl hydrogen atom of the lattice DMF and the carboxylate oxygen atom of the nearby ligand unit. In addition, C–H⋯N interactions are present between the solvent molecules. Details of all these interactions (Fig. 3) are summarized in Table S3 (ESI†). The phase purity of the bulk material is confirmed by the similarity of the powder X-ray diffraction pattern of 1 to that of the simulated pattern, obtained from the single-crystal data (Fig. S2, ESI†).
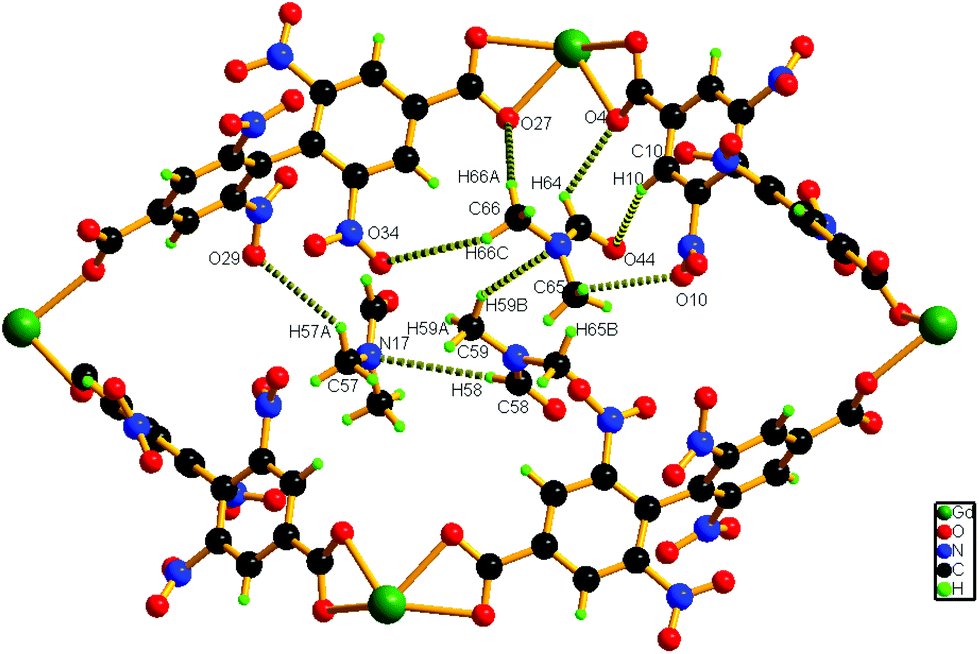 | ||
| Fig. 3 Schematic representation of the interaction of solvent molecules inside a pore of compound 1. | ||
Reversible single-crystal to single-crystal transformation of 1 to 1a
Keeping our previous findings in mind, we decided to examine the utilization of the framework 1 in hosting different aromatic aldehydes. As a starting point to elaborate the SC–SC transformation, we evaluated the encapsulation scenario of the 4-chlorobenzaldehyde molecule. A crystal of 1 was kept in a DCM solution of 4-chlorobenzaldehyde (Scheme 1) at room temperature for 5 days. Single crystal X-ray analysis reveals that the encapsulated water molecules in channel B are completely replaced by 4-chlorobenzaldehyde molecules to afford the daughter framework, {[Gd2(L)3(DMF)4]·(4-ClPhCHO)·(4DMF)}n (1a) (Fig. 4). Structural investigation also shows that the overall framework, including carboxylate connectivities to the Gd2 SBU, remains the same (vide supra) as that of 1.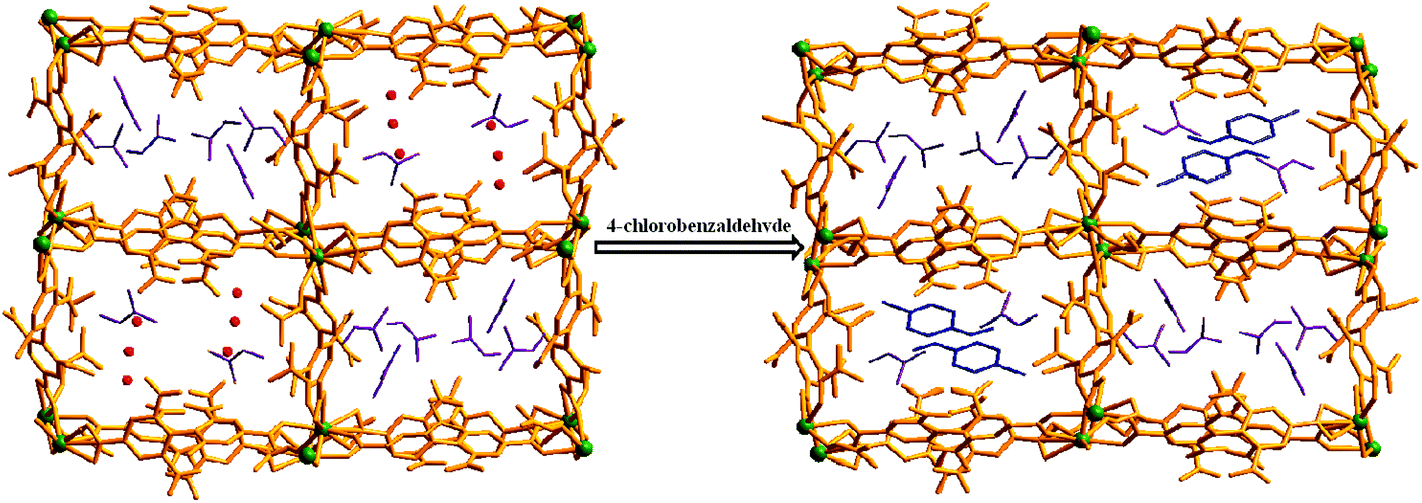 | ||
| Fig. 4 Single-crystal to single-crystal guest-exchange process in compound 1 (hydrogen atoms have been removed for clarity). | ||
A close inspection of the structure of 1a depicts that the phenyl rings in L2− are rotated, which causes an expansion of channel B (to 3.145 × 8.735 Å2) and facilitates accommodation of the guest 4-chlorobenzaldehyde molecules approximately in the middle of the channel (Fig. 4). Several weak host–guest interactions are observed in 1a (Table S4, ESI†), which include: (a) C–H⋯O interactions (2.6176–3.182 Å) between the methyl H atom of the coordinated DMF molecule and the O (–CHO) atom; (b) interactions between the O (–NO2) atom and the H (–CHO) atom, (c) C–H⋯Cl interactions (3.233 Å) between the Cl atom of the guest aldehyde and the H atom of the carboxylate ligand. All these interactions (Fig. 5) assist in stabilizing the 4-chlorobenzaldehyde guests inside the framework. Similar to the mother framework, here also, the DMF solvent and aldehyde guest molecules are disordered. The IR spectrum of 1a exhibits (Fig. S3, ESI†) a sharp peak at 1700 cm−1, corresponding to the aldehyde stretching vibration. The bulk phase purity of 1a is confirmed by the similarity of the PXRD pattern to that of the simulated pattern (Fig. S4, ESI†).
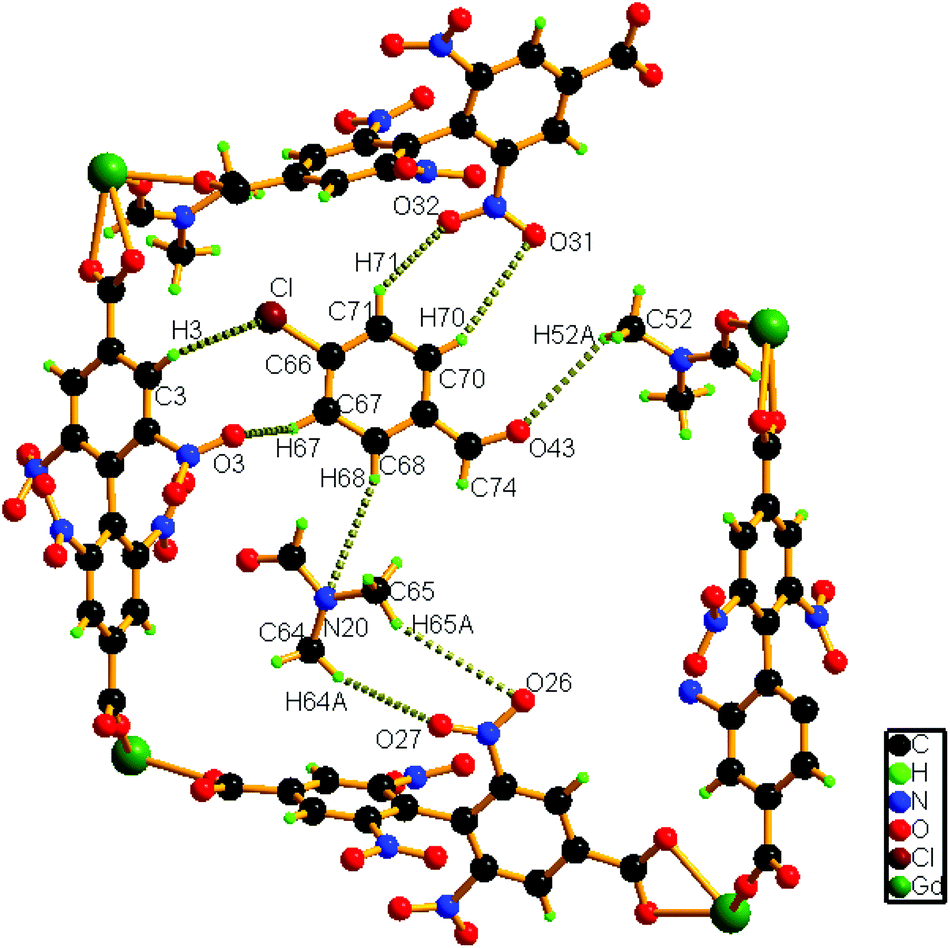 | ||
| Fig. 5 Schematic representation of the interactions of the guest 4-chlorobenzaldehyde molecule with the host framework as well as with solvent molecules in 1a. | ||
Independent synthesis to achieve 1a, by mixing 4-chlorobenzaldehyde with Gd(III) and H2L in an appropriate stoichiometric ratio, proved unsuccessful, indicating that SC–SC transformation is the only route towards its formation. Thermogravimetric analysis of 1a shows a gradual weight loss of 12.8% (calculated, 12.7%) up to 235 °C, accounting for the removal of four lattice DMF guest molecules. Complete decomposition of the framework occurs beyond that temperature (Fig. S5, ESI†), due to expulsion of the coordinated DMF molecule.
With a quest to probe the reversibility of the SC–SC transformation, i.e. whether the as-synthesized framework can be regenerated from its guest-loaded framework, the crystal of 1a was dipped in a DMF solution for 4 days. To our delight, the cell parameters from single crystal X-ray diffraction data exactly matched with those of 1, affording a structure identical to that of the mother crystal, corroborating complete reverse transformation. The unaltered crystal morphology during the entire process (Fig. S6, ESI†) confirms that no dissolution of 1a occurs in DMF. In the regenerated framework, the absence of any aldehyde guest is also corroborated by the lack of a peak at 1700 cm−1 in the IR spectrum (Fig. S7, ESI†). Moreover, the similarity of the PXRD patterns (Fig. S8, ESI†) of 1 and DMF soaked 1a supports the bulk phase transformation.
Reversible single-crystal to single-crystal transformation of 1 to 1b or 1c
Encouraged by the successful entrapment of the 4-chlorobenzaldehyde guest by 1, we further checked its potential utility in hosting 4-fluorobenzaldehyde and 4-methylbenzaldehyde guests (Scheme 1). Crystals of 1 do not show any loss of single crystallinity when kept separately in vapors of the abovementioned aldehydes at room temperature for 5 days. The possibility of dissolution of 1 in aldehyde vapor, followed by recrystallization at the surface and growth of a new phase, is excluded by photographs of the mother crystals (Fig. S6, ESI†) at different time intervals, which show no change in size, color, morphology and transparency during the entire process. Individual single crystal-X-ray diffraction measurements reveal that although the crystal systems remain unaltered, the space group changes to C2/c (Table S2, ESI†), producing two new daughter frameworks {[Gd(L)1.5(DMF)(H2O)3]·(4-FPhCHO)·(DMF)·(3H2O)}n (1b) (Fig. 6) and {[Gd(L)1.5(DMF)(H2O)3]·(4-MePhCHO)·(2DMF)·(H2O)}n (1c).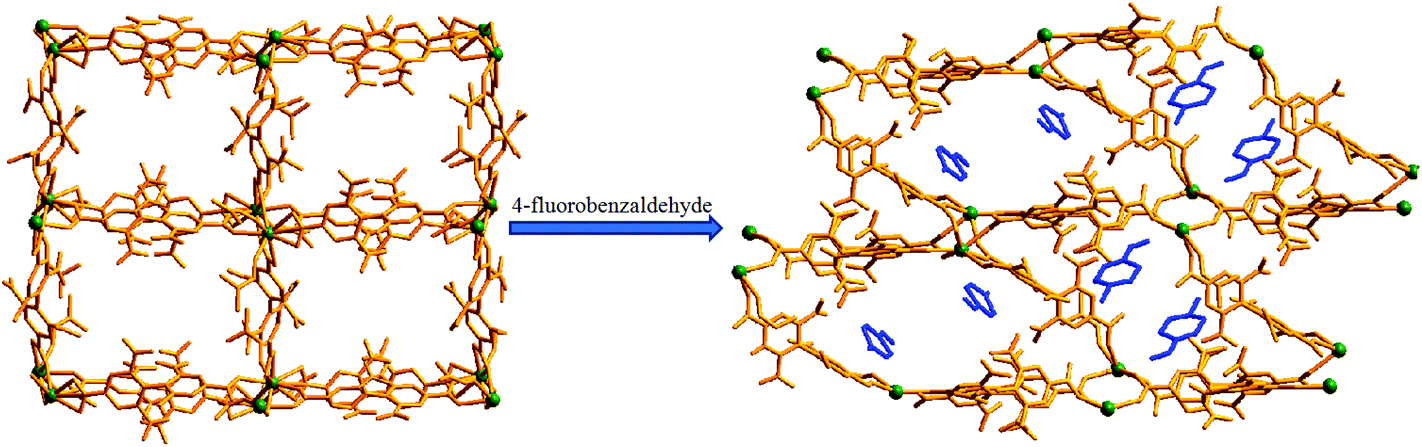 | ||
| Fig. 6 Single-crystal to single-crystal loading of the 4-chlorobenzaldehyde guest in compound 1, forming the daughter compound 1b. A noticeable rearrangement of the framework channels is depicted. DMF and water molecules have been removed for clarity. | ||
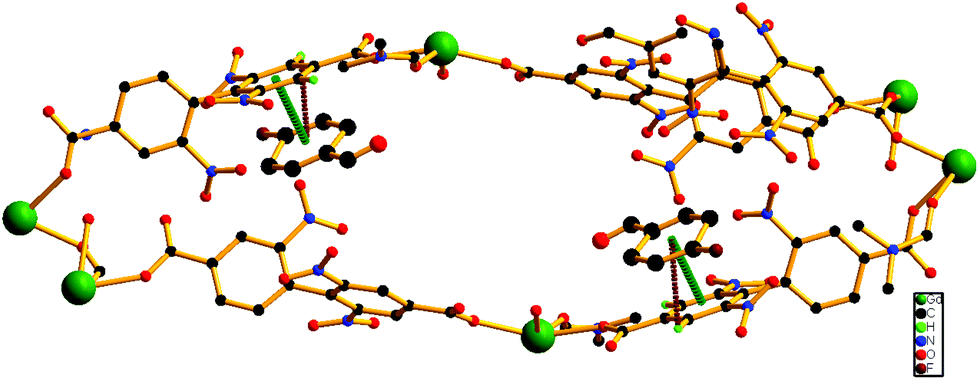
Since 1b and 1c have similar structures, only the structure of the former is described here. The structure of 1b is quite different from its mother framework or 1a, with several bond transformations. To our surprise, the lattice water molecule moves to the metal center. In this modus operandi, the η2-bridging carboxylate in the mother crystal undergoes a pronounced ‘carboxylate shift’ and transforms into mono-dentate η1-carboxylate (Fig. 7). This carboxylate shift also causes a significant increase in the Gd⋯Gd separation from 4.048 (11) Å to 5.031(11) Å (Fig. 7). Moreover, the terminal chelating carboxylate group, attached to each Gd(III) centre in 1 opens up to mono-dentate η1-ligation mode in 1b. Consequently, the coordination number around each metal center of Gd2 SBU diminishes to eight with coordination from two syn carboxylates, two η1-carboxylates, three water molecules and one DMF molecule (Fig. 7).
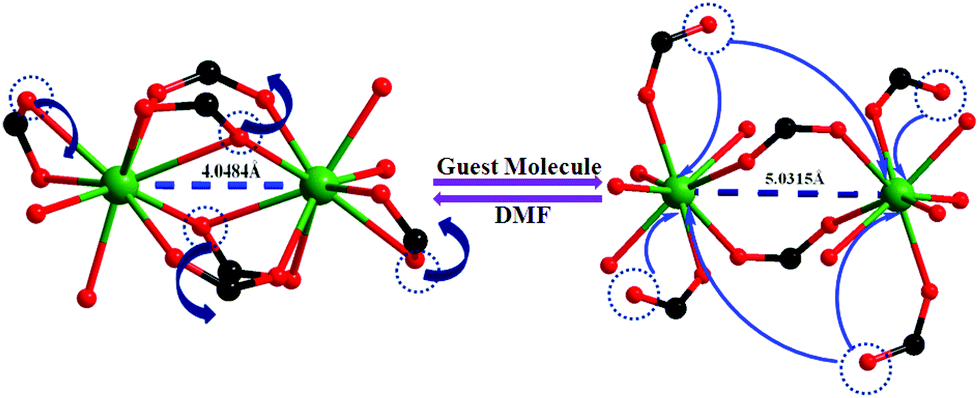 | ||
| Fig. 7 The carboxylate shift process at the metal centre, occurring through a reversible SC–SC transformation from 1 to 1b and vice versa. | ||
It should be pointed that although a number of SC–SC transformations are reported in the literature, those involving changes in the first coordination sphere are comparatively fewer in number.19 Moreover, while rearrangements in the carboxylate coordination modes, i.e. the “carboxylate shift”, are more commonly reported20 in biological systems, this phenomenon is observed21 in only a handful of examples of coordination polymers. The O atoms of dissociated terminal chelating carboxylate, which was earlier bound to the Gd(III) centre in 1, are rotated towards a neighboring guest aldehyde molecule to form intermolecular hydrogen bonds (Fig. S9, ESI†) in 1b. The overall structure of 1b is also unique in the sense that unlike 1 or 1a, the two channels (A and B) undergo substantial distortion during the guest encapsulation process (Fig. 8). Thus, the revised channel dimensions in 1b can be approximated to be 3.40 × 12.19 Å2 (channel A) and 2.95 × 6.60 Å2 (channel B).
 | ||
| Fig. 8 Perspective view of the channel rearrangement during reversible SC–SC transformation from 1 to 1b and vice versa. | ||
As depicted in Fig. 6, unlike in 1a, both the channels here contain aldehyde molecules, where the guest 4-fluorobenzaldehyde molecules are not positioned in the middle of the channels; rather they are slightly shifted towards the edge (Fig. 9), exhibiting C–H⋯π and π⋯π stacking interactions (for detailed interaction, see Fig. S9 and Table S5, ESI†) with the nearby benzene ring of L2−. We speculate that this different positioning of the guest aldehyde molecules in 1b leads to such a pronounced distortion in framework channels.
 | ||
| Fig. 9 Schematic representation of the interaction of the guest 4-fluorobenzaldehyde molecule with the host framework inside the pore of compound 1b. | ||
The other daughter compound 1c has a comparable structure to that of 1b, involving: (a) the coordination environment and the coordination number around each metal center (Fig. S10, ESI†), (b) ligation modes of the carboxylate groups, carboxylate shifts and lattice water movement to the metal center (Fig. S10 and S11, ESI†), (c) rearrangement of the framework channels (Fig. S12, ESI†) and (d) positioning of guest 4-methylbenzaldehyde (Fig. S12, ESI†) molecules inside the channels (for detailed interaction, see Fig. S13 and Table S6, ESI†).
We failed to synthesize 1b or 1c independently by mixing 4-fluorobenzaldehyde or 4-methylbenzaldehyde with Gd(III) and H2L separately in an appropriate stoichiometry ratio, indicating that SC–SC transformation is the only route towards their formation.
The bulk phase purity of both 1b and 1c is corroborated by the concurrence of their individual PXRD patterns (Fig. S14 and S15, ESI†) with those of the simulated patterns, obtained from respective single crystal data. The IR spectra of compounds 1b and 1c exhibit sharp peaks at 1706 cm−1 and 1695 cm−1, respectively (Fig. S16 and S17, ESI†), that correspond to the aldehyde stretching vibration. Thermogravimetric analysis of 1b shows 4.5% (calculated, 4.6%) weight loss at 80 °C due to loss of three water molecules from the cavity. A further 17.1% (calculated, 16.9%) weight loss in the temperature range of 90–205 °C attenuates the loss of one 4-fluorobenzaldehyde molecule and one DMF molecule from the cavity. Beyond that temperature the coordinated DMF molecule is lost and decomposition of the framework occurs (Fig. S18, ESI†). Thermogravimetric analysis of 1c shows a gradual weight loss of 24% (calculated, 23.7%) in the temperature range of 60–240 °C, corresponding to the loss of one water, two DMF and one 4-methylbenzaldehyde molecules from the cavity (Fig. S19, ESI†).
Interestingly, when the daughter crystals 1b and 1c are soaked in DMF for 4 days, the unit cell parameters change to those of the mother crystal 1, giving an identical structure of the mother crystal, corroborating reverse SC–SC transformation. This observation is further shown in IR measurements (Fig. S20 and S21, ESI†), which show no peaks for the aldehyde molecules. The similarity between PXRD patterns of respective DMF soaked daughter crystals and 1 supports the bulk transformation (Fig. S8, ESI†). Thus, the mother framework represents a dynamic porous coordination polymer in the true sense, where guest shape-responsive fitting and guest release inside the channels are realized in a complete single-crystal-to-single-crystal manner. This unique property renders framework 1 a deliverable container for aromatic guest aldehydes under ambient conditions. We further searched for the SC–SC transformation cycles for individual guest loaded frameworks 1a, 1b and 1c. In each case, the PXRD patterns clearly demonstrate that the structural integrity is maintained up to two cycles, after which the framework collapses (Fig. S22–S24, ESI†). However, cross exchange of guest aldehyde molecules in the cavity is not possible between the daughter crystals even after long-term exposure. We were also unsuccessful in loading other aromatic aldehydes like 4-nitrobenzaldehyde, 4-hydroxybenzaldehyde, 4-methoxybenzaldehyde and 4-bromobenzaldehyde inside 1, which reflects a substantial affinity of the host framework to the above discussed aldehydes (Scheme 1) only.
Conclusion
In conclusion, by rationally choosing the flexible ligand H2L, we have synthesized a flexible Gd(III) framework (1), containing two different channels. The lattice water molecules of channel B can be completely replaced by the 4-chlorobenzaldehyde guest to afford the guest encapsulated framework 1avia SC–SC transformation. The overall framework of 1a, including carboxylate connectivities to the Gd(III) centre, and dimensionalities of channels remain the same as that of the mother framework. On the other hand, two new daughter frameworks are formed when crystals of 1 are separately exposed to the vapors of 4-fluorobenzaldehyde (framework 1b) and 4-methylbenzaldehyde (framework 1c). In sharp contrast to 1a, both the later transformations involve several “carboxylate shift” processes, accompanied by substantial changes in the ligand conformation and the metal coordination environment. Additionally, the later SC–SC transformations accompany substantial opening of channel A and closing of channel B. We assume that such a pronounced channel distortion is a consequence of different positioning of the guest aldehyde molecules inside framework voids. The understanding of such a non-coordinating guest-driven, anomalous structural transition, followed by molecular motion, offers ways to explain various dynamic aspects of coordination polymers, besides being potentially used in fabricating molecular devices. More significantly, all the host–guest complexes revert back to the mother crystal (1) in a single-crystal-to-single-crystal manner when kept in DMF, rendering framework 1 a flexible and deliverable container for the aromatic aldehydes. Given the immense importance of realizing the “guest shape-responsive fitting” or dynamic guest accommodation of the voids, the present research paper allows the prospect of trapping even very reactive species that are unstable outside the restricted environment. We are currently working along these lines.Acknowledgements
We gratefully acknowledge the financial support received from the Department of Science and Technology, New Delhi, India (J. C. Bose National Fellowship to PKB) and the SRF from the CSIR to TKP and DD. SN thanks IIT Kanpur for a post-doctoral fellowship.Notes and references
- (a) R. Kitaura, K. Fujimoto, S. Noro, M. Kondo and S. Kitagawa, Angew. Chem., Int. Ed., 2002, 41, 133 CrossRef CAS; (b) C. Janiak, Dalton Trans., 2003, 2781 RSC; (c) R. Kitaura, G. Onoyama, H. Sakamoto, R. Matsuda, S. Noro and S. Kitagawa, Angew. Chem., Int. Ed., 2004, 43, 2684 CrossRef CAS; (d) B. Chen, F. R. Fronczek and A. W. Maverick, Inorg. Chem., 2004, 43, 8209 CrossRef CAS PubMed; (e) P. A. Maggard, B. Yan and J. Luo, Angew. Chem., Int. Ed., 2005, 44, 2 CrossRef PubMed; (f) R. Custelcean and M. G. Gorbunova, J. Am. Chem. Soc., 2005, 127, 16362 CrossRef CAS PubMed; (g) S. H. Cho, B. Ma, S. T. Nguyen, J. T. Hupp and T. E. Albrecht-Schmitt, Chem. Commun., 2006, 2563 RSC; (h) Y. Yoshida, K. Inoue and M. Kurmoo, Inorg. Chem., 2009, 48, 267 CrossRef CAS PubMed; (i) Y. Inokuma, T. Arai and M. Fujita, Nat. Chem., 2010, 2, 780 CrossRef CAS PubMed; (j) M. J. Manos, E. J. Kyprianidou, G. S. Papaefstathiou and A. J. Tasiopoulos, Inorg. Chem., 2012, 51, 6308 CrossRef CAS PubMed.
- (a) S. Kitagawa and K. Uemura, Chem. Soc. Rev., 2005, 34, 109 RSC; (b) D. Bradshaw, J. B. Claridge, E. J. Cussen, T. J. Prior and M. J. Rosseinsky, Acc. Chem. Res., 2005, 38, 273 CrossRef CAS PubMed.
- (a) Y. Inokuma, T. Arai and M. Fujita, Nat. Chem., 2010, 2, 780 CrossRef CAS PubMed; (b) R. Matsuda, R. Kitaura, S. Kitagawa, Y. Kubota, T. C. Kobayashi, S. Horike and M. Takata, J. Am. Chem. Soc., 2004, 126, 14064 Search PubMed.
- (a) D. J. Tranchemontagne, J. L. Mendoza-Cortés, M. O'Keeffe and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1257 RSC; (b) L. Ma, C. Abney and W. Lin, Chem. Soc. Rev., 2009, 38, 1248 RSC; (c) J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450 RSC; (d) M. D. Allendorf, C. A. Bauer, R. K. Bhakta and R. J. T. Houk, Chem. Soc. Rev., 2009, 38, 1330 RSC.
- (a) T. K. Maji, G. Mostafa, R. Matsuda and S. Kitagawa, J. Am. Chem. Soc., 2003, 127, 17152 CrossRef PubMed; (b) K. Hanson, N. Calin, D. Bugaris, M. Scancella and S. C. Sevov, J. Am. Chem. Soc., 2004, 126, 10502 CrossRef CAS PubMed; (c) N. L. Toh, M. Nagarathinam and J. J. Vittal, Angew. Chem., Int. Ed., 2005, 44, 2237 CrossRef CAS PubMed; (d) C.-D. Wu and W. Lin, Angew. Chem., Int. Ed., 2005, 44, 1958 CrossRef CAS PubMed; (e) D. Bradshaw, J. E. Warren and M. J. Rosseinsky, Science, 2007, 315, 977 CrossRef CAS PubMed; (f) T. Haneda, M. Kawano, T. Kawamichi and M. Fujita, J. Am. Chem. Soc., 2008, 130, 1578 CrossRef CAS PubMed; (g) S. Sen, S. Neogi, K. Rissanen and P. K. Bharadwaj, Chem. Commun., 2015, 51, 3173 RSC.
- (a) E. Y. Lee and M. P. Suh, Angew. Chem., Int. Ed., 2004, 43, 2798 CrossRef CAS PubMed; (b) C.-L. Chen, A. M. Goforth, M. D. Smith, C.-Y. Su and H.-C. zur Loye, Angew. Chem., Int. Ed., 2005, 44, 6673 CrossRef CAS PubMed; (c) Q. Chu, D. C. Swenson and L. R. MacGillivray, Angew. Chem., Int. Ed., 2005, 44, 3569 CrossRef CAS PubMed; (d) J.-P. Ma, Y.-B. Dong, R.-Q. Huang, M. D. Smith and C.-Y. Su, Inorg. Chem., 2005, 44, 6143 CrossRef CAS PubMed; (e) J. Y. Lee, S. Y. Lee, W. Sim, K.-M. Park, J. Kim and S. S. Lee, J. Am. Chem. Soc., 2008, 130, 6902 CrossRef CAS PubMed; (f) Z. Wang and S. M. Cohen, J. Am. Chem. Soc., 2007, 129, 12368 CrossRef CAS PubMed.
- (a) Y.-B. Dong, Q. Zhang, L.-L. Liu, J.-P. Ma, B. Tang and R.-Q. Huang, J. Am. Chem. Soc., 2007, 129, 1514 CrossRef CAS PubMed; (b) Y.-F. Han, W.-G. Jia, Y.-J. Lin and G.-X. Jin, Angew. Chem., Int. Ed., 2009, 34, 6234 CrossRef PubMed.
- (a) B. Moulton and M. Zaworotko, Chem. Rev., 2001, 101, 1629–1658 CrossRef CAS PubMed; (b) O. Delgado-Friedrichs, M. O'Keeffe and O. M. Yaghi, Phys. Chem. Chem. Phys., 2007, 9, 1035 RSC.
- (a) P. N. Turowski, A. Bino and S. J. Lippard, Angew. Chem., Int. Ed. Engl., 1990, 29, 811 CrossRef; (b) R. L. Rardin, A. Bino, P. Poganiuch, W. B. Tolman, S. Liu and S. J. Lippard, Angew. Chem., Int. Ed. Engl., 1990, 29, 812 CrossRef; (c) B. Burger, S. Dechert, C. Grose, S. Demeshko and F. Meyer, Chem. Commun., 2011, 47, 10428 RSC.
- R. K. Das, A. Aijaz, M. K. Sharma, P. Lama and P. K. Bharadwaj, Chem. – Eur. J., 2012, 18, 6866 CrossRef CAS PubMed.
- E. Y. Lee, S. Y. Jang and M. P. Suh, J. Am. Chem. Soc., 2005, 127, 6375 Search PubMed.
- International Tables for X-Ray Crystallography, Kynoch Press, Birmingham, England, 1952, vol. III Search PubMed.
- SAINT+, version 6.02, Bruker AXS, Madison, WI, 1999 Search PubMed.
- G. M. Sheldrick, SADABS, Empirical Absorption Correction Program, University of Gottingen, Gottingen, Germany, 1997 Search PubMed.
- XPREP, 5.1 ed, Siemens Industrial Automation Inc., Madison, WI, 1995 Search PubMed.
- G. M. Sheldrick, SHELXTL Reference Manual, version 5.1, Bruker AXS, Madison, WI, 1997 Search PubMed.
- G. M. Sheldrick, SHELXL-97, Program for Crystal Structure Refinement, University of Gottingen, Gottingen, Germany, 1997 Search PubMed.
- A. T. Nielsen, W. P. Norris, R. L. Atkins and W. R. Vuono, J. Org. Chem., 1983, 48, 1056 CrossRef CAS.
- (a) M. C. Das and P. K. Bharadwaj, J. Am. Chem. Soc., 2009, 131, 10942 CrossRef CAS PubMed; (b) M. C. Das and P. K. Bharadwaj, Chem. – Eur. J., 2010, 16, 5070 CrossRef CAS PubMed; (c) P. K. Allan, B. Xiao, S. J. Teat, J. W. Knight and R. E. Morris, J. Am. Chem. Soc., 2010, 132, 3605 CrossRef CAS PubMed; (d) S. Neogi, S. Sen and P. K. Bharadwaj, CrystEngComm, 2013, 9239 RSC.
- (a) R. L. Rardin, W. B. Tolman and S. J. Lippard, New J. Chem., 1991, 15, 417 CAS; (b) D. D. LeCloux, A. M. Barrios, T. J. Mizoguchi and S. J. Lippard, J. Am. Chem. Soc., 1998, 120, 9001 CrossRef CAS; (c) M. Arnold, D. A. Brown, O. Degg, W. Errington, W. Haase, K. Herlihy, T. J. Kemp, H. Nimir and R. Werner, Inorg. Chem., 1998, 37, 2920 CrossRef CAS; (d) S. F. Sousa, P. A. Fernandes and M. J. Ramos, J. Am. Chem. Soc., 2007, 129, 1378 CrossRef CAS PubMed.
- (a) D.-X. Xue, W.-X. Zhang, X.-M. Chen and H.-Z. Wang, Chem. Commun., 2008, 1551 RSC; (b) R. Singh, J. Mrozinski and P. K. Bharadwaj, Cryst. Growth Des., 2014, 14, 3623 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Several spectroscopic data and thermogravimetric analyses, powder X-ray diffraction of all the compounds and photographs of all the single crystals. X-ray crystallographic data for 1, 1a, 1b and 1c in CIF format. CCDC 843413 and 1032264–1032266. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qi00193a |
| This journal is © the Partner Organisations 2015 |