
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhosphate binding by a novel Zn(II) complex featuring a trans-1,2-diaminocyclohexane ligand. Effective anion recognition in water†
Oscar
Francesconi
a,
Matteo
Gentili
a,
Francesco
Bartoli
a,
Andrea
Bencini
*a,
Luca
Conti
a,
Claudia
Giorgi
a and
Stefano
Roelens
*b
aDipartimento di Chimica “Ugo Schiff”, Università di Firenze, Polo Scientifico e Tecnologico, I-50019 Sesto Fiorentino, Firenze, Italy. E-mail: andrea.bencini@unifi.it; Tel: +39 0554573371
bCNR, Istituto di Metodologie Chimiche (IMC), Università di Firenze, Polo Scientifico e Tecnologico, I-50019 Sesto Fiorentino, Firenze, Italy. E-mail: stefano.roelens@unifi.it; Tel: +39 0554573546
First published on 3rd December 2014
Abstract
In this work we have investigated the binding properties of a new synthetic receptor for phosphate anions that combines metal ion coordination with electrostatic and H-bonding interactions. The described receptor is obtained by assembling an iminodiacetic (IDA) fragment, as a Zn(II) binding site, with a polyamine macrocyclic portion containing two trans-1,2-diaminocyclohexane (DAC) units and a pyrrole ring, as a cationic binding site, into an adaptive structure appropriately spanning the length of di- and tridentate phosphates. Potentiometric measurements together with 1H and 31P NMR investigation showed that, in a wide pH range including values of physiological interest, the Zn(II) complex of the receptor binds di- and triphosphates, such as ADP, ATP, pyrophosphate (PP) and triphosphate (TP), far better than monophosphate (MP), and that TP is poorly bound by methyliminodiacetate (MIDA) as a model for the Zn(II) binding site. Besides the excellent selectivity over other phosphates, the affinity for TP is the largest reported to date for Zn(II) complexes in water.
Introduction
There is intense interest in the development of molecular systems capable of binding phosphate anions, due to their relevance in areas as diverse as biology, medicine, catalysis and environment.1,2 For many applications, particularly in the areas of biology and medicine, artificial receptors must be able to strongly bind anions in aqueous media. When compared to metal ions, anionic species, including inorganic nucleotide phosphates, show larger sizes, a variety of shapes, higher hydration energies, a wide scale of hydrophobicity and, in some cases, a limited pH range of existence, due to their propensity to undergo protonation processes in water. All these features complicate enormously the design of abiotic receptors for the selective recognition of anionic species.3–19 So far, two main approaches have been followed to obtain effective receptors for phosphate binding in water: (i) development of metal-free polyammonium cations interacting with phosphate anions via multiple charge–charge and H-bonding interactions; (ii) synthesis of metal complexes, mostly transition metal complexes, in which the metal ion is the anchoring point for anionic species. In the first strategy, the binding ability must rely upon weak intermolecular forces like H-bonding, π-stacking interactions, electrostatic interactions, and hydrophobic effects, cooperatively competing with water; in most cases, the selectivity is determined mainly by charge–charge and H-bonding interactions via topological complementarity.3–10 The second strategy for anion recognition takes advantage of coordination to one or two metal ions bearing vacant coordination sites for anionic guest binding. While a number of different metal ions have been used, including main group, transition, and lanthanide metal ions, one of the most commonly employed for this purpose is Zn(II), particularly when the investigated guests are anions of biological relevance, such as phosphate anions.3–5,10–38 This receptor design is inspired by metalloenzymes, for which phosphates act as substrates or inhibitors by reversibly coordinating to one or more Zn(II) ions in the enzymatic pocket.1 Although these strategies have been widely exploited in recent literature, a synergetic combination of the two approaches is still largely unexplored.In this context, we have synthesized receptor 1 featuring an iminodiacetate (IDA) moiety appended to a polyamine macrocycle containing two trans-1,2-diaminocyclohaexane (DAC) units and a pyrrole ring. Apart from a H-bonding pyrrole unit, this receptor features two potential binding sites for metal cations, the IDA subunit and the DAC-based macrocyclic moiety. Both IDA and DAC are indeed well known chelating agents for metal cations.39 Because DAC is also strongly basic, due to its ability to “chelate” the proton between the two amine groups,39,40 it is expected that the macrocyclic unit will be protonated in a wide pH range, in which coordination of amine to Zn(II) would be inhibited. These characteristics could make receptor 1 capable of binding Zn(II) ions to one or both binding sites depending on the pH of the solution. Even in the presence of the protonated macrocycle, however, Zn(II) mediated bridging of the two binding sites of the receptor by phosphate anions may occur through electrostatic and H-bonding interactions with ammonium ions and cooperatively enhance recognition in a wide range of pH. With this in mind, we have investigated the protonation and Zn(II) binding properties of 1, as well as the binding ability of the resulting Zn(II) complexes toward both inorganic phosphate anions (monophosphate, pyrophosphate and triphosphate, hereafter indicated as MP, PP and TP, respectively, independently from their protonation state) and the ATP and ADP nucleotides.
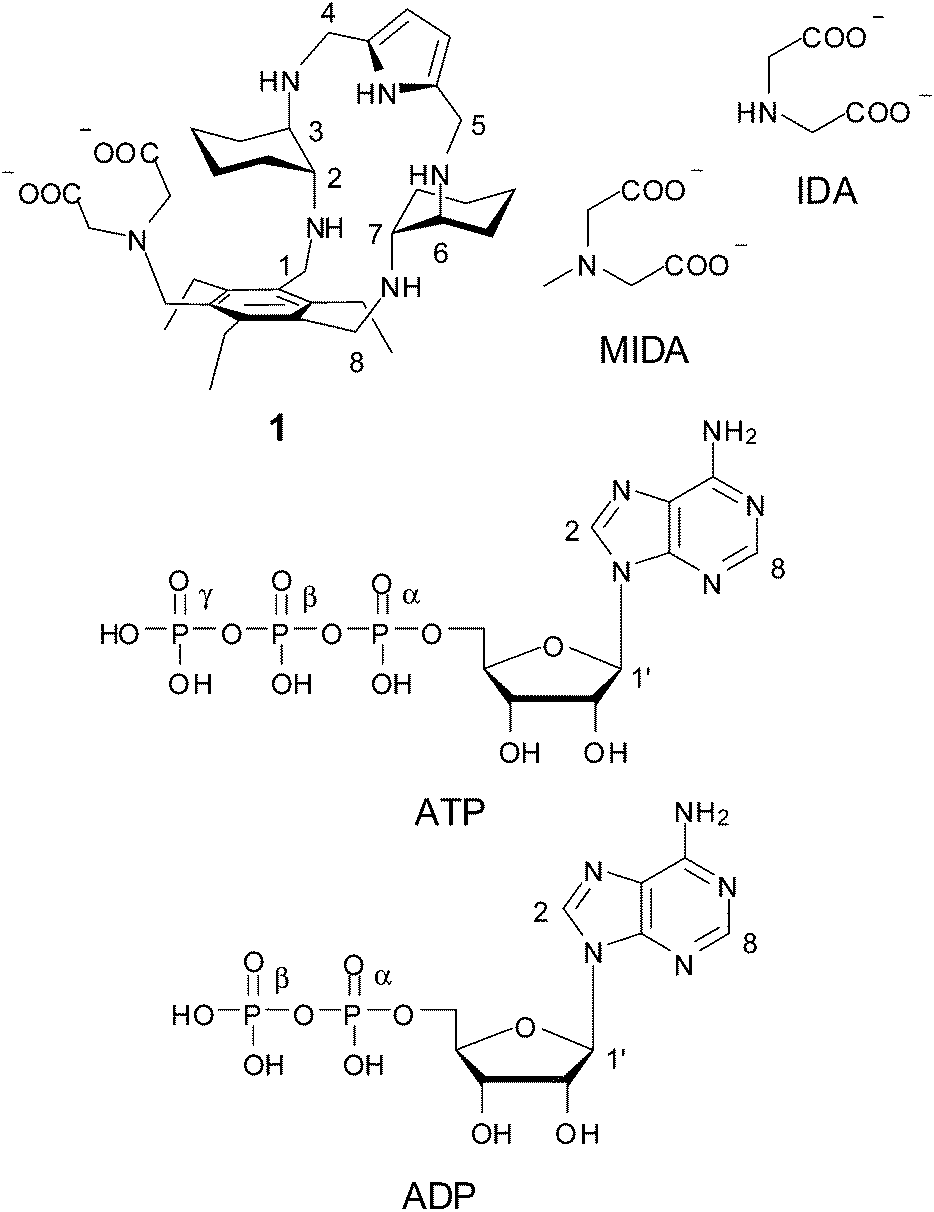
Results and discussion
Synthesis of the receptor
In the course of our molecular recognition studies we have recently described a new family of tripodal aminopyrrolic receptors based on a benzenic scaffold that show significant binding affinity for monosaccharides, and in particular for mannosides, in competitive organic solvents.41,42 This class of synthetic receptors is characterized by three binding arms containing DAC and pyrrole units. With the aim of obtaining a synthetic receptor freely soluble in water, we have appended an IDA group as a third binding arm to the cyclic structure of the two remaining arms of the parent architecture, leading to receptor 1. While testing the binding properties towards carbohydrates in water is still in progress, receptor 1 displays the features of a ditopic receptor for metal cations, through both the IDA moiety and the polyamino macrocycle. The synthesis of receptor 1 is reported in Scheme 1. The dialdehyde 2, prepared according to literature procedures,42 was condensed with neopentylglycol to obtain the acetal-protected compound 3. Reduction of the azido group afforded the amine 4, which was dialkylated to 5 with ethylbromoacetate to introduce the IDA moiety. Acetal deprotection, followed by condensation with mono-BOC-protected (R,R)-trans-1,2-diaminocyclohexane and in situ reduction of the resulting imine, afforded the BOC-protected compound 6. Deprotection with TFA led to the aminic intermediate 7, which was cyclized with 2,5-pyrroledicarbaldehyde and the resulting imine was reduced to the corresponding amine. Finally, alkaline hydrolysis of ethyl esters afforded the desired receptor 1.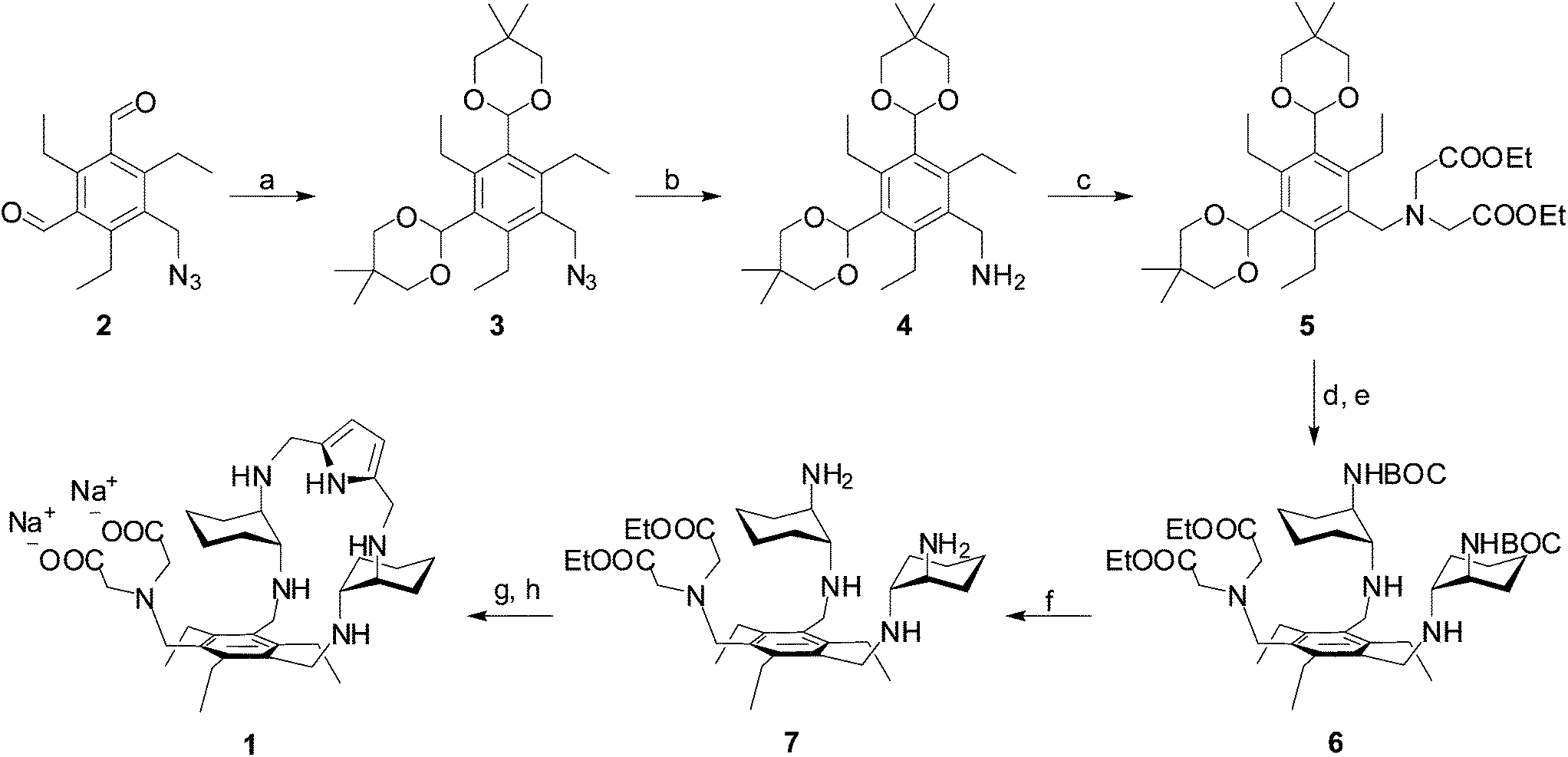 | ||
Scheme 1 Synthesis of receptor 1. Reagents and conditions: (a) neopentylglycol, p-TsOH cat., toluene, reflux, overnight, 87%; (b) LiAlH4, THF, r.t., 2.5 h, 92%; (c) ethyl bromoacetate, KHCO3, DMF, r.t. overnight, 83%; (d) HCl 1 M, THF, overnight; (e) N-BOC-(1R, 2R)-1,2-trans-diaminocyclohexane, CHCl3, reflux, overnight, then NaBH4 suspension in MeOH, r.t., 1 h, 62%; (f) TFA, CH2Cl2, r.t., 2 h, 79%; (g) pyrrole-2,5-dicarbaldehyde, CHCl3, r.t., then NaBH4 suspension in MeOH, r.t., 1 h, 70%; (h) NaOH, H2O/EtOH 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, r.t., overnight, 81%. :1, r.t., overnight, 81%. | ||
Binding measurements
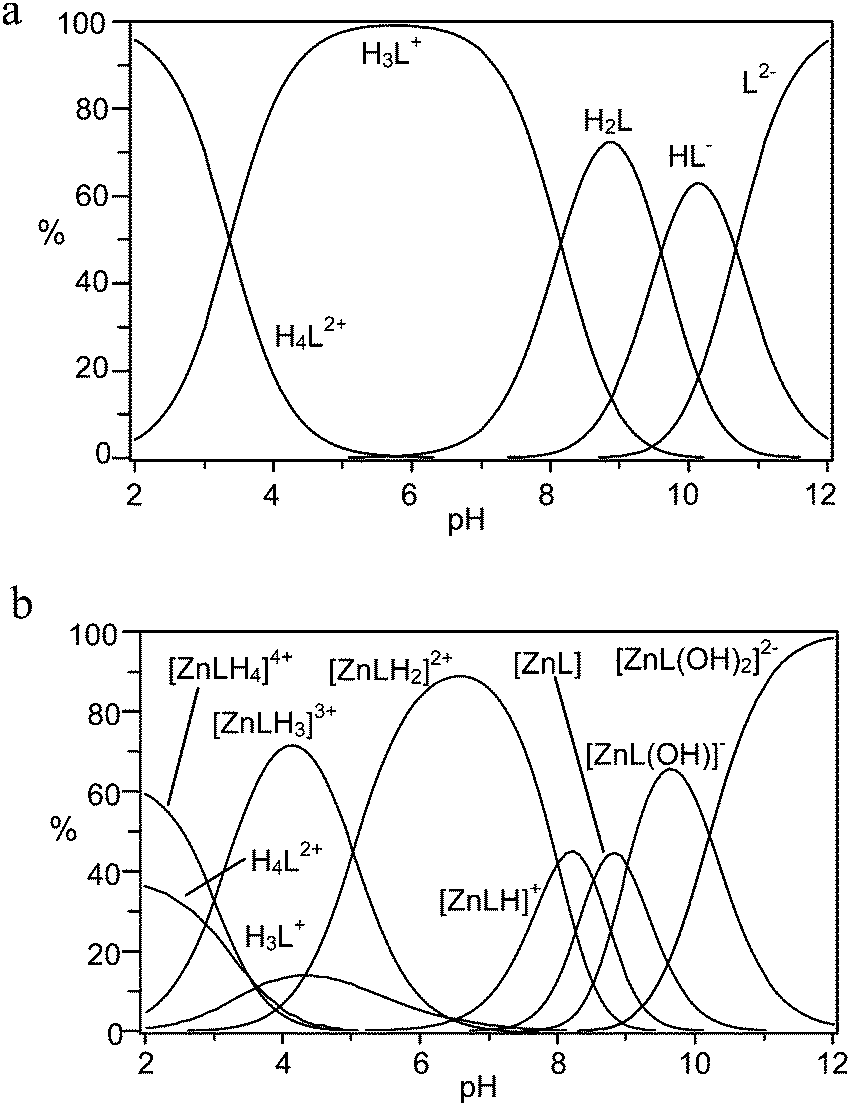 | ||
| Fig. 1 Distribution diagrams of (a) the protonated species, and (b) the Zn(II) complexes formed by receptor 1 (L2−, NMe4Cl, 0.1 M, 308 K). | ||
| Equilibrium | LogK |
|---|---|
| a Measured at 308 K in 0.1 M NMe4Cl. | |
| H+ + L2− = HL− | 10.67(5) |
| H+ + HL− = H2L | 9.60(6) |
| H+ + H2L = H3L+ | 8.15(1) |
| H+ + H3L+ = H4L2+ | 3.37(1) |
| Zn2+ + L2− = [ZnL] | 11.53(3) |
| [ZnL] + H+ = [ZnLH]+ | 8.50 (3) |
| [ZnLH]+ + H+ = [ZnLH2]2+ | 8.24(3) |
| [ZnLH2]2+ + H+ = [ZnLH3]3+ | 6.52(3) |
| [ZnLH3]3+ + H+ = [ZnLH4]4+ | 3.51(8) |
| [ZnL] + OH− = [ZnL(OH)]− | 5.45(2) |
| [ZnL(OH)]− + OH− = [ZnL(OH)2]2− | 3.61(3) |
Concerning protonation of 1, L2− and HL− anionic species are present in the alkaline pH region, as expected for polyamine carboxylate ligands (Fig. 1a).39 The corresponding protonation constants are unusually high, suggesting that the two DAC moieties of the macrocyclic subunit of the receptor are involved in the first two steps. Indeed, the basicity of DAC (logK = 9.96) is higher than that of MIDA (logK = 8.49).39,43 The third protonation step most likely occurs on the tertiary amine of the IDA subunit because the second protonation constant of DAC is significantly smaller (logK = 6.47).39,43 Finally, the last protonation step at acidic pH values can occur either on the DAC unit or on a carboxylate moiety.
Focusing on Zn(II) coordination, despite the presence of two potential binding sites for metal cations, the formation of dinuclear metal complexes was never observed, even in the presence of an excess of Zn(II). Rather, several mononuclear Zn(II) complexes are formed through a number of protonation–deprotonation steps occurring on the ligand or on the metal, respectively (Table 1 and Fig. 1b). Because DAC39 and the fully deprotonated MIDA (Table S2†) form strong complexes with Zn(II), ([Zn(DAC)]2+, logK = 7.74; [Zn(MIDA)], logK = 8.64), one would expect that receptor 1, featuring both binding groups in the structure, would form an exceptionally strong complex. Counterintuitively this is not observed, as the logK value of 11.53 for the formation of the [ZnL] complex is smaller than that could be intuitively anticipated, and even smaller than the values reported for linear or cyclic tetraamines.43 On a closer inspection, however, this result may not be anomalous, when considering that conformational restrictions may hinder coordination of the metal ion to the macrocycle and that competition between the two binding sites for the same ligand may weaken the individual group interactions. As a consequence, the metal ion likely features a low coordination number in the complex and, therefore, its coordination sphere is not fulfilled by receptor donors, leaving ‘free’ binding sites available for exogenous substrate anchoring. This conclusion is confirmed by the formation of hydroxo-complexes ([ZnL(OH)]− and [ZnL(OH)2]2−) at high pH values, showing that the [ZnL] complex can accept additional ligands in its coordination sphere.
The most interesting finding that can be inferred from Table 1 is the marked protonation tendency of the [ZnL] complex, giving [ZnLHx]x+ species, where the first two protonation constants are notably large (logK = 8.50 and 8.24, respectively). The consequence of this tendency, which can be appreciated from the distribution plot of Fig. 1b, is that the species largely prevalent at physiological pH, where binding of phosphate is relevant, is the diprotonated complex [ZnLH2]2+, suggesting that under these conditions the DAC moieties would give little or no contribution to metal ion binding. In other words, at physiological pH, Zn(II) would essentially bind to the MIDA moiety and would therefore be available for binding to phosphate.
To test this hypothesis, 1H NMR spectra of the receptor were recorded, in the presence and in the absence of Zn(II), at a pH value such that the L2− and [ZnL] species, or the H2L and [ZnLH2]2+ species, were the prevalent species in solution, i.e. at pH 12, 8.9, 9, and 6.6, respectively. Because the fast-exchange regime on the NMR spectroscopy timescale was consistently observed in NMR experiments, the chemical shift variations of the single set of signals were monitored. In Fig. 2 are reported the chemical shift differences (CSD) observed in the above spectra for the protonated and the unprotonated species. It can be easily appreciated that negligible random CSDs are exhibited by the protonated species upon addition of Zn(II), whereas marked downfield CSDs are induced by the metal ion on the macrocycle signals for the unprotonated species. This evidence strongly supports binding of Zn(II) to the DAC moieties of the macrocycle in the unprotonated species, which is substantially inhibited when the DAC moieties are protonated, in agreement with the above hypothesis.
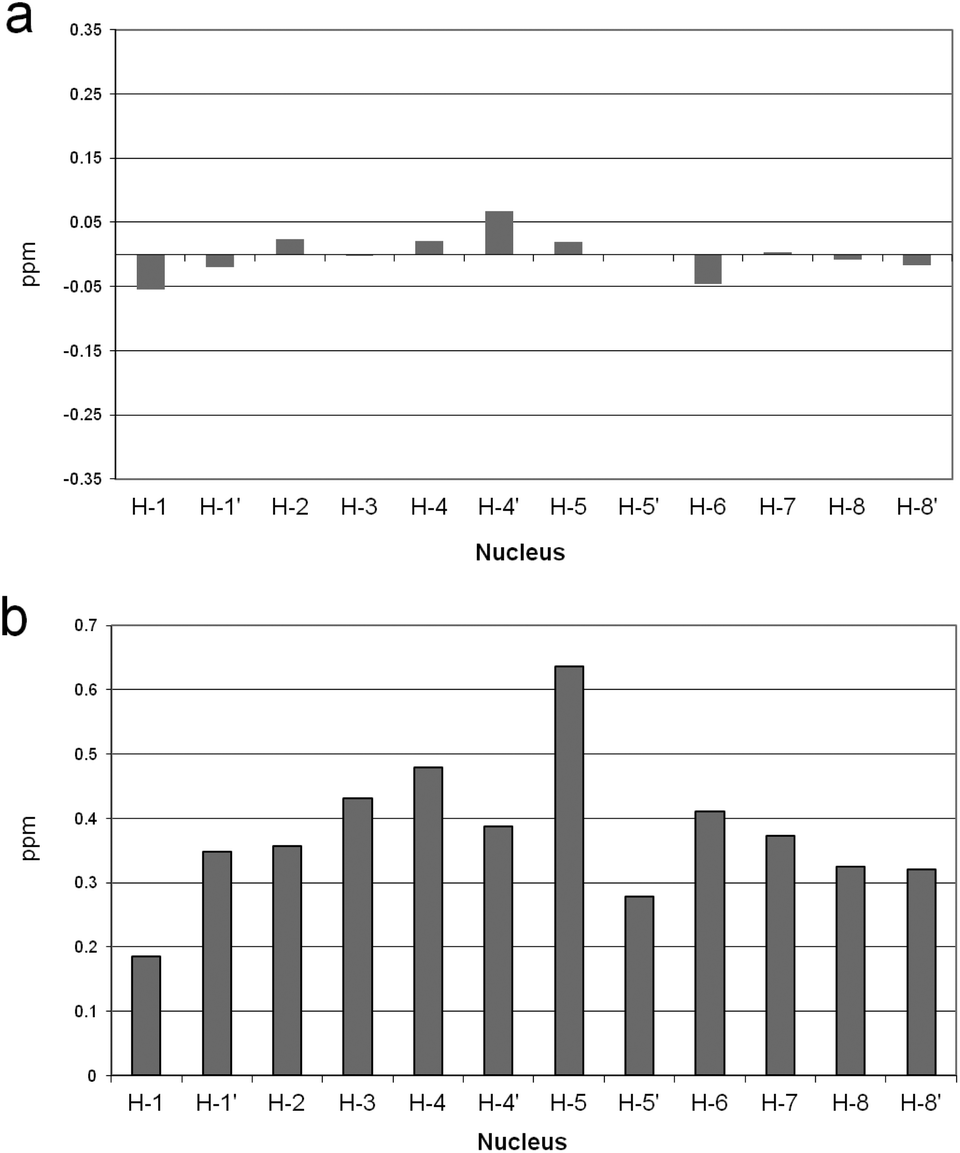 | ||
| Fig. 2 Plots of the chemical shift differences (CSDs) between receptor 1 in the presence and in the absence of Zn(II), at 500 MHz in D2O at T = 298 K. The signals of the macrocyclic moiety are reported in plots: (a) diprotonated; (b) unprotonated. For the numbering scheme, see the chart. | ||
:1 adducts between the metal-free receptor or the Zn(II) complex and the phosphate anions, with no evidence of adducts of higher stoichiometry, in agreement with polyamine-based receptors.3,4
Potentiometric measurements showed that free receptor 1 can hardly bind to phosphate anions. MP is not bound at any pH value, whereas, with the other anions, only the H3L+ form of the receptor gave adducts of modest affinity. The corresponding association constants are reported in Table 2. The low affinities observed can be understood considering that only the monocharged (H3L+), weakly interacting, form is populated above pH 4, whereas below pH 4, where the discharged form (H4L2+) is present, anionic guests are protonated as well. The observed behavior is in sharp contrast to that of polyamine receptors, which give highly charged, strongly binding polyammonium cations3–10 at neutral or slightly acidic pH values.
| Equilibrium | LogK |
|---|---|
| a Measured at 308 K in 0.1 M NMe4Cl. | |
| H3L+ + P2O74− = [H3LP2O7]3− | 3.21(3) |
| H3L+ + HP2O73− = [H4LP2O7]2− | 3.05(3) |
| H3L+ + P3O105− = [H3LP3O10]4− | 3.48(2) |
| H3L+ + HP3O104− = [H4LP3O10]3− | 3.18(3) |
| H3L+ + ADP3− = [H3LADP]2− | 3.04(2) |
| H3L+ + HADP2− = [H4LADP]− | 3.12(2) |
| H3L+ + HATP3− = [H4LATP]2− | 3.41(1) |
| H3L+ + H2ATP2− = [H5LATP]− | 3.35(1) |
The presence of Zn(II) coordinated to the receptor dramatically enhanced the binding affinity for all the investigated anions, giving rise to several adducts with the Zn complex in different protonation states. The results of potentiometric measurements are reported in Table 3, where the addition constants of TP to the Zn(II) complex of MIDA are also reported for comparison.
| Equilibrium | LogK |
|---|---|
| a Measured at 308 K in 0.1 M NMe4Cl. | |
| ZnL + HPO42− = [ZnLHPO4]2− | 3.55(7) |
| ZnLH+ + HPO42− = [ZnLH2PO4]− | 3.98(7) |
| ZnLH22+ + HPO42− = [ZnLH3PO4] | 4.05(8) |
| ZnLH33+ + HPO42− = [ZnLH4PO4]+ | 4.15(8) |
| ZnL + P2O74− = [ZnLP2O7]4− | 4.75(5) |
| ZnL + HP2O73− = [ZnLHP2O7]3− | 4.45(5) |
| ZnLH+ + HP2O73− = [ZnLH2P2O7]2− | 5.54(5) |
| ZnLH22+ + HP2O73− = [ZnLH3P2O7]− | 6.23(8) |
| ZnLH22+ + H2P2O72− = [ZnLH4P2O7] | 6.08(8) |
| ZnLH33+ + H2P2O72− = [ZnLH5P2O7]+ | 6.20(8) |
| ZnL + P3O105− = [ZnLP3O10]5− | 5.53(4) |
| ZnLH+ + P3O105− = [ZnLHP3O10]4− | 6.84(4) |
| ZnLH22+ + P3O105− = [ZnLH2P3O10]3− | 8.02(5) |
| ZnLH22+ + HP3O104− = [ZnLH3P3O10]2− | 7.77(6) |
| ZnLH33+ + HP3O104− = [ZnLH4P3O10]− | 7.92(6) |
| ZnL + ADP3− = [ZnLADP]3− | 4.09(6) |
| ZnL + HADP2− = [ZnLHADP]2− | 3.98(6) |
| ZnLH+ + HADP2− = [ZnLH2ADP]− | 4.52(6) |
| ZnLH22+ + HADP2− = [ZnLH3ADP] | 5.15(8) |
| ZnLH33+ + HADP2− = [ZnLH4ADP]+ | 5.75(8) |
| ZnL + ATP4− = [ZnLATP]4− | 5.2(1) |
| ZnLH+ + ATP4− = [ZnLHATP]3− | 6.1(1) |
| ZnLH22+ + ATP4− = [ZnLH2ATP]2− | 6.84(8) |
| ZnLH22+ + HATP3− = [ZnLH3ATP]− | 6.93(7) |
| ZnLH33+ + HATP3− = [ZnLH4ATP] | 7.04(7) |
| Zn(MIDA) + P3O105− = [Zn(MIDA)P3O10]5− | 4.45(2) |
| Zn(MIDA) + HP3O104− = [Zn(MIDA)HP3O10]4− | 4.35(3) |
| Zn(MIDA) + H2P3O103− = [Zn(MIDA)H2P3O10]3− | 4.07(4) |
Because the exclusive formation of 1:1 adducts was revealed in all cases by data treatment, an independent confirmation was sought by recording 31P NMR spectra of anionic substrates by addition of increasing amounts of the Zn(II) complex at pH 7. As in proton spectra, the fast-exchange regime on the NMR spectroscopy timescale was consistently observed in 31P NMR spectra, so that the chemical shift variations of the averaged signals were monitored. As shown in Fig. 3 for TP, the addition induced an upfield shift of both 31P resonances of the anion, which increased linearly up to 0.9:1 anion/complex mole ratio and reached a plateau above 1.1:1 ratio. Analogous behavior was found for the other anionic substrates (ESI,† Fig. S3–S6).
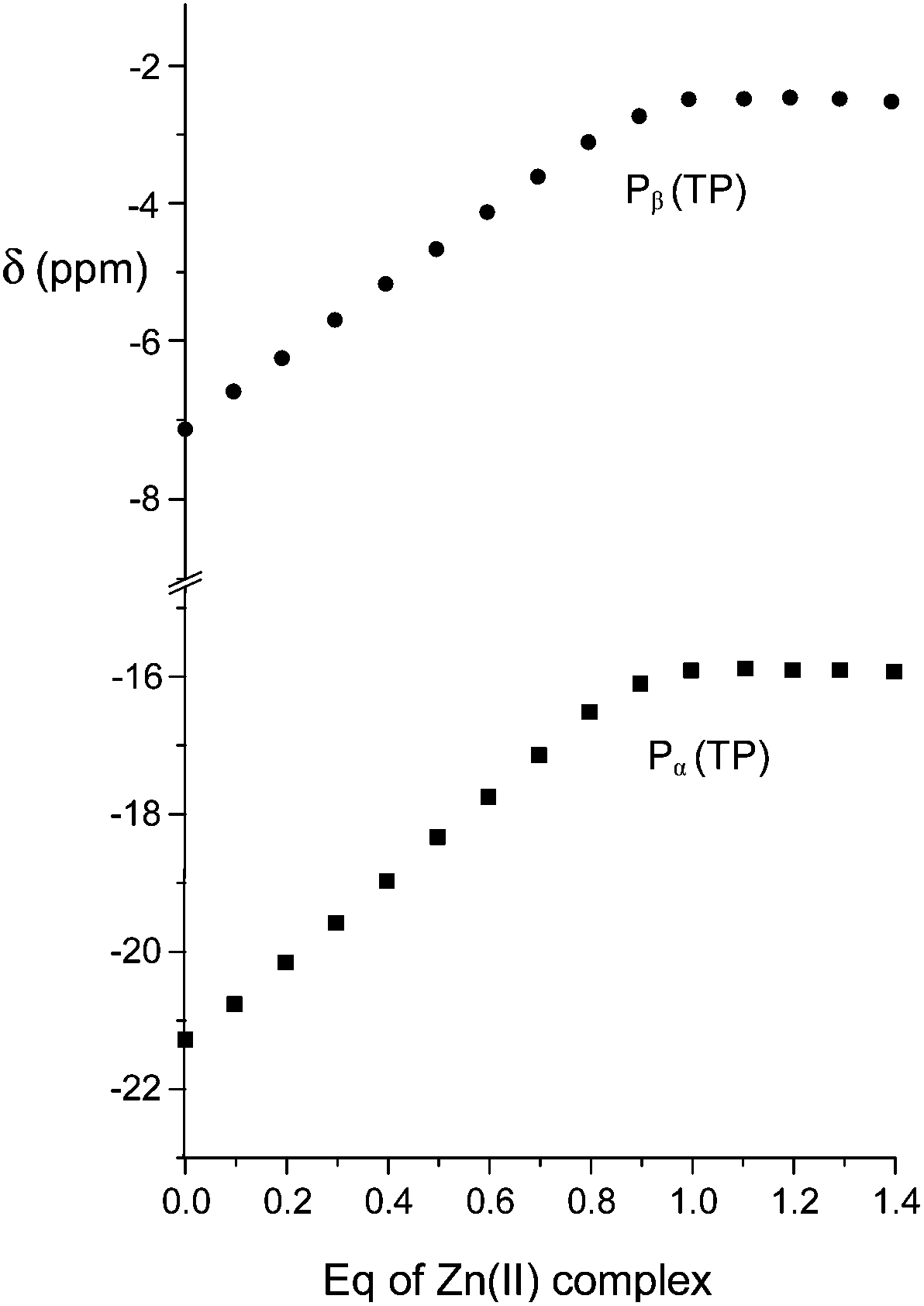 | ||
| Fig. 3 Plot of the chemical shifts of the terminal (Pβ) and central (Pα) phosphorous nuclei of TP in the presence of an increasing amount of the Zn(II) complex of 1 at 162 MHz, pH 7 and 308 K. | ||
Comparison of Tables 2 and 3 clearly shows that the anionic substrates form much more stable adducts with the Zn(II) complexes than with the zinc-free polyammonium cations of the same charge. For example, the addition constant of P3O105− to the [ZnLH]+ complex is ca. 3 orders of magnitude larger than that for the addition to the H3L+ cation. Analogous enhancements have been observed for PP, ATP and ADP.
The higher binding affinity for phosphate anions displayed by the protonated Zn(II) complexes with respect to the unprotonated Zn(II) complexes can be ascribed to the presence of the metal center, which can behave as the anchoring point for the phosphate unit(s), and to the protonated DAC units, which provide electrostatic interactions with the anion charge, possibly reinforced by H-bonding to the pyrrolic ring. Bridging of the two binding sites by phosphate would therefore enhance the affinity for the anionic guest in a synergetic fashion. This effect is particularly evident in the case of TP, for which the addition constants of TP in its fully deprotonated form (P3O105−) to the ZnL, [ZnLH]+ and [ZnLH2]2+ species are 5.35, 6.84 and 8.02 log units, respectively, with an increase larger than 2.5 log units from the [ZnL] to the [ZnLH2]2+ complex. Likewise, the addition constants of HP2O73− and ATP4− to the [ZnLH2]2+ complex are ca. 1.8 and 1.6 log units, respectively, larger than those found for the addition to [ZnL]. Clearly, the above differences are too large to be ascribed exclusively to the increased charge of the receptor. As a matter of fact, with the exception of MP, the anions are so strongly bound that their adducts represent the major species in the pH range 4–9.5, as shown by the distribution diagrams of the complexes formed by PP, TP and ATP in Fig. 4 (Fig. S7 and S8† for the distribution diagrams of the adducts formed by MP, ADP and Fig. S9† for that of the adducts formed by TP with the Zn(II) complex of MIDA).
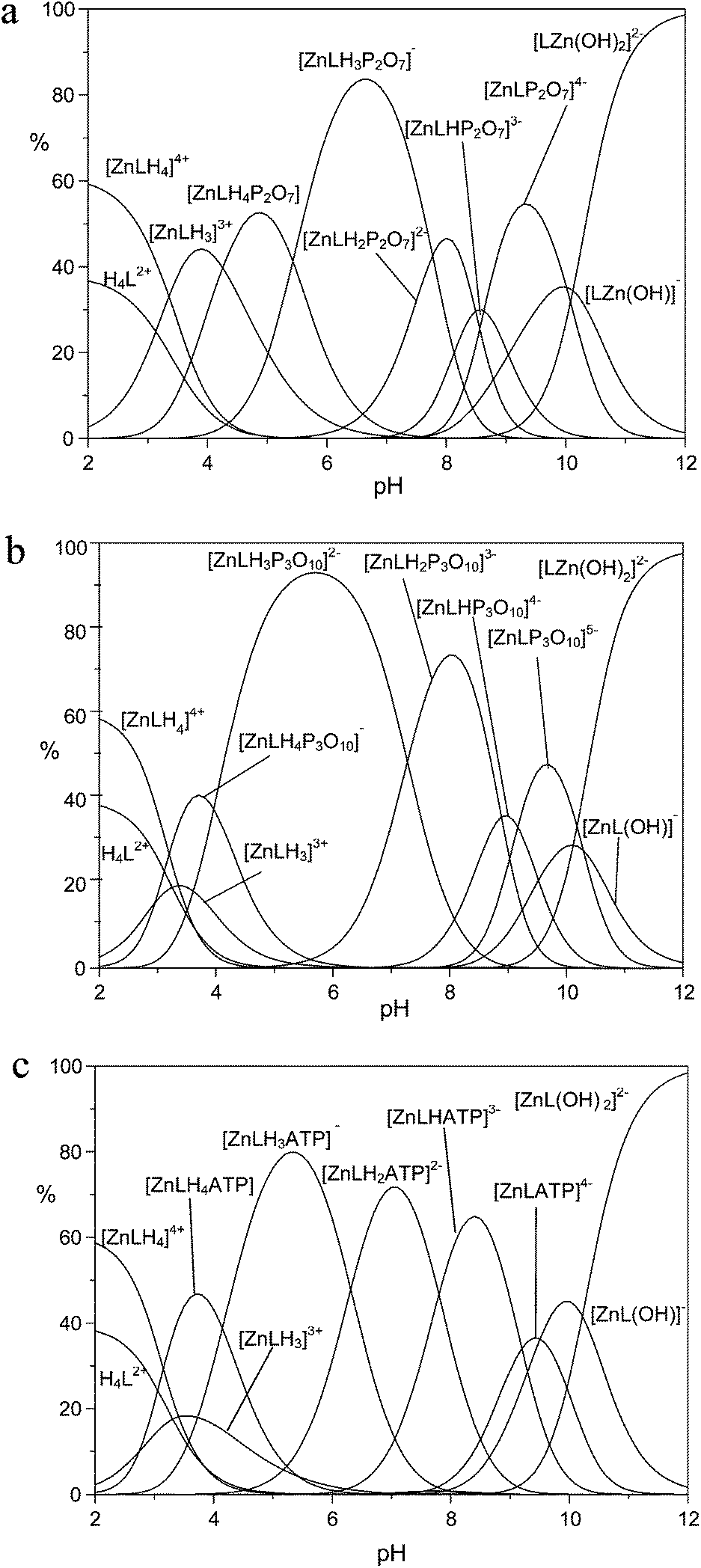 | ||
| Fig. 4 Distribution diagrams of the adducts formed by (a) PP, (b) TP, and (c) ATP with the Zn(II) complexes of receptor 1. Conditions: [1] = [Zn2+] = [PP] = [TP] = [ATP] = 1 × 10−3 M, NMe4Cl 0.1 M, 308 K. | ||
At pH > 9.5, the formation of the hydroxo-complexes [ZnL(OH)x)]x− (x = 1, 2) effectively competes with phosphate anion binding, while at pH < 4 the formation of ternary adducts is inhibited by the presence of protonated anions, and/or metal decomplexation. As a support to our hypothesis, in agreement with the view that the affinity enhancement is due to the concomitant bridging of binding sites, the monodentate MP, which is incapable of binding to both sites, shows weak binding scarcely dependent on protonation (Fig. S7†). Likewise, binding of TP to the Zn(II) complex of MIDA, which lacks the protonated macrocycle site, clearly shows modest affinities compared to those observed for receptor 1.
At first glance, the data in Table 3 seem to indicate that TP forms the most stable complexes. However, the interpretation of the stability constants is complicated by the different acid–base characteristics of the substrates as well as by the presence of multiple simultaneous equilibria, which make it difficult to compare the binding ability of the receptor at a given pH and to evaluate selectivity patterns. This issue can be addressed by considering a competitive system containing the receptor and equimolecular amounts of each anionic substrate, and calculating the percentages of the complexed anions over a wide pH range.44Fig. 5 displays the plot obtained for a competitive system containing the Zn(II) complex of 1 and five phosphate anions.
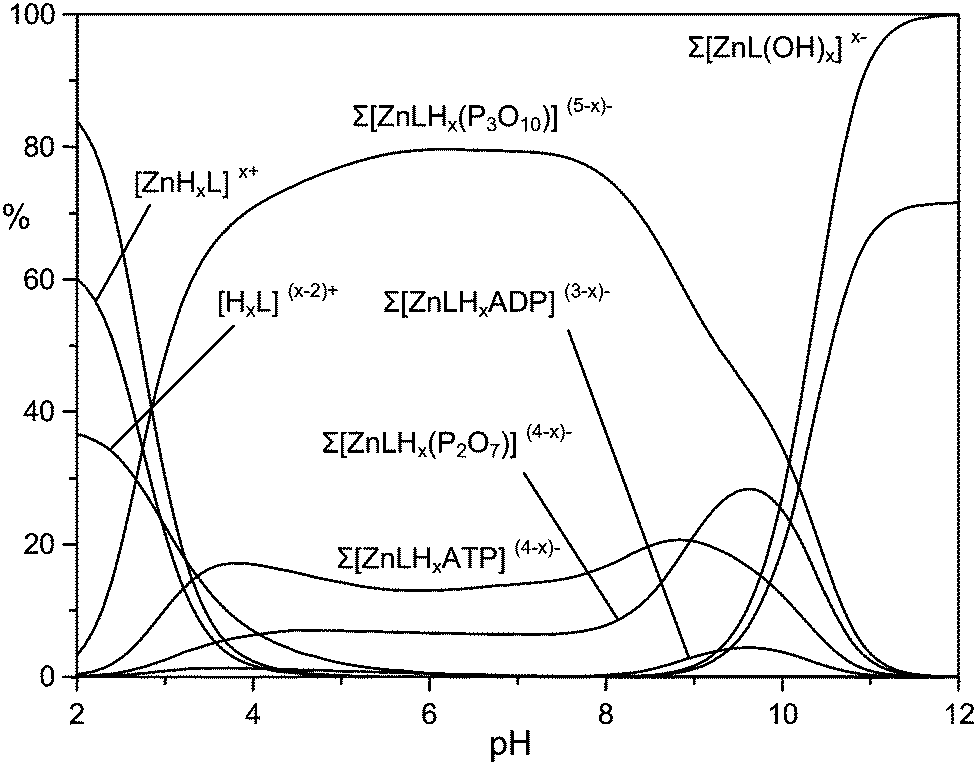 | ||
| Fig. 5 Overall percentages of the adducts between the Zn(II) complex of 1 and the phosphate anions in a competing system containing Zn(II), 1, MP, DP, TP, ADP and ATP in equimolecular amounts. Conditions: [Zn2+] = [1] = [MP] = [PP] = [TP] = [ADP] = [ATP] = 1 × 10−3 M, NMe4Cl 0.1 M, 308 K. | ||
Fig. 5 clearly shows that the Zn(II) receptor preferentially binds to TP over all other nucleotide and inorganic phosphate anions in the pH range of 3 to 10. At neutral pH, TP, ATP and PP are complexed in 80%, 13% and 7%, respectively. The ternary adduct with ADP is present in only minor amounts (<5%) at alkaline pH values, whereas the adduct with MP is not formed at any pH. The selectivity for TP over both PP and ATP is somewhat unexpected, because TP and PP show a similar binding ability for Zn(II) and its complexes with polyamine ligands.39 Furthermore, in most cases nucleotide anions, in particular ATP, form more stable complexes than inorganic phosphates, due to the interaction with positively charged hosts not only through the triphosphate chain, but also through the nucleobase, which shows H-bonding and/or π-stacking interactions with the receptor.
To elucidate the binding mode of inorganic and nucleotide anions to the Zn(II) receptor, we recorded 1H and 31P NMR spectra as a function of pH on solutions containing the Zn(II) complex of 1 and the nucleotides in 1:1 molar ratio. Fig. 6 shows the chemical shifts of the 31P signals of TP, ATP, PP and ADP, in the presence and in the absence of the metal complex (for the MP plot, see Fig. S10†).
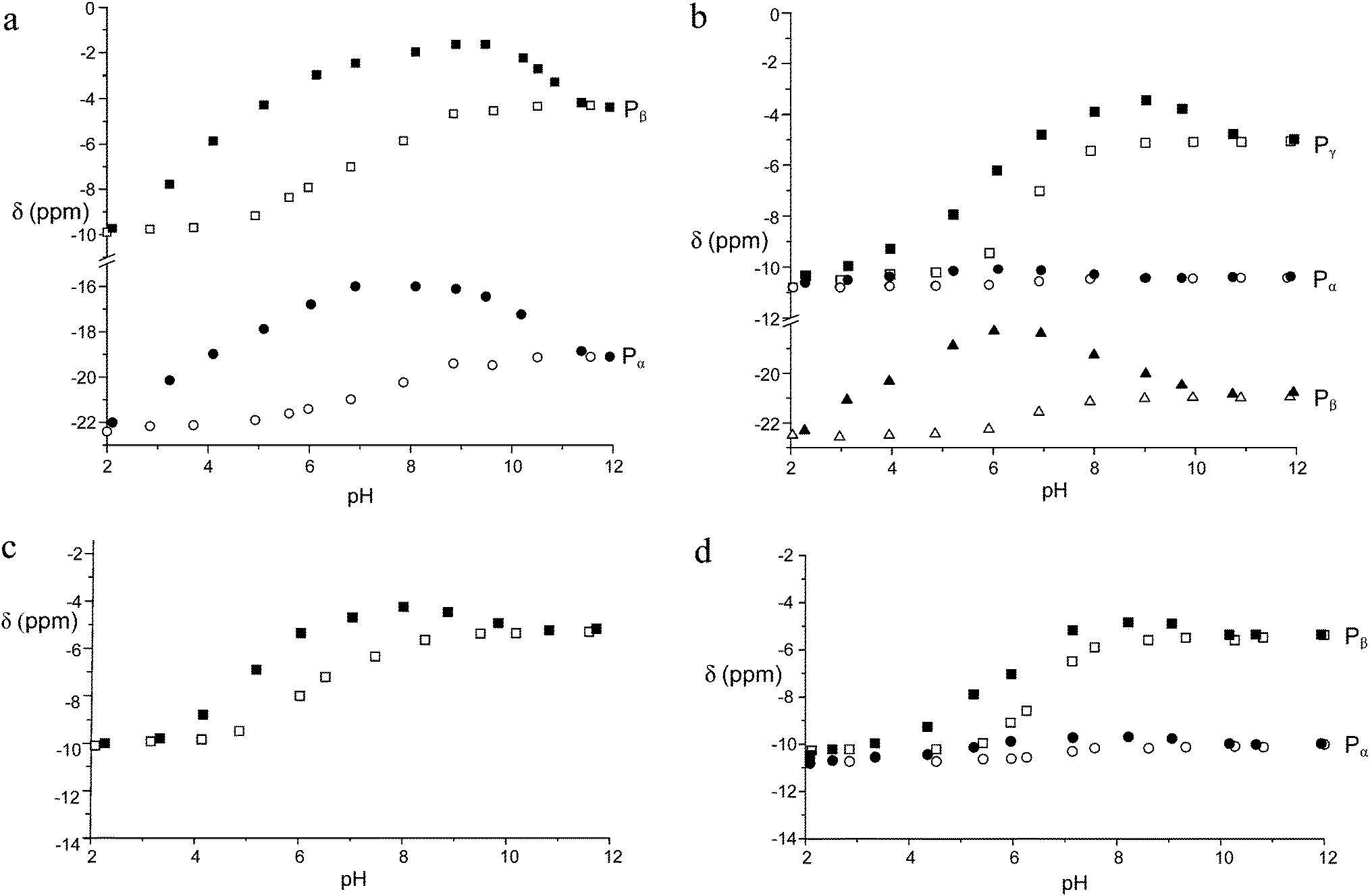 | ||
| Fig. 6 pH dependence of the 31P NMR signals (162 MHz) of (a) TP, (b) ATP, (c) PP, and (d) ADP in the presence (filled symbols) and in the absence (empty symbols) of the Zn(II) complex of 1. Conditions: [1] = [Zn2+] = [TP] = [PP] = [ADP] = [ATP] = 5 × 10−3 M, 308 K. | ||
In all cases, a strong dependence of chemical shifts on pH and on binding to the Zn(II) receptor can be easily appreciated. The latter induced a marked and systematic downfield shift of the 31P signals with respect to the uncomplexed phosphate. Notably, the CSD between the free and the complexed phosphates tends to peak in the pH range between 6 and 7, that is in the range where the receptor shows the largest binding ability and in which the protonated complex is predominant. It is also worth noting that the stronger the binding the larger the shift, suggesting that the proximity of phosphate to the Zn ion, as detected by the 31P NMR chemical shift, follows the same trend of the binding affinity for the five anions under scrutiny, because stronger binding provides larger amounts of phosphate complexes in the weighted average of species under the 31P signal. Interestingly, the chemical shifts of the Pα of both ATP and ADP are essentially insensitive to metal coordination, indicating that the phosphorous adjacent to the adenosine moiety is not involved in binding, most likely because of the hindrance of the adenosine moiety against the macrocyclic portion of the receptor, as observed for polyammonium hosts.3,4 Furthermore, the Pβ signal of ATP shows a maximum CSD of 3.8 ppm at pH 6, whereas the Pγ and the Pβ of ADP exhibit a much shallower CS trend between pH 9 and 5. This evidence suggests that the terminal phosphates Pγ of ATP and Pβ of ADP bind to the metal ion all through this pH range, whereas the Pβ of ATP comes into play on interaction with the doubly protonated macrocycle. This explains not only the larger binding ability of ATP with respect to ADP, but also the larger stability of the TP adduct, which does not suffer from the steric hindrance between the macrocycle and the adenosine moiety, thus showing the analogous trend of both 31P signals.
The contribution to binding of the nucleoside portion of ATP and ADP is revealed by the 1H NMR spectra as a function of pH. In Fig. 7, the plot of the anomeric H1′ and the adenine H2 and H8 protons is reported for ATP binding to the free and the Zn(II) complexed receptor (see Fig S11† for ADP). As can be seen, the corresponding CSDs are essentially null, indicating that little or no contribution to binding comes from the nucleoside residue, in contrast to the active role in stabilization often observed with positively charged hosts,3,10 in particular polyammonium cations, through H-bonding, π-stacking and hydrophobic interactions.
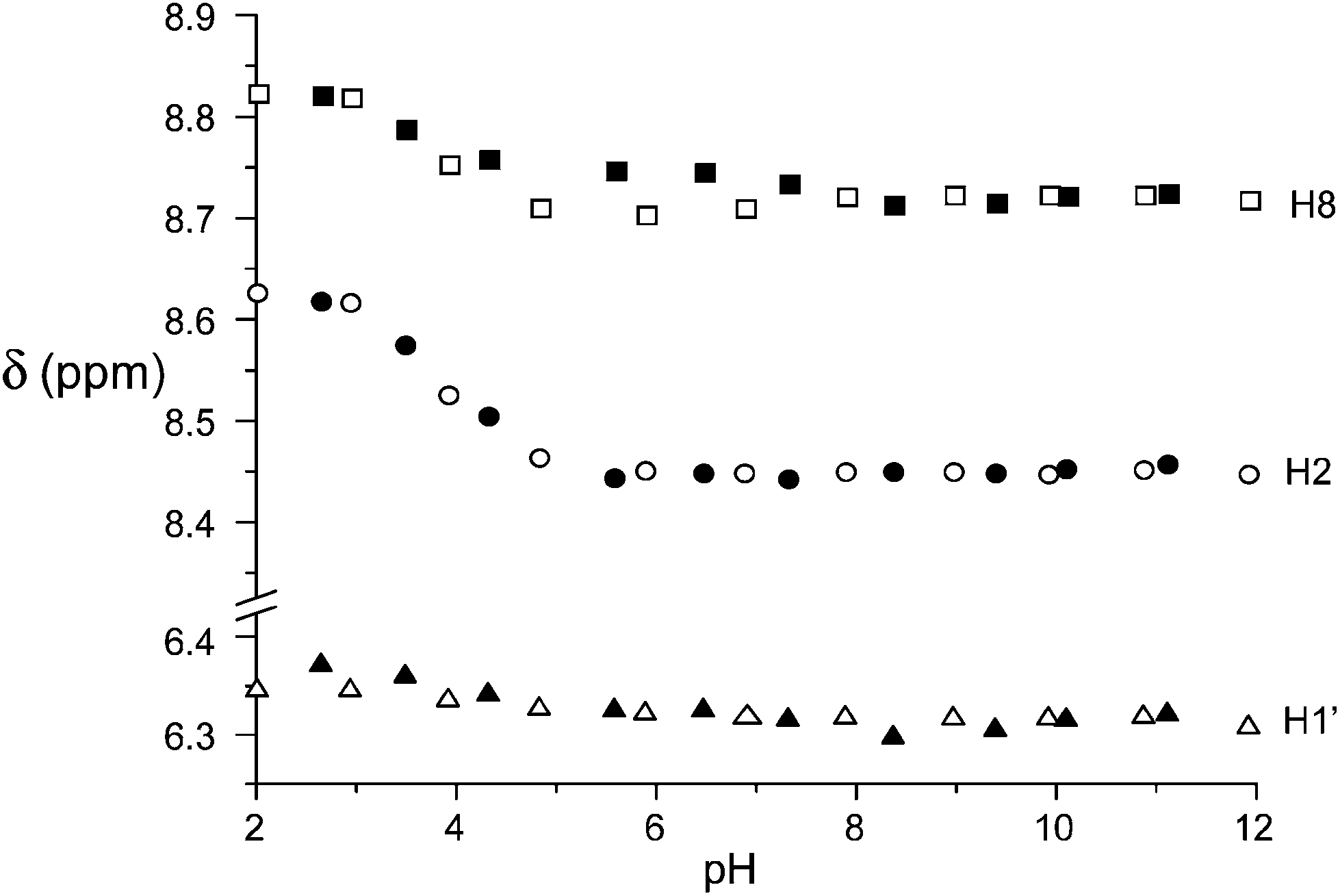 | ||
| Fig. 7 pH dependence of the 1H NMR signals (400 MHz) of the H8 (top) and H2 (middle) adenine protons and of the anomeric H1′ (bottom) proton of the sugar moiety of ATP in the presence (filled symbols) and in the absence (empty symbols) of the Zn(II) complex of 1. Conditions: [1] = [Zn2+] = [ATP] = 5 × 10−3 M, 308 K. | ||
Finally, although experimental data do not allow a direct assessment, the pyrrolic group of the macrocyclic portion of the receptor may likely contribute to reinforce the binding interaction through H-bonding to the oxygen atoms of the phosphate anions.
All the experimental evidence seems to demonstrate that concerted phosphate binding to the Zn(II) ion and to the protonated DAC macrocycle significantly enhances the binding ability of receptor 1 toward di- and triphosphates in the pH range of physiological interest. A sketch of the proposed receptor–Zn(II)–phosphate adduct of the ternary complex [ZnLH3P2O7]−, in agreement with experimental data, is depicted in Fig. 8. To the best of our knowledge, the present Zn(II)-containing receptor, in its protonated form, shows the highest binding affinity for TP in water among the Zn(II) complexes reported to date.
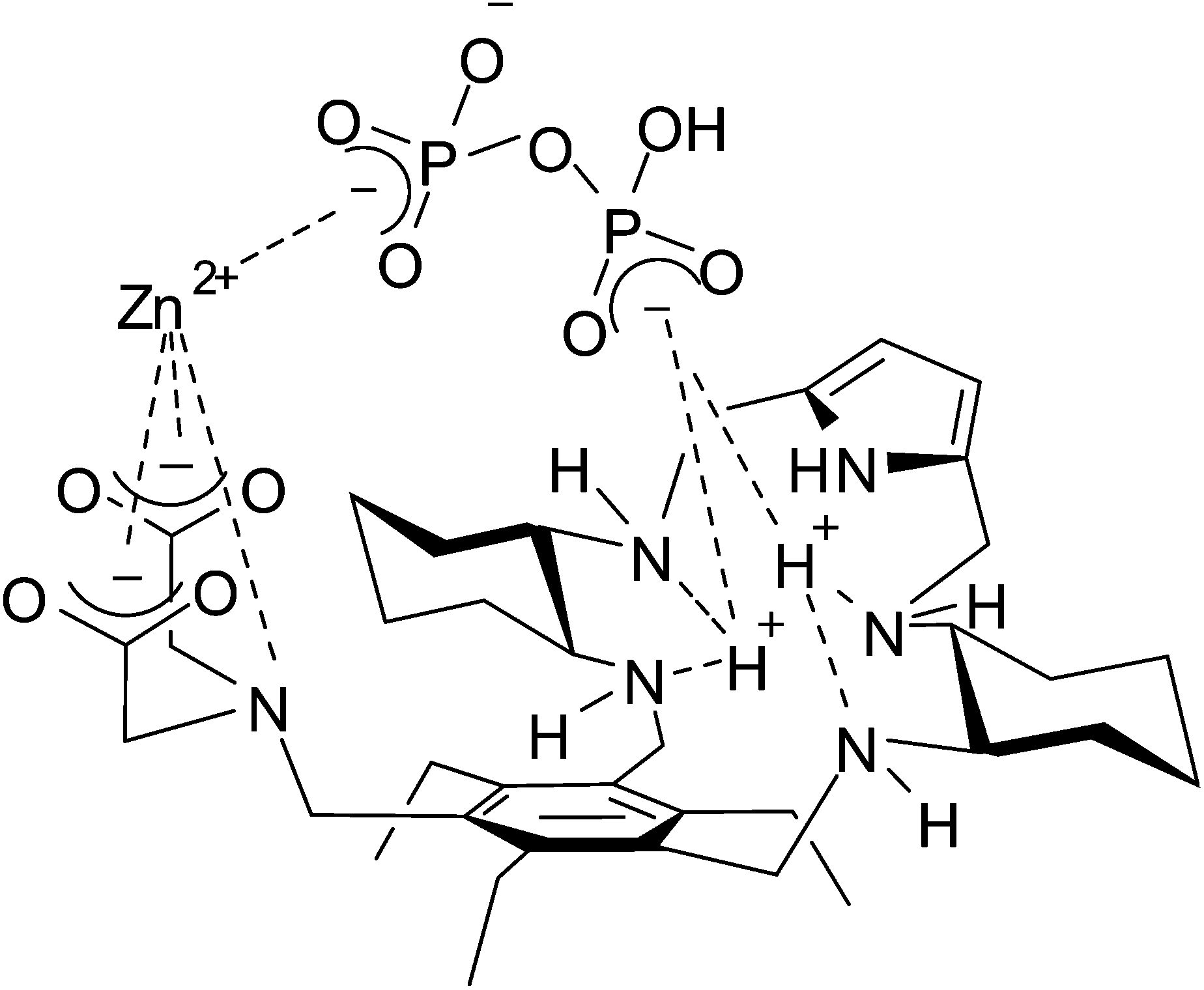 | ||
| Fig. 8 Sketch of the ternary complex [ZnLH3P2O7]− featuring the Zn(II)-mediated PP bridging the two binding sites of diprotonated receptor 1. | ||
Conclusions
In this work we have shown that combination of the two main approaches usually exploited for phosphate binding, that is (i) development of metal-free polyammonium receptors interacting with phosphate anions via multiple charge–charge and H-bonding interactions, and (ii) synthesis of metal complexes, in which the metal ion is the anchoring point for anionic species, into a single ditopic receptor is an effective strategy that can lead to a synergetic boosting of the di- and triphosphate recognition ability. The key to the binding enhancement lies in the ability of the di- or triphosphate anions to concertedly bridge both the metal ion site and the polyammonium macrocyclic site, with the possible participation in H-bonding of the pyrrole ring, whenever the correct geometry is allowed by the receptor architecture. The adaptive nature of receptor 1 may play a crucial role in this respect. To the best of our knowledge, the receptor described in this work features the highest binding affinity for triphosphates reported to date for Zn(II) complexes in water.Notes and references
- (a) A. K. H. Hirsch, F. R. Fischer and F. Diederich, Angew. Chem., Int. Ed., 2007, 46, 338–352 CrossRef CAS PubMed; (b) H. Dugas, Bioorganic Chemistry: a Chemical Approach to Enzyme Action, 1996 Search PubMed; (c) A. M. L. Davidson, E. Dassa, C. Orelle and J. Chen, Microbiol. Mol. Biol. Rev., 2008, 72, 317–364 CrossRef CAS PubMed; (d) G. R. Alton and E. A. Lunney, Expert Opin. Drug Discovery, 2008, 3, 595–605 CrossRef CAS PubMed; (e) J. A. Lewis, E. P. Lebois and C. W. Lindsley, Curr. Opin. Chem. Biol., 2008, 12, 269–280 CrossRef CAS PubMed; (f) B. E. Turk, Curr. Opin. Chem. Biol., 2008, 12, 4–10 CrossRef CAS PubMed; (g) K. Hollenstein, R. J. P. Dawson and K. P. Locher, Curr. Opin. Struct. Biol., 2007, 17, 412–418 CrossRef CAS PubMed; (h) J. R. Morrow, T. L. Amyes and J. P. Richard, Acc. Chem. Res., 2008, 41, 539–548 CrossRef CAS PubMed.
- (a) C. F. Mason, Biology of Freshwater Pollution, Longman, New York, 1991 Search PubMed; (b) H. Tiessen, Phosphorous in the Global Environment: Transfer, Cycles, and Management, Wiley, New York, 1995 Search PubMed.
- (a) K. Bowman-James, A. Bianchi and E. García-España Anion, Coordination Chemistry, Wiley-VCH, New York, 2012 Search PubMed.
- J. L. Sessler, P. A. Gale and W. S. Cho, Anion Receptor Chemistry, The Royal Society of Chemistry, Cambridge, UK, 2006 Search PubMed.
- A. E. Hargrove, S. Nieto, T. Zhang, J. L. Sessler and E. V. Anslyn, Chem. Rev., 2011, 111, 6603–6782 CrossRef CAS PubMed.
- (a) L. Fabbrizzi, M. Licchelli, G. Rabaioli and A. Taglietti, Coord. Chem. Rev., 2000, 205, 85–108 CrossRef CAS; (b) V. Amendola, M. Bonizzoni, D. Esteban-Gomez, L. Fabbrizzi, M. Licchelli, F. Sancenon and A. Taglietti, Coord. Chem. Rev., 2006, 250, 1451–1470 CrossRef CAS PubMed; (c) V. Amendola and L. Fabbrizzi, Chem. Commun., 2009, 513–531 RSC.
- E. Garcia España, P. Diaz, J. M. Llinares and A. Bianchi, Coord. Chem. Rev., 2006, 250, 2952–2980 CrossRef PubMed.
- A. Schaly, R. Belda, E. Garcia-Espana and S. Kubik, Org. Lett., 2013, 15, 6238–6241 CrossRef CAS PubMed.
- C. Caltagirone and P. A. Gale, Chem. Soc. Rev., 2009, 38, 520–563 RSC.
- (a) C. Bazzicalupi, A. Bencini and V. Lippolis, Chem. Soc. Rev., 2010, 39, 3709 RSC; (b) A. Bencini and V. Lippolis, Coord. Chem. Rev., 2012, 256, 149–169 CrossRef CAS PubMed.
- S. K. Kim, D. H. Lee, J. Hong and J. Yoon, Acc. Chem. Res., 2009, 42, 23–31 CrossRef CAS PubMed.
- (a) H. T. Ngo, X. Liu and K. A. Jolliffe, Chem. Soc. Rev., 2012, 41, 4928–4965 RSC; (b) J. V. Carolan, S. J. Butler and K. A. Jolliffe, J. Org. Chem., 2009, 74, 2992–2996 CrossRef CAS PubMed.
- S. O. Kang, M. A. Hossain and K. Bowman-James, Coord. Chem. Rev., 2006, 250, 3038–3052 CrossRef CAS PubMed.
- T. Gunnlaugsson, M. Glynn, G. M. Tocci, P. E. Kruger and F. M. Pfeffer, Coord. Chem. Rev., 2006, 250, 3094–3117 CrossRef CAS PubMed.
- M. D. Lankshear and P. D. Beer, Coord. Chem. Rev., 2006, 250, 3142–3160 CrossRef CAS PubMed.
- N. Gimeno and P. D. Vilar, Coord. Chem. Rev., 2006, 250, 3161–3189 CrossRef CAS PubMed.
- (a) M. Formica, V. Fusi, L. Giorgi and M. Micheloni, Coord. Chem. Rev., 2012, 256(1–2), 170–192 CrossRef CAS PubMed; (b) G. Ambrosi, M. Formica, V. Fusi, L. Giorgi, A. Guerri, E. Macedi, M. Micheloni, P. Paoli, R. Pontellini and P. Rossi, Inorg. Chem., 2009, 48, 5901–5912 CrossRef CAS PubMed; (c) G. Ambrosi, M. Formica, V. Fusi, L. Giorgi, A. Guerri, E. Macedi, M. Micheloni, P. Paoli, R. Pontellini and P. Rossi, Chem. – Eur. J., 2011, 17, 1670–1682 CrossRef CAS PubMed.
- S. Kubik, Chem. Soc. Rev., 2009, 38, 585–605 RSC.
- (a) M. J. Chmielewski, J. J. Davis and P. D. Beer, Org. Biomol. Chem., 2009, 7, 415–424 RSC; (b) P. D. Beer and S. R. Bayly, Top. Curr. Chem., 2005, 255, 125–162 CAS.
- F. P. Schmidtchen, Coord. Chem. Rev., 2006, 250, 2918–2928 CrossRef CAS PubMed.
- J. W. Steed, Chem. Soc. Rev., 2009, 38, 506–519 RSC.
- E. J. O'Neil and B. D. Smith, Coord. Chem. Rev., 2006, 250, 3068–3080 CrossRef PubMed.
- (a) E. Kimura, T. Shiota, T. Koike, M. Shim and M. Kodama, J. Am. Chem. Soc., 1990, 112, 5805–5811 CrossRef CAS; (b) E. Kimura, S. Aoki, T. Koike and M. Shiro, J. Am. Chem. Soc., 1997, 119, 3068–3076 CrossRef CAS.
- L. Rodriguez, J. C. Lima, A. J. Parola, F. Pina, R. Meitz, R. Aucejo, E. Garcia-España, J. M. Llinares, C. Soriano and J. Alarcon, Inorg. Chem., 2008, 47, 6173–6183 CrossRef CAS PubMed.
- Z. Zeng, A. A. J. Torriero, A. M. Bond and L. Spiccia, Chem. – Eur. J, 2010, 16, 9154–9163 CrossRef CAS PubMed.
- P. Mahato, A. Ghosh, S. K. Mishra, A. Shrivastav, S. Mishra and A. Das, Inorg. Chem., 2011, 50, 4162–4170 CrossRef CAS PubMed.
- M. Strianese, S. Milione, A. Maranzana, A. Grassia and C. Pellecchia, Chem. Commun., 2012, 48, 11419–11421 RSC.
- P. Hu, S. Yang and G. Feng, Org. Biomol. Chem., 2014, 12, 3701–3706 CAS.
- M. Bhuyan, E. Katayev, S. Stadlbauer, H. Nonaka, A. Ojida, I. Hamachi and B. König, Eur. J. Org. Chem., 2011, 2807–2817 CrossRef CAS.
- J. H. Lee, J. Park, J. Chin and J.-I. Hong, Org. Lett., 2007, 9, 3729–3731 CrossRef CAS PubMed.
- W. H. Chen, Y. Xing and Y. Pang, Org. Lett., 2011, 13, 1362–1365 CrossRef CAS PubMed.
- T. Kurishita, T. Kohira, A. Ojida and I. Hamachi, J. Am. Chem. Soc., 2010, 132, 13290–13299 CrossRef PubMed.
- C. Bazzicalupi, A. Bencini, S. Puccioni, B. Valtancoli, P. Gratteri, A. Garau and V. Lippolis, Chem. Commun., 2012, 139–142 RSC.
- R. G. Hanshaw, S. M. Hilkert, H. Jiang and B. D. Smith, Tetrahedron Lett., 2004, 45, 8721–8724 CrossRef CAS PubMed.
- D. H. Lee, S. Y. Kim and J.-I. Hong, Angew. Chem., Int. Ed., 2004, 43, 4777–4780 CrossRef CAS PubMed.
- D. H. Lee, J. H. Im, S. U. Son, Y. K. Chung and J.-I. Hong, J. Am. Chem. Soc., 2003, 125, 7752–7753 CrossRef CAS PubMed.
- L. J. Liang, X. J. Zhao and C. Z. Huang, Analyst, 2012, 137, 953–958 RSC.
- J. Seo, S. Kim and S. Y. Park, J. Am. Chem. Soc., 2004, 126, 11154–11155 CrossRef CAS PubMed.
- A. E. Martell and R. M. Smith, NIST Standard Reference Database, NIST, Gaithersburg, USA, 2004, 46 Search PubMed.
- A. Bencini, A. Bianchi, E. Garcia-España, M. Micheloni and J. A. Ramirez, Coord. Chem. Rev., 1999, 188, 97–156 CrossRef CAS.
- (a) A. Ardá, F. J. Cañada, C. Nativi, O. Francesconi, G. Gabrielli, A. Ienco, J. Jiménez-Barbero and S. Roelens, Chem. – Eur. J., 2011, 17, 4821–4829 CrossRef PubMed; (b) C. Nativi, O. Francesconi, G. Gabrielli, A. Vacca and S. Roelens, Chem. – Eur. J., 2011, 17, 4814–4820 CrossRef CAS PubMed; (c) A. Ardá, C. Venturi, C. Nativi, O. Francesconi, G. Gabrielli, F. Javier Cañada, J. Jiménez-Barbero and S. Roelens, Chem. – Eur. J., 2010, 16, 414–418 CrossRef PubMed.
- O. Francesconi, C. Nativi, G. Gabrielli, M. Gentili, M. Palchetti, B. Bonora and S. Roelens, Chem. – Eur. J., 2013, 19, 11742–11752 CrossRef CAS PubMed.
- P. Amico, R. P. Bonomo, R. Calì, V. Cucinotta, P. G. Daniele, G. Ostacoli and E. Rizzarelli, Inorg. Chem., 1989, 28, 3555–35661 CrossRef CAS.
- A. Bianchi and E. Garcia-España, J. Chem. Educ., 1999, 76, 1727–1732 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ob02321h |
| This journal is © The Royal Society of Chemistry 2015 |