
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence17.6% stabilized efficiency in low-temperature processed planar perovskite solar cells†
Chen
Tao
a,
Stefanie
Neutzner
ab,
Letizia
Colella
cd,
Sergio
Marras
e,
Ajay Ram
Srimath Kandada
a,
Marina
Gandini
ab,
Michele De
Bastiani
a,
Giuseppina
Pace
a,
Liberato
Manna
e,
Mario
Caironi
a,
Chiara
Bertarelli
ac and
Annamaria
Petrozza
*a
aCenter for Nano Science and Technology@PoliMi, Istituto Italiano di Tecnologia, via Giovanni Pascoli 70/3, 20133, Milan, Italy. E-mail: annamaria.petrozza@iit.it
bDipartimento di Fisica, Politecnico di Milano, Piazza L. da Vinci, 32, 20133 Milano, Italy
cDipartimento di Chimica, Materiali e Ing. Chimica “G. Natta”, Politecnico di Milano, Piazza L. Da Vinci 32, 20133 Milano, Italy
dINAF, Osservatorio Astronomico di Brera, via Bianchi 46, 23807 Merate, Italy
eDepartment of Nanochemistry, Istituto Italiano di Tecnologia, via Morego, 30, 16163 Genova, Italy
First published on 19th June 2015
Abstract
We present here a planar perovskite solar cell with a stabilized power conversion efficiency (PCE) of 17.6% at the maximum power point and a PCE of 17% extracted from quasi-static J–V with an open-circuit voltage of 1.11 V. Such excellent figures of merit can be achieved by engineering a solution-processed electron buffer layer that does not require high temperature steps. A compact thin film of perovskite absorber is grown onto a PCBM-based electron extraction layer by implementing a novel two-step procedure which preserves the soluble organic interlayer during the deposition of successive layers. We demonstrate that efficient charge extraction is the key for high steady state efficiency in perovskite solar cells with a highly integrable architecture.
Broader contextOne of the most promising approaches to boost efficiency in photovoltaics is to make tandem solar cells. The challenge is to find a way to make this approach economically viable, by reducing the production costs. Over the past few years, organic–inorganic lead halide perovskites have emerged in the PV technology scenario. The rapid progress in the field has led to certified solar energy conversion efficiencies above 20%, which brings this technology to the cohort of highly performing solar cells, while keeping the promise of low cost energy production. All-perovskite-based tandem solar cells may represent the ultimate goal considering the relatively easy tunability of the semiconductor's bandgap. However, on a shorter term, the realization of hybrid systems which couple perovskite devices with market-leading technologies is highly attractive. For achieving these targets we need to obtain perovskite solar cells with high steady state efficiency (and thus reliable) and to deliver a device architecture and fabrication process that enables multi-layered structures. |
Hybrid lead halide perovskite solar cells (PSCs) are experiencing a drastic boost in solar energy conversion performances, with reported efficiencies approaching crystalline silicon solar cells.1 Methylammonium lead iodide CH3NH3PbI3 or its analogue, chlorine-doped CH3NH3PbI3, is typically employed in order to achieve good light harvesting, with an absorption onset close to 800 nm and an open circuit voltage close to 1 V. Stemming from the field of dye-sensitized solar cells, the first device architecture deployed CH3NH3PbX3 (X = Br, I) as sensitizers on a mesoscopic TiO2 layer.2 Since then, the demonstration of ambipolar charge transport within the perovskite layer,3 with diffusion lengths of photo-carriers exceeding 1 μm,4,5 has led to the development of planar structures. The mesoscopic TiO2 has been replaced only by a flat TiO2 layer, working as an electron-extracting layer (EEL), on top of which the perovskite semiconductor is deposited. Finally an organic semiconductor is generally used as a p-type hole-extracting layer (HEL) [e.g., 2,2′,7,7′-tetrakis(N,N-di-p-methoxyphenylamine)-9-9′-spirobifluorene (spiro-MeOTAD), poly(triarylamine) (PTAA), poly(3-hexylthiophene) (P3HT)].6 This architecture, where the perovskite is grown on the EEL, i.e. the transparent electrode works as an anode, is indicated as a standard structure. The planar architecture is definitely advantageous from a fabrication point of view. This is particularly true when aiming at the promising implementation of perovskite-based solar cells in tandem structures.7 However, the TiO2-based flat device, though showing efficiencies surpassing 19%,8 is also affected by important shortcomings. One primary limitation is the necessity of a 500 °C sintering step to form a crystalline TiO2 layer, precluding its application on temperature sensitive substrates, including plastic foil, silicon solar cells, and second perovskite solar cells. Although low-temperature solution-processed TiO2, ZnO nanoparticles and cesium carbonate have been occasionally reported as EELs,8–10 the notorious hysteretic behaviour has so far prevented the correct evaluation of their potential.11,12
To address these issues, indium tin oxide (ITO)/poly(3,4-ethylenedioxythiophene)–poly(styrenesulfonate) (PEDOT:PSS)/perovskite/fullerene derivative/aluminium devices (indicated as an inverted structure) have been developed with simple low temperature processing. Efficiencies of up to ∼16% have been reported for low-temperature processed polycrystalline films.13–15 The degradation of the underlying perovskite layer in such a structure is prevented by processing fullerene derivatives, i.e., [6,6]-phenyl-C61-butyric acid methyl ester (60-PCBM), from orthogonal solvents. On the other hand, the realization of a structure where the electron selective layer is used as a bottom layer for PSCs with low-temperature processing would make the device architecture much more versatile, both because it would be opened to a large library of p-type layers that could be used on top of the perovskite layer and because it would simplify the realization of tandem perovskite structures.16 Wojciechowski et al. employed a self-assembled fullerene monolayer to modify the TiO2 surface.17 This solution leads to an improvement of the steady state operation of the devices (a power conversion efficiency of approximately 13% is demonstrated with a scan rate of 0.0015 V s−1). However, it still needs high temperature processing. Ryu et al. succeeded in the fabrication of a standard architecture at low temperature (∼100 °C), i.e., FTO/PEI/60-PCBM/CH3NH3PbI3/PTAA/Au, but such a device still suffers from large hysteresis.18
In this communication we propose the use of a low-temperature solution-processed titanium oxide (TiOX)/60-PCBM layer that conformably covers the FTO as an EEL on top of which the hybrid perovskite can be directly deposited. Moreover, we demonstrate that by implementing a novel two-step procedure for growing the perovskite films we are able to form a compact, uniform layer that preserves the soluble 60-PCBM interlayer even upon spin coating of the organic hole-extracting component spiro-MeOTAD. This has a broader impact on the design and optimization of future EELs as their choice will not be limited anymore by the processing of the top layers. Remarkably, we demonstrate a planar junction with a standard architecture that is able to achieve a 17.6% stabilized power conversion efficiency.
TiOX is synthesized starting from titanium(IV) isopropoxide and deposited directly through spin coating onto FTO substrates in order to get a nearly conformal coverage of the rough FTO (see the ESI† for details of the synthetic procedure). An extremely thin layer of 60-PCBM is subsequently spin cast on top of TiOX. The TiOX/60-PCBM layer serves as an EEL (see the ESI† for the characterization of its surface work function, Table S1 (ESI†), and morphology, Fig. S1, ESI†), which shows comparable optical transmittance as TiO2 deposited according to the procedures used in state-of-the-art planar heterojunction perovskite solar cells (see Fig. S2 in the ESI†).17,19–23 To fabricate the CH3NH3PbI3 film the traditional one-step method is unsuccessful, since any combinations of dimethylformamide (DMF), dimethyl sulfoxide (DMSO), or γ-butyrolactone (GBL) are detrimental to the very thin underlying 60-PCBM layer. Similarly, the direct application of known two-step methods using such solvents is prohibitive. Thus, we developed a new two-step process compatible with 60-PCBM that combines a thermally evaporated PbI2 layer followed by MAI deposition by spin coating from 2-propanol, a bad solvent for 60-PCBM. Following MAI deposition, the light yellow PbI2 films turn slightly darker, indicating the interfacial reaction between PbI2 and MAI, hence the formation of a very thin CH3NH3PbI3 layer. The remaining dense PbI2 layer together with the already transformed CH3NH3PbI3 layer would prevent further MAI penetration at room temperature.24 However, upon thermal annealing at 100 °C the film turns totally dark, indicating an extended transformation of PbI2 into CH3NH3PbI3 perovskite crystals. To check the quality of the polycrystalline thin film and the presence of any unconverted PbI2 precursor, we measured the XRD patterns of the hybrid perovskite deposited on both TiOX/60-PCBM and TiO2 layers, spun cast onto FTO substrates. Fig. 1a shows, in the bottom panel, the XRD patterns of thin layers of PbI2 evaporated on the amorphous (TiOX/60-PCBM) and the crystalline (TiO2) substrates. They both look identical to the one obtained from solution-processed films,24 exhibiting a preferential orientation along the c axis accompanied by the appearance of four diffraction peaks, corresponding to the (001), (002), (003), and (004) lattice planes. The top panel of Fig. 1a shows the XRD patterns of the hybrid perovskite films grown on these two substrates. The absence of any PbI2 diffraction peaks indicates the full conversion in both cases. The perovskite shows the typical peaks of the tetragonal phase. However, it is interesting to note that while the crystalline TiO2 substrate induces a preferential orientation of the perovskite grains mainly along the [220] crystallographic direction,25 the amorphous substrate induces the preferential orientation of the grains also in the [004] crystallographic direction [see the inset in Fig. 1(a)]. In Fig. 1(b) we show a top-view SEM image of the perovskite films grown on these two substrates. Generally, the uncontrolled precipitation of perovskite using one-step spin casting from a mixture of PbX2 and CH3NH3X (X = Cl, I) in a common solvent produces large morphological variations, in particular numerous pinholes in planar PSCs. We show that our novel two-step hybrid procedure allows for better control of the thin film uniformity. A closer inspection of the surface images suggests that the presence of the amorphous substrate allows for an even better coverage. This may be induced by the growth of the crystals along different planes, possibly allowing denser packing.
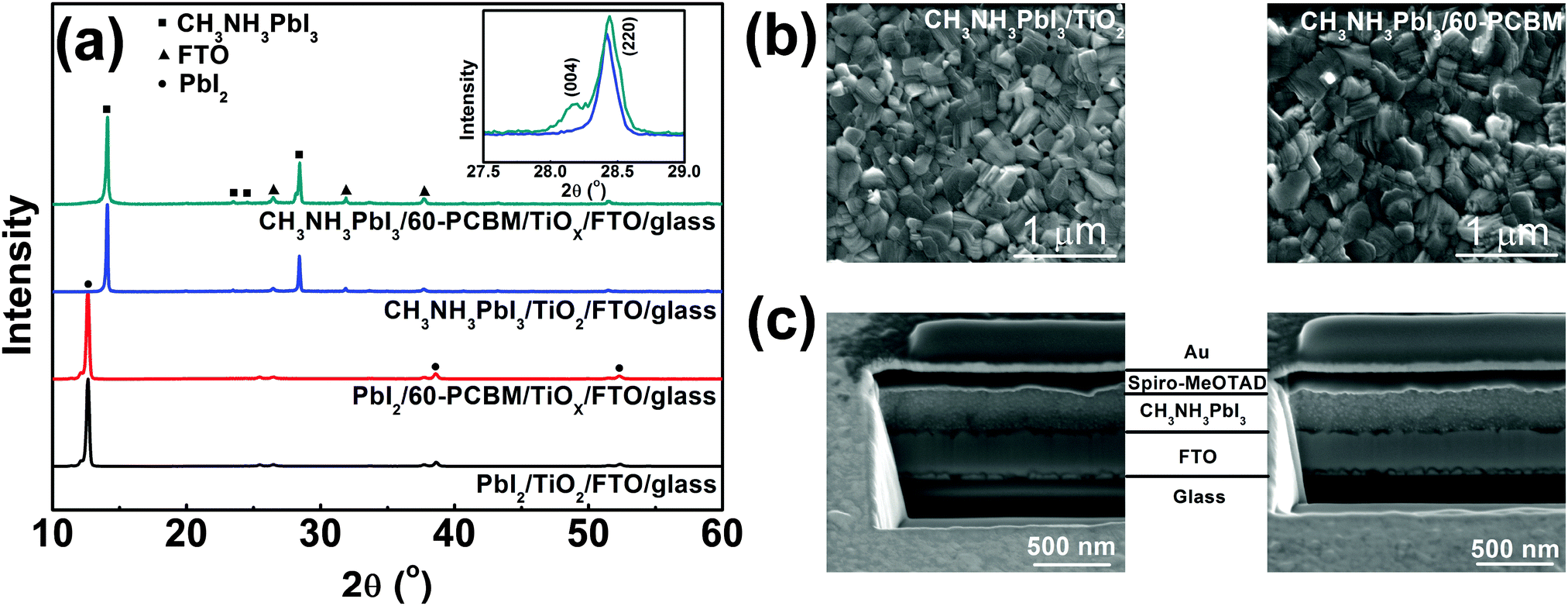 | ||
| Fig. 1 (a) XRD patterns of PbI2 on glass/FTO/TiO2 and glass/FTO/TiOX/60-PCBM before and after the deposition. The inset shows the enlarged CH3NH3PbI3 XRD pattern between 25° and 30° on two substrates. (b) SEM top-view of CH3NH3PbI3 grown on TiO2/FTO/glass (left) and 60-PCBM/TiOX/FTO/glass (right). (c) SEM cross section of complete photovoltaic devices: glass/FTO/TiO2/CH3NH3PbI3/spiro-MeOTAD/Au (left) and glass/FTO/TiOX/60-PCBM/CH3NH3PbI3/spiro-MeOTAD/Au (right). Note that the thin TiO2 and TiOX/60-PCBM layers present between the FTO and CH3NH3PbI3 are not resolved in the SEM images. | ||
To complete the device, we deposited by spin coating a spiro-MeOTAD layer as HELs followed by gold via thermal evaporation. The ability to create a uniform and compact CH3NH3PbI3 film is essential for the deposition of spiro-MeOTAD that is dissolved in chlorobenzene (CB). The presence of pin holes would leave free paths for CB penetration through the perovskite films, re-dissolving the 60-PCBM layer. In Fig. 1(c) we show a cross section of the full device employing extremely thin layers of crystalline TiO2 and TiOX/60-PCBM EELs. The total thickness of these two devices is almost the same, with very uniform perovskite and HELs. The image also confirms that the EELs, hardly distinguishable, simply form a conformable layer that covers the FTO surface.
Fig. 2(a) and (b) show representative current density versus voltage (J–V) characteristics of optimized planar devices with TiO217,19–23 and TiOX/60-PCBM as EELs, measured under air mass 1.5 global (AM 1.5G) conditions. To check for possible hysteresis phenomena, which are known to strongly influence the device testing for many perovskite solar cells reported so far in the literature,11,12 we show the J–V curves as a function of the scan rate, in forward and reverse scan directions. Table 1 summarizes the main figures of merit of the tested samples, i.e., short-circuit current density (JSC), open-circuit voltage (VOC), fill factor (FF) and power conversion efficiency (PCE). Fig. 2(c) shows the transient behaviour of JSC for both devices. The differences are striking and clear. The TiO2-based device shows performances which are strongly dependent on the scan rate and direction. As already reported in ref. 12, high PCE (14.4%) can be achieved only upon a fast scanning of the device from open-circuit to short-circuit conditions (reverse scan). However, such apparent PCE drastically drops when the scan rate slows down (8.8%), and is even lower when the scan direction is inverted (6.9% and 2.3% for fast and slow scan rates, respectively). It should be noted that no pre-polarization conditioning has been performed, differently from what is generally done in most of the published work; this may explain the lower figures of merit with respect to the literature value only when scanning the device from short-circuit to open-circuit conditions (forward scan).17,19–23 This seems to indicate that the TiO2-based device needs a sort of a latent, pre-charging treatment, which may also lead to erroneous PCE estimation if J–V characteristics are not judiciously reported. In agreement, JSC shows a drastic reduction over time, dropping from 21.0 mA cm−2 to 6.8 mA cm−2 within 38.5 s. This indeed leads to a very poor steady state (0.043 V s−1 scan rate) PCE of about 2%.
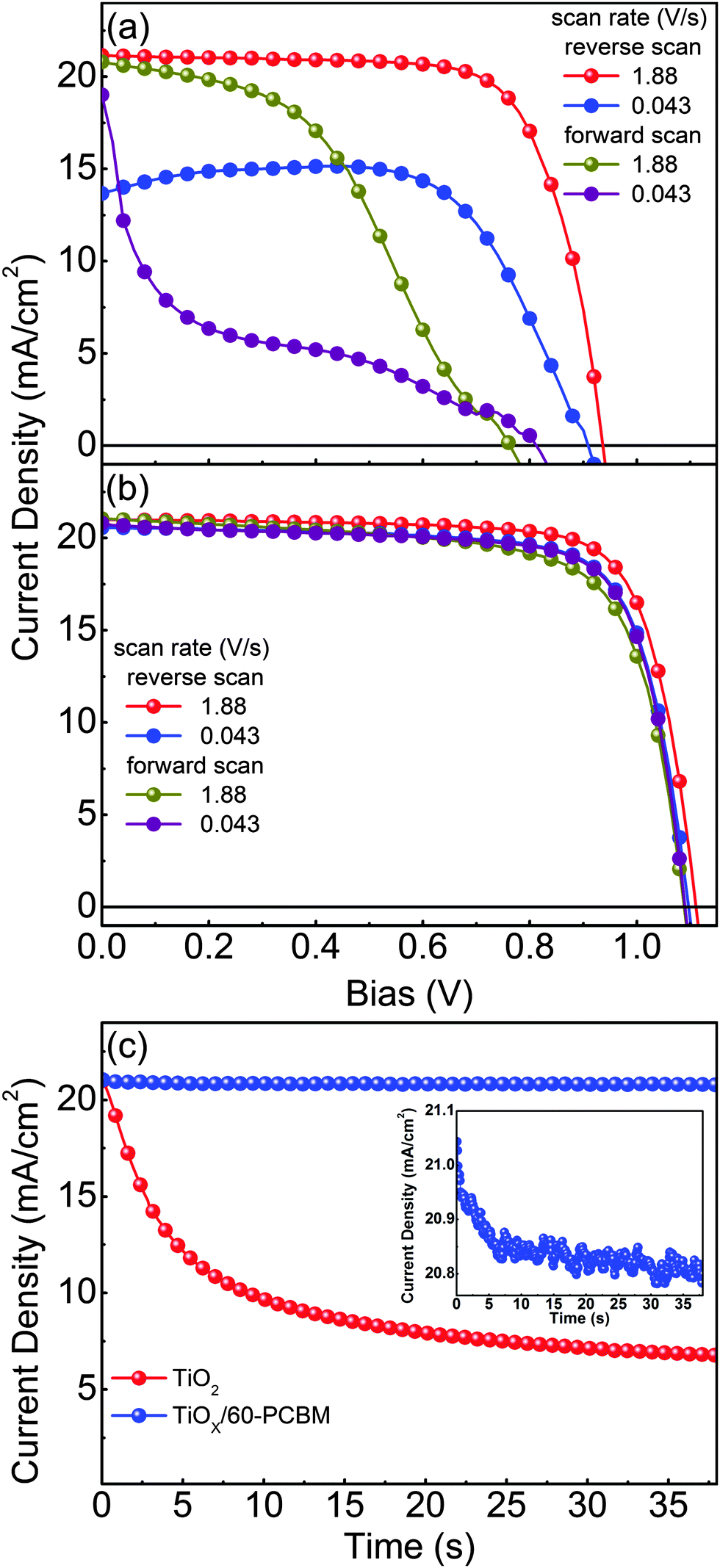 | ||
| Fig. 2 J–V curves for photovoltaic devices FTO/TiO2/CH3NH3PbI3/spiro-MeOTAD/Au (a) and FTO/TiOX/60-PCBM/CH3NH3PbI3/spiro-MeOTAD/Au (b) measured with different scan rates (V s−1) and different scan directions under AM 1.5G white light illumination with an intensity of approximately 100 mW cm−2. Note that the devices were exposed for 10 s to white light before testing. No pre-polarization has been performed. (c) Corresponding short-circuit current density transients during illumination. | ||
| Device | Scan rate (V s−1) | V OC (V) | J SC (mA cm−2) | FF (%) | PCE (%) | |
|---|---|---|---|---|---|---|
| TiO2 | Reverse scan | 1.88 | 0.94 | 21.1 | 72.5 | 14.4 |
| 0.043 | 0.91 | 13.8 | 69.9 | 8.8 | ||
| Forward scan | 1.88 | 0.76 | 20.8 | 43.9 | 6.9 | |
| 0.043 | 0.81 | 19.0 | 14.7 | 2.3 | ||
| TiOX/60-PCBM | Reverse scan | 1.88 | 1.11 | 21.0 | 76.6 | 17.9 |
| 0.043 | 1.10 | 20.6 | 75.2 | 17.0 | ||
| Forward scan | 1.88 | 1.09 | 21.1 | 70.8 | 16.2 | |
| 0.043 | 1.09 | 20.7 | 74.6 | 16.9 | ||
The TiOX/60-PCBM device delivers instead a PCE under steady state conditions of 17.0%, with VOC = 1.10 V, JSC = 20.6 mA cm−2, FF = 75.2% (see Table 1). The integrated EQE current densities also agree within 10% of the measured white light JSC (see Fig. S3, ESI†). The high VOC demonstrates that the presence of the amorphous TiOX does not open any detrimental recombination/trapping paths as the thin layer of 60-PCBM is enough to drive the optimal device behaviour (note that we also find a linear dependence of the JSC as a function of the incident light intensity, see Fig. S7 in the ESI†). We also tested planar devices with only amorphous TiOX as an EEL, and TiO2/60-PCBM-based solar cells. The TiOX-only devices show very poor performances (see Fig. S4 in the ESI†). The TiO2/60-PCBM-based devices behave similarly to those embodying TiOX/60-PCBM (see Fig. S5 in the ESI†), highlighting the role of the thin-60-PCBM/perovskite interface.
The steady state operation of the TiOX/60-PCBM-based devices gives full credit for the actual implementation of this architecture. However, it is fair to notice that the TiOX/60-PCBM-based devices still show a transient behaviour. The JSC transient shows that there is a loss of ca. 0.2 mA cm−2 (from 21 to 20.8 mA cm−2, see the inset in Fig. 2(c)). A very mild variation in VOC and/or FF is also found depending on the scan direction. In fact, an enhancement of the PCE can be observed slowing down the scan rate and operating the device with a forward scan, i.e., from 16.2% at a scan rate of 1.88 V s−1 to 16.9% at a scan rate of 0.043 V s−1. On the other hand, a reduction of the PCE can be observed when slowing down the scan rate and operating the device with a reverse scan, i.e., from 17.9% at a scan rate of 1.88 V s−1 to 17.0% at a scan rate of 0.043 V s−1. We have measured the dependence of the transient photocurrent density on the applied bias (Fig. S8 of the ESI†) and we can safely say that a scan rate 0.043 V s−1 is enough to test the steady state behaviour of a solar cell (see also Fig. S6, S7 and S9 of the ESI† for statistics on devices' performances and stability studies). For a direct comparison with the literature values (ref. 6 and 17), we present the stabilized power output close to the maximum power point in Fig. 3, showing that the PCE reaches a stable value as high as 17.6%.
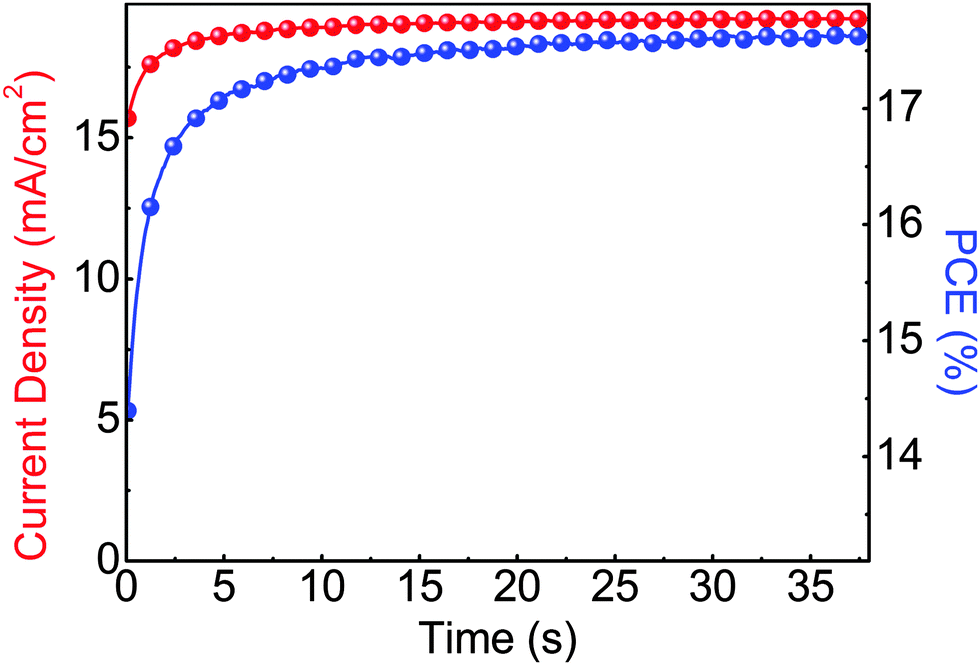 | ||
| Fig. 3 Stabilized current density and power output measured close to the maximum power point (∼0.92 V) for a photovoltaic device of glass/FTO/TiOX/60-PCBM/CH3NH3PbI3/spiro-MeOTAD/Au. | ||
Clearly such behaviours are the result of a transient internal field formation in a time scale of seconds26,27 and that the presence of 60-PCBM has a particularly beneficial role in keeping the steady state performances of the device extremely high. Perovskite grain boundaries passivation by 60-PCBM has been recently suggested for hysteresis mitigation in 60-PCBM-based devices with an inverted architecture.28 However, the extremely thin thickness of the 60-PCBM bottom layer with respect to the perovskite layer (a few tens versus a few hundreds) makes it difficult to consider the passivation as the only dominant mechanism. Liang et al. have also recently highlighted an interesting correlation between the device performances and the EEL electron mobilities when different fullerene derivatives are used as a top contact in inverted planar devices.28 Again, this cannot be directly transferred to our system as the extremely thin thickness of the amorphous oxide, covered by a much thin 60-PCBM layer, is not expected to present carrier mobilities higher than those from the same flat, crystalline TiO2.29,30
To get a deeper insight into the working mechanism of these systems we performed femtosecond (fs)-resolved transient absorption spectroscopy on working devices. In Fig. 4 we show the dynamics of the photo-bleaching (PB) bands (see Fig. S10 in the ESI† for the entire spectral analysis) belonging to the photo-excited perovskite in FTO/TiO2/CH3NH3PbI3/spiro-MeOTAD/Au and FTO/TiOX/60-PCBM/CH3NH3PbI3/spiro-MeOTAD/Au devices when working under short-circuit conditions. Such device architectures allow us, for the first time, to directly compare electron extraction at the TiO2/perovskite and 60-PCBM/perovskite interface. We recall that the PB band dynamics will follow the electron and hole population dynamics in the hybrid perovskite semiconductor as it originates from the transparency induced at the onset of the optical absorption after population of the bottom of the conduction band and top of the valence band by photogenerated electrons and holes, respectively.4 A faster decay of the PB band occurs in TiOX/60-PCBM-based devices with a lifetime of approximately 1.3 ns. Since both devices employ the same HEL – spiro-MeOTAD, we assign this to a more efficient charge transfer at the 60-PCBM/CH3NH3PbI3 interface. Interestingly, when the TiO2-based device is kept at 1 V for a few seconds, and then the perovskite PB dynamic is probed again under short-circuit conditions, its lifetime becomes shorter and matches the lifetime of the PB band in TiOX/60-PCBM-based devices (see Fig. 4), suggesting that the electron transfer is suddenly activated. On the other hand, electron injection at the 60-PCBM interface is hardly affected (see Fig. S11 in the ESI†). This indicates that, upon polarization, the TiO2/perovskite interface is modified and such a modification is needed to allow for an efficient charge transfer (note that during the revision of this manuscript a theoretical work has been published by Jon Azpiroz et al., which suggests a similar scenario).31 This is in full agreement with the fact that the TiO2-based devices generally show a much higher JSC value when the device is pre-polarized.12 In particular, we highlight that the TiOX/60-PCBM-based device shows a JSC of approximately 21 mA cm−2 under short-circuit conditions, with virtually no dependence on the device polarization history (Fig. 2b). On the other hand, the TiO2-based device shows a lower JSC value, unless it is reversely scanned with a very fast scan rate. In such a case the JSC is again ∼21 mA cm−2. This matches the trend observed by TA spectroscopy on the electron transfer dynamics. Recently there has been various experimental evidence for the formation of n and p regions in the perovskite films upon ion migration.32 Importantly, this is independent of the charge extracting layers used.26,27 If there is already an optimal energy alignment, such phenomena will have a minimal effect on the steady state performances of the device, as we suggest in the presence of 60-PCBM. On the other hand, the pre-biasing history can temporarily improve the extraction in devices with poor contact quality. Our results suggest that the compact TiO2 falls in the latter case.
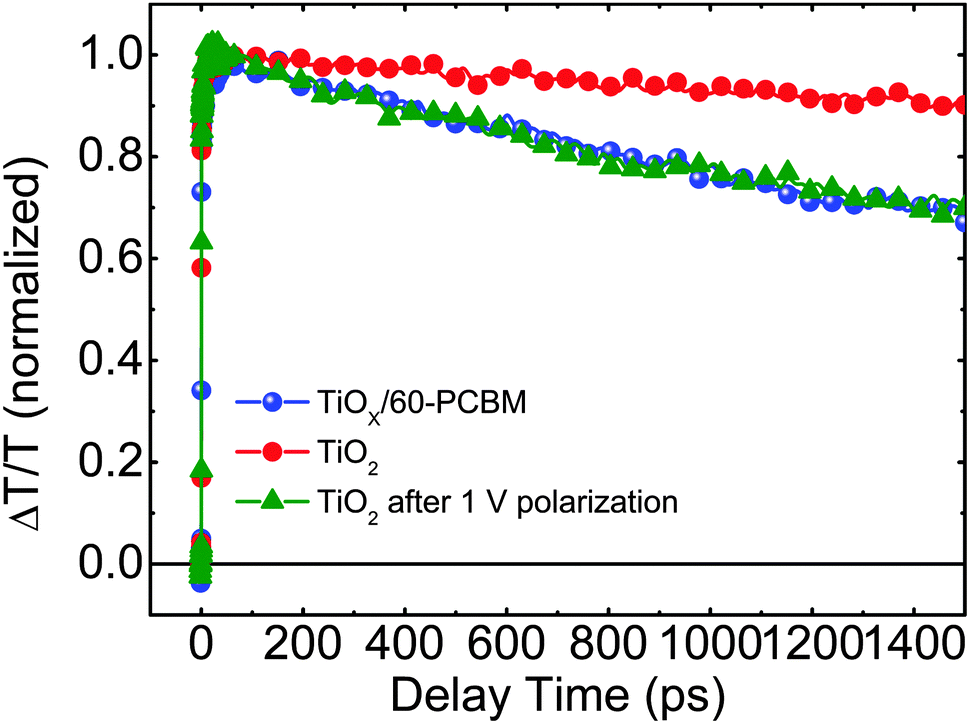 | ||
| Fig. 4 The dynamics of the photo-bleaching bands for the photo-excited perovskites in working devices under short-circuit conditions. | ||
Conclusions
We presented the integration of a low-temperature, solution-processed, transparent EEL, the TiOX/60-PCBM, for planar perovskite solar cells with a standard structure. We have also introduced a novel, hybrid deposition method for hybrid perovskites, which produces good quality polycrystalline thin film independently of the nature of beneath layers, thus opening the solar cell design to a large library of EELs. The TiOX/60-PCBM-based devices reach a stabilized PCE of 17.6% close to the maximum power point and a PCE of 17% extracted from quasi-static J–V. Such solar cells exhibit negligible hysteresis, which has been a huge obstacle in developing conventional planar perovskite solar cells so far. We have evidenced the role of charge extraction in reducing the detrimental effects related to slow transient phenomena. Efficient charge extraction achieved with the use of 60-PCBM as EEL makes the device much less sensitive to the device polarization, thus producing an inherently more stable device. On the other hand, in the presence of a flat TiO2 layer, the electron extraction is strongly dependent on the pre-polarization conditions, thus undermining device stability. Overall, the solution we here propose will foster great advancements in the establishment of a hybrid perovskite-based PV technology thanks to its high level of integrability in both single junction and tandem structures.Acknowledgements
The research leading to these results has received funding from the European Union Horizon 2020 under grant agreement no. 604032 of the Synchornics project and from Fondazione Cariplo (project GREENS no. 2013-0656)”. The authors thank Dr Mario Malerba for support in the experimental procedures.Notes and references
- http://www.nrel.gov/ncpv/images/efficiency_chart.jpg, accessed 24 November 2014.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050 CrossRef CAS PubMed.
- G. Xing, N. Mathews, S. Sun, S. S. Lim, Y. M. Lam, M. Grätzel, S. Mhaisalkar and T. C. Sum, Science, 2013, 342, 344 CrossRef CAS PubMed.
- S. D. Stranks, G. E. Eperon, G. Grancini, C. Menelaou, M. J. P. Alcocer, T. Leijtens, L. M. Herz, A. Petrozza and H. J. Snaith, Science, 2013, 342, 341 CrossRef CAS PubMed.
- D. Shi, V. Adinolfi, R. Comin, M. Yuan, E. Alarousu, A. Buin, Y. Chen, S. Hoogland, A. Rothenberger, K. Katsiev, Y. Losovyj, X. Zhang, P. A. Dowben, O. F. Mohammed, E. H. Sargent and O. M. Bakr, Science, 2015, 347, 519 CrossRef CAS PubMed.
- J. H. Heo, S. H. Im, J. H. Noh, T. N. Mandal, C.-S. Lim, J. A. Chang, Y. H. Lee, H.-j. Kim, A. Sarkar, M. K. Nazeeruddin, M. Grätzel and S. I. Seok, Nat. Photonics, 2013, 7, 486 CrossRef CAS PubMed.
- M. A. Green and T. Bein, Nat. Mater., 2015, 14, 559 CrossRef PubMed.
- H. Zhou, Q. Chen, G. Li, S. Luo, T. B. Song, H. S. Duan, Z. Hong, J. You, Y. Liu and Y. Yang, Science, 2014, 345, 542 CrossRef CAS PubMed.
- D. Liu and T. L. Kelly, Nat. Photonics, 2014, 8, 133 CrossRef CAS PubMed.
- Q. Hu, J. Wu, C. Jiang, T. Liu, X. Que, R. Zhu and Q. Gong, ACS Nano, 2014, 8, 10161 CrossRef CAS PubMed.
- H. J. Snaith, A. Abate, J. M. Ball, G. E. Eperson, T. Leijtens, N. K. Noel, S. D. Stranks, J. T.-W. Wang, K. Wojciechowski and W. Zhang, J. Phys. Chem. Lett., 2014, 5, 1511 CrossRef CAS.
- E. L. Unger, E. T. Hoke, C. D. Bailie, W. H. Nguyen, A. R. Bowring, T. Heumüller, M. G. Christoforo and M. D. McGehee, Energy Environ. Sci., 2014, 7, 3690 CAS.
- Z. Xiao, C. Bi, Y. Shao, Q. Dong, Q. Wang, Y. Yuan, C. Wang, Y. Gao and J. Huang, Energy Environ. Sci., 2014, 7, 2619 CAS.
- Q. Lin, A. Armin, R. C. R. Nagiri, P. L. Burn and P. Meredith, Nat. Photonics, 2015, 9, 106 CrossRef CAS PubMed.
- O. Malinkiewicz, A. Yella, Y. H. Lee, G. M. Espallargas, M. Grätzel, M. K. Nazeeruddin and H. J. Bolink, Nat. Photonics, 2014, 8, 128 CrossRef CAS PubMed.
- C. D. Bailie, M. G. Christoforo, J. P. Mailoa, A. R. Bowring, E. L. Unger, W. H. Nguyen, J. Burschka, N. Pellet, J. Z. Lee, M. Grätzel, R. Noufi, T. Buonassisi, A. Salleo and M. D. McGehee, Energy Environ. Sci., 2015, 8, 956 CAS.
- K. Wojciechowski, S. D. Stranks, A. Abate, G. Sadoughi, A. Sadhanala, N. Kopidakis, G. Rumbles, C.-Z. Li, A. K.-Y. Jen and H. J. Snaith, ACS Nano, 2014, 8, 12701 CrossRef CAS PubMed.
- S. Ryu, J. Seo, S. S. Shin, Y. C. Kim, N. J. Jeon, J. H. Noh and S. I. Seok, J. Mater. Chem. A, 2015, 3, 3271 CAS.
- J. M. Ball, M. M. Lee, A. Hey and H. J. Snaith, Energy Environ. Sci., 2013, 6, 1739 CAS.
- P. Docampo, F. C. Hanusch, S. D. Stranks, M. Döblinger, J. M. Feckl, M. Ehrensperger, N. K. Minar, M. B. Johnston, H. J. Snaith and T. Bein, Adv. Energy Mater., 2014, 4, 1400355 Search PubMed.
- A. H. Ip, L. N. Quan, M. M. Adachi, J. J. McDowell, J. Xu, D. H. Kim and E. H. Sargent, Appl. Phys. Lett., 2015, 106, 143902 CrossRef PubMed.
- W. Zhang, M. Saliba, D. T. Moore, S. K. Pathak, M. T. Hörantner, T. Stergiopoulos, S. D. Stranks, G. E. Eperson, J. A. Alexander-Webber, A. Abate, A. Sadhanala, S. Yao, Y. Chen, R. H. Friend, L. A. Estroff, U. Wiesner and H. J. Snaith, Nat. Commun., 2015, 6, 6142 CrossRef PubMed.
- Q. Chen, H. Zhou, Z. Hong, S. Luo, H.-S. Duan, H.-H. Wang, Y. Liu, G. Li and Y. Yang, J. Am. Chem. Soc., 2014, 136, 622 CrossRef CAS PubMed.
- J. Burschka, N. Pellet, S.-J. Moon, R. Humphry-Baker, P. Gao, M. K. Nazeeruddin and M. Grätzel, Nature, 2013, 499, 316 CrossRef CAS PubMed.
- E. Mosconi, E. Ronca and F. De Angelis, J. Phys. Chem. Lett., 2014, 5, 2619 CrossRef CAS.
- Z. Xiao, Y. Yuan, Y. Shao, Q. Wang, Q. Dong, C. Bi, P. Sharma, A. Gruverman and J. Huang, Nat. Mater., 2015, 14, 193 CrossRef CAS PubMed.
- Y. Zhao, C. Liang, H. Zhang, D. Li, D. Tian, G. Li, X. Jing, W. Zhang, W. Xiao, Q. Liu, F. Zhang and Z. He, Energy Environ. Sci., 2015, 8, 1256 CAS.
- Y. Shao, Z. Xiao, C. Bi, Y. Yuan and J. Huang, Nat. Commun., 2014, 5, 5784 CrossRef CAS PubMed.
- P.-W. Liang, C.-C. Chueh, S. T. Williams. and A. K.-Y. Jen, Adv. Energy Mater., 2015, 5, 1402321 Search PubMed.
- H. Tang, K. Prasad, R. Sanjinès, P. E. Schmid and F. Lévy, J. Appl. Phys., 1994, 75, 2042 CrossRef CAS PubMed.
- J. M. Azpiroz, E. Mosconi, J. Bisquert and F. De Angelis, Energy Environ. Sci., 2015, 8, 2118 Search PubMed.
- Y. Zhang, M. Liu, G. E. Eperon, T. C. Leijtens, D. McMeekin, M. Saliba, W. Zhang, M. de Bastiani, A. Petrozza, L. M. Herz, M. B. Johnston, H. Lin and H. J. Snaith, Mater. Horiz., 2015, 2, 315 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ee01720c |
| This journal is © The Royal Society of Chemistry 2015 |