
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Record activity and stability of dealloyed bimetallic catalysts for proton exchange membrane fuel cells†
Binghong
Han
ac,
Christopher E.
Carlton
ac,
Anusorn
Kongkanand
*d,
Ratandeep S.
Kukreja
d,
Brian R.
Theobald
e,
Lin
Gan
f,
Rachel
O'Malley
e,
Peter
Strasser
*f,
Frederick T.
Wagner
d and
Yang
Shao-Horn
*abc
aDepartment of Materials Science and Engineering, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA. E-mail: shaohorn@mit.edu
bDepartment of Mechanical Engineering, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA
cElectrochemical Energy Laboratory, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA
dElectrochemical Energy Research Lab, General Motors Powertrain, Honeoye Falls, NY 14472, USA. E-mail: anusorn.kongkanand@gm.com
eJohnson Matthey Technology Centre, Blount's Court, Sonning Common, Reading, RG4 9NH, UK
fThe Electrochemical Energy, Catalysis and Materials Science Laboratory, Department of Chemistry, Chemical Engineering Division, Technical University Berlin, Berlin 10623, Germany. E-mail: pstrasser@tu-berlin.de
First published on 12th September 2014
Abstract
We demonstrate the unprecedented proton exchange membrane fuel cell (PEMFC) performance durability of a family of dealloyed Pt–Ni nanoparticle catalysts for the oxygen reduction reaction (ORR), exceeding scientific and technological state-of-art activity and stability targets. We provide atomic-scale insight into key factors controlling the stability of the cathode catalyst by studying the influence of particle size, the dealloying protocol and post-acid-treatment annealing on nanoporosity and passivation of the alloy nanoparticles. Scanning transmission electron microscopy coupled to energy dispersive spectroscopy data revealed the compositional variations of Ni in the particle surface and core, which were combined with an analysis of the particle morphology evolution during PEMFC voltage cycling; together, this enabled the elucidation of alloy structure and compositions conducive to long-term PEMFC device stability. We found that smaller size, less-oxidative acid treatment and annealing significantly reduced Ni leaching and nanoporosity formation while encouraged surface passivation, all resulting in improved stability and higher catalytic ORR activity. This study demonstrates a successful example of how a translation of basic catalysis research into a real-life device technology may be done.
Broader contextReduction of costly Pt usage in proton exchange membrane fuel cell electrodes is one of the major challenges towards the development and commercialization of fuel cell vehicles. In order for fuel cell vehicles to be economically viable, the US Department of Energy (DOE) has set specific performance targets for initial fuel cell catalyst activity and fuel cell durability. While a number of different low-Pt catalyst concepts have met the initial-activity target using preliminary and idealized disk electrode screening tests, few to none have met the initial-activity requirements in a realistic fuel cell device. No catalyst material has ever met the demanding fuel cell durability targets. Here we report previously unachieved catalyst activity and device durability in fuel cells measured under automotive testing conditions. The performance durability of this new fuel cell catalyst meets and exceeds the official 2017 DOE targets. This step-change in fuel cell catalyst performance was made possible through a uniquely effective DOE-funded team of academia and industrial catalyst and MEA developers. This enabled a swift translation of fundamental atomic-scale insights on catalyst synthesis, activity and degradation of dealloyed core–shell nanoparticles into industrial catalyst material scale-up coupled to unprecedented device performance. |
1. Introduction
Proton exchange membrane fuel cells (PEMFCs) represent a means of converting renewable chemical energy into electrical energy with high theoretical efficiency and good enough power density for it to be of great interest to automotive applications. Due to the sluggishness of the oxygen reduction reaction (ORR) at the cathode of PEMFCs, Pt-based catalysts are required to achieve reasonable overpotentials at realistic current densities.1 Although Pt nanoparticles supported on high surface area carbon have traditionally been used as the ORR catalyst, high Pt loadings needed to achieve sufficient power densities for automotive demands make them unsuitable for commercialization due to the preciousness and limited supply of Pt.1,2 Studies of bimetallic Pt–M surfaces and nanoparticles, where M is a transition metal, have demonstrated several times higher intrinsic ORR activity than pure Pt.3–17 This catalytic enhancement has been attributed to optimizing the oxygen absorption energy of surface Pt through strain5,10,11,18–20 and ligand6–8,11,21–23 effects in a Pt sandwich segregation structure induced by annealing3,4,7,24,25 or a Pt skeleton structure induced by acid leaching of transition metal.3,4,7,18,26,27Although supported Pt–M nanoparticles have shown greater stability than supported Pt nanoparticles,5–9,17,27,28 considerable losses are noted in the specific and mass activities of Pt–M nanoparticle catalysts during fuel cell operation.28–32 The activity loss can result from the dissolution and loss of Pt from the cathode,33,34 reduction of strain and ligand effects associated with the dissolution of transition metal,28,32,35,36 re-deposition of dissolved Pt on the particle surface,13,28,33,35,36 and Erlebacher-like37–41 dealloying. Previous work has shown that the Pt–M catalyst stability is highly dependent on the composition, size and morphologies of catalyst nanoparticles.4,16,26–28,35,42–47 Of significance, previous work has shown that having Ni or Co compositions greater than 50 at.% in PtxNi or PtxCo nanoparticles could lead to the formation of spongy particles with nano-scale porosity due to severe metal leaching26,28 while small particles below certain critical size could avoid porosity formation.16,42,43 However, the effects of catalyst preparation such as the acid-leaching protocol and post-acid-leaching heat treatment, on metal leaching and surface porosity formation are still poorly understood. Although recent studies show that de-alloyed Pt-based catalysts can exhibit remarkably high activities,48–50 these catalysts have been examined using rotating disk electrode (RDE) measurements but not by membrane electrode assembly (MEA) testing, which is critical to the development of efficient, durable MEAs for PEMFCs.
In this work, we have systematically investigated the influence of precursor morphology, acid-leaching conditions and post-acid-treatment annealing on nanoporosity and surface passive layer formation of Pt–Ni nanoparticles, as well as the consequent evolution of their compositional, electrochemical and morphological properties during PEMFC operation. What sets this work apart from previous studies7,9,14,24,48–50 is the combination of systematic changes in the catalyst preparation conditions, MEA testing and durability data with detailed transmission electron microscopy (TEM) characterization before and after PEMFC testing. This combination made a step-change in fuel cell catalyst performance and enabled a swift translation of fundamental atomic-scale insights on dealloyed core–shell nanoparticles into industrial catalyst material scale-up coupled to unprecedented device performance. Our results show that smaller particle size, non-oxidative acid treatment and post-acid-treatment annealing can reduce transition metal leaching from catalyst nanoparticles, and suppress the nanoporosity formation. This provides insights into the design of Pt–M nanoparticle catalysts with enhanced stability and activity. As a result, we demonstrate record PEMFC cathode activity and durability exceeding scientific and technological targets.51,52
2. Experimental
2.1 Catalyst and electrode preparation
Two PtNi3 catalyst precursors (P1 and P2) supported on high-surface-area carbon were developed using highly scalable synthesis routes and provided by Johnson Matthey Fuel Cells Ltd (UK), which showed very similar shapes and Pt![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ni ratios but significantly different particle size distributions (PSDs)53 having P2 with smaller particles size (∼5 nm) with a narrower PSD, compared to P1 with larger particles of ∼8 nm (Fig. S1†). Here PtNi3 was chosen as the precursor for a number of reasons. First, the initial 75 at.% Ni content is safely above the parting limit of Pt–Ni solid solution alloys and thus ensures the existence of a critical dealloying potential at which Ni selectively leaches from the precursor particles. Consistent with earlier studies,26,44 this dealloying process results in the formation of high surface area core shell nanoparticle catalysts with compact Pt skin. A series of different acid-leaching conditions and post-dealloying treatments were applied to these precursors at GM, as shown in Fig. 1. The precursors were either treated in (1) 1 M nitric acid at 70 °C for 24 h in air (marked as P1-NA and P2-NA) or (2) 0.5 M sulfuric acid at 80 °C for 24 h in nitrogen (P2-SA). The resultant catalysts were then thoroughly washed with DI water and dried in nitrogen at room temperature. After the dealloying, some P2-SA catalysts were thermally annealed at 400 °C for 4 h in 5% H2 and 95% N2,54 marked as P2-SA-AN. For comparison, 46 wt% Pt supported on Vulcan carbon from TKK (Tanaka Kikinzoku K.K., Japan) was used.
:Ni ratios but significantly different particle size distributions (PSDs)53 having P2 with smaller particles size (∼5 nm) with a narrower PSD, compared to P1 with larger particles of ∼8 nm (Fig. S1†). Here PtNi3 was chosen as the precursor for a number of reasons. First, the initial 75 at.% Ni content is safely above the parting limit of Pt–Ni solid solution alloys and thus ensures the existence of a critical dealloying potential at which Ni selectively leaches from the precursor particles. Consistent with earlier studies,26,44 this dealloying process results in the formation of high surface area core shell nanoparticle catalysts with compact Pt skin. A series of different acid-leaching conditions and post-dealloying treatments were applied to these precursors at GM, as shown in Fig. 1. The precursors were either treated in (1) 1 M nitric acid at 70 °C for 24 h in air (marked as P1-NA and P2-NA) or (2) 0.5 M sulfuric acid at 80 °C for 24 h in nitrogen (P2-SA). The resultant catalysts were then thoroughly washed with DI water and dried in nitrogen at room temperature. After the dealloying, some P2-SA catalysts were thermally annealed at 400 °C for 4 h in 5% H2 and 95% N2,54 marked as P2-SA-AN. For comparison, 46 wt% Pt supported on Vulcan carbon from TKK (Tanaka Kikinzoku K.K., Japan) was used.
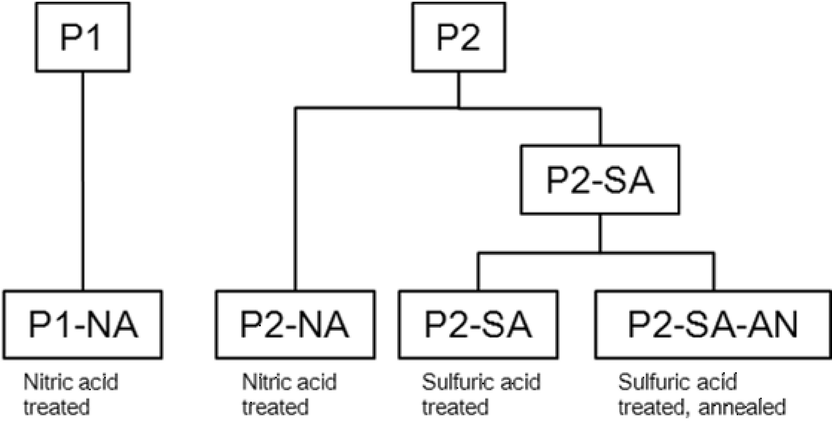 | ||
| Fig. 1 Schematic showing different catalysts studied in this paper. The precursors were either treated in (1) 1 M nitric acid at 70 °C for 24 h in air (P1-NA and P2-NA) or (2) 0.5 M sulfuric acid at 80 °C for 24 h in nitrogen (P2-SA). After the dealloying, some P2-SA catalysts were thermally annealed at 400 °C for 4 h in 5% H2 and 95% N2, marked as P2-SA-AN. | ||
Catalyst-coated membranes (CCMs) of 50 cm2 were used in fuel cell testing, which were prepared using the decal transfer method, with a loading of 0.1 mgPt cm−2 in the cathode and 0.05 mgPt cm−2 in the anode. Nafion® D2020 ionomer, with an equivalent weight of 950 g equiv.−1, was utilized with an ionomer to carbon weight ratio of 0.95 and 0.6 in the cathode and anode, respectively. Catalysts derived from P1 and P2 precursors were studied in the cathode while 20 wt% Pt/C was used as the anode catalyst. Nafion® NRE211 (25 μm thick) was used as the membrane. The gas diffusion medium used was carbon fiber paper backing (∼200 μm) coated with a microporous layer (MPL, ∼30 μm). The detailed procedure for the fabrication of electrodes and MEA can be found in previous work.1
2.2 Electrochemical measurements
The MEAs were conditioned in a fuel cell for about 7 h followed by a series of electrochemical measurements to give a conditioned MEA after a total run time of 17 h. To accelerate the degradation of the cathode catalyst, some MEAs were then subjected to voltage cycling tests which were performed in 200/50 standard cubic cm per min of H2/N2 at 80 °C, 100% RH and 101 kPaabs for up to 30000 (30k) cycles at a scan rate of 50 mV s−1 between 0.6 and 1.0 V vs. RHE (30k-cycled). Averages from at least 4 MEAs with standard deviations are reported.
At each aging stage, electrochemical properties such as mass and area-specific ORR activities, and Pt electrochemically active surface area (ECSA) were measured. The mass and specific activities of oxygen reduction reaction at the cathode were calculated at 0.9 V vs. the reversible hydrogen electrode (RHE), which was obtained from the H2/O2 polarization curve collected at 80 °C, 100% relative humidity (RH) and 150 kPaabs after high frequency resistance-correction.1 Polarization curves in H2/air were obtained at 80 °C, 100% RH, and 170 kPaabs. Here we assume that the Pt loading did not change during electrochemical measurements, and the initial Pt loading in the MEA was used in the mass activity calculation. The ECSAs were obtained from the H adsorption/desorption (HAD) peaks using cyclic voltammetry.55 Although it has been shown in single-crystal electrode studies that the hydrogen adsorption charge per surface area (the conversion constant used in the HAD method) would decrease with increasing Co or Ni content in Pt–Co or Pt–Ni alloys due to a modified electronic structure,56–58 meanwhile the electrochemical CO stripping might give a different ECSA than those measured by HAD,48 both methods gave comparable results for all of our MEA studies with CO giving a 5–20% higher value than HAD does.57 Therefore, only ECSAs measured by H adsorption/desorption were reported in this work, ignoring the influence of Ni content change on the HAD conversion constant. The evolution of ECSA before and after 30k cycles is shown in Fig. S2 in the ESI.†
2.3 Electron probe micro-analysis
Cross-sectioned MEA samples were studied by electron probe micro-analysis (EPMA) using an SX100 electron probe micro-analyzer from CAMECA Instruments, Inc. Small pieces of the sample, approximately 4 × 10 mm, were cut from the center of the MEAs and mounted in brominated epoxy under vacuum. After allowing the epoxy to harden, the samples were polished to a mirror finish manually using standard polishing papers followed by lapping using 1-micron diamond suspension. A thin layer (∼3 nm) of Au–Pd alloy was then sputter coated immediately after the final lapping to provide surface electron conductivity needed for EMPA. The cross-sectioned MEAs were analyzed with an electron probe of 15 kV and 10 nA. An area of 75 × 75 μm covering the anode MPL, anode, membrane, cathode and cathode MPL, was scanned with a step size of 0.3 μm at 10 ms per step to generate elemental maps of Pt and Ni. Wavelength dispersive spectrometers were used to collect X-rays of Pt-Mα and Ni-Kα lines using the thallium acid phthalate crystal and large lithium fluoride crystal, respectively. At least three such maps were generated from different locations on each sample. X-ray intensities from pure Pt and Ni were determined to calculate k-ratios from X-ray intensities in band profiles constructed from X-ray maps, thus allowing conversion to weight/atomic percent concentration profiles. The concentration profiles thus generated were used for quantification and distribution analyses of Pt and Ni atomic percentages. Inductively coupled plasma-atomic emission spectrometry on the non-active-area membranes was also performed to quantify the amount of Pt and Ni that migrated away from the active area.2.4 Transmission electron microscopy
TEM samples of cathode catalysts in the MEA were prepared by using a razor to remove the carbon paper diffusion layer, carefully cleaning the MPL off of the membrane and scraping the membrane directly into a scintillation vial. The cathode scrapings were sonicated briefly in ethanol (Sigma-Aldrich, r 99.5%), and then applied to TEM grids (Electron Microscopy Science, lacy carbon/copper film 200 mesh). All TEM studies were performed on a JEOL 2010F microscope equipped with an ultrahigh resolution polepiece, having a point resolution of 0.19 nm. For each sample, over 100 catalyst nanoparticles under bright-field-diffraction-contrast imaging were used to determine the PSDs. The bright field diffraction contrast images were formed by aligning a 20 μm aperture with the transmitted beam, and captured by a CCD camera. The digital images were analyzed using Gatan Digital Micrograph v2.01 (Gatan Inc.) and ImageJ v1.44p (National Institute of Health, USA). High-resolution TEM (HRTEM) images were obtained without an objective aperture and were analyzed using a Gatan Digital Micrograph. High angle annular dark field (HAADF) scanning TEM (STEM) images were formed by aligning the Ronchigram with optic axis and a 50 μm condenser aperture with the center of the Ronchigram. An electron beam probe size of 0.5 nm and a camera length of 40 cm were used to minimize the effect of diffraction contrast on the HAADF images. Parallel-beam and STEM energy dispersive spectroscopy (EDS) data were collected from individual catalyst particles and analyzed using the Inca (Oxford Instruments) software. The Ni and Pt atomic compositions were determined from the Ni-K series and the Pt-L series using the INCA software as described in previous work.35 For parallel-beam EDS measurements, three different spots with a diameter of ∼200 nm (totally containing over 100 particles) were used to collect the average atomic percentage of Ni for each sample. The uncertainty in the EDS-determined atomic percentage is ∼10% of the measured value.3. Results and discussion
3.1 PtxNi PEM single cell performance activity and durability
PEMFC polarization curves of cathodes with P1-NA or P2-NA are compared with that of a commercial Pt/C catalyst (from TKK) in Fig. 2a. Despite the fact that P1-NA and P2-NA cathodes (0.1 mgPt cmgeo.−2) had a Pt loading which is four times lower than that of the commercial Pt/C (0.4 mgPt cmgeo.−2), P1-NA and P2-NA showed comparable voltage profiles as a function of geometric current density. After 30k cycles (0.6–1.0 V vs. RHE) of accelerated voltage cycle testing, both P1-NA and P2-NA showed noticeable voltage loss (dashed lines), with smaller voltage loss for the P2-NA cathode compared to P1-NA after cycling, which indicates lower mass activity loss. It is interesting to note that the voltage loss at high current densities can be greatly mitigated by lowering the upper voltage limit to which the voltage cycling was performed from 1.0 V to 0.925 V despite more extensive cycles (Fig. S3†). The smaller loss may be attributed to decreases in Pt surface area loss, mass activity loss, Ni and Pt dissolution, and carbon corrosion.59 The specific ORR activity of the P1-NA cathode was reduced by ∼1/3 after 10k cycles (Fig. 2b), which was further reduced from 10k to 30k cycles. In contrast, the specific activities of P2-NA and the other P2-derived cathodes were found to increase with PEMFC cycling, where activities greater than 1.7 mA cmpt−2 were obtained after 30k cycles. Moreover, the mass activity of the P1-NA catalyst reduced considerably during PEMFC cycling (Fig. 2c), where ∼1/2 and 2/3 of the activity found in the conditioned MEA was lost after 10k and 30k cycles, respectively. On the other hand, the P2-NA cathode as well as the other P2-derived catalysts showed much smaller loss in mass activity than P1-NA during cycling. Of significance, all P2-derived catalysts surpassed the US Department of Energy 2017 PEMFC cathode catalyst target (initial activity of conditioned MEA >0.44 A mgPt−1 and <40% activity loss after 30k voltage cycling),51 which has not yet been reported for any catalysts and sets the record for MEA catalyst mass activity and stability, as shown in Fig. 2d.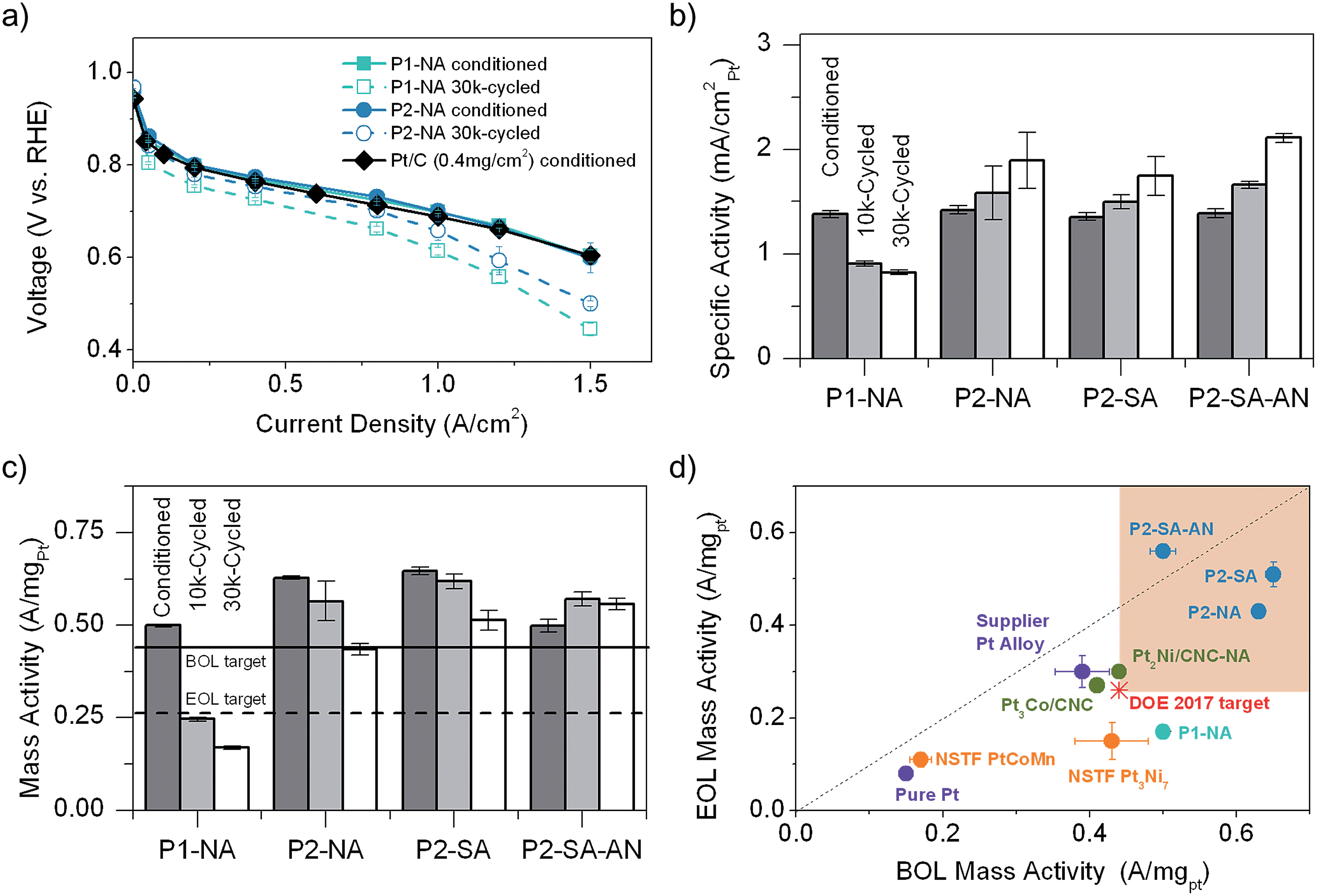 | ||
| Fig. 2 (a) Fuel cell polarization curves of MEAs with P1-NA and P2-NA cathodes at different aging stages (conditioned and 30k-cycled), and of conditioned Pt as a reference. The catalyst loadings were 0.1 and 0.4 mgPt cm−2 for the dealloyed PtNi and commercial Pt catalysts, respectively. Testing conditions of the anode/cathode were H2/air, 100/100% RH, 170/170 kPaabs back pressure, and stoichiometric coefficient of 1.5/2, respectively. (b) Specific activity and (c) mass activity of the catalysts made using P1 and P2. The activities were measured at 0.9 V (vs. RHE) from the H2/O2 polarization curve collected at 80 °C, 100% relative humidity (RH) and 150 kPaabs after high frequency resistance-correction, where dark grey, light grey and white represent conditioned, 10k-cycled and 30k-cycled MEA, respectively. From (a) to (c) the error bars represent the standard deviations of activity measurements among at least 4 MEA samples. (d) The end-of-life (EOL, 30k-cycled MEA) vs. beginning-of-life (BOL, conditioned MEA) mass activity for catalysts from this work and from previous work on MEAs: 3 M NSTF PtCoMn, NSTF Pt3Ni7,61 Pt3Co/CNC and Pt2Ni/CNC,62 Supplier Pt Alloy and pure Pt.52,63 The solid and dash horizontal lines in (c) and the red star and the green square regions represent the DOE 2017 PEMFC cathode catalyst targets for BOL mass activity (0.44 A mgPt−1) and EOL mass activity (0.26 A mgPt−1),51 respectively. | ||
Previous studies have shown that the reduced specific activity loss of Pt-alloy catalysts can be attributed to transition metal dissolution28,32,35,36 while mass activity loss can result from loss in the specific activity and electrochemically active surface area due to Pt dissolution,33,60 coarsening34,60 and coalescence of Pt nanoparticles.34,35 We will examine and discuss differences in the Ni atomic percentage change in the MEA, particle size, morphologies and Ni atomic percentage distribution within individual particles of P1-NA and P2-derived catalysts before and after PEMFC cycling in order to explain their markedly different stability in PEMFCs.
3.2 Catalyst composition changes inside the MEA cathode after PEMFC testing
Examining the average Ni atomic percentage of catalyst particles in CCMs using EMPA, we found that the P1-NA catalyst had much less Ni than P2-derived catalysts in the pristine CCMs, as shown in Fig. 3a. In addition, sulfuric acid appeared to remove less Ni from the P2-derived catalysts than nitric acid. PEMFC conditioning testing further reduced the Ni atomic percentage in these catalysts, where the largest loss of Ni was found for P1-NA. Moreover, PEMFC cycling led to significant Ni loss in all catalysts, where P2-SA-AN was found to have the highest Ni percentage (∼15 at.%) while P1-NA had the least Ni percentage (∼5 at.%) after 30k cycles. These various Ni leaching results are different from a previous Pt–Co study, where the remaining Co at.% reached a constant value after HClO4 treatment irrespective of the initial composition, implying that the different particle size, acid-treatment and annealing conditions have a greater influence than initial Pt content on metal leaching.64 As shown in Fig. 3b, the mass activities of P1- and P2-based catalysts before and after cycling were found to largely correlate with the Ni atomic percentage in the cathode. The P2-derived catalysts showed the maximum mass activities with ∼30 at.% of Ni, which is consistent with previous studies.65 However, the decrease in mass activities after 30k cycles was largely governed by the decrease in Pt surface area (Pt surface area decreased from ∼45 to ∼25 m2 gPt−1 for the conditioned and 30k-cycled samples regardless of the catalysts, as shown in Fig. S2†). In contrast, the specific activities (the activity per surface area of Pt) of these catalysts were found largely independent of Ni atomic percentage in the MEA, as shown in Fig. 3c, which highlights the importance of the near surface composition on the catalytic activity.26 Here it is important to note that the estimation of ECSA using the HAD method may be affected by the leaching of Ni,56 and, therefore, Fig. 3c highlights the trends of activity change after cycling. Moreover, synchrotron X-ray absorption studies of these catalysts showed that the composition of the subsurface Ni could greatly affect the distribution of different adsorbed oxygen species and hence the ORR activity.66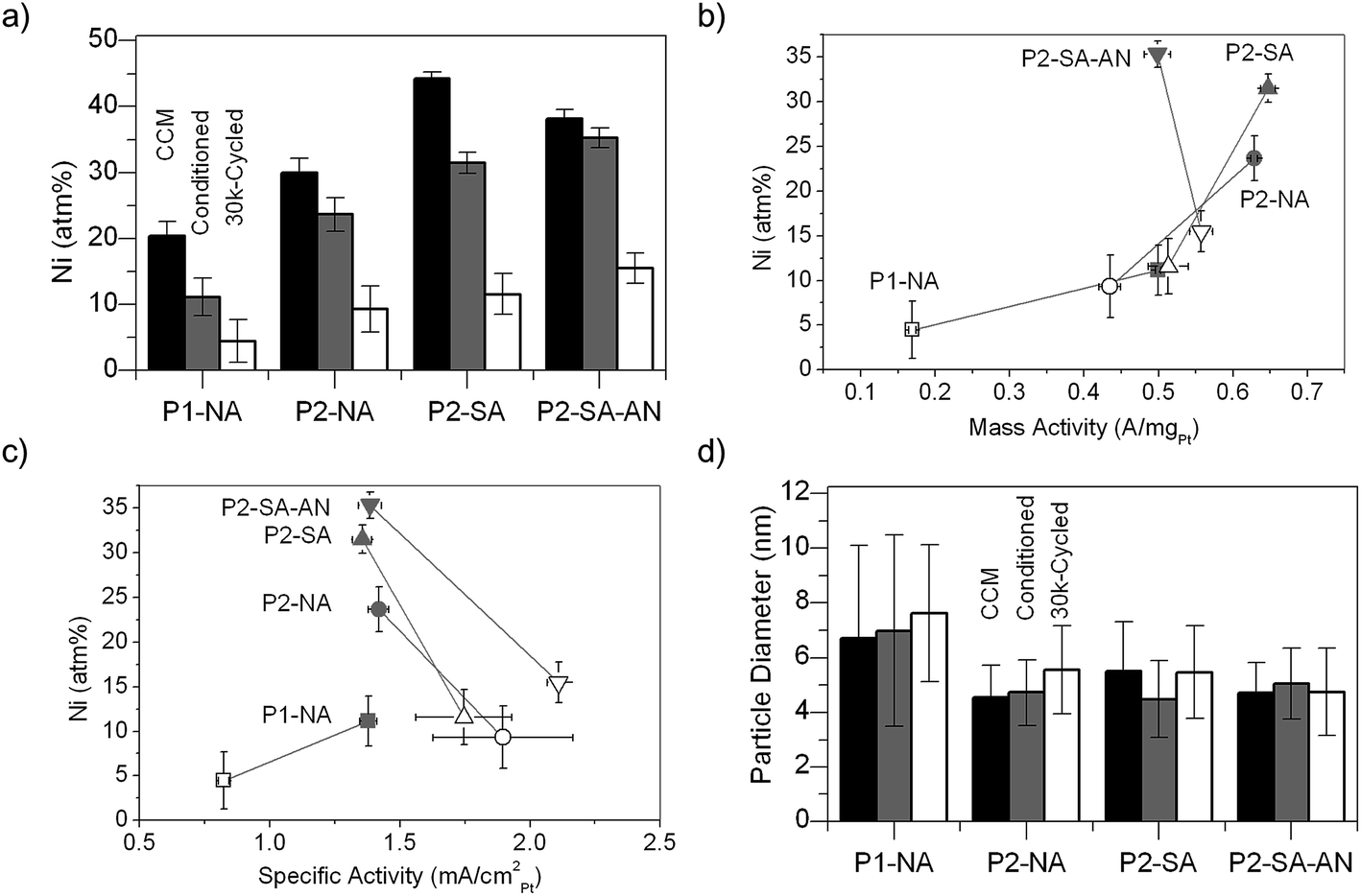 | ||
| Fig. 3 (a) EPMA-determined Ni atomic percentages, where black, dark grey, and white represent pristine CCM, conditioned and 30k-cycled MEA, respectively. (b) and (c) The average Ni atomic percentages determined by EPMA plotted against the mass and specific activities of the MEAs, respectively (dark grey: conditioned; white: 30k-cycled). From (a) to (c), the activity error bars represent the standard deviation of activity measurements among at least 4 MEA samples, while the Ni-content error bars represent the standard deviation of EMPA measurements among at least 3 different locations. (d) TEM-determined mean particle size from different MEAs, where black, dark grey and white represent pristine CCM, conditioned MEA and 30k-cycled MEA, respectively. For each sample over 100 particles were measured to get the average size and the error bars represent the standard deviation. | ||
3.3 Correlating particle size, morphology and composition of dealloyed nanocatalysts in the CCM
P1-NA catalysts were found to have larger particle sizes (∼7 nm) with a broader particle distribution (σ ∼3 nm) than P2-NA (5 nm ± 1 nm) from TEM analysis, as shown in Fig. 3d. Here σ represents the standard deviation of PSD of more than 100 particles for each sample. At the same time, P2-NA, P2-SA and P2-SA-AN showed similar particle sizes of ∼5 nm, implying that the size effect was not the dominating factor among these particles compared with acid-treatment and annealing effects.The EDS data collected from at least 3 different locations (200 nm-diameter circular areas) totally containing over 100 nanoparticles showed that P1-NA contained less Ni (19 ± 6.9 at.%) than P2-NA (25 ± 3 at.%), which is in good agreement with the average Ni atomic percentages obtained from EPMA results in Fig. 3a. In addition, while both P1-NA and P2-NA consist of both solid and porous particles, P1-NA exhibits a higher degree of nanoporosity than P2-NA, which has been observed in different Pt–M nanoparticles and different acid-leaching conditions in previous studies as “spongy particles”.16,28,35,67 The formation of these percolated nanoparticles is typically explained by dealloying with a critical size, which is defined as the lower limitation of particle size for nanoporosity formation under given leaching conditions and particle composition.35,37,38 In P1-NA, the porosity could be found from particles as large as 20 nm and as small as 6 nm (see the top right inset of Fig. 4a), indicating a critical size of ∼6 nm, which was smaller than the critical size reported by previous work16,42,43 ranging from ∼10–30 nm. These findings further confirm that P1-NA experienced more extensive Ni leaching than P2-NA, which may result from the presence of larger particles. The atomic origin of the porosity formation in alloy particles above the critical size is associated with the competition between the relative rates of metal dissolution and Pt surface diffusion, which favors Rayleigh surface instabilities resulting in bulk dealloying.37–41 Large particles that are above the Rayleigh critical length will have an unstable surface and become more vulnerable to acid leaching. Assuming this mechanism, we would expect the critical particle size decrease as the acid leaching conditions become more oxidative, because this would increase the Ni dissolution rate. The observation of these small porous particles in this study can be attributed to the fact that we used aerated hot oxidative acid (HNO3), which is more oxidative compared to room temperature non-oxidative acid (HClO4 or H2SO4) in previous studies.16,42,43 The lower nanoporosity in P2-NA might be attributed to most P2-NA nanoparticles having an average size of ∼5 nm, below the critical size for nanoporosity formation provided that ∼6 nm was the critical size.
 | ||
| Fig. 4 Bright-field diffraction contrast TEM images with PSDs, Ni atomic percentages determined from parallel-beam EDS (left) and HAADF STEM images with point-mode EDS (right) from catalyst particles taken from pristine CCMs. (a) and (b) P1-NA, (c) and (d) P2-NA, (e) and (f) P2-SA, (g) and (h) P2-SA-AN. Here σ represents the standard deviation of PSD from at least 100 measurements and the standard deviation of EDS results among 3 different locations. The insert on the top right corner is the HRTEM image of a nanoparticle with a diameter of ∼5 nm that clearly shows porosity from CCM. | ||
We also found that leaching in H2SO4 in N2, which is substantially milder in oxidation than the nitric acid, could facilitate the development of a Pt-rich skin near the surface3,4,16,28,35 and reduce the nickel leaching. P2-SA in the CCM showed a much higher Ni content than P2-NA in the CCM (Fig. 4c and e). In addition, P2-SA had nanoparticles with dense core–shell structures with Pt-rich surface layers in the HAADF STEM image for all observed particles, which is supported by EDS analysis, as shown in Fig. 4f. Such Pt-rich surface layers were also observed in P2-SA-AN particles (Fig. 4g and h). In contrast, such Pt-rich shells near the particle surface were not as noticeable in the P2-NA.
3.4 Particle size, morphological and compositional changes upon PEMFC conditioning and cycling
A small increase of ∼1 nm in the average particle size was found for P1-NA and P2-derived catalysts after PEMFC conditioning and cycling (except for P2-SA whose size first decreased after conditioning and then increased after cycling), as shown in Fig. 4 and 5 and S2–S4,† but the small size changes were still within experimental uncertainty (the standard deviation of particle sizes is ∼1 to 3 nm). Area EDS analysis revealed significant Ni loss from P1-NA and little Ni remained in catalyst particles after PEMFC conditioning and cycling (Fig. S4a† and 5a), which is in good agreement with EPMA results discussed previously. P2-NA was found to exhibit less Ni loss from area EDS analysis during PEMFC conditioning and after 30k cycles. Remarkably, P2-SA and P2-SA-AN retained considerable Ni after conditioning and even after 30k cycling, with atomic percentages of ∼24 ± 7% and ∼31 ± 5%, respectively (Fig. 5e and g). | ||
| Fig. 5 Bright-field diffraction contrast TEM images with PSDs, Ni atomic percentages determined from parallel-beam EDS (left) and HAADF STEM images with point-mode EDS (right) for catalyst particles taken from 30k-cycled MEAs. (a) and (b) P1-NA, (c) and (d) P2-NA, (e) and (f) P2-SA, (g) and (h) P2-SA-AN. Here σ represents the standard deviation of PSD from at least 100 measurements and the standard deviation of EDS results among 3 different locations. | ||
Pronounced particle morphology changes were observed in P1-NA upon conditioning, where the big spongy nanoparticles shown in Fig. 4a disappeared and were replaced by donut-like nanoparticles shown in Fig. S4a.† Similar spongy-particle transformation has been found in previous work,16,28,35,67 which might be caused by the dissolution of highly porous particles and Pt redeposition to form Pt-enriched-shell particles. As the intensity in the HAADF images is proportional to the product of the thickness and Z2 (Z being the atomic number), the EDS data in Fig. S4b† demonstrate that the center and edge have similar Pt:Ni ratios, the lower brightness near the particle center shown in Fig. S4b† can be attributed to the formation of donut-shaped particles. In contrast, combined HAADF analysis of conditioned (Fig. S4d†) and 30k-cycled P2-NA particles (Fig. 5d) and accompanied point EDS analysis of Pt and Ni atomic percentages (having a lower Pt concentration in the center) revealed that the core thickness of cycled P2-NA could be greater than particle surface regions, and thus the contrast shown in the HAADF image (Fig. S4d†) could be attributed to Pt-rich shells rather than a donut-like shape. The formation of Pt-rich shells can result from bulk dealloying,37,38 where leaching of transition metal is confined to particle surface regions.
P1-NA exhibited much worse durability in PEMFC cycling than P2-NA, which can be attributed to extensive porosity found in P1-NA. First, P2-NA maintained a higher Ni content which is twice that of P1-NA both after conditioning and after 30k cycles (Fig. S4a and S4c,†Fig. 5a and c). Second, P1-NA showed only ∼1/3 of its original mass activity and ∼1/2 of its original specific activity after 30k cycles while P2-NA showed almost no change in mass and specific activity considering the measurement uncertainty (Fig. 2b and c). The much reduced activity of P1-NA in PEMFCs could be attributed to a greater Ni loss near the surface regions, which reduced the surface electronic structure modifications of Pt atoms induced by transition metal alloying.5–8,10,11,19–23 The development of a Pt-rich passivation layer is expected to improve the resistance to base metal leaching and activity loss, which might be another reason for the higher durability of P2-NA than P1-NA.
Point EDS analysis revealed that P2-SA-AN nanoparticles retained considerably more Ni near surface regions than P2-SA after conditioning and 30k cycles (Fig. S4e and S4g,†Fig. 5e and g), where no noticeable changes in the Ni atomic percentages near surface regions after conditioning and 30k cycles were observed. The increase in stability with the thermal annealing observed in MEAs in our study is in good agreement with previous RDE work by Wang et al.68 It is proposed that the exceptional stability and high mass and specific activities of P2-SA-AN in PEMFCs can be attributed to the ability of surface regions to retain Ni, which might be critical to maintain modified electronic structures of surface Pt atoms associated with Ni necessary to provide enhanced mass and specific activities relative to pure Pt nanoparticle catalysts. This hypothesis is supported by HAADF in conjunction with electron energy loss spectroscopy on P2-SA and P2-SA-AN after conditioning, which revealed that Pt shell thicknesses (1.2 ± 0.6 and 1.3 ± 0.3 nm, respectively) were nearly unchanged (∼1.6 nm) after 30k cycles, as shown in Fig. S5.† Further support, from an X-ray absorption technique that could differentiate Pt shell structures on these samples, will be published elsewhere. This again proves the importance of a surface shell to the activity and stability of dealloyed Pt-alloy nano-catalysts, and indicates that annealing could be an effective process to improve the stability of Pt–metal core–shell catalysts.
Lastly, it is useful to note that when a PtCo3 precursor was prepared using an analogous synthesis method to P2 (PtNi3), we were able to demonstrate an active and durable dealloyed catalyst comparable to those described in this study.53 Considering this, other binary and ternary systems may also be effective. This illustrates the versatility of the dealloying approach and allows one to weigh in other selection criteria such as supply chain, manufacturability, and the interaction with other fuel cell components when developing a new catalyst.
Conclusions
We have demonstrated unprecedented PEMFC device performance durability employing a family of dealloyed PtNi3 nanoparticle catalysts, exceeding scientific and technological state-of-art activity and stability targets. We find that for dealloyed catalysts larger particle size could strongly promote the formation of nanoporosity, which is associated with the rapid reduction in Ni content. On the other hand, reducing the particle size below a certain critical value can reduce porosity generation and promote the formation of a Pt-rich passivation particle shell layer. We also find that non-porous core–shell nanostructures can be strongly encouraged by using a less oxidative acid leaching protocol, further improving base metal retention and leading to exceptional activity durability. Finally, post-acid-treatment annealing can cause the formation of comparatively dense-Pt shells, which can provide excellent resistance to Ni leaching, promote particle size stability, and can help to improve both specific and mass activities during electrochemical cycling. These results highlight the critical role of acid treatments, an aspect of catalyst design that has not been rigorously controlled to date, in creating Pt-rich passivation layers to prevent base metal leaching and improve catalytic activity.Acknowledgements
This work was supported by the U.S. Department of Energy, Office of Energy Efficiency and Renewable Energy under grant DE-EE0000458. The authors thank Eric L. Thompson, Jeanette E. Owejan, Thomas E. Moylan, Travis Downs, Peter Harvey, and Mike Flanagan of GM for engineering support. The research made use of the Shared Experimental Facilities supported by the MRSEC Program of the National Science Foundation under award number DMR 08-019762. P. S. acknowledges partial financial support by the German National Science Foundation (Deutsche Forschungsgemeinschaft) under grant STR 596/5-1.References
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Appl. Catal., B, 2005, 56, 9–35 CrossRef CAS PubMed.
- C. Jaffary, in Handbook of Fuel Cells: Fundamentals, Technology and Application, ed. A. L. W.Vielstich and H. A.Gasteiger, John Wiley and Sons, Chichester, 2003 Search PubMed.
- S. Chen, P. J. Ferreira, W. Sheng, N. Yabuuchi, L. F. Allard and Y. Shao-Horn, J. Am. Chem. Soc., 2008, 130, 13818–13819 CrossRef CAS PubMed.
- S. Chen, W. Sheng, N. Yabuuchi, P. J. Ferreira, L. F. Allard and Y. Shao-Horn, J. Phys. Chem. C, 2008, 113, 1109–1125 Search PubMed.
- A. Nilekar, Y. Xu, J. Zhang, M. Vukmirovic, K. Sasaki, R. Adzic and M. Mavrikakis, Top. Catal., 2007, 46, 276–284 CrossRef CAS.
- V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. Mayrhofer, C. A. Lucas, G. Wang, P. N. Ross and N. M. Markovic, Nat. Mater., 2007, 6, 241–247 CrossRef CAS PubMed.
- V. R. Stamenkovic, B. Fowler, B. S. Mun, G. Wang, P. N. Ross, C. A. Lucas and N. M. Marković, Science, 2007, 315, 493–497 CrossRef CAS PubMed.
- V. R. Stamenkovic, B. S. Mun, K. J. J. Mayrhofer, P. N. Ross and N. M. Markovic, J. Am. Chem. Soc., 2006, 128, 8813–8819 CrossRef CAS PubMed.
- F. Hasché, M. Oezaslan and P. Strasser, J. Electrochem. Soc., 2011, 159, B24–B33 Search PubMed.
- J. Zhang, F. H. B. Lima, M. H. Shao, K. Sasaki, J. X. Wang, J. Hanson and R. R. Adzic, J. Phys. Chem. B, 2005, 109, 22701–22704 CrossRef CAS PubMed.
- Y. Xu, A. V. Ruban and M. Mavrikakis, J. Am. Chem. Soc., 2004, 126, 4717–4725 CrossRef CAS PubMed.
- J. K. Norskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1, 37–46 CrossRef CAS PubMed.
- D. Wang, Y. Yu, H. L. Xin, R. Hovden, P. Ercius, J. A. Mundy, H. Chen, J. H. Richard, D. A. Muller, F. J. DiSalvo and H. D. Abruña, Nano Lett., 2012, 12, 5230–5238 CrossRef CAS PubMed.
- Y. S. Kim, S. H. Jeon, A. Bostwick, E. Rotenberg, P. N. Ross, V. R. Stamenkovic, N. M. Markovic, T. W. Noh, S. Han and B. S. Mun, Adv. Energy Mater., 2013, 3, 1257–1261 CrossRef CAS.
- C. Chen, Y. Kang, Z. Huo, Z. Zhu, W. Huang, H. L. Xin, J. D. Snyder, D. Li, J. A. Herron and M. Mavrikakis, Science, 2014, 343, 1339–1343 CrossRef CAS PubMed.
- L. Gan, M. Heggen, R. O'Malley, B. Theobald and P. Strasser, Nano Lett., 2013, 13, 1131–1138 CrossRef CAS PubMed.
- S. V. Selvaganesh, P. Sridhar, S. Pitchumani and A. K. Shukla, J. Electrochem. Soc., 2013, 160, F49–F59 CrossRef CAS PubMed.
- P. Strasser, S. Koh, T. Anniyev, J. Greeley, K. More, C. Yu, Z. Liu, S. Kaya, D. Nordlund, H. Ogasawara, M. F. Toney and A. Nilsson, Nat. Chem., 2010, 2, 454–460 CrossRef CAS PubMed.
- S. Mukerjee and S. Srinivasan, J. Electroanal. Chem., 1993, 357, 201–224 CrossRef CAS.
- S. Mukerjee, S. Srinivasan, M. P. Soriaga and J. Mcbreen, J. Phys. Chem., 1995, 99, 4577–4589 CrossRef CAS.
- T. Toda, H. Igarashi, H. Uchida and M. Watanabe, J. Electrochem. Soc., 1999, 146, 3750–3756 CrossRef CAS PubMed.
- S. Mukerjee, S. Srinivasan, M. P. Soriaga and J. Mcbreen, J. Electrochem. Soc., 1995, 142, 1409–1422 CrossRef CAS PubMed.
- R. R. Adzic, J. Zhang, K. Sasaki, M. B. Vukmirovic, M. Shao, J. X. Wang, A. U. Nilekar, M. Mavrikakis, J. A. Valerio and F. Uribe, Top. Catal., 2007, 46, 249–262 CrossRef CAS.
- V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. J. Mayrhofer, C. A. Lucas, G. Wang, P. N. Ross and N. M. Markovic, Nat. Mater., 2007, 6, 241–247 CrossRef CAS PubMed.
- R. Subbaraman, D. Strmcnik, A. P. Paulikas, V. R. Stamenkovic and N. M. Markovic, ChemPhysChem, 2010, 11, 2825–2833 CrossRef CAS PubMed.
- L. Gan, M. Heggen, S. Rudi and P. Strasser, Nano Lett., 2012, 12, 5423–5430 CrossRef CAS PubMed.
- M. Oezaslan and P. Strasser, J. Power Sources, 2011, 196, 5240–5249 CrossRef CAS PubMed.
- S. Chen, H. A. Gasteiger, K. Hayakawa, T. Tada and Y. Shao-Horn, J. Electrochem. Soc., 2010, 157, A82–A97 CrossRef CAS PubMed.
- M. F. Mathias, R. Makharia, H. A. Gasteiger, J. J. Conley, T. J. Fuller, C. J. Gittleman, S. S. Kocha, D. P. Miller, C. K. Mittelsteadt, T. Xie, S. G. Yan and P. T. Yu, Electrochem. Soc. Interface, 2005, 14, 24–35 CAS.
- S. C. Ball, S. L. Hudson, B. R. Theobald and D. Thompsett, ECS Trans., 2007, 11, 1267–1278 CAS.
- F. T. Wagner, H. A. Gasteiger, R. Makharia, K. C. Neyerlin, E. L. Thompson and S. G. Yan, ECS Trans., 2006, 3, 19–29 CAS.
- K. J. J. Mayrhofer, K. Hartl, V. Juhart and M. Arenz, J. Am. Chem. Soc., 2009, 131, 16348–16349 CrossRef CAS PubMed.
- P. J. Ferreira, G. J. la O', Y. Shao-Horn, D. Morgan, R. Makharia, S. Kocha and H. A. Gasteiger, J. Electrochem. Soc., 2005, 152, A2256–A2271 CrossRef PubMed.
- Y. Shao-Horn, W. C. Sheng, S. Chen, P. J. Ferreira, E. F. Holby and D. Morgan, Top. Catal., 2007, 46, 285–305 CrossRef CAS PubMed.
- C. E. Carlton, S. Chen, P. J. Ferreira, L. F. Allard and Y. Shao-Horn, J. Phys. Chem. Lett., 2011, 3, 161–166 CrossRef.
- H. L. Xin, J. A. Mundy, Z. Liu, R. Cabezas, R. Hovden, L. F. Kourkoutis, J. Zhang, N. P. Subramanian, R. Makharia, F. T. Wagner and D. A. Muller, Nano Lett., 2011, 12, 490–497 CrossRef PubMed.
- J. Erlebacher, M. J. Aziz, A. Karma, N. Dimitrov and K. Sieradzki, Nature, 2001, 410, 450–453 CrossRef CAS PubMed.
- J. Erlebacher, J. Electrochem. Soc., 2004, 151, C614–C626 CrossRef CAS PubMed.
- J. Rugolo, J. Erlebacher and K. Sieradzki, Nat. Mater., 2006, 5, 946–949 CrossRef CAS PubMed.
- J. Erlebacher, Phys. Rev. Lett., 2011, 106, 225504 CrossRef CAS.
- K. Sieradzki, N. Dimitrov, D. Movrin, C. McCall, N. Vasiljevic and J. Erlebacher, J. Electrochem. Soc., 2002, 149, B370–B377 CrossRef CAS PubMed.
- M. Oezaslan, M. Heggen and P. Strasser, J. Am. Chem. Soc., 2012, 134, 514–524 CrossRef CAS PubMed.
- J. Snyder, I. McCue, K. Livi and J. Erlebacher, J. Am. Chem. Soc., 2012, 134, 8633–8645 CrossRef CAS PubMed.
- L. Gan, C. Cui, S. Rudi and P. Strasser, Top. Catal., 2014, 57, 236–244 CrossRef CAS.
- C. Cui, L. Gan, M. Neumann, M. Heggen, B. Roldan Cuenya and P. Strasser, J. Am. Chem. Soc., 2014, 136, 4813–4816 CrossRef CAS PubMed.
- L. Gan and P. Strasser, in Electrocatalysis in Fuel Cells, ed. M. Shao, Springer London, 2013, vol. 9, ch. 18, pp. 533–560 Search PubMed.
- L. Gan, S. Rudi, C. Cui and P. Strasser, ChemCatChem, 2013, 5, 2691–2694 CrossRef CAS.
- C. Cui, L. Gan, M. Heggen, S. Rudi and P. Strasser, Nat. Mater., 2013, 12, 765–771 CrossRef CAS PubMed.
- C. Chen, Y. Kang, Z. Huo, Z. Zhu, W. Huang, H. L. Xin, J. D. Snyder, D. Li, J. A. Herron, M. Mavrikakis, M. Chi, K. L. More, Y. Li, N. M. Markovic, G. A. Somorjai, P. Yang and V. R. Stamenkovic, Science, 2014, 343, 1339–1343 CrossRef CAS PubMed.
- S.-I. Choi, S. Xie, M. Shao, J. H. Odell, N. Lu, H.-C. Peng, L. Protsailo, S. Guerrero, J. Park, X. Xia, J. Wang, M. J. Kim and Y. Xia, Nano Lett., 2013, 13, 3420–3425 CrossRef CAS PubMed.
- The US Department of Energy (DOE), Energy Efficiency and Renewable Energy, 2012, http://www1.eere.energy.gov/hydrogenandfuelcells/mypp/pdfs/fuel_cells.pdf Search PubMed.
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef CAS PubMed.
- A. Kongkanand and F. Wagner, Annual Merit Review DOE Hydrogen and Fuel Cells and Vehicle Technologies Programs, Washington, DC, 2013, http://www.hydrogen.energy.gov/pdfs/review13/fc087_kongkanand_2013_o.pdf Search PubMed.
- C. Wang, M. Chi, D. Li, D. Strmcnik, D. van der Vliet, G. Wang, V. Komanicky, K.-C. Chang, A. P. Paulikas, D. Tripkovic, J. Pearson, K. L. More, N. M. Markovic and V. R. Stamenkovic, J. Am. Chem. Soc., 2011, 133, 14396–14403 CrossRef CAS PubMed.
- R. N. Carter, S. S. Kocha, F. Wagner, M. Fay and H. A. Gasteiger, ECS Trans., 2007, 11, 403–410 CAS.
- M. Wakisaka, Y. Hyuga, K. Abe, H. Uchida and M. Watanabe, Electrochem. Commun., 2011, 13, 317–320 CrossRef CAS PubMed.
- D. F. van der Vliet, C. Wang, D. Li, A. P. Paulikas, J. Greeley, R. B. Rankin, D. Strmcnik, D. Tripkovic, N. M. Markovic and V. R. Stamenkovic, Angew. Chem., Int. Ed., 2012, 51, 3139–3142 CrossRef CAS PubMed.
- M. Wakisaka, S. Morishima, Y. Hyuga, H. Uchida and M. Watanabe, Electrochem. Commun., 2012, 18, 55–57 CrossRef CAS PubMed.
- A. Kongkanand, Annual Merit Review DOE Hydrogen and Fuel Cells and Vehicle Technologies Programs, Washington, DC, 2014, http://www.hydrogen.energy.gov/pdfs/review14/fc087_kongkanand_2014_o.pdf Search PubMed.
- E. F. Holby, W. Sheng, Y. Shao-Horn and D. Morgan, Energy Environ. Sci., 2009, 2, 865–871 CAS.
- A. Steinbach, Annual Merit Review DOE Hydrogen and Fuel Cells and Vehicle Technologies Programs, Washington, DC, 2014, http://www.hydrogen.energy.gov/pdfs/review14/fc104_steinbach_2014_o.pdf Search PubMed.
- B. N. Popov, Annual Merit Review DOE Hydrogen and Fuel Cells and Vehicle Technologies Programs, Washington, DC, 2014, http://www.hydrogen.energy.gov/pdfs/review14/fc088_popov_2014_o.pdf Search PubMed.
- F. T. Wagner, S. G. Yan and P. T. Yu, in Handbook of Fuel Cells, John Wiley & Sons, Ltd, 2010 Search PubMed.
- H. Yano, J. M. Song, H. Uchida and M. Watanabe, J. Phys. Chem. C, 2008, 112, 8372–8380 CAS.
- T. Toda, H. Igarashi and M. Watanabe, J. Electrochem. Soc., 1998, 145, 4185–4188 CrossRef CAS PubMed.
- Q. Jia, K. M. Caldwell, J. M. Ziegelbauer, A. Kongkanand, F. T. Wagner, S. Mukerjee and D. E. Ramaker, J. Electrochem. Soc., 2014, 161, F1323–F1329 CrossRef CAS PubMed.
- Z. Liu, H. Xin, Z. Yu, Y. Zhu, J. Zhang, J. A. Mundy, D. A. Muller and F. T. Wagner, J. Electrochem. Soc., 2012, 159, F554–F559 CrossRef CAS PubMed.
- D. Wang, H. L. Xin, R. Hovden, H. Wang, Y. Yu, D. A. Muller, F. J. DiSalvo and H. D. Abruña, Nat. Mater., 2013, 12, 81–87 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ee02144d |
| This journal is © The Royal Society of Chemistry 2015 |