Anticancer metallodrugs of glutamic acid sulphonamides: in silico, DNA binding, hemolysis and anticancer studies†
Imran Ali*a,
Waseem A. Wania,
Kishwar Saleema and
Ming-Fa Hsiehb
aDepartment of Chemistry, Jamia Millia Islamia (Central University), New Delhi - 110025, India. E-mail: drimran_ali@yahoo.com; drimran.chiral@gmail.com; Fax: +91-11-26985507; Tel: +91-9211458226
bDepartment of Biomedical Engineering, Chung Yuan Christian University, 200, Chung Pei Rd., Chung Li, Taiwan
First published on 16th June 2014
Abstract
In response to an increased demand for effective anticancer drugs, a series of disodium sulphonamides of L-glutamic acid (L1–L3) was synthesized. Sulphonamides were complexed with copper(II), nickel(II) and ruthenium(III) ions, separately and respectively. The sulphonamides and their complexes were characterized by various physico-chemical, analytical and spectroscopic techniques. Solution stability studies indicated the robust nature of the complexes in PBS at pH 7.4. DNA binding constants (Kb) revealed good binding (0.7 × 103 to 5.24 × 104 mol−1) capacities of the reported compounds. Complexes bound to DNA more efficiently as compared to their ligands. In silico studies supported DNA binding of the reported ligands. Cumulative evidence from the results of in silico and DNA binding studies indicated that the polarizing and non-polarizing effects of chloro and methyl groups significantly affected the DNA binding ability of the compounds. The compounds were less toxic towards rabbit RBCs as compared to the well-known anticancer drug doxorubicin. All the compounds had good anticancer activities (131–153% viability) on MCF-7 (wild type) cell lines.
Introduction
Cancer has been a big threat to human beings for a long period. The number of cancer patients is increasing continuously. Many factors contribute to increasing cancer genesis, among which the modernization of our society is one of the major contributing factors.1,2 Despite a number of anticancer drugs now being available in the market, cancer kills millions of people each year worldwide. Furthermore, the available anticancer drugs pose serious side effects, which limit their uses considerably.3–5 Therefore, the demand for an ideal anticancer drug that can control this disease even at the late stages with no or fewer side effects still continues.Glutamine (a glutamic acid derivative) is an essential growth component for proliferating tumor cells. It is probably the most abundant free amino acid in the human body, essential for the growth of normal and neoplastic cells. Tumors are known to produce great changes in the host glutamine metabolism. Overall, glutamine promotes the rapid growth and multiplication of cancer cells. The effects of glutamine on cancer cell growth and multiplication suggested the possibility of a close association between glutamine, glutamic acid and cancer.6 In addition, the re-introduction of thalidomide (a synthetic glutamic acid derivative) in clinical trials for the treatment of various malignant tumors firmly supported the association of glutamic acid and glutamine with cancer.7,8 As a result of these findings, it was realized that certain structural variants and analogues of glutamic acid and glutamine might be opposing the effects produced by glutamine in proliferating cancer cells.9 In this context, a few glutamic acid and glutamine derivatives were synthesized and screened for their antiproliferative effects.10–13 It was observed that the reported glutamic acid and glutamine derivatives showed fair anticancer activities.
The interesting pharmacology of glutamic acid and its derivatives has been known for a long time. In 1948, Farber and co-workers14 reported the clinical results of the temporary remissions in acute leukemia in children treated with 4-aminopteroyl-glutamic acid (aminopterin) (Fig. 1), a folic acid antagonist. Later on, Lederle Laboratories (Pearl River, New York) in the United States marketed aminopterin from 1953 to 1964 for the treatment of pediatric leukemia. However, Lederle Laboratories simultaneously marketed methotrexate (MTX or amethopterin) (Fig. 1), which later led to the discontinuation of aminopterin due to its toxic side effects. Methotrexate is currently being used either alone or in combination with other agents for the treatment of a range of malignancies, including those of the breast, head and neck, leukemia, lymphoma, lung cancer, osteosarcoma, bladder cancer and trophoblastic neoplasms. The main side effects associated with treatment with methotrexate include ulcerative stomatitis, low white blood-cell count, nausea, abdominal pain, fatigue, fever, dizziness, acute pneumonitis and, in rare cases, pulmonary fibrosis.15 To combat some of the side effects caused by the therapy with methotrexate, folinic acid (Fig. 1) (another congener of glutamic acid) is administered at the appropriate time following methotrexate medication, which rescues bone marrow and gastrointestinal mucosa cells from methotrexate.16 Moreover, folinic acid is used in combination chemotherapy with 5-fluorouracil for the treatment of colon cancer. Folinic acid enhances the effect of 5-fluorouracil by the inhibition of thymidylate synthase.
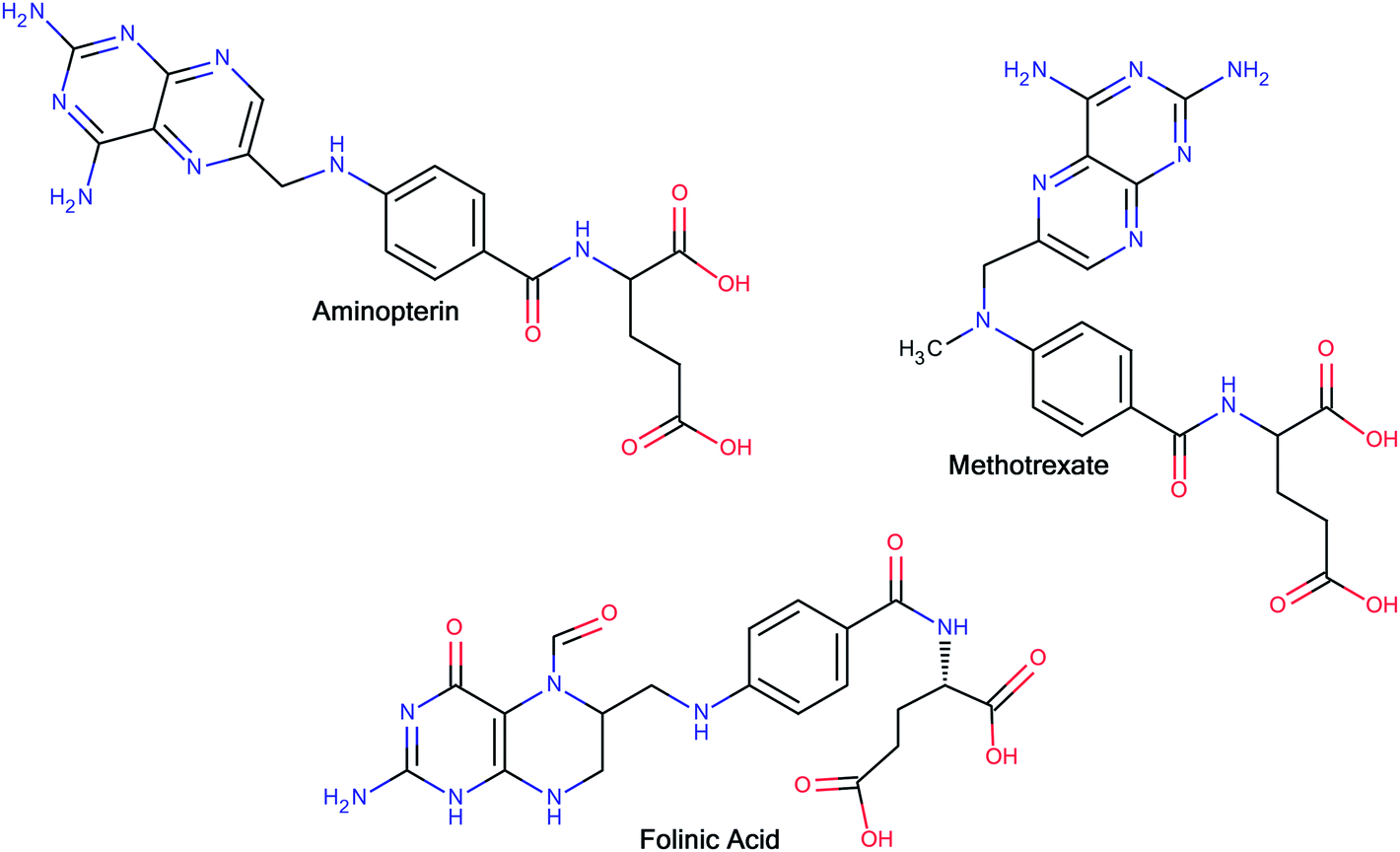 | ||
| Fig. 1 Glutamic acid derivatives with a rich history as anticancer agents. | ||
Sulphonamides are an important class of drugs. Some sulphonamides and their derivatives have been observed to exhibit substantial in vitro and in vivo antitumor activity.17 The antitumor action of sulphonamides has been attributed to several mechanistic phenomena, including carbonic anhydrase inhibition, cell cycle arrest in the G1 phase, microtubule assembly disruption and angiogenesis.17 Some compounds with basic sulphonamide motifs have been investigated as possible anticancer agents. The results indicated good anticancer activities on different cell lines.18–21
Appropriate selection of ligands may be used to develop active metallodrugs with various advantages over the organic-based drugs. This is due to good affinities of different metals towards DNA. Copper complexes are known for their reputation in cancer chemotherapy and, therefore, are being extensively investigated for the treatment of various cancers.22–25 Nickel is considered a weak carcinogen. However, reports have suggested that nickel interacts with DNA and DNA-binding proteins, and a few reports on the anticancer potentials of nickel complexes are available in the literature.26–29 Ruthenium anticancer drugs have been extensively explored during the last 20 years and two of them have entered into clinical trials. Ruthenium complexes are, generally, less toxic and capable of overcoming platinum drug resistance in cancer cells;30 they cause selective antimetastasis and have lower systemic toxicity. Moreover, they penetrate reasonably well into tumor cells and are therefore able to bind effectively to DNA.31 In addition, they exhibit ligand exchange kinetics similar to those of platinum(II) antitumor drugs. Presently, ruthenium complexes are being extensively synthesized and screened for their anticancer activities and quite interesting results are being obtained.32–35
In view of these findings, in the work reported here, a series of disodium sulphonamides of L-glutamic acid was synthesized. The sulphonamides were complexed with Cu(II), Ni(II) and Ru(III) ions, separately and respectively. The binding of ligands and their complexes with Ct-DNA was studied by UV-vis absorption spectrophotometry. Hemolysis assays were carried out on rabbit red blood cells (RBCs) and the anticancer profiles were determined on MCF-7 (wild-type) cell lines. The results of DNA binding of the ligands were verified by in silico studies. The results of these findings are presented herein.
Experimental
Materials and methods
All the reagents were of A.R. grade and used without further purification. L-Glutamic acid was purchased from K.C. Biological, Lenexa, KS, USA. Benzenesulphonyl chloride, p-chlorobenzenesulphonyl chloride and p-toluenebenzenesulphonyl chloride were purchased from S.D. Fine Chemicals, Mumbai, India. RuCl3·3H2O was procured from Avarice Lab. Pvt. Ltd., G.B. Nagar, India. CuCl2·2H2O, NiCl2·6H2O, DMF, ethanol, methanol and hexane were supplied by E. Merck, Mumbai. Pre-coated aluminium silica gel 60 F254 thin layer plates were purchased from E. Merck, Germany. The disodium salt of Ct-DNA and tris-(hydroxymethyl) aminomethane were supplied by Sisco Research Lab., Mumbai. Human breast cancer cell lines (MCF-7) were collected from the School of Pharmacy, College of Medicine, National Taiwan University. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) was purchased from Sigma-Aldrich (St Louis, MO, USA). Dulbecco's modified eagle's medium (DMEM) and antibiotics/antimycotics were purchased from GIBCO (NY, USA). Fetal bovine serum (FBS) was obtained from HyClone (Utah, USA).The percentages of C, H, N and S were determined by a Vario EL elemental analyzer (EL-III). UV-vis spectra were recorded on a Perkin Elmer Lambda 40 UV-vis spectrometer (CT 06859 USA). FT-IR spectra were recorded on a Perkin Elmer RXIFT system spectrometer (LR 64912C) in the range of 4000–400 cm−1 using KBr discs. 1H NMR spectra were recorded on a Bruker 300 MHz instrument (DPX 300). ESI-mass spectra were recorded on a micrOTOF-Q II spectrometer (10262). The reactions were monitored by thin-layer chromatography using a UV Cabinet for visualization. Molar conductivities were recorded on a Decibel conductivity meter (DB-1038). A pH meter of Control Dynamics (APX 175 E/C) was used to record the pH of solutions. Melting points were recorded on a Veego instrument (REC-22038 A2). Double-distilled water was prepared using a Millipore Milli-Q (Bedford, MA, USA) water purification system. Molecular modelling studies were carried out by the use of a semi-empirical method PM3 as implemented in hyperchem 8.0 (Hypercube, Inc., USA). In silico studies were carried out using an AutoDock 4.2 (Scripps Research Institute, USA) on an Intel® core™ i3 CPU (3.2 GHz) with a Windows XP operating system. An incubator for cell culture (MCO-15AC, Sanyo), a centrifuge (CN2060, Hsiangtai Co.) and a microplate photometer (Multiskan FC, Thermo Scientific) were used for carrying out the hemolysis and anticancer assays of the developed compounds.
Synthesis of ligands (L1–L3)
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30, v/v). The product solution was reduced to one-third of its volume on a rotary evaporator and kept in a refrigerator overnight. A white solid (L1) precipitated out and was collected by filtration on a Buchner funnel. Finally, the product was washed with cold methanol and hexane followed by drying in a vacuum dessicator over fused calcium chloride.
:30, v/v). The product solution was reduced to one-third of its volume on a rotary evaporator and kept in a refrigerator overnight. A white solid (L1) precipitated out and was collected by filtration on a Buchner funnel. Finally, the product was washed with cold methanol and hexane followed by drying in a vacuum dessicator over fused calcium chloride.Yield: 83.0%, mol. wt 331.31 Da, decomposed over 230 °C, white powder, anal. calc. Na2C11H11SNO6 (%): calculated C (39.85), H (3.32), N (4.22), S (9.66); found C (39.82), H (3.39), N (4.28), S (9.77); I.R. (KBr pellets, cm−1): 1178 (νS![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)sym, 908 (νS–N), 1375 (νOCO)sym, 1598 (νOCO)asym, 3488 (νN–H); UV-vis (H2O, nm): 208–219 (n–σ*), 243–272 (π–π*); 1H NMR (d6-DMSO): 7.770 (s, SO2NH, 1H), 7.626 (d, aromatic, 2H), 7.332 (d, aromatic, 3H), 3.592 (t, 1H), 2.082 (m, 2H), 2.310 (t, 2H); ESI-MS: m/z = 663.97 [2M + H+]+, 372 [M + K+ + 2H]+, 352.08 [M + NH4+ + 3H+]+, 324.05 [M − Na+ + NH4+ − 2H+]+, 202.97 [M − 2(COONa) + H+]+.
O)sym, 908 (νS–N), 1375 (νOCO)sym, 1598 (νOCO)asym, 3488 (νN–H); UV-vis (H2O, nm): 208–219 (n–σ*), 243–272 (π–π*); 1H NMR (d6-DMSO): 7.770 (s, SO2NH, 1H), 7.626 (d, aromatic, 2H), 7.332 (d, aromatic, 3H), 3.592 (t, 1H), 2.082 (m, 2H), 2.310 (t, 2H); ESI-MS: m/z = 663.97 [2M + H+]+, 372 [M + K+ + 2H]+, 352.08 [M + NH4+ + 3H+]+, 324.05 [M − Na+ + NH4+ − 2H+]+, 202.97 [M − 2(COONa) + H+]+.
The ligands (L2 and L3) were prepared by a similar procedure using p-toluenebenzenesulphonyl chloride and p-chlorobenzenesulphonyl chloride, separately and respectively.
L2. Yield: 80%, mol. wt 345.07 Da, decomposed over 212 °C, white powder, anal. calc. Na2C12H13SNO6 (%): calculated C (41.73), H (3.76), N (4.05), S (13.05); found C (41.75), H (3.79), N (4.11), S (13.12); I.R. (KBr pellets, cm−1): 1187.0 (νS
O)sym, 900.0 (νS–N), 1414.1 (νOCO)sym, 1582.5 (νOCO)asym, 3381.7 (νN–H); UV-vis (H2O, nm): 215–240 (n–σ*), 250–269 (π–π*); 1H NMR (d6-DMSO): 7.520 (s, SO2NH, 1H), 7.637 (d, aromatic, 2H), 7.140 (d, aromatic, 2H), 3.667 (t, 1H), 2.06–2.5 (m, 2H), 3.4 (t, 2H), 3.26 (s, CH3–, 3H); ESI-MS: m/z = 410.99 [M + Na+ + K+ + 3H]+, 360.04 [M + NH4+ − 3H]+, 346.03 [M + H]+, 338.06 [M + Na+ + NH4+ − 2H]+, 324.05 [M − Na+ + 2H]+, 320.05 [M − Na+ − 2H]−, 216.99 [M − 2(COONa) + 5H]+.
L3. Yield: 75%, mol. wt 365.5 Da, m.p. > 270 °C, white powder, anal. calc. Na2C11H10SNO6Cl (%): calculated C (36.11), H (2.73), N (3.83), S (8.75); found C (36.19), H (2.67), N (3.89), S (8.65); I.R. (KBr pellets, cm−1): 1188 (νS
O)sym, 903.0 (νS–N), 1330 (νOCO)sym, 1600 (νOCO)asym, 1042 (νAr–Cl), 3466.5 (νN–H); UV-vis (H2O, nm): 220–235 (n–σ*), 245–283 (π–π*); 1H NMR (d6-DMSO): 7.870 (s, SO2NH, 1H), 7.723 (d, aromatic, 2H), 7.422 (d, aromatic, 2H), 3.622 (t, 1H), 2.282 (m, 2H), 2.420 (t, 2H); ESI-MS: m/z = 427.95 [M + Na+ + K+]+, 389.95 [M + Na+ + H]+, 384.90 [M + NH4+ + H]+, 383.90 [M + NH4+]+, 382.89 [M + NH4+ − H]+, 362.04 [M − 3H]+, 238.93 [M − Cl − (COONa) − Na+ − 2H]+, 236.9 [M − 2(COONa) + 5H]+.
Synthesis of complexes CuL1 to RuL3
Nickel and ruthenium complexes of L1, as well as copper, nickel and ruthenium complexes of L2 and L3, were prepared by a similar procedure. The complexes obtained were kept in a vacuum dessicator over fused calcium chloride.
CuL1. Yield: 72.0%, mol. wt 679.48 Da, decomposed over 120 °C, light green solid, anal. calc. Na2[Cu(C11H11SNO6)2](%): calculated C (38.84), H (3.22) N (4.12), S (9.4); found C (38.78), H (3.25), N (4.16), S (9.38); I.R. (KBr pellets, cm−1): 1199.6 (νS
O)sym, 902 (νS–N), 1396 (νOCO)sym, 1621.8 (νOCO)asym, 3497 (νN–H)str, 569.3 (νCu–O), 488 (νCu–N); UV-vis (H2O, nm): 205–223 (n–σ*), 247–271 (π–π*), 276–355 (charge transfer band), 788–809 (2T2g → 2Eg); ˄M (1 × 10−3 M, H2O): 183 Ω−1 cm2 mol−1 (1:2 electrolyte); ESI-MS: m/z = 747.78 [M + 3Na − H]+, 387.81 {M − Na2[C9H11SNO2] − 2COO}, 207.82 {M − Na2[Cu(C11H11SNO6) − 2(COO)] + 5H}+.
NiL1. Yield: 63.0%, mol. wt 674.63 Da, decomposed over 260 °C, light sky blue solid, anal. calc. Na2[Ni(C11H11SNO6)2](%): calculated C (39.12), H (3.26) N (4.14), S (9.48); found C (39.09), H (3.30), N (4.19), S (9.52); I.R. (KBr pellets, cm−1): 1198.4 (νS
O)sym, 899 (νS–N), 1401 (νOCO)sym, 1623 (νOCO)asym, 3409.1 (νN–H)str, 569.7 (νNi–O), 486 (νNi–N); UV-vis (H2O, nm): 210–222 (n–σ*), 252–275 (π–π*), 513–527 (3T1g(F) → 3A2g(F)), 879–899 (3T2g(F) → 3A2g(F); ˄M (1 × 10−3 M, H2O): 171 Ω−1 cm2 mol−1 (1:2 electrolyte); ESI-MS: m/z = 742.93 [M + 3Na − H+]+, 382.96 {M − Na2[C9H11SNO2] − 2COO}, 202.97 {M − Na2[Ni(C11H11SNO6) − 2(COO)] + 5H+}+.
RuL1. Yield: 55.0%, mol. wt 694.01 Da, m.p. > 275 °C, drab olive solid, anal. calc. Na[Ru(C11H11SNO6)2](%): calculated C (38.02), H (3.16) N (4.02), S (9.22); found C (38.07), H (3.22), N (3.98), S (9.17); I.R. (KBr pellets, cm−1): 1199.6 (νS
O)sym, 901 (νS–N), 1386 (νOCO)sym, 1625 (νOCO)asym, 3442 (νN–H)str, 569.3 (νRu–O), 492 (νRu–N); UV-vis (H2O, nm): 250–270 (n–σ*), 272–282 (π–π*), 286–335 (n–π*), 530–730 (6A1g → 4T1g); ˄M (1 × 10−3 M, H2O): 131 Ω−1 cm2 mol−1 (1:1 electrolyte); ESI-MS: 762.01 [M + 3Na − H+]+, 386.05 {M − Na[C9H11SNO2] − 2COO}, 201.24 {M − Na[Ru(C11H11SNO6) − 2(COO)] + 5H}+.
CuL2. Yield: 47.0%, mol. wt 707.54 Da, decomposed over 160 °C, green amorphous solid, anal. calc. Na2[Cu(C12H13SNO6)2](%): calculated C (40.70), H (3.66) N (3.94), S (9.04); found C (40.74), H (3.60), N (3.88), S (9.07); I.R. (KBr pellets, cm−1): 1190.2 (νS
O)sym, 905 (νS–N), 1405.5 (νOCO)sym, 1598.2 (νOCO)asym, 3375.5 (νN–H)str, 566.1 (νCu–O), 493 (νCu–N): UV-vis (H2O, nm): 224–245 (n–σ*), 253–275 (π–π*), 284–361 (charge transfer band), 796–812 (2T2g → 2Eg); ˄M (1 × 10−3 M, H2O): 161 Ω−1 cm2 mol−1 (1:2 electrolyte); ESI-MS: m/z = 662.95 [M − 2Na+ + H+]−, 644.82 [M − 2Na+ − CH3 − 2H]−, 633.99 [M − 2Na+ − 2CH3 + 2H], 410.99 [M − 2Na+ − (C10H13SNO2) − COO− + 4H]+, 360.05 [M − 2Na+ − (C10H13SNO2) − 2COO− − 2H]−, 216.99 {M − 2Na+ − [Cu(C11H16SNO2)] − 4COO− + NH4+ + 3H]+.
NiL2. Yield: 55.0%, mol. wt 702.69 Da, decomposed over 230 °C, aero blue amorphous solid, anal. calc. Na2[Ni(C12H13SNO6)2](%): calculated C (40.98), H (3.7) N (3.98), S (9.1); found C (40.93), H (3.67), N (3.95), S (9.14); I.R. (KBr pellets, cm−1): 1189.0 (νS
O)sym, 902 (νS–N), 1416.7 (νOCO)sym, 1620.3 (νOCO)asym, 3418.1 (νN–H)str, 569.7 (νNi–O), 515 (νNi–N); UV-vis (H2O, nm): 225–237 (n–σ*), 239–251 (π–π*), 255–273 (n–π*); 498–509 (3T1g(F) → 3A2g(F), 875–896 (3T2g(F) → 3A2g(F)); ˄M (1 × 10−3 M, H2O): 168 Ω−1 cm2 mol−1 (1:2 electrolyte); ESI-MS: m/z = 662.95 [M − 2Na+ + H]−, 355.53 [M − 2Na+ − (C10H13SNO2) − 2COO− − 2H]−, 212.34{M − 2Na+ − [Ni(C11H16SNO2)] − 4COO− + H2O + 3H]+.
RuL2. Yield: 63.0%, mol. wt 722.07 Da, m.p. > 275 °C, black amorphous solid, anal. calc. Na[Ru(C12H13SNO6)2](%): calculated C (39.88), H (3.6) N (3.86), S (8.86); found C (39.85), H (3.63), N (3.81), S (8.89); I.R. (KBr pellets, cm−1): 1186.5 (νS
O)sym, 903 (νS–N), 1417 (νOCO)sym, 1619 (νOCO)asym, 3440 (νN–H)str, 568.3 (νCu–O), 503 (νRu–N); UV-vis (H2O, nm): 225–233 (n–σ*), 253–273 (π–π*), 308–338 (n–π*), 600–890 (6A1g → 4T1g); ˄M (1 × 10−3 M, H2O): 129 Ω−1 cm2 mol−1 (1:1 electrolyte); ESI-MS: m/z = 700.21 [M − Na+ + H+]−, 398.92 [M − Na+ − (C10H13SNO2) − 2COO− − 2H+]−, 217.14{M − Na+ − [Ru(C11H16SNO2)] − 4COO− + H2O + 3H}+.
CuL3. Yield: 68.0%, mol. wt 748.48 Da, decomposed over 130 °C, citron amorphous solid, anal. calc. Na2[Cu(C11H10SNO6Cl)2](%): calculated C (35.26), H (2.66) N (3.74), S (8.58); found C (35.31), H (2.71), N (3.69), S (8.55). I.R. (KBr pellets, cm−1) 1176.2 (νS
O)sym, 911 (νS–N), 1396.3 (νOCO)sym, 1621.9 (νOCO)asym, 3510 (νN–H)str, 1044.1 (νAr–Cl), 575.9 (νCu–O), 492.4 (νCu–N): UV-vis (H2O, nm): 219–242 (n–σ*), 258–271 (π–π*), 288–382 (charge transfer band), 798–806 (2T2g → 2Eg); ˄M (1 × 10−3 M, H2O): 177 Ω−1 cm2 mol−1 (1:2 electrolyte); ESI-MS: m/z = 787.51 [M + K+]+, 771.64 [M + Na+]+, 701.22 [M − 2Na+], 668.89 [M − 2Na+ − Cl + 2H+]+, 241.32 {M − 2Na+ − [Cu(C9H10SNO2Cl2)] − 3COO− + H+}+.
NiL3. Yield: 83.0%, mol. wt 743.63 Da, decomposed over 260 °C, bud green amorphous solid, anal. calc. Na2[Ni(C11H10SNO6Cl)2](%): calculated C (35.5), H (2.68) N (3.76), S (8.60); found C (35.48), H (2.65), N (3.69), S (8.65); I.R. (KBr pellets, cm−1) 1190 (νS
O)sym, 916 (νS–N), 1396.4 (νOCO)sym, 1624.6 (νOCO)asym, 3382.9 (νN–H)str, 1040.4 (νAr–Cl), 568.8 (νNi–O), 486.3 (νNi–N); UV-vis (H2O, nm): 232–242 (n–σ*), 253–279 (π–π*), 507–519 (3T1g(F) → 3A2g(F)), 862–889 (3T2g(F) → 3A2g(F)); ˄M (1 × 10−3 M, H2O): 194 Ω−1 cm2 mol−1 (1:2 electrolyte); ESI-MS: m/z = 784.74 [M + K+ + 2H]+, 782.75 [M + K+]+, 766.79 [M + Na+]+, 724.79 [M − Na+ + 4H]+,722.79 [M − Na+ + 2H]+, 697.66 [M − 2Na+], 666.83 [M − 2Na+ − Cl + 4H]+, 664.83 [M − 2Na+ − Cl + 2H]+, 236.93 {M − 2Na+ − [Ni(C9H10SNO2Cl2)] − 3COO− − 4H}+.
RuL3. Yield: 70.0%, mol. wt 763.01 Da, m.p. > 280 °C, black olive amorphous solid, anal. calc. Na[Ru(C11H10SNO6Cl)2](%): calculated C (34.58), H (2.62) N (3.66), S (8.38); found C (34.54), H (2.66), N (3.65), S (8.34); I.R. (KBr pellets, cm−1) 1186.4 (νS
O)sym, 896 (νS–N), 1355 (νOCO)sym, 1625 (νOCO)asym, 3428.3 (νN–H)str, 1046 (νAr–Cl) 503.6 (νRu–O), 495 (νRu–N); UV-vis (H2O, nm): 222–238 (n–σ*), 250–280 (π–π*), 280–348 (n–π*), 630–865 (6A1g → 4T1g); ˄M (1 × 10−3 M, H2O): 123 Ω−1 cm2 mol−1 (1:1 electrolyte); ESI-MS: m/z = 802.54 [M + K+]+, 786.32 [M + Na+]+, 740.21 [M − Na+], 706.23 [M − Na+ − Cl + 2H]+.
Solution stability
A qualitative insight into the stability of the complexes at physiological pH was obtained by monitoring their UV-vis spectra in PBS solution at 7.4 pH over a period of 24 h. 10−4 M solutions of the complexes were prepared in PBS at pH 7.4. The hydrolysis and aquation profiles of the complexes were assessed by recording their electronic spectra over a 24 h time period at 25 °C.Molecular modelling
Molecular modelling was carried out with a semi-empirical PM3 force field as implemented in the HyperChem 8.0 software.36,37 It is a graphics program with features of structure building, minimum energy geometry optimization and quick molecular display. Polak-Ribiere was chosen as the minimization algorithm with an RMS gradient of 0.1 kcal (Å−1 mol−1) and 250 energy calculations were carried out.DNA binding
UV-vis absorption spectrophotometry was used to study the interactions of ligands and their complexes with Ct-DNA at 7.4 pH in double-distilled water containing tris-(hydroxymethyl)-amino methane (Tris, 10−2 M). The concentration of the freshly prepared Ct-DNA solution was determined spectrophotometrically at 260 nm (ε = 6600 M−1 cm−1).38 The binding experiments were carried out by adding increasing concentrations of DNA (0.5 × 10−4 to 1.4 × 10−4 M) to a fixed concentration of ligands and their complexes (1.6 × 10−4 M). First of all, λmax and absorbance values of pure DNA, ligands and their metal complexes in buffer solutions were recorded. 2.0 mL of each solution of DNA and ligand or metal complex were mixed together and their λmax and absorbance values were recorded. The absorption spectra were recorded after each addition of different concentrations of DNA solution (2.0 mL).In silico studies
Docking studies of the ligands were performed by Intel® dual CPU (1.86 GHz) with Windows XP operating system. The 3D structures of the ligands were drawn using Marwin sketch. The 3D structures so obtained were converted to the pdb file format. Ligand preparation was done by assigning Gastegier charges, merging non-polar hydrogens, and saving in a PDBQT file format using AutoDock Tools (ADT) 4.2.39 The X-ray crystal structure of DNA (PDB ID: 1BNA) was obtained from the Protein Data Bank.40 Using AutoDock Tools (ADT) 4.2, DNA was saved in a PDB file format leaving hetero-atoms (water). Gastegier charges were assigned to DNA and saved in a PDBQT file format using ADT. Preparation of parameter files for grid and docking was carried out using ADT. Docking was performed with AutoDock 4.2 (Scripps Research Institute, USA), considering all the rotatable bonds of ligand as rotatable and the receptor as rigid.41 A grid box size of 60 × 80 × 110 Å with 0.375 Å spacing was used that included the whole DNA. Docking to the macromolecule was performed using an empirical-free energy function and Lamarckian Genetic Algorithm, with an initial population of 150 randomly placed individuals, a maximum number of 2500000 energy evaluations, a mutation rate of 0.02, and a crossover rate of 0.80. Fifty independent docking runs were performed for each ligand and DNA–ligand adduct for the lowest free energy of binding conformation from the largest cluster and saved in PDBQT format. Docking results were analysed using UCSF Chimera42 for possible polar and hydrophobic interactions.
Cytotoxicity profiles
Drug significance of the developed compounds was evaluated by investigating their hemolysis profiles and anticancer activities. Hemolysis behaviour of the compounds was evaluated on rabbit RBCs. In addition, their anticancer profiles were determined on MCF-7 cancer cell lines. Doxorubicin was used as the reference drug. Both hemolysis and anticancer assays were carried out in triplicate. These studies were carried out as described below.Hemolytic assays
The experimental procedure for evaluating the hemolysis behaviour of the compounds is an adjustment of ASTM standard F-756-00,43 which is based on colorimetric detection of Drabkin's solution. 1.5 mL of test compounds was incubated in 0.214 mL of dilute blood (0.1 mL rabbit whole blood mixed with 0.9 mL PBS) at 37 °C for 3 h. The hemoglobin in as-harvested plasma of rabbit blood was found to be less than 220 μg mL−1 (basal level for hemolysis test), confirming that fresh rabbit blood was used in the test. Following incubation, the solution was centrifuged at 3800 rpm for 15 min. To determine the supernatant hemoglobin, 0.8 mL of Drabkin's solution was added to 0.2 mL of supernatant and the sample was allowed to stand for 15 min. The amount of cyanmethemoglobin in the supernatant was measured by absorbance measurement at 540 nm and then compared to a standard curve (hemoglobin concentrations ranging from 32 to 1068 mg mL−1). The percent hemolysis refers to the hemoglobin concentration in the supernatant of a blood sample not treated with test compounds to the obtained percentage of test compound-induced hemolysis. Additionally, the absorption of the test compounds was determined at 540 nm in order to eliminate the effect of absorption of test compounds. Finally, saline solution and double-distilled water were used as negative and positive controls, respectively.Anticancer assays
The in vitro anticancer profiles were determined by testing L1–L3 and CuL1 to RuL3 against a human breast cancer cell line; MCF-7, by a cell viability assay (MTT assay).44 DMEM (low glucose), 10% fetal bovine serum and antibiotics/antimycotics formed the main constituents of the culture medium. MCF-7 cells were seeded in a 96-well plate at a density of 2 × 103 cells per well and were incubated at 37 °C under a humidified atmosphere containing 5% CO2 for 24 h before assay. After that, the cells were further incubated in media containing various concentrations of the test compounds. After 24 h, the medium was removed and washed with PBS. About 20 μL of MTT solution was added to each well followed by 4 h incubation at 37 °C. Subsequently, the medium was removed and 200 μL of DMSO were added. After shaking slowly twice for 5 s, the absorbance of each well was determined at 570 nm. The cell viability (%) was calculated as the ratio of the number of surviving cells in the test-compound treated samples to that in the control.Results and discussion
The analytical and spectroscopic data of L1–L3 and CuL1 to RuL3 supported their proposed structures. All the compounds were solids and were stable to air. In addition, all the compounds were readily soluble in water, DMSO and DMF. Water solubility of the compounds is an added advantage for therapeutic applications, since all biochemical reactions are based on small molecules that dissolve in aqueous media.45 The compounds were purified by washing with cold methanol and hexane.Glutamic acid was neutralized with aqueous sodium hydroxide to facilitate the condensation of its –NH2 group with the –SO2Cl group of benzenesulphonyl chlorides. The excess sodium hydroxide neutralized the HCl produced during the condensation of benzenesulphonyl chlorides. The condensation and the subsequent reaction mechanism involved in the formation of ligands are shown in Scheme 1. The ligands (L1–L3) complexed with copper(II), nickel(II) and ruthenium(III) ions to form the complexes CuL1 to RuL3, as shown in Scheme 2.
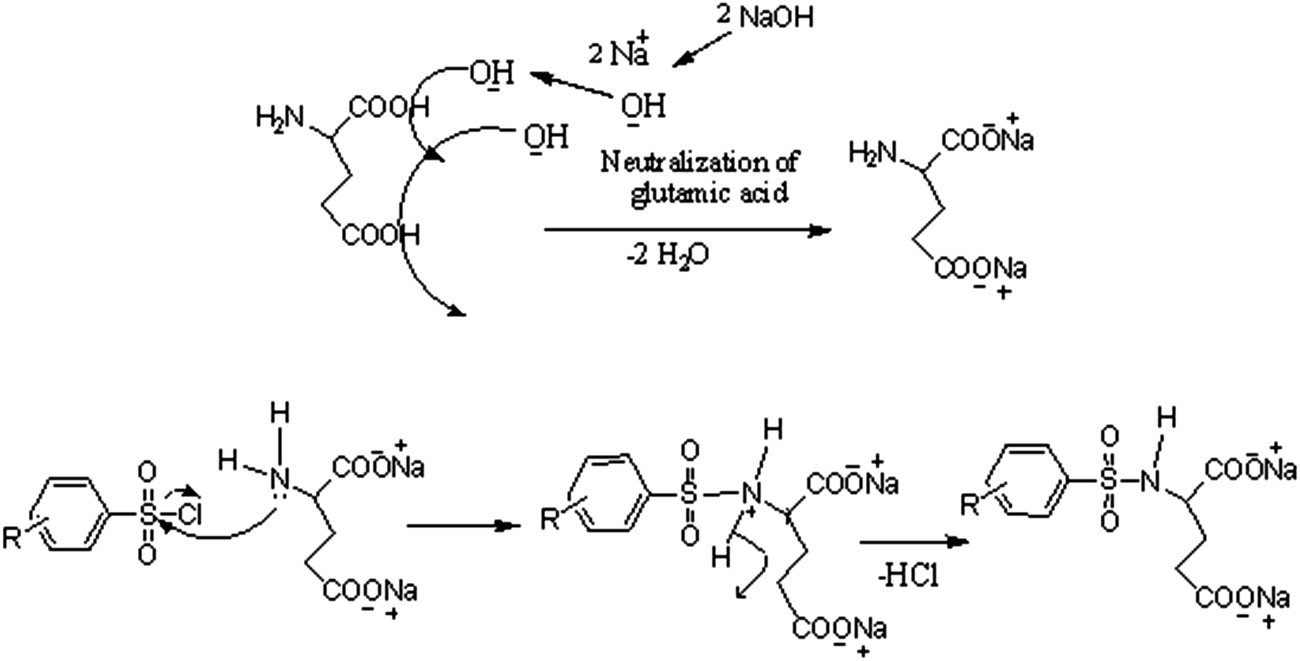 | ||
| Scheme 1 Mechanistic representation of the formation of ligands (L1, L2 and L3). | ||
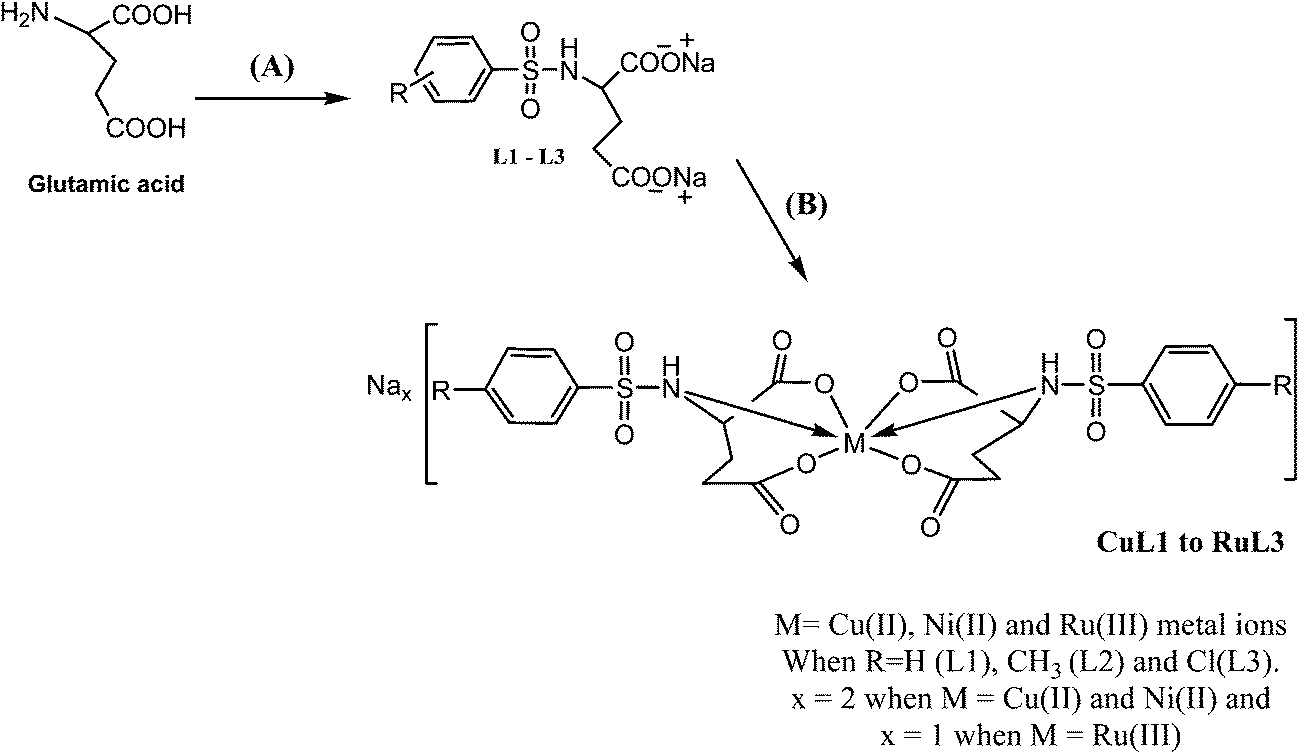 | ||
| Scheme 2 Schematic depiction of the syntheses of ligands L1–L3 and their complexes (CuL1 to RuL3). Reagents and conditions: (A) solid sodium hydroxide, benzenesulphonyl chlorides, reflux at 70 °C for 8 h, (B) hydrated metal chlorides. | ||
The compositions of all the compounds were ascertained on the basis of elemental analysis and ESI-MS spectra. Copper(II) and nickel(II) complexes had molar conductance in the ranges 161–183 and 168–194 Ω−1 cm2 mol−1, respectively, indicating their 1:2 electrolytic nature. However, ruthenium complexes had molar conductance in the range of 123–131 Ω−1 cm2 mol−1, which indicated their 1:1 electrolytic nature.46 It might be inferred from the molar conductance data that the two units of excess negative charge of the copper and nickel complexes were balanced by two sodium cations existing outside their coordination spheres. However, only one sodium cation was needed to balance the one unit of extra negative charge in ruthenium complexes. Finally, it can be assumed from the results of elemental analysis and ESI-MS spectra that 1:2 metal to ligand complexes resulted, wherein metal ions in nickel and ruthenium complexes are in an octahedral environment and those in copper complexes are in a tetragonal environment, which was further supported by their UV-vis spectra.
The formation of L1–L3 and CuL1 to RuL3 was confirmed by the appearance of peaks due to S–N stretching vibrations in the range of 896–916 cm−1. In addition, the peaks due to the symmetric and asymmetric vibrations of carboxylate groups were found in the ranges 1330–1417 and 1582.5–1625 cm−1, respectively. The monodentate-type coordination of the carboxylate groups to the metal ions in the complexes was confirmed by the prominent shifts in both the symmetric and asymmetric vibrations of (–OCO–) groups in the spectra of the metal complexes.47–50 Furthermore, the stretching frequencies for metal to oxygen and metal to nitrogen (Cu–O, Cu–N, Ni–O, Ni–N, Ru–O and Ru–N) bonds were observed in the spectra of all the complexes; suggesting the coordination of metal ions with the ligand donor atoms. 1H NMR spectra of the ligands showed the signals of the –NH protons of –SO2NH group in the range 7.520–7.870 ppm. In addition, the aromatic protons were observed in the region 7.140–7.723 ppm. The proton signals in the spectra of the complexes were slightly shifted due to the coordinating effect of the metal ions.51,52 Mass spectra of L1, L2 and L3 showed peaks at m/z values 372.01, 346.03 and 382.89, corresponding to the moieties [Na2C11H11SNO6 + K+ + 2H]+, [Na2C12H13SNO6 + H]+ and [Na2C11H10SNO6Cl + NH4+ − H]+, respectively. Mass spectra of NiL1, CuL2 and NiL3 showed peaks at m/z values 382.96 & 742.93, 644.82 & 662.95 and 766.79 & 782.75, corresponding to {Na2[Ni(C11H11SNO6)2] − Na2[C9H11SNO2] − 2COO}+ & {Na2[Ni(C11H11SNO6)2] + 3Na − H}+, {Na2[Cu(C12H13SNO6)2] − 2Na+ − CH3 − 2H}+ & {Na2[Cu(C12H13SNO6)2] − 2Na+ + H}− and {Na2[Ni(C11H10SNO6Cl)2] + Na+}+ & {Na2[Ni(C11H10SNO6Cl)2] + K+}+, respectively. In addition, several fragmentation peaks were observed in the spectra of the compounds, in accordance with their proposed structures. UV-vis spectra of the ligands showed absorption bands in the regions 208–240 and 243–283 nm corresponding to n–σ* and π–π* high energy transitions, respectively. The spectra of complexes were characterized by the presence of additional absorption bands due to metal-originated d–d transitions characteristic of their geometries. CuL1 to CuL3 showed absorption bands in the ranges 276–382 and 788–812 nm assigned to charge transfer bands and 2T2g ← 2Eg transitions, respectively, indicating their tetragonally distorted octahedral geometries.53,54 NiL1 to NiL3 absorption bands in the ranges 498–527 and 862–899 nm were assigned to 3T1g(F) → 3A2g(F) and 3T2g(F) → 3A2g(F) transitions, respectively, indicating their octahedral geometries.55–57 RuL1 to RuL3 showed absorption bands in the range 530–890 nm, assigned to the 6A1g → 4T1g transition, indicating their octahedral geometry.58–60
Solution stability
Aquation is an important step for the proper and safe functioning of many therapeutically active drugs, including the well-known KP1019 and NAMI-A.61,62 Therefore, the solution stabilities of CuL1 to RuL3 in PBS at physiological pH were assessed by UV-vis spectrophotometry. From Fig. 2, it is clear that UV-vis spectra recorded for the fresh solutions of CuL1 showed no intra-ligand band shifts after 24 h; however, mild shifts in intensity were observed. In addition, UV-vis spectra of NiL1 to RuL3 (Fig. S1†) exhibited similar behavior after 24 h. Moreover, the solutions of the complexes did not precipitate during this time. All these results indicated the robust nature of the complexes.63–65 Therefore, the complexes under study did not undergo hydrolysis.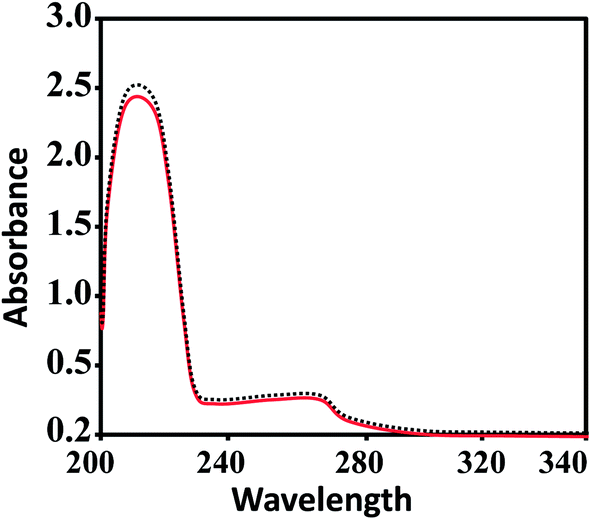 | ||
| Fig. 2 UV-vis spectra of CuL1 in PBS at 7.4 pH. The solid red and dashed black lines indicate the spectra of fresh solutions and the spectra of solutions after 24 h, respectively. | ||
Molecular modelling
Basically, molecular modelling exploits theoretical methods and computational techniques to mimic the behavior of molecules. In the absence of crystal structure data of the complexes (CuL1 to RuL3), a molecular modelling approach was used to gain insight into their structural characteristics. Energy-minimized configurations of complexes are achieved by the application of molecular mechanics, which therefore has become a tool of increasing utility for the structural investigation of metal complexes.66,67 Generally, energy minimization methods are used to obtain the equilibrium configuration of molecules. Molecular systems in their stable states correspond to global and local minima on their potential energy surface. Energy minimization exploits the mathematical procedure of optimization and moves atoms to reduce the net forces (the gradients of potential energy) on the atoms till they are negligible. These studies were carried out by the use of the semi-empirical method PM3 as implemented in hyperchem 8.0, using the Polak-Ribiere (conjugate gradient) algorithm and keeping a RMS gradient of 0.01 kcal Å−1 mol−1. The ball and stick models of CuL1 to RuL3 are shown in Fig. 3. The total energies and heats of formation calculated for CuL1 to RuL3 ranged from −747295.87 to −846239.10 and from 6.15 to −885.71 kJ mol−1, respectively (Table 1). In addition, the surface areas and volumes of the modelled molecules were also calculated and found to lie within the ranges 279.03–688.89 Å2 and 1302.11–1468.76 Å3, respectively, for CuL1 to RuL3 (Table 1). | ||
| Fig. 3 A perspective view of the ball and stick models of the energy-minimized structures of CuL1 to RuL3 created through molecular modelling. Lone pairs have been excluded for clarity. Nitrogen (blue), sulphur (yellow), carbon (cyan), oxygen (red), chlorine (green), copper, nickel and ruthenium (orange). Sodium cations outside the coordination spheres have been ignored for the energy minimization procedures. | ||
| Compounds | % hyperchromism | Kb (M−1) | Total energy (kJ mol−1) | Heat of formation (kJ mol−1) | Surface area (Å2) | Volume (Å3) |
|---|---|---|---|---|---|---|
| L1 | 41.50 | 0.7 × 103 | ||||
| CuL1 | 34.37 | 1.8 × 103 | −800604.21 |
323.59 | 469.23 | 1405.69 |
| NiL1 | 30.22 | 3.9 × 103 | −787633.81 |
−885.71 | 483.82 | 1468.76 |
| RuL1 | 41.93 | 0.97 × 104 | −747295.87 |
539.77 | 554.18 | 1328.13 |
| L2 | 50.01 | 1.8 × 103 | ||||
| CuL2 | 39.65 | 4.3 × 103 | −829733.22 |
6.15 | 464.64 | 1302.11 |
| NiL2 | 55.55 | 2.4 × 103 | −816114.30 |
−553.62 | 489.27 | 1329.37 |
| RuL2 | 38.98 | 1.32 × 104 | −776194.76 |
453.00 | 619.63 | 1426.46 |
| L3 | 17.14 | 2.1 × 103 | ||||
| CuL3 | 27.63 | 8.1 × 103 | −846239.10 |
244.55 | 279.03 | 1374.30 |
| NiL3 | 44.44 | 8.5 × 103 | −845214.02 |
−356.35 | 578.89 | 1411.12 |
| RuL3 | 51.11 | 5.24 × 104 | −805411.63 |
529.15 | 688.89 | 1465.17 |
DNA binding
The majority of anticancer drugs specifically target DNA and, therefore, DNA binding is one of the most critical steps for the functioning of a large number of metallo anticancer drugs. DNA offers several binding modes (outer-sphere non-covalent binding, metal coordination to nucleobases and phosphate backbone interactions) for anticancer drugs.68 DNA binding is often investigated by electronic absorption spectroscopy, by recording the changes in the absorbance and shifts in wavelength.69 The different spectral absorbances of DNA with complexes are indications of their interactions.70,71The ratio of absorbance of the stock solution of Ct-DNA in buffer at 260 nm and 280 nm was greater than 1.80 (A260/A280 > 1.80), indicating the protein-free nature of DNA.72 The spectra depicting the interactions of DNA with L1 and CuL1 are given in Fig. 4 and 5, respectively. In addition, the spectra of the interactions of L2, L3 and NiL1 to RuL3 are shown in Figs. S2–S11.† Addition of DNA solutions of increasing concentrations, 0.5 × 10−4, 0.8 × 10−4, 1.1 × 10−4 and 1.4 × 10−4 M, separately to the ligand and metal–complex solutions (1.6 × 10−4 M) resulted in hyperchromic shifts in the range 17.14–55.55% (Table 1).
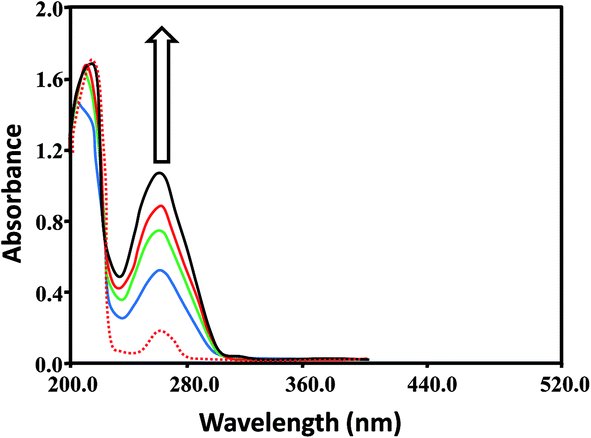 | ||
| Fig. 4 Absorption spectra of L1 (1.6 × 10−4 M) in the absence (red dashed line) and presence (solid lines) of increasing DNA concentrations; 0.5 × 10−4 M (blue), 0.8 × 10−4 M (green), 1.1 × 10−4 M (red) and 1.4 × 10−4 M (black). Arrow indicates the hyperchromic shifts with increasing DNA concentrations (0.5–1.4 × 10−4). | ||
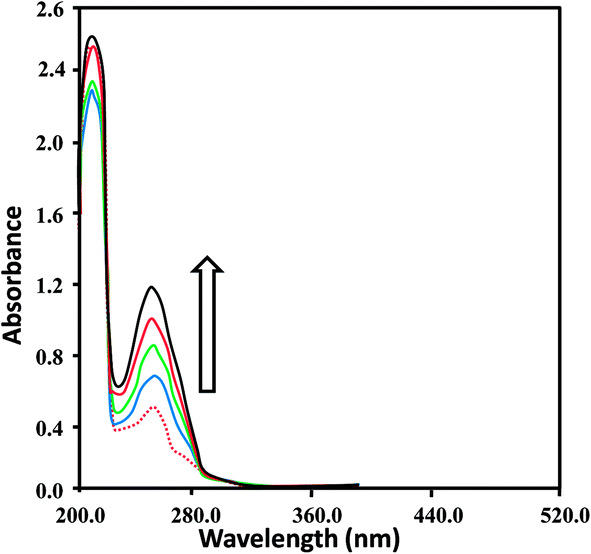 | ||
| Fig. 5 Absorption spectra of CuL1 (1.6 × 10−4 M) in the absence (red dashed line) and presence (solid lines) of increasing DNA concentrations; 0.5 × 10−4 M (blue), 0.8 × 10−4 M (green), 1.1 × 10−4 M (red) and 1.4 × 10−4 M (black). Arrow indicates the hyperchromic shifts with increasing DNA concentrations (0.5–1.4 × 10−4 M). | ||
These spectral changes indicated that the complexes bound to DNA via non-covalent interactions or simply to the uncoiled DNA double helix and exposed more DNA bases.73 The non-covalent interactions may include hydrogen bonding between the base pairs (accessible in the minor grooves) and the nitrogen and oxygen atoms of ligands and their complexes. In addition, van der Waal's attractive forces may also be involved in causing the binding of the ligands and complexes. Hyperchromism and hypochromism are indications of the interaction of compounds with the DNA helix. Hyperchromic shifts revealed the changes in DNA structure and conformation that occurred after the compounds bound to DNA, leading to structural damage to the DNA helix.74,75
For the quantitative determination of DNA binding potentials of L1–L3 and CuL1 to RuL3, their binding constants (Kb) were obtained by monitoring the changes in absorbance of the π → π* spectral band (239–296 nm) with increasing concentrations of DNA, using the following equation:76
| [DNA]/(εa − εf) = [DNA]/(εb − εf) + 1/Kb(εb − εf) |
DNA binding constants (Kb) calculated for L1–L3 and CuL1 to RuL3 ranged from 0.7 × 103 to 2.1 × 103 M−1 and 1.8 × 103 to 5.24 × 104 M−1 (Table 1), respectively. These values indicated good binding of ligands and their complexes with DNA. The order of DNA binding of the compounds is RuL3 > RuL2 > RuL1 > NiL3 > CuL3 > CuL2 > NiL1 > NiL2 > L3 > CuL1 = L2 > L1. Metal complexes are generally known to bind to DNA more strongly as compared to their free ligands, due to the presence of an additional charge on the central metal ion core and vacant d-orbitals.77 Ruthenium complexes displayed higher binding constants as compared to copper and nickel complexes, due to their uninegative charge.78 Furthermore, ruthenium complexes exhibited similar binding constants, as seen in the single report on the DNA binding studies of anionic ruthenium complexes.78 The higher binding constants of CuL2 to RuL3 as compared to CuL1 to RuL1 might be attributed to the polarizing and non-polarizing effects of –CH3 and –Cl groups in these complexes.
In silico studies
Combinatorial chemistry and virtual screening are well reputed, possibly due to their reduction of the extremely time-consuming steps of organic and inorganic synthesis and biological screening. Molecular docking is a very useful tool for the prediction of the interactions of drugs with various biological macromolecules at a supramolecular level.79 B-DNA, the most prevalent form of DNA, has a deep and wide major groove and a deep and narrow minor groove. It is the base pairing between the two strands of DNA that gives rise to the distinct hydrogen bond acceptor/donor patterns in the major and minor grooves. The rigid molecular DNA docking of the ligands has been carried out using AutoDock 4.2 to find out the possible sites of the interactions of DNA with the ligands. The docking studies of ligands were performed with DNA dodecamers d(CGCGAATTCGCG)2 (PDB ID: 1BNA). The molecularly docked models of the ligand–DNA adducts of L1, L2 and L3 are shown in Fig. 6, 7 and 8, respectively. It is clear from these figures that the ligands interacted with DNA through the minor groove. Besides, L1, L2 and L3 formed two, one and six hydrogen bonds with DNA, respectively. The docking energies of the ligands were in the order L2 > L1 > L3 (Table 2). In addition, the van der Waal's energies of the ligands were in the order L2 > L1 > L3 (Table 2), the same as the order of their docking energy. The greater van der Waal's energy of L2 in comparison to L1 and L3 might be attributed to the presence of a methyl group on the aromatic ring of L2. Lower van der Waal's energy of L3 as compared to L1 might be due to the hydrophilicity of the chloro-substituted aromatic ring. Furthermore, it might be said that the van der Waal's forces have played a more significant role than that played by the number of hydrogen bonds in causing the binding of the ligands to DNA. The maximum number of hydrogen bonds formed by L3 might be attributed to the assistance of the chloro-substituted aromatic ring in bringing the hydrogen bond forming portions of L3 in close proximity to the hydrogen bond forming portions of DNA. In the case of L2, the methyl substitution on the aromatic ring might be causing steric hindrance to its hydrogen bonding portions. Overall, the order of the docking energy of the ligands is the same as the order of their DNA binding constants and, therefore, in silico studies verified the DNA binding of the ligands.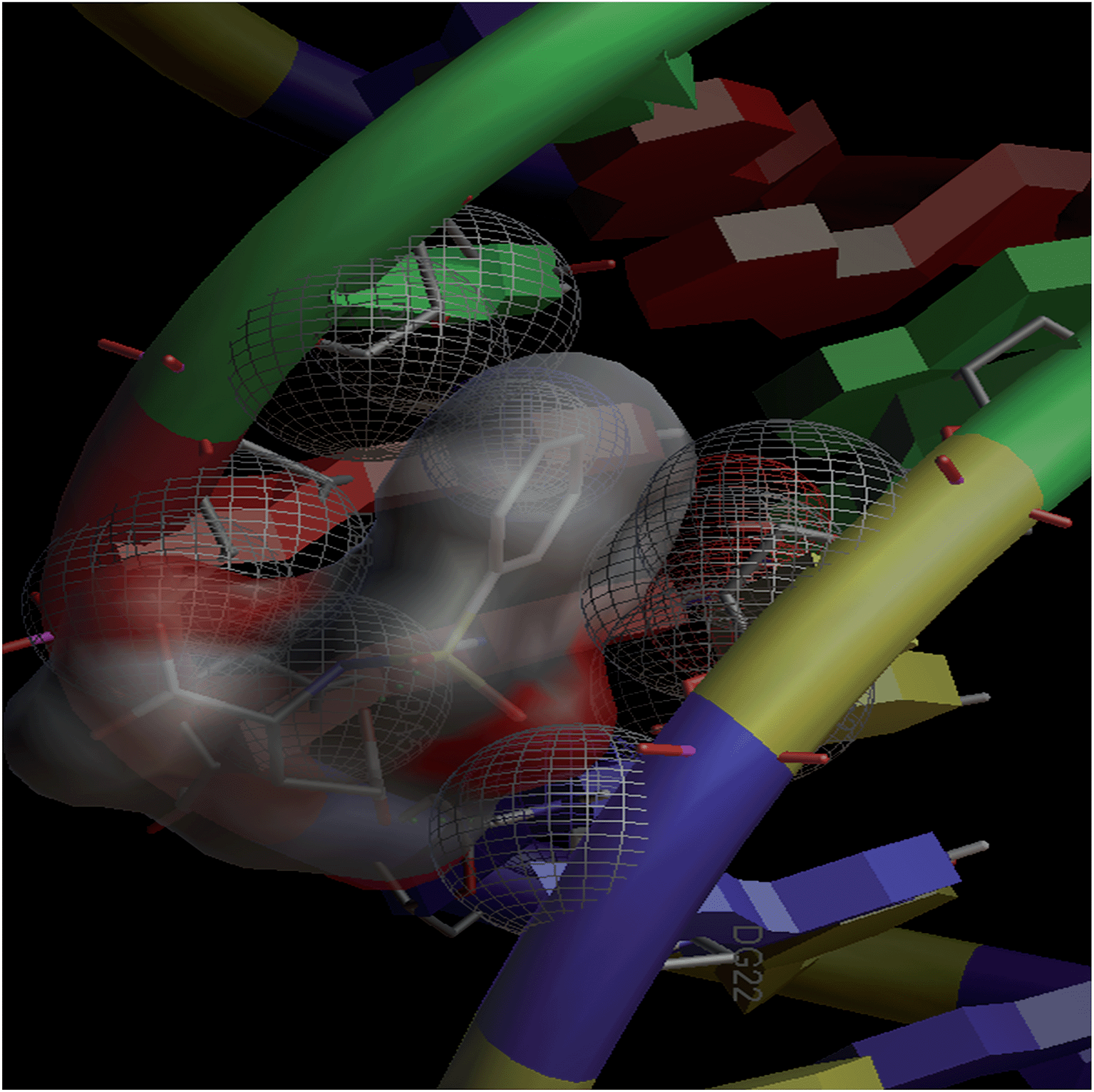 | ||
| Fig. 6 Docking image showing the ligand (L1) forming two hydrogen bonds with DNA. The van der Waal's interaction of the ligand with the hydrophobic chains of DNA can also be visualized. | ||
 | ||
| Fig. 7 Docking image showing the ligand (L2) forming one hydrogen bond with DNA. The van der Waal's interaction of the ligand can also be visualized. | ||
 | ||
| Fig. 8 Docking image showing the ligand (L3) forming six hydrogen bonds with DNA. The van der Waal's interaction of the ligand can also be visualized. | ||
| Compounds | No. of hydrogen bounds with DNA | Residues involved in hydrogen bonding | Docking energy (kJ mol−1) | van der Waal's energy (kJ mol−1) |
|---|---|---|---|---|
| L1 | 2 | A: DG4:H22 and A: DA5:H3 | −12.09 | −21.67 |
| L2 | 1 | A: DA5:H3 | −15.06 | −24.97 |
| L3 | 6 | A: DA6:H7, A: DA6:H61, B: DA17:H7, B: DA17:H6, B: DA18:H7, B: DA18:H62 | −9.70 | −19.20 |
Hemolysis assays
During the entry of drugs into the animal body, they interact with blood components, particularly RBCs (oxygen-carrying blood cells). Therefore, it is quite important to assess the effects of the newly developed drugs on RBCs. In vitro hemolysis assay is a widely accepted screening tool for predicting in vivo toxicity to host cells.80 In vitro toxicities of the developed compounds were compared with the standard anticancer drug, doxorubicin. The hemolysis results for the ligands and their complexes are shown in Fig. 9. A perusal of this figure indicates that L1, CuL1, NiL1, L2, CuL2, NiL2, L3 and CuL3 were the least toxic (5% hemolysis) at a concentration of 100 μg mL−1. NiL3 was slightly toxic (6% hemolysis). RuL1, RuL2 and RuL3 were more toxic, with 12, 17 and 8% hemolysis at the same concentration. On the other hand, doxorubicin exhibited 42% hemolysis at 100 μg mL−1 concentration. Therefore, the reported compounds are much less toxic towards RBCs in comparison to doxorubicin.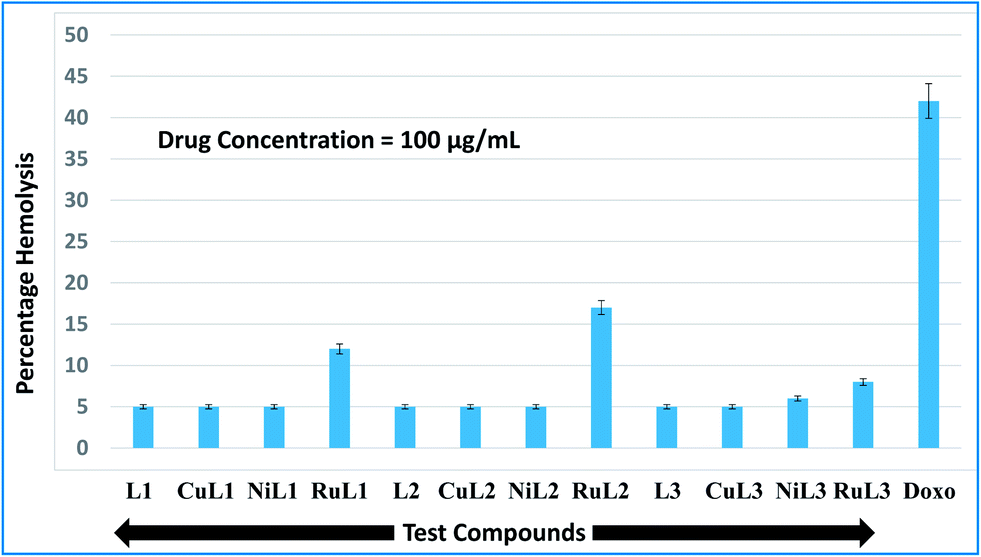 | ||
| Fig. 9 Percentage hemolysis potentials of L1–L3 and CuL1 to RuL3 against rabbit RBCs at 100 μg mL−1 concentration with respect to doxorubicin (used as standard). | ||
Anticancer profiles
Anticancer profiles of L1–L3 and CuL1 to RuL3 were assessed from percentage viabilities. The effects of the reported compounds on MCF-7 cells were evaluated at 0.0001–1.0 μg mL−1 concentration ranges with a 10× dilution factor. The percentage viabilities for the compounds are given in Fig. 10. A perusal of the figure indicates that for all the compounds viability was maintained in the range of 86–131% at 0.0001 μg mL−1 concentration. At a higher concentration (0.001 μg mL−1), the greatest inhibitions were exhibited by L2 (90% viability) > CuL3 (95% viability) > RuL3 (97% viability) = CuL1 (97% viability). Interestingly, at 0.01 μg mL−1 concentration, L1 (77% viability) and RuL1 (80% viability) exhibited better anticancer effects than doxorubicin (87% viability). Besides, the maximum antiproliferative effects at 0.1 μg mL−1 concentration were exhibited by CuL1 (73% viability) and L1 (86% viability). Moreover, NiL2 (53% viability), L1 (60% viability), CuL1 (68% viability) and CuL2 (68% viability) showed maximum activities among the tested compounds at 1.0 μg mL−1 concentration. The anticancer activities of the reported ligands and metal complexes depend on the structure and activity relationship leading to different binding modes with DNA. There was no trend in the anticancer activities of ligands and their metal complexes. However, it was observed that metal complexes of the ligands showed higher anticancer activities than their ligands. This may be due to the fact that metal complexes have greater affinities for DNA than ligands. Among all the tested concentrations, the maximum inhibition (53% viability) was observed with NiL2 at 1.0 μg mL−1.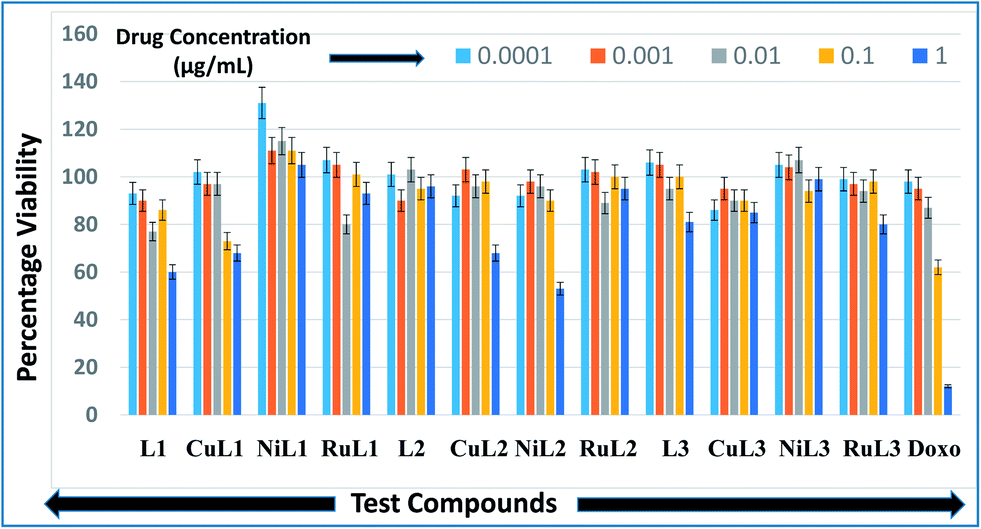 | ||
| Fig. 10 Anticancer activities of L1–L3 and CuL1 to RuL3 with respect to doxorubicin in the concentration range 0.0001–1.0 μg mL−1. Anticancer activities are expressed as percentage viabilities. | ||
Conclusion
This paper describes the facile syntheses of copper(II), nickel(II) and ruthenium(III) complexes of a series of disodium sulphonamides of L-glutamic acid. Ligands and their complexes were freely soluble in water, which is one of the most important requirements for a suitable drug. The complexes were robust and resisted hydrolysis in PBS at 7.4 pH. DNA binding constants indicated good binding of the compounds. Complexes bound to DNA more strongly as compared to their ligands. As per in silico studies, the ligands preferred to enter into the minor groove of DNA. DNA–ligand adducts were mainly stabilized by hydrogen bonding and van der Waal's attractions. All the compounds were significantly less toxic to RBCs as compared to the standard drug, doxorubicin. Furthermore, the compounds showed good anticancer activities on MCF-7 cell lines. In a nutshell, the compounds showed encouraging therapeutic properties and, therefore, have a promising future.Conflict of interest
There are no conflicts of interest to declare.Acknowledgements
The authors are thankful to the UGC (University Grants Commission) New Delhi for providing a UGC-BSR Meritorious research fellowship to Mr Waseem A. Wani.References
- I. Ali, W. A. Wani and K. Saleem, Cancer Ther., 2011, 8, 56 Search PubMed.
- I. Ali, Rahis-ud-din, Kishwar Saleem, H. Y. Aboul-Enein and A. Rather, Cancer Ther., 2011, 8, 6 Search PubMed.
- I. Ali, K. Rahis-ud-din, Kishwar Saleem, M. A. Rather, W. A. Wani and A. Haque, Curr. Cancer Drug Targets, 2011, 11, 135 CrossRef CAS.
- I. Ali, W. A. Wani, K. Saleem and A. Haque, Anti-Cancer Agents Med. Chem., 2013, 13, 296 CrossRef CAS.
- N. Shah and D. S. Dizon, Future Oncol., 2009, 5, 33 CrossRef CAS PubMed.
- M. A. Medina, J. Nutr., 2001, 131, 2539S CAS.
- I. Ali, W. A. Wani, K. Saleem and A. Haque, Curr. Drug Ther., 2012, 7, 13 CrossRef CAS.
- S. Kumar, T. E. Witzig and S. V. Rajkumar, J. Cell Mol. Med., 2002, 6, 160 CrossRef CAS PubMed.
- I. Ali, W. A. Wani, A. Haque and K. Saleem, Future Med. Chem., 2013, 5, 961 CrossRef CAS PubMed.
- C. L. Vishwanathan, S. Deb, A. Jain, T. Lokhande and A. Juvekar, Indian J. Pharm. Sci., 2008, 70, 245 CrossRef CAS PubMed.
- C. Cui, Y. Zhang, L. Wang, H. Liu and G. Cui, J. Pharm. Pharmacol., 2009, 61, 1353 CrossRef CAS.
- S. Samanta, S. M. Alam, P. Panda and T. Jha, Eur. J. Med. Chem., 2009, 44, 70 CrossRef CAS PubMed.
- S. Samanta, K. Srikanth, S. Banerjee, B. Debnath, S. Gayen and T. Jha, Bioorg. Med. Chem., 2004, 12, 1413 CrossRef CAS PubMed.
- S. Farber, L. K. Diamond, R. D. Mercer, R. F. Sylvester and J. A. Wolf, N. Engl. J. Med., 1948, 238, 787 CrossRef CAS PubMed.
- Drug information online. Methotrexate side effects. http//:www.drugs.com/sfx/www.drugs.com/sfx/methotrexate-side-effects.html.
- Z. Ortiz, B. Shea, M. E. Suarez-Almazor, D. Moher, G. A. Wells and P. Tugwell, J. Rheumatol., 1998, 25, 36 CAS.
- A. Scozzafava, T. Owa, A. Mastrolorenzo and C. T. Supuran, Curr. Med. Chem., 2003, 10, 925 CrossRef CAS.
- S. G. Kucukguzel, I. Coskun, S. Aydın, G. Aktay, S. Gursoy, O. Cevik, O. B. Ozakpınar, D. Ozsavcı, A. Sener, N. Kaushik-Basu, A. Basu and T. T. Talele, Molecules, 2013, 18, 3595 CrossRef CAS PubMed.
- E. E. Gurdal, M. Yarim, I. Durmaz and R. Cetin-Atalay, Drug Res., 2013, 63, 121 CrossRef CAS PubMed.
- M. S. Al-Dosari, M. M. Ghorab, M. S. Al-Said and Y. M. Nissan, Chem. Pharm. Bull., 2013, 61, 50 CrossRef CAS PubMed.
- N. Abbassi, H. Chicha, M. Rakib el, A. Hannioui, M. Alaoui, A. Hajjaji, D. Geffken, C. Aiello, R. Gangemi, C. Rosano and M. Viale, Eur. J. Med. Chem., 2012, 57, 240 CrossRef CAS PubMed.
- S. J. Tan, Y. K. Yan, P. P. F. Lee and K. H. Lim, Future Med. Chem., 2010, 2, 1591 CrossRef CAS PubMed.
- K. Saleem, W. A. Wani, A. Haque, M. N. Lone, M. F. Hsieh, M. A. Jairajpuri and I. Ali, Future Med. Chem., 2013, 5, 135 CrossRef CAS PubMed.
- M. Ganeshpandian, R. Loganathan, S. Ramakrishnan, A. Riyasdeen, M. A. Akbarsha and M. Palaniandavar, Polyhedron, 2013, 52, 924 CrossRef CAS PubMed.
- I. Ali, W. A. Wani, K. Saleem and D. Wesselinova, Med. Chem., 2013, 9, 11 CrossRef CAS.
- I. Ali, W. A. Wani, K. Saleem and M. F. Hseih, Polyhedron, 2013, 56, 134 CrossRef CAS PubMed.
- F. Zhang, Q. Y. Lin, X. L. Zheng, L. L. Zhang, Q. Yang and J. Y. Gu, J. Fluoresc., 2012, 22, 1395 CrossRef CAS PubMed.
- M. Sobiesiak, I. P. Lorenz, P. Mayer, M. Wozniczka, A. Kufelnicki, U. Krajewska, M. Rozalski and E. Budzisz, Eur. J. Med. Chem., 2011, 46, 5917 CrossRef CAS PubMed.
- A. Buschini, S. Pinelli, C. Pellacani, F. Giordani, M. B. Ferrari, F. Bisceglie, M. Giannetto, G. Pelosi and P. Tarasconi, J. Inorg. Biochem., 2009, 103, 666 CrossRef CAS PubMed.
- A. Bergamo and G. Sava, Dalton Trans., 2011, 40, 7817 RSC.
- I. Kostova, Curr. Med. Chem., 2006, 13, 1085 CrossRef CAS.
- I. Ali, K. Saleem, D. Wesselinova and A. Haque, Med. Chem. Res., 2013, 22, 1386 CrossRef CAS.
- E. M. Nagy, A. Pettenuzzo, G. Boscutti, L. Marchio, L. Dalla Via and D. Fregona, Chem.–Eur. J., 2012, 18, 14464 CrossRef CAS PubMed.
- E. Corral, A. C. G. Hotze, H. Dulk, A. Leczkowska, A. Rodger, M. J. Hannon and J. Reedijk, J. Biol. Inorg. Chem., 2009, 14, 439 CrossRef CAS PubMed.
- D. Griffith, S. Cecco, E. Zangrando, A. Bergamo, G. Sava and C. J. Marmion, J. Biol. Inorg. Chem., 2008, 13, 511 CrossRef CAS PubMed.
- P. R. Reddy and N. Raju, Polyhedron, 2012, 44, 1 CrossRef CAS PubMed.
- HyperChem Version 8.0 Hypercube, Inc., USA.
- M. F. Reichmann, S. A. Rice, C. A. Thomas and P. Doty, J. Am. Chem. Soc., 1954, 76, 3047 CrossRef CAS.
- M. F. Sanner, J. Mol. Graphics Modell., 1999, 17, 57 CAS.
- Protein Data Bank, http://www.rcsb.org/pdb.
- G. M. Morris, D. S. Goodsell, R. S. Halliday, R. Huey, W. E. Hart, R. K. Belew and A. J. Olson, J. Comput. Chem., 1998, 19, 1639 CrossRef CAS.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605 CrossRef CAS PubMed.
- ASTM F756-00, Standard Practice for Assessment of Hemolytic Properties of Materials; ASTM International, West Conshohocken, PA, USA, 2000, DOI:10.1520/F0756-00.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55 CrossRef CAS.
- T. Nogrady, Medicinal Chemistry, A Biochemical Approach, Oxford University press, New York, 1985 Search PubMed.
- I. Ali, W. A. Wani and K. Saleem, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2013, 43, 1162 CrossRef CAS.
- T. A. Stephenson, S. M. Morehouse, A. R. Powell, J. P. Heffer and G. Wilkinson, J. Chem. Soc., 1965, 3632 RSC.
- T. A. Stephenson and G. Wilkinson, J. Inorg. Nucl. Chem., 1967, 29, 2122 CrossRef CAS.
- K. Nakamoto, Infrared Spectra of Inorganic and Co-ordination Compounds, John Wiley & Sons, New York, 1970 Search PubMed.
- H. A. Laila and P. B. Luigi, Polyhedron, 1996, 15, 327 CrossRef.
- A. Chaudhary and R. V. Singh, Ind. J. Chem., 2004, 43, 2529 Search PubMed.
- W. Kemp, Organic spectroscopy, Macmillan Press Ltd., London, 1975 Search PubMed.
- C. H. Krushna, C. M. Mahapatra and A. K. Dash, J. Inorg. Nucl. Chem., 1977, 39, 1253 CrossRef.
- N. N. Greenwood and A. Earnshaw, Chemistry of the Elements, Pergamon Press, New York, USA, 1984 Search PubMed.
- R. Atkins, G. Brewer, E. Kokot, G. M. Mocler and E. Sinn, Inorg. Chem., 1985, 24, 127 CrossRef CAS.
- B. N. Figgis and J. Lewis, Modern Coordination Chemistry, Interscience, New York, USA, 1960 Search PubMed.
- O. M. Adly, Spectrochim. Acta, Part A, 2012, 95, 483 CrossRef CAS PubMed.
- A. B. P. Lever, Inorganic Electronic Spectroscopy, Elsevier, Amsterdam, The Netherlands, 2nd edn, 1984 Search PubMed.
- M. M. T. Khan, D. Srinivas, R. I. Khureshy and N. H. Khan, Inorg. Chem., 1990, 29, 2320 CrossRef CAS.
- S. N. Pal and S. Pal, Eur. J. Inorg. Chem., 2003, 2003, 4244 CrossRef.
- M. Groessl, C. G. Hartinger, P. J. Dyson and B. K. Keppler, J. Inorg. Biochem., 2008, 102, 1060 CrossRef CAS PubMed.
- M. Bacac, A. C. G. Hotze, K. vander Schilden, J. G. Haasnoot, S. Pacor, E. Alessio, G. J. Sava and J. Reedijk, J. Inorg. Biochem., 2004, 98, 402 CrossRef CAS PubMed.
- F. Arjmand, A. Jamsheera and D. K. Mohapatra, J. Photochem. Photobiol., B, 2013, 121, 75 CrossRef CAS PubMed.
- C. Tan, S. Hu, J. Liu and L. Ji, Eur. J. Med. Chem., 2011, 46, 1555 CrossRef CAS PubMed.
- F. Arjmand, M. Aziz and M. Chauhan, J. Inclusion Phenom. Macrocyclic Chem., 2008, 61, 265 CrossRef CAS.
- M. A. El-ghamry, A. A. Saleh, S. M. Khalil and A. A. Mohammed, Spectrochim. Acta, Part A, 2013, 110, 205 CrossRef CAS PubMed.
- S. Chandra, S. Bargujar, R. Nirwal, K. Qanungo and S. K. Sharma, Spectrochim. Acta, Part A, 2013, 113, 164 CrossRef CAS PubMed.
- S. Mathur and S. Tabassum, Biometals, 2008, 21, 299 CrossRef CAS PubMed.
- S. Parveen and F. Arjmand, Spectrochim. Acta, Part A, 2012, 85, 53 CrossRef CAS PubMed.
- Z. D. Xu, H. Liu, S. L. Xiao, M. Yang and X. H. Bu, J. Inorg. Biochem., 2002, 90, 79 CrossRef CAS.
- Y. Wang and Z. Y. Yan, Transition Met. Chem., 2005, 30, 902 CrossRef CAS.
- J. Marmur, J. Mol. Biol., 1961, 3, 208 CrossRef CAS.
- P. J. Cox, G. Psomas and C. A. Bolos, Bioorg. Med. Chem., 2009, 17, 6054 CrossRef CAS PubMed.
- J. K. Barton, A. T. Danishefsky and J. M. Goldberg, J. Am. Chem. Soc., 1984, 106, 2172 CrossRef CAS.
- C. S. Chow and J. K. Barton, Methods Enzymol., 1992, 212, 219 CAS.
- A. Wolfe, G. H. J. Shimer and T. Meehan, Biochemistry, 1987, 26, 6392 CrossRef CAS.
- M. Chauhan and F. Arjmand, Chem. Biodiversity, 2006, 3, 660 CAS.
- N. Ljubijankic, A. Zahirovic, E. Turkusic and E. Kahrovic, Croat. Chem. Acta, 2013, 86, 215 CrossRef CAS.
- I. Ali, A. Haque, K. Saleem and M. F. Hsieh, Bioorg. Med. Chem., 2013, 21, 3808 CrossRef CAS PubMed.
- C. M. Sayes, K. L. Reed and D. B. Warheit, Toxicol. Sci., 2007, 97, 163 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra02570a |
| This journal is © The Royal Society of Chemistry 2014 |