 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Early amyloid β-protein aggregation precedes conformational change†
Bogdan
Barz
a,
Olujide O.
Olubiyi
a and
Birgit
Strodel
*ab
aForschungszentrum Jülich GmbH, Institute of Complex Systems: Structural Biochemistry (ICS-6), 52425 Jülich, Germany. E-mail: b.strodel@fz-juelich.de; Fax: +49 2461 618766; Tel: +49 2461 613670
bInstitute of Theoretical and Computational Chemistry, Heinrich Heine University Düsseldorf, 40225 Düsseldorf, Germany
First published on 15th January 2014
Abstract
The aggregation of amyloid-β protein (1–42) is studied at experimental concentrations using all-atom molecular dynamics simulations. We observe a fast aggregation into oligomers without significant changes in the internal structure of individual proteins. The aggregation process is characterized in terms of transition networks.
Oligomers of amyloid-β protein (Aβ) are considered one of the main causes of neurotoxicity and are thus highly associated with the onset of Alzheimer's disease.1 Experimental methods are able to identify some characteristics of aggregating proteins such as the oligomer size distribution or cellular toxicity,2 but due to the fast conversion of oligomers into fibrils, the elucidation of their structure at the molecular level is challenging. Computational methods have the advantage of atomistic detail but generally lack the size and time scales available in experiments. While there is a plethora of molecular dynamics (MD) simulations describing the structure of the full-length Aβ monomers, dimers and small oligomers at both atomistic3–6 and coarse grained levels,7,8 the aggregation from a large number of spatially separated monomers has been mainly studied by coarse-grained simulations with an implicit solvent.9 The studies of monomers and dimers are very insightful, but the structure of small Aβ oligomers observed in experiments during early aggregation might be considerably different due to their interaction with other oligomeric species. In addition, the structure of oligomers could also be influenced by the solute concentration due to different lifetimes of oligomeric species at different concentrations. Generally, in computational studies the solute concentration is one or even two orders of magnitude higher than in experiments, which will influence the aggregation process. Here we report the early assembly of Aβ42 proteins at experimental concentrations using all-atom MD simulations in an implicit solvent, which was initiated from 20 isolated Aβ42 monomers.10 To describe the assembly process we derived a maximum flow transition network (MTN) based on aggregation states defined by N1|N2|N3, where N1 represents the oligomeric size, N2 is the average number of hydrogen-bonds between individual chains from the oligomer, and N3 is the average number of amino acids in the β-strand conformation per peptide in the oligomer. Detailed information about the methods are provided in the ESI.†
The MTN shown in Fig. 1 displays a complex aggregation process in which initial monomers assemble into oligomers up to 18-mers for 200 ns. The aggregation states with N1 = 1 are distributed linearly with a gradual increase in the β-strand content from right to left. The monomers with more β-strands more readily aggregate than the others: state 1|0|17 is the main connection node to oligomers 2|2|15, 3|4|16, 4|6|13, 5|6|17, 12|7|13 and 17|8|13. These states are also the central connection to the other states with the same N1 value. In addition, state 1|0|20 is in direct contact with state 18|9|13 and thus to the rest of the 18-mer cluster. A representative snapshot of the 18-mer at the end of the simulation indicates an elongated conformation rather than a globular one as observed for the 8-mer (Fig. 1). The central aggregation states for hexamer and heptamers are 6|9|15 and 7|7|16, respectively, which are preferentially formed from trimers. Dimers are directly connected to aggregation state 8|7|12 and thus to the other N1 = 8 states, indicating that octamers are largely formed by the addition of dimers to either tetramers or hexamers. The oligomer mass distribution shown in Fig. 2a reveals a higher population for dimers, tetramers, hexamers, octamers, 12-mers and 18-mers. Previous computational9 and experimental studies2 of Aβ aggregation report significant peaks in pentamers/hexamers and 12/13-mers, in agreement with our results.
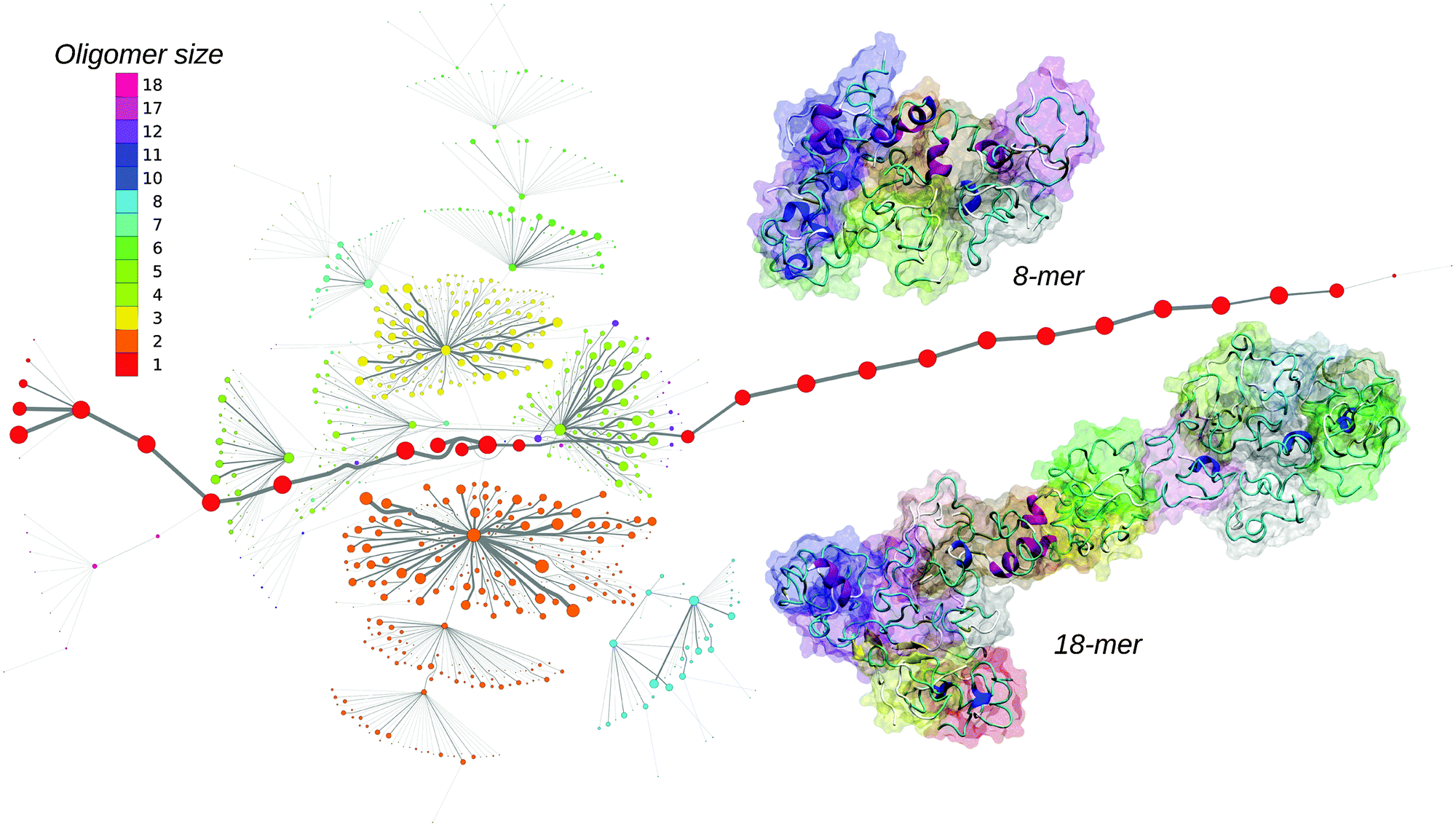 | ||
| Fig. 1 Maximum flow transition network. The node area is proportional to the state population and the color corresponds to the oligomer size. The edge thickness correlates with the number of transitions. Two representative oligomer structures for the octamer and 18-mer are shown in the cartoon and surface representation. | ||
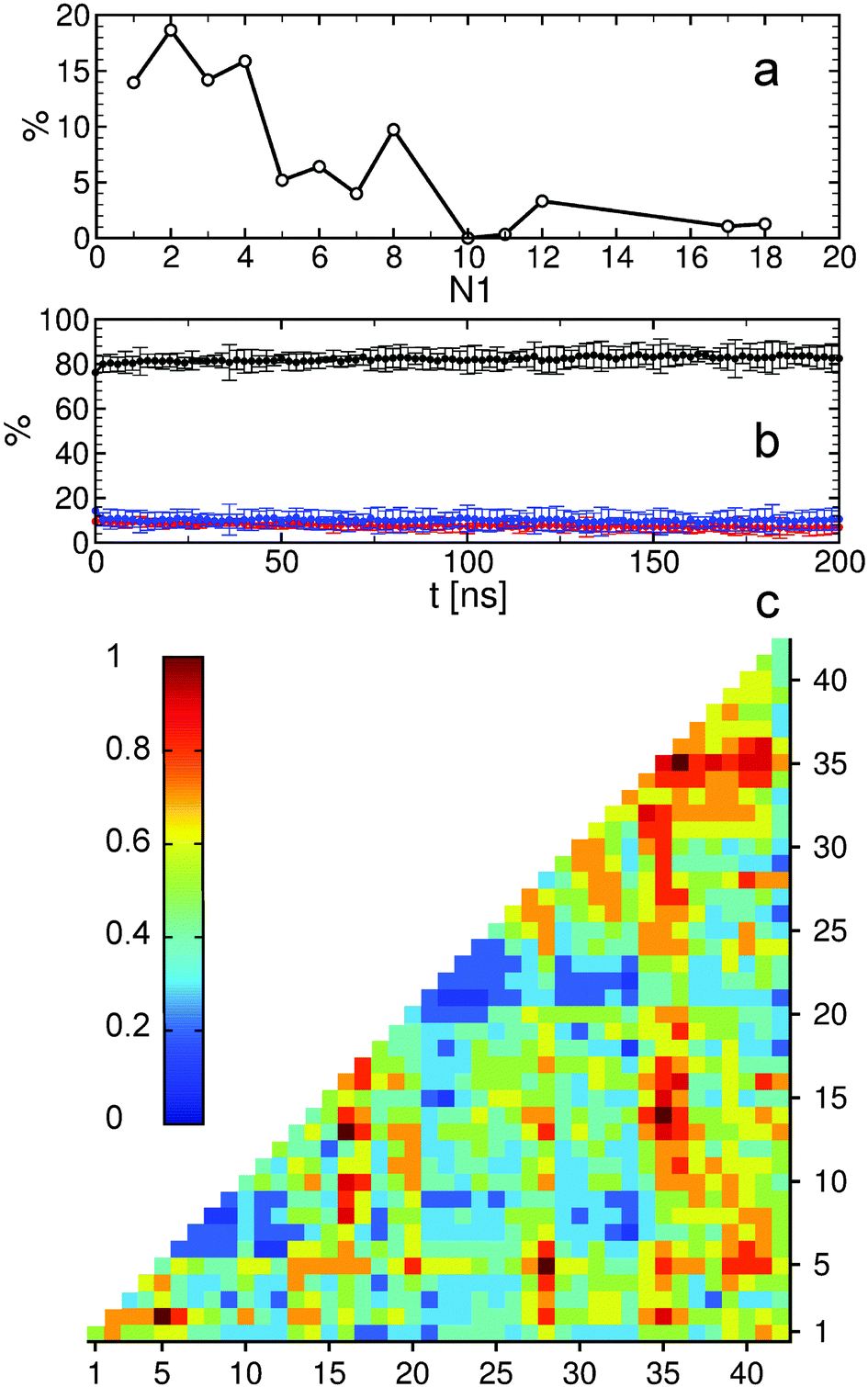 | ||
| Fig. 2 Analysis of Aβ oligomers. (a) Relative oligomer mass distribution during aggregation. The probability was determined as the population of N1 multiplied by N1. (b) Secondary structure propensity as a function of time. Black corresponds to coil, red to β-strand, and blue to helix. (c) Inter-molecular contact map for all oligomers based on a cutoff distance of 0.75 nm. Color coding corresponds to the normalized number of contacts. | ||
To describe structural changes during the assembly process we derived the time evolution of the secondary structure propensities (Fig. 2b). The initial β-strand propensity (∼10%) decreases slightly throughout the simulation and has an average of 7.6 ± 2.2%. The average helical propensity is around 10.1 ± 4.0% and the average coil propensity starts around 80% and increases slightly, having an average value of 82.2 ± 4.3%. This indicates little change in the overall secondary structure. Coarse-grained computational studies of Aβ aggregation showed an increase in the β-strand propensity upon dimer formation from ∼10% to ∼15%.9 However, when converted to all-atom models, these values change to 6–7% for both monomers and dimers.5 Experimental studies2 indicate 13–20% β-sheet for Aβ42 and Aβ40 while a more recent study shows a jump from ∼25% to ∼45% when converting from monomers to tetramers of Aβ40.11 Another experimental study reports Aβ42 oligomers without a β-sheet structure, in close agreement with our findings, which are on-pathway intermediates for fibril formation.12 It is important to mention the high turn propensity of residues V36 and G37 (67.1 ± 0.1% and 50.5 ± 0.1%, respectively), which has been recently shown to play an important role in the increased toxicity of Aβ42 relative to Aβ40 oligomers.13 In addition to the secondary structure analysis we calculated the RMSD of individual monomers throughout the simulation with respect to their conformations at the end of the simulation (Fig. S1, ESI†). This quantity reveals that besides an initial change in the monomeric conformation most of the proteins were quite stable following oligomerization with RMSD values below 0.5 nm. This result in combination with the small changes in the secondary structure suggests an aggregation process with few structural changes of individual peptides due to oligomer formation. To investigate the key amino acids involved in the assembly process we calculated contact maps for the interface between any two proteins that are part of an oligomer (Fig. 2c). An important result from the inter-molecular contact map is the proximity of hydrophobic regions from the C-terminus including I31–A42. In addition, region L34–V36 is in close proximity to region L17–F20. On the basis of solid-state NMR spectroscopy it has been shown that the C-terminal region is buried inside disc-shaped oligomers (pentamers and 10-mers) with strong contacts between F19 and L34.12 The 8-mer shown in Fig. 1 matches the description of the low-order oligomers in the non-β-sheet secondary structure observed by Ahmed et al.12 The increased interaction between the C-termini observed in Fig. 2c is in agreement with the study of Urbanc et al.,9 who proposed that oligomer formation of Aβ42 proceeds through interactions between the C-termini while for Aβ40 through interactions between the central hydrophobic regions (L17–A21). In addition, the 18-mer from Fig. 1 resembles the elongated protofibrillar assemblies (28-mer) reported by Urbanc et al. To gain additional information regarding the effect of the amino acids I41–A42 on the aggregation process we performed an additional 200 ns simulation with 20 Aβ40 peptides and computed the difference between Aβ42 and Aβ40 contact maps (Fig. S2, ESI†). We observe that Aβ42 forms more contacts between the C-termini than Aβ40, and exhibits many interactions between the charged C-terminus at A42 and positively charged residues, which in Aβ40 are formed with V40 instead. The extension of Aβ42 by I41 and A42 increases the hydrophobicity of V39–I41 compared to Aβ40 as the C-terminal charge is shifted by two residues, explaining the increased number of contacts between the C-termini in Aβ42. Electrostatic interactions contribute to oligomer formation by strong contacts formed by amino acid K16 with region S8–Q15 containing the negatively charged residue E11, and by K28 with region D1–D7 containing the negatively charged residues D1, E3, and D7.
Our results suggest that early Aβ aggregation precedes β-sheet formation. This is an important observation considering the low Aβ concentration in the current study (∼0.8 mM), close to experimental concentrations (∼30 μM–1 mM),2,12 and smaller than concentrations obtained from other computational studies of 3.4 mM.9 At the same time, the structurally stable oligomers and the small changes in their secondary structure indicate a fast assembly process relative to the time scale corresponding to their conformational reorganization. Whether the oligomers observed here during a very early aggregation stage are similar to the toxic species observed in experiments is difficult to assess. While we find similarities with experimentally observed Aβ42 oligomers in terms of size and structure,9,12 there is still a large debate regarding which Aβ oligomers are the toxic ones.14 Some groups consider small oligomers with a high content of β-sheet as toxic species,11 others suggest a second nucleation process where amyloid fibrils are present with small oligomer species as the source for toxic oligomers,15 and others propose that toxic Aβ oligomers have a cross-β structure.16 In our future studies we plan to follow the further growth and structural conversion of the oligomers obtained here and study their interactions with membranes. The aim should be that experiments probe at the same time the size, secondary structure and toxicity of low-order oligomers, allowing us to directly relate simulation and experimental results.
References
- W. L. Klein, G. A. Krafft and C. E. Finch, Trends Neurosci., 2001, 24, 219–224 CrossRef CAS.
- G. Bitan, M. D. Kirkitadze, A. Lomakin, S. S. Vollers, G. B. Benedek and D. B. Teplow, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 330–335 CrossRef CAS PubMed.
- A. Baumketner, S. L. Bernstein, T. Wyttenbach, G. Bitan, D. B. Teplow, M. T. Bowers and J.-E. Shea, Protein Sci., 2006, 15, 420–428 CrossRef CAS PubMed.
- T. Takeda and D. Klimov, J. Phys. Chem. B, 2009, 113, 6692–6702 CrossRef CAS PubMed.
- B. Barz and B. Urbanc, PLoS One, 2012, 7, e34345 CAS.
- B. Urbanc, L. Cruz, D. B. Teplow and H. E. Stanley, Curr. Alzheimer Res., 2006, 3, 493–504 CrossRef CAS.
- S. Côté, R. Laghaei, P. Derreumaux and N. Mousseau, J. Phys. Chem. B, 2012, 116, 4043–4055 CrossRef PubMed.
- A. Melquiond, X. Dong, N. Mousseau and P. Derreumaux, Curr. Alzheimer Res., 2008, 5, 244–250 CrossRef CAS.
- B. Urbanc, M. Betnel, L. Cruz, G. Bitan and D. B. Teplow, J. Am. Chem. Soc., 2010, 132, 4266–4280 CrossRef CAS PubMed.
- O. O. Olubiyi and B. Strodel, J. Phys. Chem. B, 2012, 116, 3280–3291 CrossRef CAS PubMed.
- K. Ono, M. M. Condron and D. B. Teplow, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14745–14750 CrossRef CAS PubMed.
- M. Ahmed, J. Davis, D. Aucoin, T. Sato, S. Ahuja, S. Aimoto, J. I. Elliott, W. E. Van Nostrand and S. O. Smith, Nat. Struct. Mol. Biol., 2010, 17, 561–567 CAS.
- R. Roychaudhuri, M. Yang, A. Deshpande, G. M. Cole, S. Frautschy, A. Lomakin, G. B. Benedek and D. B. Teplow, J. Mol. Biol., 2013, 425, 292–308 CrossRef CAS PubMed.
- I. Benilova, E. Karran and B. De Strooper, Nat. Neurosci., 2012, 15, 349–357 CrossRef CAS PubMed.
- S. I. A. Cohen, S. Linse, L. M. Luheshi, E. Hellstrand, D. A. White, L. Rajah, D. E. Otzen, M. Vendruscolo, C. M. Dobson and T. P. J. Knowles, Proc. Natl. Acad. Sci. U. S. A., 2013, 201218402 Search PubMed.
- J. C. Stroud, C. Liu, P. K. Teng and D. Eisenberg, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7717–7722 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Methods section and additional figures. See DOI: 10.1039/c3cc48704k |
| This journal is © The Royal Society of Chemistry 2014 |