Visible-light excited red emitting luminescent nanocomposites derived from Eu3+-phenathrene-based fluorinated β-diketonate complexes and multi-walled carbon nanotubes†
V.
Divya
and
M. L. P.
Reddy
*
Chemical Sciences and Technology Division, CSIR-Network of Institutes for Solar Energy, National Institute for Interdisciplinary Science & Technology (NIIST), CSIR, Thiruvananthapuram-695 019, India. E-mail: mlpreddy55@gmail.com
First published on 18th October 2012
Abstract
A novel β-diketone ligand, 4,4,5,5,5-pentafluoro-3-hydroxy-1-(phenanthren-3-yl)pent-2-en-1-one (Hpfppd), which contains a polyfluorinated alkyl group, as well as a conjugated phenanthrene unit, has been synthesized and utilized for the construction of two new europium coordination compounds [Eu(pfppd)3(H2O)2] 1 and [Eu(pfppd)3(tpy)] 2 (where tpy = 2,2′:6,6′′-terpyridine). The designed europium compounds were well characterized and their solid-state photoluminescence (PL) properties were examined. Notably, the introduction of a highly conjugated phenanthrene moiety into the 3-position of the β-diketonate ligand remarkably extends the excitation window of the corresponding Eu3+ complexes towards the visible region (up to 500 nm). The PL study demonstrated that the replacement of high-energy oscillators O–H (water molecules) in 1 with an ancillary ligand terpyridine leads to an impressive enhancement in both overall quantum yield (from 31 to 75%) and 5D0 lifetime (from 0.51 to 1.04 ms) values. To the best of our knowledge, the solid-state quantum yield of 2 is found to be the highest so far reported in the literature under blue-light excitation (415 nm). To gain a wider applicability and also to overcome the poor thermal, photochemical and mechanical stabilities of Eu3+–β-diketonate luminophores, in the present study, a nanocomposite material (1@oxMWCNTs) has been fabricated by coupling Eu3+ complex 1 onto an oxidized multi-walled carbon nanotube (oxMWCNTs) scaffold. Characterization of the new luminescent nanocomposite material was accomplished by means of FT-IR, FT-Raman, XPS, TGA, SEM, TEM, AFM, EDAX and photoluminescence spectroscopic techniques. The designed nanocomposites 1@oxMWCNTs exhibit high thermal stability and luminescence efficiency. Furthermore, the high dispersibility of luminescent 1@oxMWCNTs in a polymer matrix (PMMA) and intense red emissions make it a promising luminophore for applications in polymer optical fibers as well as in light emitting devices.
Introduction
The unique photoluminescence properties of trivalent lanthanide ions have aroused tremendous interest for decades owing to their potential applications in medical diagnostics,1 cell imaging,2 and development of lanthanide-based organic light-emitting diodes (OLEDs).3 The attractive features of lanthanide ions as luminescent materials include their line like emission, high quantum yield, long luminescence lifetime (μs–ms range) and low long-term toxicity.4 Recently, challenges in the chemistry of the lanthanide ions have been to develop luminescent europium complexes that can be sensitized by visible-light and to determine the energy-transfer mechanisms in these systems. Thus, this field has become much more important because of the demand for less-harmful labeling reagents in the life sciences and low-voltage-driven pure-red emitters in optoelectronic technology.5 Generally there are two approaches essentially followed to solve this problem. One of the approaches is to introduce a 4d- or 5d-transition metal ion such as Ir(III) into the Eu(III) complex molecule. Unfortunately, photoluminescence efficiency of this kind of europium complexes based on the 3MMLCT (metal–metal-to-ligand charge transfer) or 3MLCT (metal-to-ligand charge transfer) was always very low.6 Another method is to modify the ligand molecule with a suitable expanded π-conjugated system to shift the excitation band of its europium complex to the visible region, and this expanded π-conjugated system cannot be either too small to absorb visible light or too big to increase the triplet state (singlet state in some complexes) of the ligand to a degree higher than 5D0, the lowest excited state energy level of the central Eu3+ ion.7 Accordingly, several longer-wavelength-sensitized europium complexes have been developed by several groups through the usual triplet pathway mechanism, with the use of antenna chromophoric groups, which have a smaller energy gap between the lowest singlet excited state and the triplet excited state, or the singlet pathway mechanism.8 However, some of the europium adducts reported exhibit poor quantum yields.8,9 This has inspired us to design a novel β-diketonate ligand, namely 4,4,5,5,5-pentafluoro-3-hydroxy-1-(phenanthren-3-yl)pent-2-en-1-one (Hpfppd), and utilize it for the development of two new europium β-diketonate coordination compounds [Eu(pfppd)3(H2O)2] 1 and [Eu(pfppd)3(tpy)] 2 (where tpy = 2,2′:6,6′′-terpyridine) in order to shift the excitation wavelength to the visible region. The designed Eu3+-fluorinated β-diketonate complexes have been characterized by standard spectroscopic techniques, and their solid-state photophysical properties were investigated.The β-diketone ligand class represents one of the important “antennas” for facilitating efficient ligand to Eu3+ ion energy transfers, thus generating high harvest emissions.10 However, the low thermal and photochemical stability, together with their poor mechanical and conducting properties and tendency to aggregate, represent important limitations of Eu3+–β-diketonate complexes toward their practical applications.11 In order to overcome some of the inherent weaknesses, herein efforts have been made to enhance the luminescence intensity by taking advantage of several scaffolding materials.12 It is well documented that carbon nanotubes (CNTs) are suitable platforms for the blending of luminescent Eu3+–β-diketonate complexes due to their exceptional electrical, mechanical and thermal properties.13,14 Inspired by the recent seminal works of Laura Maggini et al.,14a Wong et al.14f and Shi et al.,14e herein we described the synthesis, characterization and photophysical properties of visible-light excited red emitting nanocomposites (1@oxMWCNTs), derived from an Eu3+-phenathrene-based fluorinated β-diketonate complex and multi-walled carbon nanotubes. In the present work, efforts have also been made to isolate red emitting luminescent molecular europium plastic materials by combining the unique optical properties of nanocomposites 1@oxMWCNTs with the mechanical characteristics, thermal stability, flexibility and film-forming tendency of polymers (PMMA).15
Results and discussions
Synthesis and characterization of ligand and Eu3+ complexes 1–2
The β-diketone ligand 4,4,5,5,5-pentafluoro-3-hydroxy-1-(phenanthren-3-yl)pent-2-en-1-one (Hpfppd) was synthesized in 91% yield by a Claisen condensation reaction of 3-acetylphenanthrene with the methyl pentafluoropropionate in the presence of sodium methoxide in THF medium. The corresponding one-pot synthesis of the ligand is summarized in Scheme 1. The designed ligand has been characterized by the 1H NMR, 13C NMR (Fig. S1 and S2 of the ESI†), FT-IR and mass spectroscopic (FAB-MS) methods as well as by elemental analysis. The ligand is present mainly as enol form in CDCl3 solutions, which is evident from the 1H NMR data. In the 1H NMR spectrum of Hpfppd, a broad peak at δ 15.51 ppm corresponding to enolic –OH has been observed. Further, the absence of methyne protons at ∼δ 3.7 ppm confirms the existence of the ligand in enolic form. The synthesis routines for the Ln3+ binary and ternary complexes are shown in Schemes 2 and 3, respectively. The elemental analyses and FAB-MS studies of Eu3+ complexes (1–2) revealed that the central Eu3+ ion is coordinated to three β-diketonate ligands. In the case of complex 2, one molecule of the tridentate nitrogen donor, 2,2′:6,6′′-terpyridine (tpy), is also present in the coordination sphere. The FT-IR spectrum of complex 1 exhibits a broad absorption in the 3000–3500 cm−1 region, thereby indicating the presence of water molecules in the coordination sphere of the Eu3+ ion. On the other hand, the absence of this broad band in the 3000–3500 cm−1 region in the case of complex 2 implied that the water molecules are displaced successfully by the tridentate terpyridine ligand. The carbonyl stretching frequency of Hpfppd (1582 cm−1) was shifted to higher wave numbers in complexes 1–2 (1609 cm−1) indicating the involvement of carbonyl oxygen in the complex formation with the Eu3+ ion. Two groups of very intense bands assigned to υ(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) and υ(CC) appear at 1582–1560 and 1478–1422 cm−1 for free terpy.16 Both these bands show a ∼10 cm−1 shift to higher frequencies in 2 due to coordination.
N) and υ(CC) appear at 1582–1560 and 1478–1422 cm−1 for free terpy.16 Both these bands show a ∼10 cm−1 shift to higher frequencies in 2 due to coordination.
 | ||
| Scheme 1 Synthetic procedure for the ligand Hpfppd. | ||
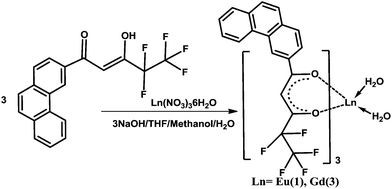 | ||
| Scheme 2 Synthetic procedure for complexes 1 and 3. | ||
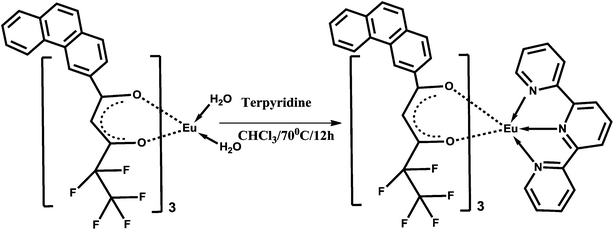 | ||
| Scheme 3 Synthetic procedure for complex Eu(pfppd)3tpy (2). | ||
Synthesis and characterization of luminescent nanocomposites (1@oxMWCNTs)
By means of the facile coordination of carboxylate of oxMWCNTs and the lanthanide metal ion, herein we report the nanocomposites of carboxylate-modified MWCNTs mediated by Eu(pfppd)3(H2O)21 resulting in nanotube assembly (Scheme 4). The surface morphologies, microstructure and physical properties of luminescent nanocomposites (1@oxMWCNTs) have been fully characterized via thermogravimetric analysis (TGA), FT-IR, FT-Raman, XPS, SEM, TEM, AFM, EDAX and photoluminescence spectroscopic techniques.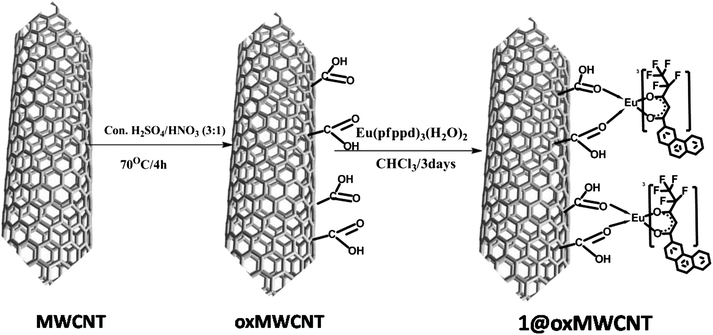 | ||
| Scheme 4 Synthesis pathway of 1@oxMWCNTs. | ||
The FT-IR spectra of MWCNTs, oxMWCNTs and 1@oxMWCNTs are displayed in Fig. 1. The infrared spectrum of oxMWCNTs showed two strong and broad absorption bands at 3445 and 1646 cm−1, corresponding to the OH stretching vibrations and the CO stretching of COOH, respectively. The CO stretching of COOH in oxMWCNTs was red shifted to 1709 cm−1. On the other hand, the OH stretching frequency of COOH was almost unaltered in 1@oxMWCNTs. The above spectral features confirm that the COOH group is coordinately bonded to the Eu3+ metal ions via the oxygen lone pair of electrons of CO and not by that of O–H.
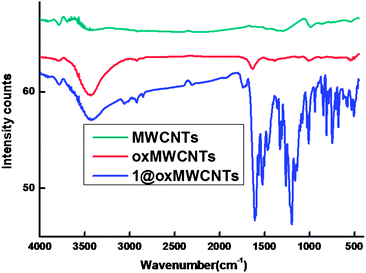 | ||
| Fig. 1 FT-IR spectra of MWCNTs, oxMWCNTs and 1@oxMWCNTs. | ||
The Raman spectra depicted in Fig. 2 revealed that MWCNTs, functionalized oxMWCNTs and 1@oxMWCNTs, all showed two characteristic bands at ∼1587 and ∼1321 cm−1, which are denoted as G bands and D bands, respectively. The G bands are related to the in-plane E2g vibration mode of the benzenoid framework of CNTs, whereas D bands are attributed to sp3 hybridized carbon atoms in the walls.17 The intensity ratio of these two bands ID/IG is typically used as an index of graphitization of carbonous materials. With side wall functionalization of MWCNTs, the ratio ID/IG is increased due to the introduction of more defects into MWCNTs. It is clearly shown that the ID/IG ratios were all increased when comparing functionalized MWCNTs with MWCNTs. The ID/IG of MWCNTs is 0.47, whereas ID/IG of oxMWCNTs and 1@oxMWCNTs are 0.76, and 0.99, respectively. These phenomena further confirm the surface functionalization of MWCNTs.
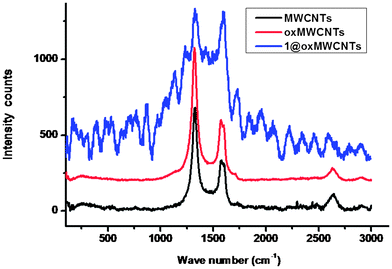 | ||
| Fig. 2 Raman spectra of MWCNTs, oxMWCNTs and 1@oxMWCNTs. | ||
In order to determine the atomic composition of the synthesized materials (MWCNTs, oxMWCNTs and 1@oxMWCNTs), X-ray photoelectron spectroscopy (XPS) elemental analysis was performed and the results are depicted in Table 1. MWCNTs and oxMWCNTs presented only the C(1s) and O(1s) signals at ∼285 and ∼535 eV, respectively (Fig. S3 and S4 of the ESI†). An increase in the O/C atomic ratio for oxMWCNTs confirms the effectiveness of the oxidative treatment. The fingerprint signals of 1, namely Eu(3d) (1136 eV) and F(1s) (689 eV), were detected in 1@oxMWCNTs (Fig. S5 of the ESI†) with considerable atomic percentages, 0.23 and 13%, respectively.
| Sample | C [%] | O [%] | F [%] | Eu [%] |
|---|---|---|---|---|
| MWCNTs | 97.63 | 2.37 | — | — |
| oxMWCNTs | 89.02 | 10.98 | — | — |
| 1@oxMWCNTs | 73.84 | 12.52 | 13.31 | 0.23 |
The thermal behaviour of complexes 1–2 and 1@oxMWCNTs was examined by means of thermogravimetric analysis (TGA) under nitrogen atmosphere and the results are shown in Fig. S6 of the ESI.† It is clear from the TGA data that complex 1 undergoes mass loss of approximately 2.78% (calcd: 2.81%) in the first step up to 170 °C, corresponding to the elimination of two coordinated water molecules. On the other hand, complex 2 and 1@oxMWCNTs are thermally stable up to 210 °C and above which both decompose in two steps. Further, no weight loss was noted in the range of 100 to 170 °C, which indicates that they are anhydrous in nature. This is in good agreement with the FT-IR spectral data. The total weight loss of all materials is compatible with the formulae proposed and the masses of the final products correspond to the complete combustion of these complexes to their corresponding europium oxy-fluorides.
AFM measurements of acetone dispersions containing a suspension of MWCNTs and 1@oxMWCNTs spin coated on mica substrates reproducibly revealed the presence of fibre-like objects, i.e., rod-like bundles (Fig. S7 and S8 in the ESI†). Although the bundles are major fractions, some single scattered MWCNTs are also observed. These results are in good agreement with those obtained from SEM and TEM measurements, which shows at most the presence of MWCNT bundles. The morphological characterization of MWCNTs and 1@oxMWCNTs has been carried out by electron microscopic techniques (Fig. 3 and 4). TEM observations reveal that 1@oxMWCNTs retain their tubular morphology of MWCNTs after loading with a luminescent Eu3+ complex. TEM analysis of 1@oxMWCNTs on the enlarged view provides evidence for the presence of the Eu3+ complex as little aggregates homogeneously dispersed along the side wall of the carbon nanotubes. EDAX analysis also demonstrates the presence of the luminescent Eu3+ complex in the nanocomposites (Fig. 4c). SEM analysis shows bigger agglomerates in both MWCNTs and 1@oxMWCNTs due to the electrostatic interactions between the tubes (Fig. S9 and S10 of the ESI†).
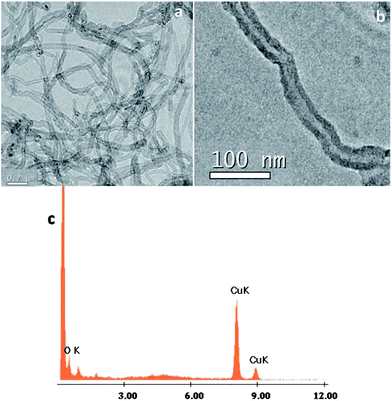
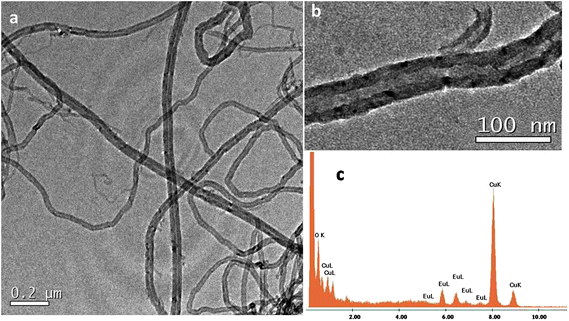
Electronic spectra of the Ln3+ complexes
The room-temperature electronic spectra of the ligands and the corresponding Eu3+ complexes 1–2 recorded in tetrahydrofuran solution (c = 1 × 10−5 M) are shown in Fig. 5. The spectral shapes of the complexes are similar to that of Hpfppd, indicating that the coordination of Eu3+ does not have significant influence on the energy of the singlet state of the β-diketone ligand.10 The ligand Hpfppd displays a composite broad band in the 300–400 nm (λmax = 365 nm) region corresponding to the singlet–singlet n–π* enolic transition assigned to the β-diketonate moiety. Furthermore, the higher energy absorption band detected in the range of 220–255 nm is attributable to the π–π* transition of the aromatic moiety of the β-diketonate ligand. The molar absorption coefficient of the ligand was found to be 0.9 × 104 L mol−1 cm−1 at λmax = 365 nm, which indicates that it has a strong ability to absorb light. The magnitudes of the molar absorption coefficient values of 1–2 (3 × 104 L mol−1 cm−1 for 1 and 2.99 × 104 L mol−1 cm−1 for 2), calculated at the respective λmax values, were found to be approximately three times higher than that of the β-diketonate ligand, and this trend is consistent with the presence of three β-diketonate ligands in each of the complexes. The spectral shapes observed in the UV-visible absorption spectra of complex 1 and nanocomposite 1@oxMWCNTs are identical, which again indicates that the electronic state of the ligand is not significantly affected by the grafting of luminescent species on carbon nanotube side walls.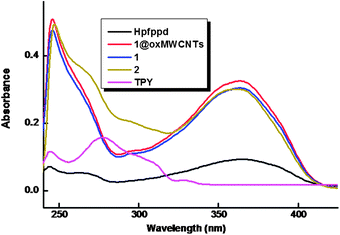 | ||
| Fig. 5 UV-vis absorption spectra of the free ligands, complexes 1–2 and 1@oxMWCNTs in THF (c = 1 × 10−5 M). | ||
According to Dexter theory, the intramolecular energy transfer from the triplet state (3ππ*) of the ligand to the emitting resonance level of the Ln3+ ion has a significant influence on lanthanide luminescence.4 The efficiency of intramolecular energy transfer depends mainly on two processes. One is the energy transfer from the lowest triplet state energy of the ligand to the emissive excited state of the Ln3+ ion and the other one is the inverse energy transition from the Ln3+ ion to the ligand by a thermal deactivation mechanism.10a,b,18 If the energy difference is too small, the inverse energy transfer will take place more easily.10a,b,18 According to Latva's empirical rule the ligand-to-metal energy transfer process for Eu3+ requires ΔE = (3ππ* − 5D4) = 2500–4000 cm−1.19 The triplet energy level (3ππ*) of the ligand has been estimated by reference to the lower wavelength emission edge (502 nm: 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 920 cm−1) of the low-temperature phosphorescence spectrum of Gd3+ complex 3 (Fig. S11 of the ESI†). It is also crucial to determine the singlet energy levels of the ligand in order to elucidate the energy migration pathways in lanthanide complexes. The singlet (1ππ*) energy level of the ligand (414 nm: 24154 cm−1) was estimated by reference to the wavelength of the UV-vis upper absorption edge of Gd3+ complex 3 (Fig. S11 of the ESI†). It is interesting to note that the triplet energy level of the newly designed β-diketonate (502 nm: 19920 cm−1) is situated well above the energies of the main emitting levels of 5D0 of Eu3+, confirming the suitability of the ligand as a sensitizer for Eu3+.20 The energy gap between the singlet and triplet states of Hpfppd, ΔE = (1ππ* − 3ππ*) is 4234 cm−1, while a value around 5000 cm−1 is commonly considered as optimum for efficient intersystem crossing in these systems.21
920 cm−1) of the low-temperature phosphorescence spectrum of Gd3+ complex 3 (Fig. S11 of the ESI†). It is also crucial to determine the singlet energy levels of the ligand in order to elucidate the energy migration pathways in lanthanide complexes. The singlet (1ππ*) energy level of the ligand (414 nm: 24154 cm−1) was estimated by reference to the wavelength of the UV-vis upper absorption edge of Gd3+ complex 3 (Fig. S11 of the ESI†). It is interesting to note that the triplet energy level of the newly designed β-diketonate (502 nm: 19920 cm−1) is situated well above the energies of the main emitting levels of 5D0 of Eu3+, confirming the suitability of the ligand as a sensitizer for Eu3+.20 The energy gap between the singlet and triplet states of Hpfppd, ΔE = (1ππ* − 3ππ*) is 4234 cm−1, while a value around 5000 cm−1 is commonly considered as optimum for efficient intersystem crossing in these systems.21
Photophysical properties
The solid-state excitation and emission spectra of Eu3+ complexes 1–2 and 1@oxMWCNTs recorded at room temperature are displayed in Fig. 6. The excitation profiles were recorded by monitoring the 5D0 → 7F2 (612 nm) transition of the Eu3+ ion. The excitation spectra of all the compounds exhibit a broad band between 275 and 500 nm (λmax = 415 nm), which can be assigned to the π–π* electronic transition of the aromatic ligands. The absence of any absorption bands due to the f–f transitions of the Eu3+ ion proves that luminescence sensitization via excitation of the ligand is effective. The substitution of a conjugated phenanthrene moiety in the 3-position of the β-diketonate ligand dramatically influences π-conjugation in the complex molecules of the Eu3+ complexes and shifts the excitation window to the visible region (λmax = 415 nm), with important applications in biomedical analysis and lighting devices.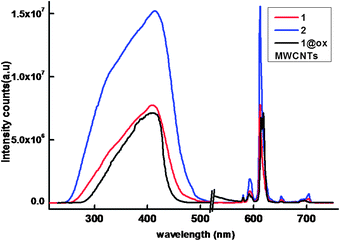 | ||
| Fig. 6 298 K excitation and emission spectra of the compounds in solid-state (λex = 415 nm and emission monitored around 612 nm). | ||
The ambient-temperature emission spectra of Eu3+ complexes 1–2 and nanocomposite 1@oxMWCNTs excited at 415 nm show characteristics of the metal ion emissions in the 550–725 nm region, and exhibit well resolved sharp peaks that are due to the transitions from the metal-centered 5D0 excited state to the 7FJ ground state multiplet (Fig. 6). Maximum peak intensities at 580, 592, 612, 652, and 702 nm were observed for the J = 0, 1, 2, 3, and 4 transitions, respectively, and the J = 2 so-called “hypersensitive” transition is intense.4,22 This very intense 5D0 → 7F2 peak points to a highly polarizable chemical environment around the Eu3+ ion and is responsible for the brilliant red emission of these complexes.22 The intensity of the 5D0 → 7F2 transition (electric dipole) is greater than that of the 5D0 → 7F1 transition (magnetic dipole), which indicates that the coordination environment of the Eu3+ ion is devoid of an inversion center. No broad emission band resulting from organic ligand molecules in the blue region can be observed, which indicates that the ligand transfers the absorbed energy effectively to the emitting level of the metal ion. Fig. 7 shows a comparison of luminescence intensity of 5D0 → 7F2 for complex 1 and 1@oxMWCNTs at different excitation wavelengths from the UV to visible range (330, 360, 390, 410, 430 and 460 nm). From these comparisons one can note that both precursor 1 and nanocomposite 1@oxMWCNT material exhibit significant luminescent efficiency at different excitation wavelengths, which demonstrates the potential utility of these materials in both light emitting diodes and bio-imaging. The bright red emission with tubular morphology can also be seen from the luminescence microscopic image (excited at 365 nm) of the nanocomposite 1@oxMWCNTs material (Fig. S12 of the ESI†).
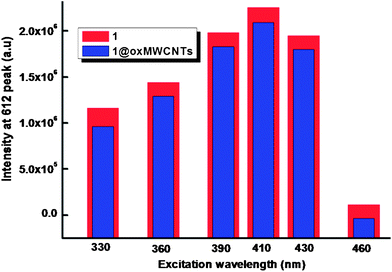 | ||
| Fig. 7 Comparison of the intensity of emission at 612 nm for complex 1 and nanocomposite 1@oxMWCNTs at different excitation wavelengths. | ||
The luminescence decay profiles (Fig. 8) of complexes 1–2 and 1@oxMWCNT material are found to be single-exponential functions, thus implying the presence of only one emissive Eu3+ center. The pertinent values are given in Table 2. The relatively shorter lifetime obtained for complex 1 may be due to dominant nonradiative decay channels associated with vibronic coupling due to the presence of solvent molecules, as is well documented for many of the hydrated Eu3+–β-diketonate complexes.7,10 The replacement of water molecules by the terpyridine moiety in complex 2 richly enhances the lifetime value due to the presence of dominant radiative decay pathways. The lifetime value of nanocomposite 1@oxMWCNTs material is considerably enhanced as compared to precursor complex 1 due to the decrease of nonradiative decay pathways. Further, it can be attributed to the decrease in vibrational relaxation of the excited states of the ligands, probably due to their proximity to the rigid structure of the MWCNT framework.14b
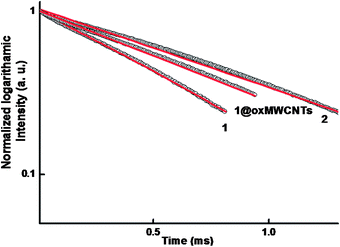 | ||
| Fig. 8 5D0 decay profiles for compounds 1, 2 and 1@oxMWCNTs (solid-state) excited at 415 nm and emission monitored around 612 nm. The straight lines are the best fits (r2 = 0.999) considering single-exponential behaviour. | ||
| Compounds | τ obs (ms) | A RAD (s−1) | A NR (s−1) | Q EuL (%) | Q EuEu (%) | η sens (%) |
|---|---|---|---|---|---|---|
| 1 | 0.51 | 802 | 1154 | 31 ± 3 | 41 | 75 |
| 2 | 1.04 | 737 | 220 | 75 ± 8 | 77 | 97 |
| 1@oxMWCNTs | 0.78 | 628 | 653 | 27 ± 3 | 49 | 55 |
To gain a better understanding of the luminescence efficiencies of the designed Eu3+ compounds, it was appropriate to analyze the emissions in terms of eqn (1), where QEuL and QEuEu represent the overall and intrinsic luminescence quantum yields of Eu3+; ηsens represents the efficiency of the ligand-to-metal energy transfer and τobs/τrad are the observed and radiative lifetimes of Eu (5D0):4,7
| QEuL = ηsens × QEuEu = ηsens × (τobs/τrad) | (1) |
The intrinsic quantum yields of Eu3+ could not be determined experimentally upon direct f–f excitation because of very low absorption intensity.4 Therefore, the radiative lifetime of Eu (5D0) has been calculated from eqn (2),4 where n represents the refractive index (1.5), AMD,0 is the spontaneous emission probability for the 5D0 → 7F1 transition in vacuo (14.65 s−1), and Itot/IMD signifies the ratio of the total integrated intensity of the corrected Eu3+ emission spectrum to the integrated intensity of the magnetic dipole 5D0 → 7F1 transition:
| 1/τrad = AMD,0 × n3 × (Itot/IMD) | (2) |
The intrinsic quantum yields for the compounds have been estimated from the ratio τobs/τrad and the pertinent values are listed in Table 2. The overall quantum yields (QEuL), radiative (AR) and nonradiative (ANR) decay rates and energy transfer efficiencies (ηsens) are also presented in Table 2. The substitution of water molecules in [Eu(pfppd)3(H2O)2] 1 by the terpyridine ligand leads to almost double the 5D0 lifetime (0.51 to 1.04 ms) and an approximately 2.4 times enhancement in the solid-state overall quantum yield (31 to 75%). Most importantly, the current results disclose that the newly developed Eu3+ complex 2 exhibits impressive quantum yield and long excited state life time values in the solid-state, which are found to be the highest so far reported for visible-light excited Eu3+–β-diketonate systems. The substitution of solvent molecules in the Eu3+–tris-β-diketonate complexes 1 by a terpyridine contributes to the overall sensitization of the Eu3+-centered luminescence in 2 and is confirmed by (i) an increase of the intrinsic quantum yields from 41% (in 1) to 77% (in 2), which results from removal of the quenching effect of the O–H vibrations, and (ii) the significant enhancement of ηsens from 75% (in 1) to 97% (in 2). When compared to precursor complex 1, the nanocomposite 1@oxMWCNTs shows a moderately lower quantum yield (27%) and sensitization efficiency (55%). However, marginally higher intrinsic quantum yields (49%) are noted, due to the minimization of nonradiative decay pathways in the nanocomposite material.
In the present work, efforts have also been made to isolate luminescent europium plastic materials by combining the unique optical properties of nanocomposite 1@oxMWCNTs with the mechanical characteristics, thermal stability, flexibility and disc forming tendency of polymers (PMMA). The polymeric composite resulted to be very luminescent and shape-persistent, even under mechanical stress, and the luminescence resulted to be homogeneous despite the low loading of the nanocomposite 1@oxMWCNTs at 415 nm excitation (Fig. 9). The luminescent intensity increases with increase of the concentration of 1@oxMWCNTs. Fig. 10 shows the photograph of the polymer disc under day light and UV light (365 nm) and found that the disc is emissive under the UV light even at very low concentration of 1@oxMWCNTs (1%).
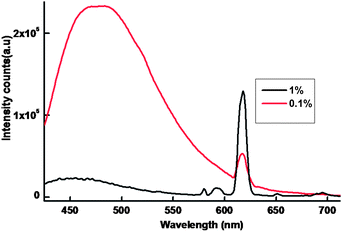 | ||
| Fig. 9 298 K emission spectra of the polymer discs in solid-state (λex = 415 nm and emission monitored around 612 nm). | ||
![Picture of the polymer composite containing the luminescent 1@MWNCNTs in 1 and 0.1% (w/w) ratio [under the day light (left panel) and UV light at 365 nm (right panel)].](/image/article/2013/TC/c2tc00186a/c2tc00186a-f10.gif) | ||
| Fig. 10 Picture of the polymer composite containing the luminescent 1@MWNCNTs in 1 and 0.1% (w/w) ratio [under the day light (left panel) and UV light at 365 nm (right panel)]. | ||
Conclusions
In conclusion, solid state photophysical properties of two efficient antenna complexes of Eu3+ supported by a β-diketone ligand, 4,4,5,5,5-pentafluoro-3-hydroxy-1-phenanthren-9-yl) pentanedione (Hpfppd) and an ancillary ligand 2,2′:6,6′′-terpyridine have been evaluated. Suitably expanded π-conjugation in the ligand molecule causes the excitation window to be red shifted to the visible region (up to 500 nm). The ternary Eu3+ complex in the presence of terpyridine as an ancillary ligand exhibits intense red emissions with an overall quantum yield of 75% under blue light excitation (λmax = 415 nm), which is the highest so far reported in the literature under visible light excitation. Thus the newly designed europium complex may find potential practical applications in bio-imaging and organic light-emitting diodes (OLEDs). Further, in the present work, the visible sensitized hydrated Eu3+ luminescent complex has been covalently immobilized onto the multi-walled carbon nanotube host through carboxylic acid functionalization. The designed luminescent nanocomposite material displays intense red emissions with an overall quantum yield of 27% under a wide excitation range from UV to visible regions (330–460 nm). Finally the high dispersibility of luminescent nanocomposites in polymer matrixes makes it a promising luminophore for possible application in OLEDs and in optical amplifiers and waveguides.Experimental section
Materials
The following chemicals were procured commercially and used without subsequent purification. Europium(III) nitrate hexahydrate, 99% (Treibacher); 3-acetylphenanthrene 98% (Aldrich); methyl pentafluoropropionate 99% (Aldrich); sodium methoxide, 95% (Aldrich); 2,2′:6,6′′-terpyridine 98% (Aldrich), polymethylmethacrylate 99% (Aldrich), Acralyn cold curing liquid (Asian Acrylates, Mumbai, India), and multi-walled carbon nanotubes (MWCNTs) 95% (Sisco Research Laboratories Pvt. Ltd). All the other chemicals used were of analytical reagent grade.Synthesis of the ligand, Hpfppd
The ligand 4,4,5,5,5-pentafluoro-3-hydroxy-1-(phenanthren-3-yl) pent-2-en-1-one (Hpfppd) was synthesized according to Scheme 1. 3-Acetylphenanthrene (1 mmol) and methyl pentafluoro methyl propionate (1 mmol) were added to 20 mL of dry tetrahydrofuran (THF) and stirred for 10 min at room temperature. To this solution, sodium methoxide (3 mmol) was added in an inert atmosphere and stirred for 30 min at room temperature followed by further stirring at 70 °C for 24 h. To the resulting solution, 2 N HCl (50 mL) was added, and extracted twice with dichloromethane (2 × 35 mL). The organic layer was separated and dried over Na2SO4, and the solvent was evaporated. The crude product thus obtained is then purified by column chromatography on silica gel with 5% chloroform–hexane as the eluent to get the product as a yellow solid. The observed results of 1H NMR and 13C NMR of the ligand are shown in Fig. S1 and S2 (of the ESI†), respectively. Yield = 91%, elemental analysis (%): calcd for C19H11F5O2 (366.67): C 62.30, H 3.03; found: C 62.36, H 3.14. 1H NMR (CDC13, 500 MHz): δ/ppm: 15.51 (broad, enol–OH), 9.36 (s, 1H), 8.78 (d, 2H), 8.17 (d, 2H), 7.96–7.66 (t, 4H), 6.86 (s, 1H). 13C NMR (125 MHz, CDCl3) δ/ppm: 185.87, 179.09, 135.72, 132.23, 130.55, 130.21, 130.01, 127.61, 127.56, 126.14, 124.25, 123.47, 122.69, 93.86, 77.27–76.77 (CDCl3). IR (KBr) νmax (cm−1): 3059, 1582, 1431, 1356, 1203, 1122, 1001, 784. m/z = 367.82 (M + H)+.Synthesis of Ln3+ complexes Eu(pfppd)3(H2O)2 (1) and Gd(pfppd)3(H2O)2 (3)
To the solution of β-diketonate ligand (6 mmol) in THF–methanol, 6 mmol of NaOH in water was added and stirred for 5 min. To this mixture, 2 mmol of the corresponding Ln(NO3)3·6(H2O) in 2 mL of water was added drop wise and stirred for 24 h at room temperature (Scheme 2). The complex formed was filtered and dried. The products were purified by recrystallization from chloroform solution.Elemental analysis (%): calcd for C57H34EuF15O8(1284.14) 1: C 53.33, H 2.67. Found: C 53.21, H 2.72. IR (KBr) νmax (cm−1): 3418, 3059, 1609, 1432, 1357, 1261, 1010, 783. m/z = 992.87 [Eu(pfppd)2 + 1]+.
Elemental analysis (%): calcd for C57H34GdF15O8(1289.26) 3: C 53.11, H 2.66. Found: C 53.24, H 2.73. IR (KBr) νmax (cm−1): 3419, 3059, 1609, 1432, 1357, 1260, 1012, 784. m/z = 1278.44 [Gd(pfppd)3 + Na + 1]+.
Synthesis of Eu3+ complex Eu(pfppd)3tpy (2)
The ternary Eu3+ complex was prepared by stirring equimolar solutions of their corresponding binary complex 1 and 2,2′:6,6′′-terpyridine in CHCl3 solution for 12 h at 70 °C (Scheme 3). The product was isolated by solvent evaporation and purified by recrystallization from a chloroform–hexane mixture. Elemental analysis (%): calcd for C71H41EuF15N3O6 (1481.39): C 58.64, H 2.90, N 2.81. Found: C 58.76, H 2.89, N 2.90. IR (KBr) νmax (cm−1): 3058, 1609, 1471, 1306, 1212, 1008, 766. m/z = 751.77 [Eu(pfppd)tpy]+.Synthesis of 1@oxMWCNTs
To obtain the desired luminescent nanocomposite 1@oxMWCNTs, the nano-extraction methodology described by Iijima et al. was employed.23 Multi-walled carbon nanotubes (MWCNTs) were oxidized by acid treatment with H2SO4–HNO3 (3:1) as an oxidizing mixture to give oxMWCNTs. An excess amount of Eu3+ complex 1 and oxMWCNTs are then dispersed in CHCl3 (10 mL) by ultrasonication for 15 min and the mixture was stirred at room temperature for 3 days (Scheme 4). Filtration on a Millipore apparatus and rinsing with CHCl3 afforded the hybrid material that was redispersed in CHCl3 and ultrasonicated for 10 min to remove all adsorbed 1 from the MWCNTs. This purification cycle was repeated several times until the washings with CHCl3 contained no trace of 1, as confirmed by emission spectroscopy. The designed material was further characterized by FT-IR, FT-Raman, thermogravimetric analysis, XPS, SEM, TEM, EDAX, AFM, etc.
Synthesis of PMMA nanocomposite discs
PMMA nanocomposite discs were prepared by dispersion of 1@oxMWCNTs (0.1% and 1 wt%) in PMMA by using acrylin solution as a binder. The thick paste was allowed to set in cylindrical molds.Instrumentation
Infrared spectra of all samples were collected on KBr pellets in the 4000–400 cm−1 region with a resolution of 4 cm−1, by accumulating 64 scans using a Perkin-Elmer Spectrum One FT-IR Spectrometer. FT-Raman spectra of the solid samples placed on a glass plate were recorded on a HR800 Lab RAM confocal Raman Spectrometer operating at 20 mW laser power using a Peltier cooled CCD detector. The laser source had an excitation wavelength of 633 nm and an acquisition time of 30 seconds using a 50× objective. Thermogravimetric analyses were performed on a TG/DTA-6200 (SII Nano Technology Inc., Japan). XPS measurements were obtained on a KRATOS-AXIS 165 instrument equipped with dual aluminum–magnesium anodes using Mg K radiation (hν = 1253.6 eV) operated at 5 kV and 15 mA with pass energy 80 eV and an increment of 0.1 eV. The samples were degassed for several hours in the XPS chamber to minimize air contamination to the sample surface. To overcome the charging problem, a charge neutralizer of 2 eV was applied and the binding energy of the C 1s core level (BE = 284.6 eV) of adventitious hydrocarbon was used as a standard. The XPS spectra were fitted using a nonlinear square method with the convolution of Lorentzian and Gaussian functions after a polynomial background was subtracted from the raw spectra. TEM and SEM were used to determine the morphology and particle size. TEM analyses were done on a JEOL 2010 (200 kV). A pinch of material was suspended in acetone and sonicated for 15 min, then dropped in a carbon-coated grid and allowed to dry at room temperature. The elemental constitution of the compounds was determined on an energy dispersive spectrometer (Technai G2 30LaB6, ST with EDAX). Scanning electron microscopy was performed on a ZEISS EVO-18 Cryo-SEM instrument. Atomic Force Microscopy images were recorded under ambient conditions using a NTEGRA (NT-MDT) operated in the tapping mode regime. Micro-fabricated TiN cantilever tips (NSG10) with a resonance frequency of 299 kHz and a spring constant of 20–80 N m−1 were used. Samples for the imaging were prepared by drop casting the samples in acetone on freshly cleaved mica at room temperature. Optical reflectance and UV absorption studies were carried out with an UV-Vis spectrometer (Shimadzu UV-2450). The excitation and emission spectra of the samples were recorded on a Spex-Fluorolog FL3 22 spectrofluorimeter equipped with a double grating 0.22 m Spex 1680 monochromator and a 450 W Xe lamp as the excitation source operating in the front face mode. The lifetime measurements were carried out at room temperature using a Spex 1040 D phosphorimeter. A luminescence image of the nanocomposites was observed by using a Leica DFC 490 polarized light optical microscope.The overall quantum yields (Φoverall) of the europium complexes were measured at room temperature using the technique for powdered samples described by Bril and De Jager-Veenis,24 through the following expression:
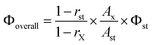 | (3) |
Acknowledgements
The authors acknowledge financial support from the CSIR, New Delhi, India (CSIR-TAPSUN project, SSL (OLEDS), NWP-55). D.V. thanks CSIR, New Delhi for the award of Senior Research Fellowship.References
- (a) L. Armelao, S. Quici, F. Barigelletti, G. Accorsi, G. Bottaro, M. Cavazzini and E. Tondello, Coord. Chem. Rev., 2010, 5–6, 487 CrossRef; (b) D. Parker and J. A. G. Williams, The Lanthanides and Their Interrelation with Biosystems, M. Dekker, Inc., NewYork, 2003, vol. 40, p. 2337 Search PubMed; (c) M. Yu, F. Li, Z. Chen, H. Hu, C. Zhan, H. Yang and C. Huang, Anal. Chem., 2009, 81, 930 CrossRef CAS; (d) J.-C. G. Bünzli, Chem. Rev., 2010, 110, 2729 CrossRef.
- (a) G. Shao, R. Han, Y. Ma, M. Tang, F. Xue, Y. Sha and Y. Wang, Chem.–Eur. J., 2010, 16, 8647 CrossRef CAS; (b) W.-S. Lo, W.-M. Kwok, G.-L. Law, C.-T. Yeung, C.-T.-L. Chan, H.-L. Yeung, H.-K. Kong, C.-H. Chen, M. B. Murphy, K.-L. Wong and W.-T. Wong, Inorg. Chem., 2011, 50, 5309 CrossRef CAS; (c) G.-L. Law, K.-L. Wong, C. W.-Y. Man, S.-W. Tsao and W.-T. Wong, J. Biophotonics, 2009, 12, 718 CrossRef; (d) S. V. Eliseeva, B. Song, C. D. B. Vandevyver, A.-S. Chauvin, J. B. Wacker and J.-C. G. Bünzli, New J. Chem., 2010, 34, 2915 RSC; (e) H.-K. Kong, F. L. Chadbourne, G.-L. Law, H. Li, H.-L. Tam, S. L. Cobb, C.-K. Lau, C.-S. Lee and K.-L. Wong, Chem. Commun., 2011, 47, 8052 RSC; (f) X. Zhang, Y. Jiao, X. Jing, H. Wu, G. He and C. Duan, Dalton Trans., 2011, 40, 2522 RSC.
- (a) F. X. Zang, Z. R. Hong, W. L. Li, M. T. Li and X. Y. Sun, Appl. Phys. Lett., 2004, 84, 2679 CrossRef CAS; (b) T. Oyamada, Y. Kawamura, T. Koyama, H. Sasabe and C. Adachi, Adv. Mater., 2004, 16, 1082 CrossRef CAS; (c) J. Kido, H. Hayase, K. Hongawa, K. Nagai and K. Okuyama, Appl. Phys. Lett., 1994, 65, 2124 CrossRef CAS; (d) J. Rocha, L. D. Carlos, F. A. Almeida Paza and D. Ananiasab, Chem. Soc. Rev., 2011, 40, 926 RSC; (e) J. Kido and Y. Okamoto, Chem. Rev., 2002, 102, 2357 CrossRef CAS.
- (a) S. V. Eliseeva and J.-C. G. Bünzli, Chem. Soc. Rev., 2010, 39, 189 RSC; (b) Y. Ma and Y. Wang, Coord. Chem. Rev., 2010, 254, 972 CrossRef CAS; (c) J.-C. G. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048 RSC.
- (a) C. Yang, L.-M. Fu, Y. Wang, J.-P. Zhang, W.-T. Wong, X.-C. Ai, Y.-F. Qiao, B.-S. Zou and L.-L. Gui, Angew. Chem., Int. Ed., 2004, 43, 5010 CrossRef CAS; (b) V. Vicinelli, P. Ceroni, M. Maestri, V. Balzani, M. Gorka and F. Vögtle, J. Am. Chem. Soc., 2002, 124, 6461 CrossRef CAS; (c) J. L. Bender, P. S. Corbin, C. L. Fraser, D. H. Metcalf, F. S. Richardson, E. L. Thomas and A. M. Urbas, J. Am. Chem. Soc., 2002, 124, 8526 CrossRef CAS.
- (a) F. Chen, Z. Bian, Z. Liu, D. Nie, Z. Chen and C. Huang, Inorg. Chem., 2008, 47, 2507 CrossRef CAS; (b) R. Ziessel, S. Diring, P. Kadjane, L. Charbonnière, P. Retailleau and C. Philouze, Chem.–Asian J., 2007, 2, 975 CrossRef CAS; (c) H.-B. Xu, L.-X. Shi, E. Ma, L.-Y. Zhang, Q.-H. Wei and Z.-N. Chen, Chem. Commun., 2006, 1601 RSC; (d) M. Ward, Coord. Chem. Rev., 2007, 251, 1663 CrossRef CAS.
- (a) V. Divya, R. O. Freire and M. L. P. Reddy, Dalton Trans., 2011, 40, 3257 RSC; (b) V. Divya, S. Biju, R. L. Varma and M. L. P. Reddy, J. Mater. Chem., 2010, 20, 5220 RSC.
- (a) D. Sykes and M. D. Ward, Chem. Commun., 2011, 47, 2279 RSC; (b) P. He, H. H. Wang, S. G. Liu, J. X. Shi, G. Wang and M. L. Gong, Inorg. Chem., 2009, 48, 11382 CrossRef CAS; (c) Y. Bretonniere, M. J. Cann, D. Parker and R. Slater, Chem. Commun., 2002, 1930 RSC; (d) P. He, H. H. Wang, H. G. Yan, W. Hu, J. X. Shi and M. L. Gong, Dalton Trans., 2010, 39, 8919 RSC; (e) C. Yang, L.-M. Fu, Y. Wang, J.-P. Zhang, W.-T. Wong, X.-C. Ai, Y.-F. Qiao, B.-S. Zou and L.-L. Gui, Angew. Chem., Int. Ed., 2004, 43, 5010 CrossRef CAS; (f) F. Xue, Y. Ma, L. Fu, R. Hao, G. Shao, M. Tang, J. Zhang and Y. Wang, Phys. Chem. Chem. Phys., 2010, 12, 3195 RSC.
- (a) M. H. V. Werts, M. A. Duin, J. W. Hofstraat and J. W. Verhoeven, Chem. Commun., 1999, 799 RSC; (b) R. V. Deun, P. Nockemann, P. Fias, K. V. Hecke, L. V. Meervelt and K. Binnemans, Chem. Commun., 2005, 590 RSC; (c) R. Ziessel, S. Diring, P. Kadjane, L. Charbonnière, P. Retailleau and C. Philouze, Chem.–Asian J., 2007, 2, 975 CrossRef CAS; (d) F. Chen, Z. Bian, Z. Liu, D. Nie, Z. Chen and C. Huang, Inorg. Chem., 2008, 47, 2507 CrossRef CAS.
- (a) J. M. Lehn, Angew. Chem., Int. Ed. Engl., 1990, 29, 1304 CrossRef; (b) N. Sabbatini, M. Guardiglia and J. M. Lehn, Coord. Chem. Rev., 1993, 123, 201 CrossRef CAS; (c) J. M. Stanley, X. Zhu, X. Yang and B. J. Holliday, Inorg. Chem., 2010, 49, 2035 CrossRef CAS; (d) N. M. Shavaleev, S. V. Eliseeva, R. Scopelliti and J.-C. G. Bünzli, Chem.–Eur. J., 2009, 15, 10790 CrossRef CAS; (e) B. Francis, D. B. Ambili Raj and M. L. P. Reddy, Dalton Trans., 2010, 39, 8084 RSC; (f) S. Biju, M. L. P. Reddy, A. H. Cowley and K. V. Vasudevan, J. Mater. Chem., 2009, 19, 5179 RSC; (g) P. N. Remya, S. Biju, M. L. P. Reddy, A. H. Cowley and M. Findlater, Inorg. Chem., 2008, 47, 7396 CrossRef CAS; (h) S. Quici, M. Cavazzini and G. Marzanni, Inorg. Chem., 2005, 44, 529 CrossRef CAS; (i) J. Zhang, P. D. Badger, S. J. Geib and S. Petoud, Angew. Chem., Int. Ed., 2005, 44, 2508 CrossRef CAS.
- T. Pagnot, P. Audebert and G. Tribillon, Chem. Phys. Lett., 2000, 322, 572 CrossRef CAS.
- (a) K. Binnemans, Chem. Rev., 2009, 109, 4283 CrossRef CAS; (b) J. C. G. Bunzli and S. V. Eliseeva, J. Rare Earths, 2010, 28, 824 CrossRef CAS; (c) K. Binnemans, in Handbook on the Physics and Chemistry of Rare Earths, ed. K. A. Gschneidner Jr, J. C. G. Bünzli and V. K. Pecharsky, North-Holland (Elsevier V.B.), Amsterdam, 2005, vol. 35, p. 107 Search PubMed; (d) L. D. Carlos, R. A. S. Ferreira, V. D. Bermudez and S. J. L. Ribeiro, Adv. Mater., 2009, 21, 509 CrossRef CAS; (e) K. Binnemans and C. Gcrller-Wallrand, Chem. Rev., 2002, 102, 2303 CrossRef CAS; (f) L. Maggini and D. Bonifazi, Chem. Soc. Rev., 2012, 41, 211 RSC.
- (a) C. N. R. Rao, B. C. Satishkumar, A. Govindaraj and M. Nath, ChemPhysChem, 2001, 2, 78 CrossRef CAS; (b) S. Srinivasan, S. S. Babu, V. K. Praveen and A. Ajayaghosh, Angew. Chem., Int. Ed., 2008, 120, 5830 CrossRef; (c) D. Bonifazi, C. Nacci, R. Marega, S. Campidelli, G. Ceballos, S. Modesti, M. Meneghetti and M. Prato, Nano Lett., 2006, 6, 1408 CrossRef CAS; (d) D. Tasis, N. Tagmatarchis, A. Bianco and M. Prato, Chem. Rev., 2006, 106, 1105 CrossRef CAS; (e) H.-C. Wu, X. Chang, L. Liu, F. Zhao and Y. Zhao, J. Mater. Chem., 2010, 20, 1036 RSC.
- (a) L. Maggini, F. M. Toma, L. Feruglio, J. M. Malicka, T. D. Ros, N. Armaroli, M. Prato and D. Bonifazi, Chem.–Eur. J., 2012, 18, 5889 CrossRef CAS; (b) X. Xin, M. Pietraszkiewicz, O. Pietraszkiewicz, O. Chernyayeva, L. Maggini, J. Mohanraj, H. Traboulsi, A. Parisini, G. Accorsi, N. Armaroli and D. Bonifazi, Chem.–Eur. J., 2011, 17, 8533 CrossRef; (c) L. Maggini, H. Traboulsi, K. Yoosaf, J. Mohanraj, J. Wouters, O. Pietraszkiewicz, M. Pietraszkiewicz, N. Armaroli and D. Bonifazi, Chem. Commun., 2011, 47, 1625 RSC; (d) G. Accorsi, N. Armaroli, A. Parisini, M. Meneghetti, R. Marega, M. Prato and D. Bonifazi, Adv. Funct. Mater., 2007, 17, 2975 CrossRef CAS; (e) D. Shi, J. Lian, W. Wang, G. Liu, P. He, Z. Dong, L. Wang and R. C. Ewing, Adv. Mater., 2006, 18, 189 CrossRef CAS; (f) T. Hemraj-Benny, S. Banerjee and S. S. Wong, Chem. Mater., 2004, 16, 1855 CrossRef CAS; (g) T. Kalwarczyk, E. Gorecka, D. Pociecha, H. Li and R. Hołyst, Carbon, 2012, 50, 436 CrossRef.
- (a) J. Kai, D. F. Parra and H. F. Brito, J. Mater. Chem., 2008, 18, 4549 RSC; (b) S. Biju, M. L. P. Reddy, A. H. Cowley and K. V. Vasudevan, J. Mater. Chem., 2009, 19, 5179 RSC; (c) D. F. Parra, A. Mucciolo and H. F. Brito, J. Appl. Polym. Sci., 2004, 94, 865 CrossRef CAS; (d) D. B. Ambili Raj, B. Francis, M. L. P. Reddy, R. R. Butorac, V. M. Lynch and A. H. Cowley, Inorg. Chem., 2010, 49, 9055 CrossRef; (e) S. Sivakumar and M. L. P. Reddy, J. Mater. Chem., 2012, 22, 10852 RSC.
- G. S. Papaefstathiou, A. Sofetis, C. P. Raptopoulou, A. Terzis, G. A. Spyroulias and T. F. Zafiropoulos, J. Mol. Struct., 2007, 837, 5 CrossRef CAS.
- J. Ma, S. Deng, X. Cheng, W. Wei and A. Hu, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3951 CrossRef CAS.
- (a) F. Aiga, H. Iwanaga and A. Amano, J. Phys. Chem. A, 2005, 109, 11312 CrossRef CAS; (b) A. Døssing, Eur. J. Inorg. Chem., 2005, 1425 CrossRef.
- (a) M. Latva, H. Takalo, V. M. Mukkala, C. Matachescu, J. C. Rodriguez-Ubis and J. Kanakare, J. Lumin., 1997, 75, 149 CrossRef CAS; (b) R. Pavithran, N. S. Saleesh Kumar, S. Biju, M. L. P. Reddy, S. Alves Jr and R. O. Freire, Inorg. Chem., 2006, 45, 2184 CrossRef CAS; (c) S. Biju, D. B. Ambili Raj, M. L. P. Reddy and B. M. Kariuki, Inorg. Chem., 2006, 45, 10651 CrossRef CAS; (d) M. Shi, F. Li, T. Yi, D. Zhang, H. Hu and C. Huang, Inorg. Chem., 2005, 44, 8929 CrossRef CAS.
- N. Armaroli, G. Accorsi, F. Barigelletti, S. M. Couchman, J. S. Fleming, N. C. Harden, J. C. Jeffery, K. L. V. Mann, J. A. McCleverty, L. H. Rees, S. R. Starling and M. D. Ward, Inorg. Chem., 1999, 38, 5769 CrossRef CAS.
- F. J. Steemers, W. Verboom, D. N. Reinhoudt, E. B. VanderTol and J. W. Verhoeven, J. Am. Chem. Soc., 1995, 117, 9408 CrossRef CAS.
- M. H. V. Werts, R. T. F. Jukes and J. W. Verhoeven, Phys. Chem. Chem. Phys., 2002, 4, 1542 RSC.
- M. Yudasaka, K. Ajima, K. Suenaga, T. Ichihashi, A. Hashimoto and S. Iijima, Chem. Phys. Lett., 2003, 380, 42 CrossRef CAS.
- A. Bril and A. W. De Jager-Veenis, J. Electrochem. Soc., 1976, 123, 396 CrossRef CAS.
- W. H. Melhuish, J. Opt. Soc. Am., 1964, 54, 183 CrossRef CAS.
- (a) C. D. Mello Donegá, S. Alves and G. F. de Sá, Chem. Commun., 1996, 1199 RSC; (b) L. D. Carlos, C. D. Mello Donegá, R. Q. Albuquerque, S. Alves Jr, J. F. S. Menezes and O. L. Malta, Mol. Phys., 2003, 101, 1037 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR, 13C NMR of the ligand, and XPS spectra for MWCNTs, oxMWCNT, 1@oxMWCNTs, TG for 1@oxMWCNTs 1 and 2, AFM and SEM for MWCNT and 1@oxMWCNTs, UV-vis absorption at 298 K and 77 K phosphorescence spectra of complex 3, and luminescence microscopic image of 1@oxMWCNTs. See DOI: 10.1039/c2tc00186a |
| This journal is © The Royal Society of Chemistry 2013 |