A microporous metal–organic framework with both open metal and Lewis basic pyridyl sites for highly selective C2H2/CH4 and C2H2/CO2 gas separation at room temperature†
Hui
Xu
ab,
Yabing
He
b,
Zhangjing
Zhang
c,
Shengchang
Xiang
c,
Jianfeng
Cai
a,
Yuanjing
Cui
a,
Yu
Yang
a,
Guodong
Qian
*a and
Banglin
Chen
*ab
aState Key Laboratory of Silicon Materials, Cyrus Tang Center for Sensor Materials and Applications, Department of Materials Science and Engineering, Zhejiang University, Hangzhou 310027, China. E-mail: gdqian@zju.edu.cn
bDepartment of Chemistry, University of Texas at San Antonio, One UTSA Circle, San Antonio, Texas 78249-0698, USA. E-mail: banglin.chen@utsa.edu; Fax: +1 210-458-7428
cCollege of Chemistry and Materials, Fujian Normal University, 3 Shangsan Road, Cangshang Region, Fuzhou, China 350007
First published on 16th October 2012
Abstract
To immobilize both open metal and Lewis basic pyridyl sites into a microporous metal–organic framework, Cu6(PDC)6·2.6H2O (UTSA-50) was solvothermally synthesized and characterized. The combination of open metal sites and Lewis basic pyridyl functional sites leads to a high acetylene adsorption enthalpy of 39.4 kJ mol−1. As a result, UTSA-50 is a very promising MOF material for the highly selective separation of C2H2/CH4 and C2H2/CO2 at room temperature with the highest C2H2/CH4 separation selectivity of 68 ever reported and moderately high acetylene uptake of 91 cm−1 g−1.
1. Introduction
Porous metal–organic frameworks (MOFs) have been rapidly emerging as very promising materials for gas storage and separation;1–38 this is because such new porous materials can have extraordinarily high surface areas which have enabled them to take up large amounts of gas molecules, particularly at high pressures and low temperatures, while the tuneable nature of the pore sizes/curvatures in porous MOFs has allowed us to maximize their size exclusive effects in which the small gas molecules can go through the pore channels while large gas molecules will be blocked. Furthermore, the pore surfaces with porous MOFs can be functionalized by the immobilization of specific sites, such as open metal sites and Lewis acidic and basic sites, to induce their stronger interactions with some gas molecules, thus reaching high storage and separation capacities and selectivity.Recently we have paid much attention to porous MOFs for the storage and separation of hydrocarbons because of their very important industrial applications, particularly acetylene storage and separation.3,24–38 Acetylene is a very important raw material for the synthesis of various industrial chemicals and consumer products, and for oxy-acetylene cutting in metal fabrication;39–41 and the high-purity acetylene is highly in need.42–44 The traditional cryogenic distillation separation technology for the separation of C2H2 from CO2 and CH4 is very energy consuming; the realization of effective new separation materials by adsorptive separation will significantly reduce the cost to produce high purity acetylene. Although some progress has been made on porous MOFs for the separation of C2H2/CH4 and C2H2/CO2 at room temperature, the MOFs with both high separation capacities and selectivity for these gas molecules have not been revealed.1–37 Maximization of both separation capacities (mainly determined by the gas storage capacities) and separation selectivity (mainly determined by the specific functional sites and size exclusive effect) is very important while very challenging. For example, a few porous MOFs exhibiting high acetylene storage capacities have low C2H2/CH4 and C2H2/CO2 separation selectivity,34–,35 while the UTSA-15 (Cu(bdc-OH)(4,4′-bipy)) has the highest C2H2/CH4 separation selectivity of 56, but low acetylene uptake of only 34 cm−1 g−1 at room temperature and 1 atm.37 In these examined MOF materials, only one of the important factors (either open metal sites, optimized pore/cage sizes, size exclusive effects or specific binding sites) has been implemented into porous MOFs to reach their acetylene separation capacities and selectivity, limiting their power to optimize both separation capacities and selectivity.3,25–38 We speculate that if those of the above mentioned important factors can be simultaneously incorporated into porous MOFs and then collaboratively utilized to maximize their acetylene separation capacities and selectivity, we should be able to realize some more promising porous MOF materials for acetylene separation with both high separation capacities and selectivity at room temperature. Herein we report the first example of such porous MOFs Cu6(PDC)6·2.6H2O (UTSA-50, PDC = 3,5-pyridine-dicarboxylate) with both open Cu2+ and Lewis basic pyridyl sites and suitable pore sizes for highly selective separation of C2H2/CH4 and C2H2/CO2 at room temperature with the highest C2H2/CH4 separation selectivity of 68 and moderately high acetylene uptake of 91 cm−1 g−1.
2. Experimental
2.1 Materials and measurements
All reagents and solvents were commercially available and used without further purification. Elemental analyses for C, H, and N were performed on an EA1112 microelemental analyzer. Fourier-transform (FT-IR) spectra were measured with a Bruker Equinox 55 FTIR spectrometer.2.2 Gas sorption measurements
A Micromeritics ASAP 2020 surface area analyzer was used to perform the gas sorption measurements.In order to remove guest solvent molecules in the framework, a freshly prepared sample of UTSA-50 was exchanged with acetone 10 times and then activated at 373 K under high vacuum for 12 h until the outgas rate was <5 μm Hg min−1 prior to measurements to get the activated UTSA-50a for gas sorption studies. The sorption measurement was maintained at 77 K with liquid nitrogen and 273 K with an ice-water bath, respectively. As the center-controlled air condition was set up at 296 K, a water bath of 296 K was used for adsorption isotherms at 296 K.
Isotherm data were analysed using the virial equation:19
| ln(n/p) = A0 + A1n + A2n2 + … |
2.3 Synthesis of UTSA-50
UTSA-50 was synthesized according to the procedure in ref. 18 in which ultrasonic dissolution with Cu(OAc)2·H2O (51.3 mg) and 3,5-PDC (85.9 mg) was performed in 9.89 ml DMF. Then 0.11 ml acetic acid was added, and the mixture was kept at 70 °C for one week. Small green crystals were obtained to give UTSA-50 Cu6(PDC)6·2.6H2O. Anal calcd for C42H29.2Cu6N6O29.6: C, 32.09; N, 5.71; H, 1.98. Found: C, 32.09; N, 5.74; H, 2.34%. IR (cm−1): 3309(w), 3045(w), 1671(s), 1627(s), 1583(s), 1446(w), 1367(s), 1334(s), 1284(s), 1093(w), 1031(w), 918(s), 823(s), 754(s), 729(s), 677(s), 571(w).3. Results and discussion
UTSA-50 was synthesized according to the procedure in ref. 18 in which Cu(NO3)2 and 3,5-PDC in DMF with addition of a small amount of acetic acid at 70 °C for 7 days gave small green crystals. The phase purity of the crystals was confirmed by Elemental Analysis (EA).As shown in the previous work,18 the 3D framework of UTSA-50 is composed of paddle wheel dinuclear Cu2 units which are connected with eight neighboring Cu2 units by PDC ligands (Fig. 1b). It is worth noting that Cu1 atoms show one open metal site by the removal of terminal waters after the thermal activation, while Cu2 atoms are coordinated with four O atoms and one N atom coming from PDCs to give a pyramid. PDCs show two different coordination modes as shown in Fig. 1a and b, in which PDC-1 has two carboxylate binding sites and one uncoordinated pyridine N site, highlighting the existence of Lewis basic pyridyl sites. Therefore, two types of pores extending along the c direction are formed, which are walled by PDC-1 and PDC-2 (Fig. 1c), respectively. The void space accounts for approximately 57.1% of the whole crystal volume (2402.0 Å3 out of the 4205.9 Å3 per unit cell volume) by PLATON analysis. The combined feature of the open metal sites, Lewis basic pyridyl sites and optimized pore size highlights the potential strong binding and high storage of gas molecules. The N2 sorption isotherm at 77 K showed that UTSA-50a displayed type-I sorption behaviour with a BET surface area of 604 m2 g−1 (Langmuir surface area, 933 m2 g−1) (Fig. 2a).
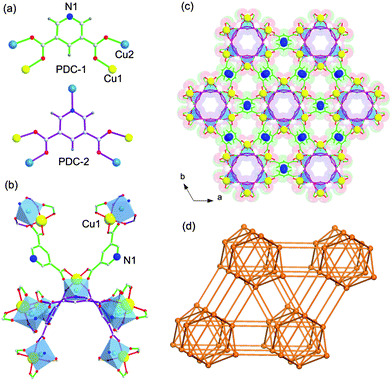 | ||
| Fig. 1 Single crystal structure of UTSA-50 indicating (a) PDCs showing two different coordination modes; (b) one paddle-wheel dinuclear Cu2 unit linking with eight Cu2 units through two PDC-1 ligands and three PDC-2 ligands; and Cu1 with open metal sites; (c) two types of pores: a large pore walled by a PDC-1 ligand of about 4 × 4 Å2 and a small pore walled by PDC-2 of about 3.6 × 3.6 Å2 (Cu1, yellow; Cu2, light blue; N, blue; O, red; C, gray; H, white); and (d) a 3D topology with Schläfli symbol {384105862}. | ||
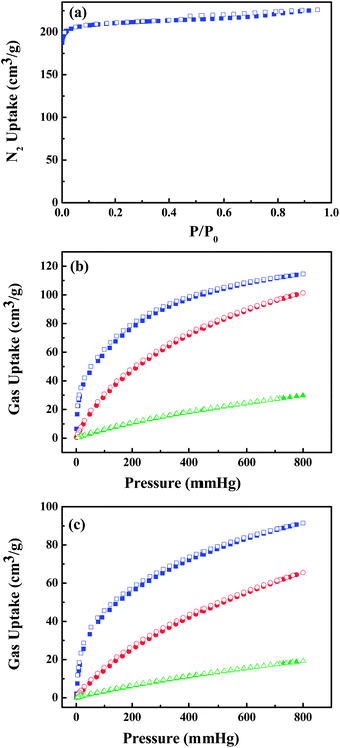 | ||
| Fig. 2 (a) N2 sorption isotherm at 77 K and C2H2 (blue), CO2 (red) and CH4 (green) sorption isotherms of UTSA-50a at (b) 273 K and (c) 296 K. Solid symbols: adsorption, open symbols: desorption. | ||
The above structural feature of UTSA-50 encourages us to examine its potential application in gas storage. As shown in Fig. 2, UTSA-50a can take the amount of C2H2 (113.9 cm3 g−1), CO2 (100.1 cm3 g−1) and CH4 (29.2 cm3 g−1) at 1 atm and 273 K; and C2H2 (90.6 cm3 g−1), CO2 (64.4 cm3 g−1) and CH4 (18.8 cm3 g−1) at 1 atm and 296 K.
The amount of absorbed C2H2 in UTSA-50a at 296 K is lower than similar structured MOFs with high density open metal sites,25,26 such as HKUST (201 cm3 g−1), NOTT-101 (184 cm3 g−1) and UTSA-20 (150 cm3 g−1). This could mainly be attributed to the lower surface area in UTSA-50a than in HKUST (1401 m2 g−1), NOTT-101 (2930 m2 g−1) and UTSA-20 (1894 m2 g−1). However, this value is comparable with the ones with small pores such as Cu2(NDC)2(DABCO) (97 cm3 g−1) and Zn2(NDC)2(DABCO) (106 cm3 g−1), which represent the best C2H2 adsorption property among the MOFs with low surface areas and small pores;20,25 but it is significantly higher than the ones with large pores such as MOF-5 (26 cm3 g−1) and ZIF-8 (25 cm3 g−1) with large surface areas.25 It is worth noting that the storage density of adsorbed acetylene in UTSA-50a micropores is 0.30 g cm−3, which is comparable with HKUST and MOF-74-Fe,23,25 and is among the high end for MOF materials. This illustrates that the combined feature of open metal sites, Lewis basic pyridyl sites and optimized pore size do lead to the efficient adsorption of acetylene molecules in UTSA-50 micropores. The comparison of some microporous MOFs for acetylene storage is listed in Table 1.
| Material | Surface area (BET) [m2 g−1] | Pore volume [cm3 g−1] | C2H2 uptake [cm3 g−1] | Density of adsorbed C2H2a [g cm−3] |
|---|---|---|---|---|
| a Calculated density of adsorbed acetylene in micropores. b H8L = tetrakis[(3,5-dicarboxyphenoxy)methyl]methane. | ||||
| UTSA-50 | 604 | 0.35 | 91 | 0.30 |
| Cu2(ndc)2(dabco)20 | — | 0.44 | 97 | 0.26 |
| Cu2(adc)2(dabco)20 | — | 0.28 | 82 | 0.34 |
| Zn2(ndc)2(dabco)20 | — | 0.52 | 106 | 0.24 |
| Zn2(adc)2(dabco)20 | — | 0.31 | 101 | 0.38 |
| HKUST25 | 1401 | 0.76 | 201 | 0.31 |
| MOF-74-Fe23 | 1350 | 0.63 | 156 | 0.29 |
| UMCM-150 (ref. 26) | 3330 | 1.21 | 129 | 0.12 |
| PCN-16 (ref. 26) | 2810 | 1.00 | 176 | 0.20 |
| MOF-5 (ref. 25) | 3610 | — | 26 | — |
| ZIF-8 (ref. 25) | 1758 | — | 25 | — |
| NOTT-101 (ref. 26) | 2930 | 1.05 | 184 | 0.20 |
| NOTT-102 (ref. 26) | 3590 | 1.28 | 146 | 0.14 |
| UTSA-20 (ref. 26) | 1894 | 0.67 | 150 | 0.26 |
| UTSA-33a 27 | 660 | 0.37 | 84 | 0.26 |
| UTSA-34b 28 | 991 | 0.54 | 121 | 0.26 |
| UTSA-35a 29 | 743 | 0.31 | 65 | 0.24 |
| UTSA-36a 30 | 495 | 0.33 | 57 | 0.20 |
| UTSA-38a 33 | 1090 | 0.61 | 64 | 0.12 |
| Cu2(ebtc)34 | 1852 | 1.00 | 160 | 0.19 |
| Cu4Lb,35 | 1115 | 0.61 | 154 | 0.29 |
| Zn4(OH)2(1,2,4-btc)2 (ref. 36) | 408 | 0.22 | 53 | 0.28 |
| Cu(bdc-OH)(4,4′-bpy)37 | 553 | 0.28 | 35 | 0.14 |
The coverage-dependent adsorption enthalpies of UTSA-50a for the three gases were calculated based on the virial method, which is a well-established and reliable methodology fitting from their adsorption isotherms at 273 K and 296 K, as listed in Table 2 and Fig. S2–S7.† The enthalpies at zero coverage are 39.4, 27.8 and 18.6 kJ mol−1 for C2H2, CO2 and CH4, respectively. Note that the adsorption enthalpy for C2H2 is significantly higher than MOF-505 (24.7 kJ mol−1) and HKUST (30.4 kJ mol−1) with high density open metal sites.25 This indicates that the high density of Lewis basic pyridyl sites and optimized pore sizes do further enhance the affinity between the MOF surface and C2H2 molecules, presumably by the H–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–H…N (PDC) hydrogen bonding. Moreover, the adsorption enthalpy for C2H2 is more than 10 kJ mol−1 and 20 kJ mol−1 larger than that for CO2 and CH4, respectively. The most remarkable feature of UTSA-50a is the significantly higher Henry's law selectivities of 68.0 and 13.3 for C2H2/CH4 and C2H2/CO2, respectively at 296 K. To the best of our knowledge, the C2H2/CH4 separation selectivity of 68.0 is the highest one ever reported.3,25–38 The highly selectivity sorption for C2H2/CH4 and C2H2/CO2 and moderately high acetylene storage capacities highlight the promise of UTSA-50 in the practical acetylene separation.
C–H…N (PDC) hydrogen bonding. Moreover, the adsorption enthalpy for C2H2 is more than 10 kJ mol−1 and 20 kJ mol−1 larger than that for CO2 and CH4, respectively. The most remarkable feature of UTSA-50a is the significantly higher Henry's law selectivities of 68.0 and 13.3 for C2H2/CH4 and C2H2/CO2, respectively at 296 K. To the best of our knowledge, the C2H2/CH4 separation selectivity of 68.0 is the highest one ever reported.3,25–38 The highly selectivity sorption for C2H2/CH4 and C2H2/CO2 and moderately high acetylene storage capacities highlight the promise of UTSA-50 in the practical acetylene separation.
| Adsorbate | T/K | A 0 (In(mol g−1 Pa−1) | A 1 (g mol−1) | R 2 | K H (mol g−1 Pa−1) | S i/CH4 | S i/CO2 | Q st,n=0 (kJ mol−1) |
|---|---|---|---|---|---|---|---|---|
| a The Henry's law selectivity for gas component i over CH4 at the speculated temperature is calculated based on the equation Sij = KH(i)/KH(j). b The Henry's law selectivity for gas component i over CO2 at the speculated temperature is calculated based on the equation Sij = KH(i)/KH(j). | ||||||||
| C2H2 | 273 | −12.706 | −1007.788 | 0.993 | 3.033 × 10−6 | 139 | 19.8 | 39.4 |
| 296 | −14.054 | −675.568 | 0.987 | 7.878 × 10−7 | 68 | 13.3 | ||
| CO2 | 273 | −15.691 | −357.548 | 0.996 | 1.533 × 10−7 | 7.0 | 1.0 | 27.8 |
| 296 | −16.641 | −314.583 | 0.998 | 5.928 × 10−8 | 5.0 | 1.0 | ||
| CH4 | 273 | −17.642 | −455.174 | 0.996 | 2.179 × 10−8 | 1.0 | — | 18.6 |
| 296 | −18.278 | −408.011 | 0.999 | 1.153 × 10−8 | 1.0 | — | ||
4. Conclusions
In summary, we have targeted the first example of porous metal–organic frameworks UTSA-50 with open metal and Lewis basic pyridyl sites and optimized pores for their strong interactions with acetylene molecules. Such collaborative enforcement has enabled UTSA-50 to take up moderately high acetylene gas molecules of 91 cm−1 g−1 at room temperature and 1 atm, and furthermore to exhibit highly selective C2H2/CH4 and C2H2/CO2 gas separation with the Henry's law selectivity of 68.0 and 13.3, respectively, at 296 K. This work not only highlights the promise of this MOF material for the practical separation of acetylene, but more importantly demonstrates the power of collaborative immobilization of functional sites and tuning of the pore sizes/curvatures within porous MOFs for their gas storage and separation. It is expected that this work will initiate more extensive research endeavours on pore engineering of porous MOFs, thus new promising porous materials will be emerging for their industrially important gas storage and separation in the near future.Acknowledgements
This work was supported by the award AX-1730 from the Welch Foundation (B.C.). The authors gratefully acknowledge the financial support for this work from the National Natural Science Foundation of China (no. 51229201, 51272231 and 51010002). We also appreciate the support from NSFC (21273033, 21203024) and the Award ‘MinJiang ScholarProgram’ in Fujian Province, China. H. X. is grateful to the China Scholarship Council.Notes and references
- M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319 CrossRef CAS; S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334 CrossRef; D. Bradshaw, J. B. Claridge, E. J. Cussen, T. J. Priora and M. J. Rosseinsky, Acc. Chem. Res., 2005, 38, 273 CrossRef; G. Ferey, Chem. Soc. Rev., 2008, 37, 191 RSC; R. E. Morris and P. S. Wheatley, Angew. Chem., Int. Ed., 2008, 47, 4966 CrossRef; M. P. Suh, Y. E. Cheon and E. Y. Lee, Coord. Chem. Rev., 2008, 252, 1007 CrossRef; D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 9, 6058 CrossRef; O. K. Farha and J. T. Hupp, Acc. Chem. Res., 2010, 43, 1166 CrossRef; B. Chen, S. C. Xiang and G. D. Qian, Acc. Chem. Res., 2010, 43, 1115 CrossRef; H. L. Jiang and Q. Xu, Chem. Commun., 2011, 47, 3351 RSC; M. C. Das, S. Xiang, Z. Zhang and B. Chen, Angew. Chem., Int. Ed., 2011, 50, 10510 CrossRef; M. P. Suh, H. J. Park, T. K. Prasad and D. W. Lim, Chem. Rev., 2012, 112, 782 CrossRef; K. Sumida, D. L. Rogow, J. A. Mason, T. M. Mcdonald, E. D. Bloch, Z. R. Herm, T. H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724 CrossRef; J. R. Li, J. Sculley and H. C. Zhou, Chem. Rev., 2012, 112, 869 CrossRef.
- W. Morris, B. Leung, H. Furukawa, O. K. Yaghi, N. He, H. Hayashi, Y. Houndonougbo, M. Asta, B. B. Laird and O. M. Yaghi, J. Am. Chem. Soc., 2010, 132, 11006 CrossRef CAS; E. D. Bloch, D. Britt, C. J. Doonan, F. J. Uribe-Romo, H. Furukawa, J. R. Long and O. M. Yaghi, J. Am. Chem. Soc., 2010, 132, 14382 CrossRef; H. Deng, S. Grunder, K. E. Cordova, C. Valente, H. Furukawa, M. Hmadeh, F. Gandara, A. C. Whalley, Z. Liu, S. Asahina, H. Kazumori, M. O'Keeffe, O. Terasaki, J. F. Stoddart and O. M. Yaghi, Science, 2012, 336(6084), 1018 CrossRef.
- B. Chen, C. Liang, J. Yang, D. S. Contreras, Y. L. Clancy, E. B. Lobkovsky, O. M. Yaghi and S. Dai, Angew. Chem., Int. Ed., 2006, 45, 1390 CrossRef CAS; B. Chen, X. Zhao, A. Putkham, K. Hong, E. B. Lobkovsky, E. J. Hurtado, A. J. Fletcher and K. M. Thomas, J. Am. Chem. Soc., 2008, 130, 6411 CrossRef; M. Xue, Z. Zhang, S. Xiang, Z. Jin, C. Liang, G. Zhu, S. Qiu and B. Chen, J. Mater. Chem., 2010, 20, 3984 RSC; S. Xiang, W. Zhou, Z. Zhang, M. A. Green, Y. Liu and B. Chen, Angew. Chem., Int. Ed., 2010, 49, 4615 CrossRef; Z. Guo, H. Wu, S. Gadipelli, T. Liao, Y. Zhou, S. Xiang, Z. Chen, Y. Yang, W. Zhou, M. O'Keeffe and B. Chen, Angew. Chem., Int. Ed., 2011, 50, 3178 CrossRef; S. Xiang, Z. Zhang, C. Zhao, K. Hong, X. Zhao, D. Ding, M. Xie, C. Wu, M. C. Das, R. Gill, K. M. Thomas and B. Chen, Nat. Commun., 2011, 2, 204 CrossRef; M. C. Das, S. C. Xiang, Z. Zhang and B. Chen, Angew. Chem., Int. Ed., 2011, 50, 10510 CrossRef; M. C. Das, Q. Guo, Y. He, J. Kim, C. G. Zhao, K. Hong, S. Xiang, Z. Zhang, K. M. Thomas, R. Krishna and B. Chen, J. Am. Chem. Soc., 2012, 134, 8703 CrossRef.
- S. Ma, D. Sun, D. Yuan, X. S. Wang and H. C. Zhou, J. Am. Chem. Soc., 2009, 131, 6445 CrossRef CAS.
- J. B. Lin, J.-P. Zhang, W.-X. Zhang, W. Xue, D.-X. Xue and X.-M. Chen, Inorg. Chem., 2009, 48, 6652 CrossRef CAS; J.-P. Zhang and X.-M. Chen, J. Am. Chem. Soc., 2009, 131, 5516 CrossRef; J. B. Lin, J. P. Zhang and X.-M. Chen, J. Am. Chem. Soc., 2010, 132, 6654 CrossRef; J. B. Lin, W. Xue, J.-P. Zhang and X.-M. Chen, Chem. Commun., 2011, 47, 926 RSC.
- F. Wang, Y.-X. Tan, H. Yang, Y. Kang and J. Zhang, Chem. Commun., 2012, 48, 4842 RSC.
- S.-S. Chen, M. Chen, S. Takamizawa, P. Wang, G.-C. Lv and W.-Y. Sun, Chem. Commun., 2011, 47, 4902 RSC.
- K. Li, D. H. Olson, J. Seidel, T. J. Emge, H. Gong, H. Zeng and J. Li, J. Am. Chem. Soc., 2009, 131, 10368 CrossRef CAS.
- Q. Lin, T. Wu, S. T. Zheng, X. Bu and P. Feng, J. Am. Chem. Soc., 2012, 134, 784 CrossRef CAS; S. Zheng, T. Wu, F. Zuo, C. Chou, P. Feng and X. Bu, J. Am. Chem. Soc., 2012, 134, 1934 CrossRef.
- J. An and N. L. Rosi, J. Am. Chem. Soc., 2010, 132, 5578 CrossRef CAS; J. An, S. J. Geib and N. L. Rosi, J. Am. Chem. Soc., 2010, 132, 38 CrossRef.
- T. M. Mcdonald, W. R. Lee, J. A. Mason, B. M. Wiers, C. S. Hong and J. R. Long, J. Am. Chem. Soc., 2012, 134, 7056 CrossRef CAS.
- H. Wu, W. Zhou and T. Yildirim, J. Am. Chem. Soc., 2009, 131, 4995 CrossRef CAS.
- F. Nouar, J. F. Eubank, T. Bousquet, L. Wojtas, M. J. Zaworotko and M. Ed0daoudi, J. Am. Chem. Soc., 2008, 130, 1833 CrossRef CAS.
- M. L. Foo, S. Horike, Y. Inubushi and S. Kitagawa, Angew. Chem., Int. Ed., 2012, 51, 6107 CrossRef CAS; S. Bureekaew, H. Sato, R. Matsuda, Y. Kubota, R. Hirose, J. Kim, K. Kato, M. Takata and S. Kitagawa, Angew. Chem., Int. Ed., 2010, 49, 7660 CrossRef.
- P. Kanoo, A. C. Ghosh, S. T. Cyriac and T. K. Maji, Chem.–Eur. J., 2012, 18, 237 CrossRef CAS.
- O. Farha, J. Hupp, I. Eryazici, C. Willmer, B. Hauser, A. Sarjeant, R. Snurr, S. Nguyen, P. Parilla and K. O'Neill, J. Am. Chem. Soc., 2012, 134, 9860 CrossRef CAS; Y. S. Bae, C. Y. Lee, K. C. Kim, O. K. Farha, P. Nickias, J. T. Hupp, S. T. Nguyen and R. Q. Surr, Angew. Chem., Int. Ed., 2012, 51, 1857 CrossRef.
- Y. Q. Lan, H. L. Jiang, S. L. Li and Q. Xu, Adv. Mater., 2011, 23, 5015 CrossRef CAS.
- T. Furuta, I. Kanoya, H. Sakai and M. Hosoe, Polymer, 2011, 3, 2133 CAS.
- X. Lin, I. Telepeni, A. J. Blake, A. Dailly, C. M. Brown, J. M. Simmons, M. Zoppi, G. S. Walker, K. M. Thomas, T. J. Mays, P. Hubberstey, N. R. Champness and M. Schröder, J. Am. Chem. Soc., 2009, 131, 2159 CrossRef CAS.
- D. Tanaka, M. Higuchi, S. Horike, R. Matsuda, Y. Kinoshita, N. Yanai and S. Kitagawa, Chem.–Asian J., 2008, 3, 1343 CrossRef CAS.
- A. R. Millward and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 17998 CrossRef CAS.
- O. K. Farha, A. Ö. Yazaydin, I. Eryazici, C. D. Milliakas, B. G. Hauser, M. G. Kanatzidis, S. T. Nguyen, R. Q. Snurr and J. T. Hupp, Nat. Chem., 2010, 2, 944 CrossRef CAS.
- E. D. Bloch, W. L. Queen, R. Krishna, J. M. Zadrozny, C. M. Brown and J. R. Long, Science, 2012, 335, 1606 CrossRef CAS.
- S. Xiang, Y. He, Z. Zhang, H. Wu, W. Zhou, R. Krishna and B. Chen, Nat. Commun., 2012, 3, 954 CrossRef.
- S. Xiang, W. Zhou, J. M. Gallegos, Y. Liu and B. Chen, J. Am. Chem. Soc., 2009, 131, 12415 CrossRef CAS.
- Y. He, R. Krishna and B. Chen, Energy Environ. Sci., 2012, 5, 9107 CAS.
- Y. He, Z. Zhang, S. Xiang, F. R. Fronczek, R. Krishna and B. Chen, Chem.–Eur. J., 2012, 18, 613 CrossRef CAS.
- Y. He, Z. Zhang, S. Xiang, H. Wu, F. R. Fronczek, W. Zhou, R. Krishna, M. O'Keeffe and B. Chen, Chem.–Eur. J., 2012, 18, 1901 CrossRef CAS.
- M. C. Das, H. Xu, Z. Wang, G. Srinivas, W. Zhou, Y.-F. Yue, V. N. Nesterov, G. Qian and B. Chen, Chem. Commun., 2011, 47, 11715 RSC.
- Y. He, Z. Zhang, S. Xiang, F. R. Fronczek, R. Krishna and B. Chen, Chem. Commun., 2012, 48, 6493 RSC.
- Y. He, W. Zhou, R. Krishna and B. Chen, Chem. Commun., 2012, 48 10.1039/c2cc35418g.
- Y. He, S. Xiang and B. Chen, J. Am. Chem. Soc., 2011, 133, 14570 CrossRef CAS.
- M. C. Das, H. Xu, S. Xiang, Z. Zhang, H. D. Arman, G. Qian and B. Chen, Chem.–Eur. J., 2011, 17, 7817 CrossRef CAS.
- Y. Hu, S. Xiang, W. Zhang, Z. Zhang, L. Wang, J. Bai and B. Chen, Chem. Commun., 2009, 7551 RSC.
- Y.-S. Xue, Y. He, S.-B. Ren, Y. Yue, L. Zhou, Y.-Z. Li, H.-B. Du, X.-Z. You and B. Chen, J. Mater. Chem., 2012, 22, 10195 RSC.
- Z. Zhang, S. Xiang, X. Rao, Q. Zheng, F. R. Fronczek, G. Qian and B. Chen, Chem. Commun., 2010, 46, 7205 RSC.
- Z. Chen, S. Xiang, H. D. Arman, J. U. Mondal, P. Li, D. Zhao and B. Chen, Inorg. Chem., 2011, 50, 3442 CrossRef CAS.
- Z. Zhang, S. Xiang and B. Chen, CrystEngComm, 2011, 13, 5983 RSC.
- P. J. Stang and F. Diederich, Modern Acetylene Chemistry, VCH, New York, 1995 Search PubMed.
- J. C. W. Chien, Polyacetylene: Chemsitry, Physics, and Material Science, Acedemic Press, New York, 1984 Search PubMed.
- R. T. Yang, Adsorbents: Fundamentals and Applications, John Wiley & Sons, Hoboken, 2003 Search PubMed.
- S. Keskin, T. M. Van Heest and D. S. Sholl, ChemSusChem, 2010, 3, 879 CrossRef CAS.
- F. G. Kerry, Industrial Gas Handbook: Gas Seperation and Purification, CRC Press, Boca Raton, 2007 Search PubMed.
- S. Budavari, The Merck Index, Merck Research Laboratories, NJ, 12th edn, 1996, p. 16 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ta00155a |
| This journal is © The Royal Society of Chemistry 2013 |