 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fascinating chemistry or frustrating unpredictability: observations in crystal engineering of metal–organic frameworks
Maarten G. Goesten*, Freek Kapteijn and Jorge Gascon*
Catalysis Engineering/Chemical Engineering Department, Delft University of Technology, Julianalaan 136, 2628 BL Delft, The Netherlands. E-mail: m.g.goesten@tudelft.nl; j.gascon@tudelft.nl; Fax: +31 15 2785006
First published on 18th September 2013
Abstract
Reticular design is a highly attractive concept, but coordination chemistry around the tectonic units of metal–organic frameworks (MOFs) and additional interplay with anionic and solvent species provide for dazzling complexity that effectively rules out structure prediction. We can however study the chemistry around pre-existing clusters, and assemble novel materials correspondingly, using a priori information about the connectivity of an investigated metal cluster. Studies, often spectroscopic of nature, have in recent years solved many puzzles in MOF crystallization. The obtained knowledge opens new doors in crystal engineering, but more research on MOF coordination chemistry has to be carried out.
Introduction
The self-assembly of molecules into an organized network is governed by molecular forces induced by so called tectons, etymologically referring to “building units” as introduced by Jim Wuest in 1991.1 Tetrahedral SiO4 and AlO4 units, which build up zeolites, are examples of tectons. In these purely inorganic self-assembling structures, small distortions in the metal–oxygen–metal angle allow for a large number of topologies, despite the exclusively tetrahedral nature of the tectons.2,3 The success of zeolites as catalysts and sorbents is beyond discussion and considerable focus on mimicking this self-assembly in non-purely inorganic structures is a consequence. For organic structures, design of suitable tectons towards self-assembly of highly structured solids has despite several breakthroughs proven to be a serious challenge up until now, as bonding is much less strongly directed.4†It is the inorganic–organic case of metal–organic frameworks (MOFs), in essence coordination polymers, where metal–(bridging) ligand bonds extend into one, two or three dimensions, that has led to massive scientific interest into the field of self-assembled, porous structures. As a benchmark ‘first paper’ on MOFs, one may choose a work published in 1989, where Hoskins and Robson proposed that a new and potentially extensive class of solid polymeric materials with unprecedented and possibly useful properties may be afforded by linking together (metal) centers with either a tetrahedral or an octahedral array of valences by rod-like connecting units.5 After a stint of steady year-by-year increase of publishing on the topic, a wave of scientific interest can be observed towards the late 1990's.6 The popularity of MOFs as potential candidates for an array of applications much lies in the tuneability and versatility of the materials, which can be formed upon self-assembly, as potentially desired inorganic and/or organic functionalities can be implemented into a porous structure through appropriate choice of metal precursor and corresponding bridging ligand. In MOFs, tectons come in the form of metal complexes, sometimes referred to as secondary building units (SBUs) and provide virtually infinite possibilities towards connectivity and therefore structure direction. Indeed, this is perhaps the most exciting feature of MOFs, as it matters crystal engineering, the synthesis of crystals with specific and predicted properties, in MOF chemistry sometimes referred to as reticular synthesis or reticular design. ‘Reticul’ is the Latin word for ‘network’, and reticular synthesis represents the targeted retrosynthetic-like synthesis of complete networks.7
One particular example which caught attention was the use of a tetranuclear Zn4(μ4-O)L6 tecton in which octahedral connectivity leads to a primitive cubic α-Po net. Here the six ligands consist of three pairs of aryldicarboxylato syn–syn oxygen, where the aryl species can be varied to deliver a series of isoreticular frameworks, in which chemical properties can be implemented in crystals with predictable topology.8 Similar observations apply for MOF tectons with different connectivities. Apparently, it seems that if the reaction conditions that lead to the formation of a particular tecton with corresponding connectivity are figured out, then control over the self-assembly towards a desired net is possible. The ideal MOF ‘designer’ is therefore both an expert in coordination chemistry and crystallography. This attractive idea has to limited extent been realized, and MOFs have, for instance, been setting records in N2, H2, CO2 and CH4 sorption (‘storage’) through the use of dimensionally very long ligands, and display magnetically predictive behaviour through the self-assembly of specific nets.9–12 Despite these (amongst other) examples, unpredictability is still common and true design is relatively rare. This should not be a surprise, as coordination chemistry around MOF crystallisation is far from being a resolved field within its own specialist domain. The contrary is true: investigations on MOF structures have led to discoveries. An example is the trimeric Al3O(BTC)6L3 (BTC: benzenetricarboxylate, L: H2O, N) tecton within MIL-96, which will be treated in the text below, a commonly observed moiety for transition metals but a new cluster for a p-block metal.13 Clearly, if the current state of coordination chemistry does not allow us to predict chemistry around the MOF tecton, little prediction in the crystallographic domain can be provided as well, as depicted artistically in Fig. 1. Thus, MOF crystallisation ought to be investigated more, so that previously unknown events can be revealed for the sake of crystal engineering. This is also a requirement from industry – the main factor determining whether the MOF ‘hype’ will turn out to be successful – as MOF applications would require crystals ‘shaped’ towards functionality, from both morphological and chemical perspective.
 | ||
| Fig. 1 Reticular design seems hampered by unpredictability around coordination chemistry.14 | ||
This critical overview aims at giving insight in progress researchers have made revealing coordination chemistry around MOFs by selecting different examples rather than at deeply reviewing all MOF synthesis papers published to date. The manuscript is divided into three parts. The first part deals with the chemistry around the metal and ligand, the tectons they form with corresponding connectivities, and their degree of predictability in synthesis. The second part deals with the chemistry of the anion that is usually present, and its large influence on MOF crystallization. The third part deals with the even more unpredictable role of the solvent. We finish with some final outlook and remarks. We underline that this manuscript focuses on the coordination-chemical and synthetic part of crystal engineering with MOFs, an area left relatively unexposed in recent years, as we felt. For detailed theory into MOF crystallography, the reader is referred to excellent reviews published by Batten and Yaghi that, respectively, give insight into unpredictability and predictability within the crystallographic domain.15,16
1. Metal and ligand
One of the most apparent claims for reticular design is the development of ZIFs (zeolitic imidazolate frameworks), porous solids where structural binding of zeolites is mimicked in the metal–organic domain by the use of tetrahedrally coordinating ions and imidazolato bridging ligands.17 The metal ion resembles tetrahedral SiO4, the ligand displays a bridging angle resembling the Si–O–Si angle in zeolites and self-assembly leads indeed to structures possessing zeolite topologies. Even the crystallization mechanism of one of the members, ZIF-8, was shown to resemble the corresponding mechanism of some high-silica zeolites.18 This successful claim for rational design is an exception in a field where unpredictability reigns. A first observation is that one is strongly limited in decorating the MOF ligand with functional groups. These groups are either seen to coordinate to the metal themselves, like sulfonate groups, or to inhibit/promote formation of the coordination polymer for ambiguous reasons.19 For example, we have observed that one can pre-functionalise MIL-101(Cr) with nitro groups, but not with amine groups. For MIL-101(Al) the reverse is true; only the aminated ligand leads to formation of the coordination polymer.‡ We have so far been unable to fully explain this observation. The general consequence of this ligand effect is that researchers have to turn to post-synthetic functionalization of metal–organic frameworks, and this has itself grown into a large field within MOF research.20–23 It is however the complexity of the tectonic units which generates most uncertainty around crystallization of MOFs. This is actually easily understood if we observe that, as stated in the introduction as well, simple tectonic units in zeolites already allow for huge diversity in topologies. In the MOF domain, (often polynuclear) transition metal chemistry governs the self-assembly mechanism, and polynuclear transition metal chemistry is an area in which inorganic chemists, despite numerous attempts, have in general only made limited progress in successfully “designing” coordination clusters.24 As Ribas Gispert states in his coordination chemistry textbook: the vast majority of these, including the most relevant and spectacular, have been made as a result of “serendipitous self-assembly”. In this context coordination chemists have learned the reaction conditions necessary to favour the formation of large polynuclear clusters, however, it currently remains impossible to predict the structure of any new system prepared in this manner. For anyone believing new MOF networks based on new tectons can be self-assembled in predictable manner, this news should be rather demoralising. If chemists cannot predict the structure of a new polynuclear complex, then surely the MOF structure built with this complex can only be guessed.The enormous diversity of tectonic units within the field of metal–organic frameworks is first of all related with the flexible coordination environment of metal ions, often those with d10 configuration such as Zn2+, Ga3+, In3+, Ti3+, Cd2+, Hg2+, Ag+, Cu+. Zinc, in particular, is frequently used as metal ion and can show many coordination geometries besides its commonly observed tetrahedral configuration. For instance, zinc can exist in pentagonal bipyramidal configuration as reported by Kitagawa in 1997, where zinc is coordinated to three 4,4′-bpy ligands and two chelating nitrato ligands (in the next section we shall see that nitrato is on more occasions seen to promote pyramidal coordination geometries),25 but octahedral and trigonal bipyramidal coordination geometries are known as well.26,27 An interesting case present the dinuclear paddlewheel clusters, which are also observed for copper, for instance in CuBTC (HKUST-1). These binuclear clusters are tectons with square connectivity.28
For f-block elements, unpredictability around the coordination environment is even more pronounced and can lead to exotic tectons such as praesodynium-based or dysprosium square antiprisms leading to lanthanide open-framework structures, and ‘supercubanes’, respectively.29,30 These examples matter clusters of higher nuclearity, and are these clusters of high nuclearity that act as even more diverse and unpredictably appearing tectons in self-assembly towards new structures.
We will here consider aluminium-based MOF members of the MIL (Materiel Institut Lavoisier) family, who provide an excellent illustration in this regard. Aluminium is one of the smallest metals available in the periodic system; it does not form bonds with d-orbitals and often coordinates in octahedral mode, yet the chemistry around multinuclear Al clusters is extremely diverse and unpredictable. MIL-96(Al), MIL-100(Al) and MIL-110(Al) are all three aluminium benzenetricarboxylate (BTC) based MOFs, containing very different molecular tectonics.§ MIL-96 and MIL-100 contain an Al3(μ3-O)(BTC)6L3 tecton, a well-known moiety in coordination chemistry for d-block elements but here for the first time seen with aluminium. In MIL-96, this cluster and its trigonal connectivity reside within a hexagonal chain made up by μ2-OH bridged Al chains which are commonly observed in clay chemistry and are known for their high chemical stability.31,32 In MIL-100(Al), the μ3-O based cluster oligomerizes to an Al12 “super tetrahedron”, which bears striking resemblance to SiO4 tectons from zeolites. As a result, this far bigger analogue directs self-assembly to zeolite MTN topology with gargantuan cage dimensions exceeding 3 nm. MIL-110(Al) does not contain the μ3-O cluster but its make-up is fascinating nevertheless, as two types of clusters (six with terminal aqua/hydroxido ligands, two without terminal ligands) make up large Al8 octagonal tectons, which self-assemble towards large hexagonal channels. Looking at Table 1, one can conclude that the synthetic conditions do not differ much, as only small changes in pH appear to inflict large structural changes through promotion of these different complexes. This inspired Haouas et al. to perform in situ NMR experiments from which they could reconstruct the mechanism, and concluded that identical dinuclear Al–BTC complexes form early on for all three topologies, but small differences in equilibria lead to different topologies.33 MIL-100(Al) is clearly the kinetic product, and MIL-96(Al) the thermodynamic product, MIL-110(Al) falls in between. It is thus not surprising that upon prolonged heating, MIL-100(Al) can be seen to rearrange itself towards MIL-110(Al), and to MIL-96(Al) correspondingly.
| |||
|---|---|---|---|
| Synth. pH | Synth. T/t (°C h−1) | Clusters and tecton geometrya | |
| a Oxygen ligands originate from BTC ligand. Writing six oxygen ligands as (BTC)3 is in agreement with the molecular composition of the frameworks, but is essentially incorrect as BTC is μ6-bridging.b Two additional notes must be made for MIL-96: (i) the formal representation for the aluminium chain is AlO2(OH)4 bound to AlO4(OH)2, with O2 and O4 coming from bridging BTC linkers (ii) besides the μ3-O based trigonal cluster and μ2-OH based chains, another type of aluminium is present. This species is only partially attached to the framework and is not considered here. | |||
| MIL-96(Al) | 1.0–3.0 | 210/24 | Al3(μ3-O)O6L3 trigonal |
| Al2(μ2-OH)3O6 hexagon chainb | |||
| MIL-100(Al) | 0.5–0.7 | 210/3–4 | Al3(μ3-O)O6L3 trimers |
| MIL-110(Al) | 0–0.3 & 3.5–4.0 | 210/72 & 3 | Al(μ2-OH)3O2L octamer |
| Al(μ2-OH)3O3 octamer | |||
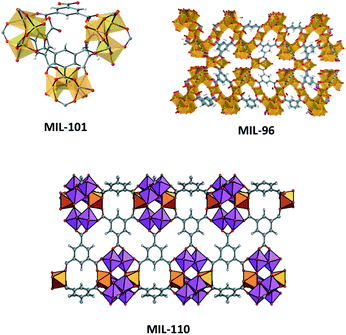
The complexity and extreme dependence on the chemical environment that MOF tectons show effectively rules out true prediction of a resulting structure. Instead, we are forced to study the coordination chemistry of tectons of reported coordination polymers, and use resulting information in the synthesis of functionalized or adapted structures. This reduced level of predictability can still be satisfactory, as functionalized or adapted frameworks have been rationally designed in this way. As we will see in the succeeding sections, even at this level of ‘design’, crystallization of MOFs is unpredictable and pervasive.
2. Influence of the anion
As MOFs are generally synthesized from metal salt precursors, anionic species will always be present in solution.¶ Influence of the anion on the final topology of the MOF is a regularly observed phenomenon.36–41 Apart from acting as potential mineralizers enhancing crystallinity, anions can act as true structure-directing agents.42 One of the earliest and perhaps still most striking example dates from 2000 where Min and Sun reported on a silver ethylenediaminetetrapropionitrile (EDTPN) MOF where the choice of precursor, Ag(NO3), Ag(CF3SO3) or Ag(ClO4) leads to self-assembly towards three completely different nets.43 EDTPN, displayed in Fig. 2, is through four cyano and two tertiary ammine ligand sites capable of μ2–6 bridging. Interestingly, solely depending on the anion, μ2 (1), μ3 (2) or μ4 (3) bridging is observed. Corresponding coordination geometries and topologies are distorted bipyramidal: linear network, tetrahedral: 2D layer, octahedral: 2D boxlike network, respectively. The EDTPN ligand coordinates to one silver ion through both its ammine ligands, and to one, two or three other silver ions through its cyano ligands.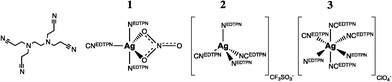 | ||
| Fig. 2 A simplified scheme around self-assembly of the silver–EDTPN networks. On the far left, the EDTPN ligand, then coordination geometries of silver in the presence of a nitrate (trigonal bipyramid), triflate (tetrahedral), or perchlorate (octahedral) anion, from left to right. | ||
The authors also report on the possibility to induce crystal-to-crystal rearrangements upon ion exchange: 1 and 2 can be reversibly transformed into each other, and both can be irreversibly transformed into 3. This illustrative example shows that the (metal salt) anion can have two structure-directional roles in MOF crystallization: it can act as an anionic terminal ligand, or less definable, as an anionic moiety stabilizing coordination geometry and resulting structure.
In the first case, it changes connectivity of the metal-precursor by blocking a coordinative site, and/or by changing coordination geometry as can be seen in structure 1, where a nitrato ligand occupies two silver coordination sites whilst promoting trigonal bipyramid coordination. EDTPN can as a result only bridge two silver ions. Depending on lability of the metal–anion bond, such blocking through anion coordination might even affect crystal morphology. One primary example is the synthesis of ZIF-7 microrod crystals through the use of ZnCl2 instead of Zn(NO3)2, in the presence of a diethyl amine modulator.44|| In an attempt at revealing the pathway of the formation of these microrods, we found that the strong Zn–Cl bond inhibits direct replacement of the two chlorido ligands by the benzimidazole bridging ligand, and dimerization of mononuclear Zn benzimidazole clusters is favoured. This rules out growth in tetrahedral direction, as normally induced by a Zn2+ tecton, but directs self-assembly towards rod-like structures (in a rather complex manner).45 As seen with the nitrato ligand in 1 ion co-coordination might also induce an alternate coordination environment by promoting (different) geometry. Surprisingly little has been published about this particular anionic effect on connectivity of the metal cluster in MOFs, whereas it is likely to have significant effect on the structure of the frameworks that crystallize. The anionic effect on connectivity is frequently observed, as we saw in the first part of this review where Zn(II) coordination polymers may display nitrato-induced pentagonal bipyramidal geometry, but for instance also isothiocyanato-induced octahedral coordination geometry (amongst a long list of other examples).46–48 Predicting this particular effect of the anion ligand on cluster connectivity and resulting framework can be a cumbersome affair that requires use of ligand field theory in combination with computational methods. With the latter, further complexities, such as anion-solvent or anion-ligand interactions might be taken into account as well.
In the second case, non-coordinate stabilization of a cluster or resulting net by an anion, chemistry is even less predictable. Referring to Fig. 2 again: 2 can rearrange into 3 upon solid-state ion exchange, yet the reverse reaction does not occur. This hints at the silver–EDTPN coordination polymer possessing greater affinity towards the perchlorate anion with respect to the triflate anion. As a matter of fact, in further experiments the authors determine the affinity to follow ClO4− > NO3− > CF3SO3− > Cl− and note that this follows the order of the Hofmeister series, a series first proposed in 1888 which ranks the relative influence of anions on macromolecules.** The Hofmeister Series are a point of discussion; whereas the anionic influence was originally related to influence on ‘water structure’ around the macromolecule, recent time-resolved and thermodynamic studies indicate otherwise. Direct ion–macromolecule interactions seem to provide a more realistic rationale for the series.49
The series is depicted in Fig. 3. Anions in the left part of the Hofmeister series are called kosmotropes, the ones to the right part chaotropes. These terms refer to the anion's ability to alter the hydrogen-bonding network of water. Kosmotropes stabilize ‘water structure’, and are seen to have a stabilizing (and salting-out) effect on proteins macromolecules. MOFs are often seen to follow the Hofmeister series in anion selectivity, and therefore the series might be used as a rough guideline towards anionic stabilization of MOFs, but nothing more than a rough guideline, as anti-Hofmeister and non-Hofmeister selectivities with MOFs are commonly observed. In an excellent review on this topic (to which the interested reader is certainly referred to), the authors state that sorely needed at this time are more quantitative studies of anion exchange and separations, including competition experiments, anion-exchange isotherms, thermodynamic and kinetic measurements, and mechanistic investigations of anion transport. Computational studies could also offer valuable information about the energetics of anion binding inside the coordination frameworks, as well as the electronic and steric requirements for optimal anion selectivity.50 We can but conclude that in this peculiar domain of anion–MOF interaction, many events are currently beyond our grasp in terms of understanding.
 | ||
| Fig. 3 The Hofmeister series. | ||
3. The ambiguous role of the solvent
Synthesis of MOFs is usually done in solvothermal media,†† and the choice of solvent is shown to be a significant parameter in kinetics of crystallization, network structure and crystal topology. The nature of structure-direction by the solvent is very versatile. Similar to the case of the anion described above, it may influence the structure of the coordination polymer acting as a ligand, or through less defined (often hydrogen-bonding) interaction with the scaffold. Yet in addition, it can promote topologies acting as a true molecular promoter, assist in post-synthetic functionalization or strongly affect crystal morphology, as we will see in this section.A typical example of solvent structure-direction through coordinating ability is the Cd(II)-4,4′-dipyridyl sulfide MOF system, in which depending on whether one chooses a DMF–dichloromethane, acetonitrile–dichloromethane or methanol–dichloromethane solvent system a 1D, 2D, or 3D network forms, respectively. In the first, 1D {[Cd(Py2S)2(DMF)2](ClO4)2}n and the second, 2D {[Cd(Py2S)2(MeCN)2](ClO4)2}n case, a pair of solvent ligands are in trans positions in octahedrally coordinated Cd(II), whereas in the 3D {[Cd2(Py2S)5(MeOH)2](ClO4)4}n, only one site of octahedral Cd(II) is occupied by MeOH solvent, resulting in a chiral 5-connected framework.52 Many analogous examples can be provided.53–56 Coordinated solvents can also exert influence over framework topology through steric effects as was shown by Noro et al. in which Lewis base solvents modify the forms of flexible chain motifs in regularity, through steric effect of coordinated solvents.57
If we proceed studying the non-coordinate structure-directing ability of solvent, we can take a look at several studies carried out by Dastidar et al. on cadmium coordination polymers. They found that only polar solvents lead to Cd(II) based coordination polymers, but less polar solvents only form complexes of lower nuclearity. They stretched this work to coordination polymers constructed from bis-pyridyl-bis-urea ligands bound to octahedral Zn(II).58 This specific ligand was chosen for its ability to form hydrogen bonds with solvent (and anionic) species. The authors successfully demonstrated the ability of the solvent to direct structure as they found that ethylene glycol promotes formation of a zig–zag coordination polymer through hydrogen bonding interactions with the urea nitrogen ligand, stabilizing its syn–syn conformation.
THF (polar aprotic), acetone (polar aprotic) and 1,4-dioxane (non-polar) were not capable of stabilising syn–syn conformation and led to crystallization of a 2D grid-like network instead. The dependence of topology on solvents is truly remarkable, and we have ourselves carried out several studies in this field, initially sparked by the competition between the NH2–MIL-53(Al) and NH2–MIL-101(Al) phases in synthesis. Both topologies carry our interest and are investigated for application in our laboratories. NH2–MIL-53(Al) is a flexible, microporous, highly stable MOF in which μ2-OH bridged aluminium chains form lozenge-shaped channels. This material presents many interesting properties, such as breathing, selective adsorption, selective separation, and was also shown to be among the first solid-state materials displaying nonlinear optical switching.59–62 NH2–MIL-101(Al) contains the μ3-O centred clusters that, like in MIL-100(Al) make up super tetrahedral tectons that self-assemble towards an MTN topology with huge cages. NH2–MIL-101(Al) is like MIL-100(Al) mesoporous, and therefore interesting for applications in catalysis, but carries an advantage over MIL-100(Al) in the 2-aminoterephthalato ligand being much more prone to post-synthetic functionalization.63‡‡ One may state that NH2–MIL-53(Al) and NH2–MIL-101(Al) are the 2-aminoterephthalato analogues of MIL-96(Al) and MIL-100(Al), respectively; NH2–MIL-53(Al) is the thermodynamic product, and owes its high stability to clay-like μ2-OH bridged Al chains. NH2–MIL-101(Al) is the kinetic product and both phases are frequently observed to be competition with each other. As was stressed in the introduction, optimising synthesis of these materials is a requirement if they are to be implemented in industry, so we decided to place these materials under the microscope.§§ In a Small/Wide Angle X-Ray Scattering (SAXS/WAXS) study, we were able to calculate kinetics of formation of both topologies in different solvent compositions, using the Gualtieri model earlier used by Millange and co-authors.65–68 The results are summarized in Table 2.
| |||
|---|---|---|---|
DMF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O molar ratio :H2O molar ratio | kg (104 s−1) | kn (104 s−1) | Product |
| 0.00:1.00 | 1.2 | 45 | NH2–MIL-53(Al) |
| 0.70:0.30 | 14 | 43 | NH2–MIL-53(Al) |
| 0.90:0.10 | 7.0 | 23 | NH2–MIL-53(Al) |
| 0.95:0.05 | 4.1 | 14 | NH2–MIL-53(Al) |
| 1.00:0.00 | 6.3 | 14 | NH2–MIL-101(Al) |
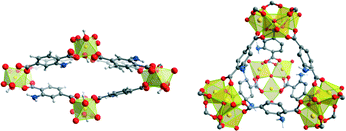
DMF inflicts a different molecular mechanism as it promotes formation of NH2–MIL-101(Al), but only when pure DMF is used as solvent, whereas it significantly enhances crystal growth rate of NH2–MIL-53(Al) in DMF:H2O mixtures. A small victory in synthetic control was obtained when we used this result to find an optimal solvent composition for the synthesis of NH2–MIL-53(Al), DMF:H2O molar ratio of 0.1:0.9, for which the yield is three times as high as in a pure H2O synthesis. This specific solvent-dependent behaviour was explained through observation of a DMF-promoted intermediate, NH2–MOF-235(Al).69 This intermediate is seen to rearrange either towards MIL-101(Al) topology (pure DMF) or to MIL-53(Al) topology (DMF-H2O mixtures). From SAXS analysis follows indeed that NH2–MOF-235(Al) crystals form very quickly in DMF containing mixtures. In further research at the molecular scale, DFT confirmed that DMF stabilizes the MOF-235 topology, which bears strong chemical kinship to the MIL-101 topology.70In situ NMR studies showed that DMF plays a kinetically promotional role in converting NH2–MOF-235(Al) to NH2–MIL-101(Al) in complexating HCl (Table 2).71 This result is highly interesting, and it is inferred that similar solvent-modulation is a factor in the promotion of many other MOF topologies (Fig. 4).
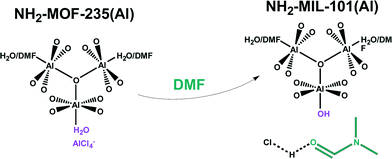 | ||
| Fig. 4 Promotional role of DMF in converting NH2–MOF-235(Al) into NH2–MIL-101(Al). The oxygen ligands originate from 2-aminoterephthalic acid linkers. | ||
Crystal topology and crystallinity of the resulting material is also highly dependent on the choice of solvent. In a study on efficiently encapsulating phosphotungstic acid (PTA) in MIL-100(Cr), we obtained a xerogel-like MOF with dual porosity, when pure DMF was used.72 A similar effect of the use of ethanol on MIL-100(Fe) was reported before.73 Also referred to as metal–organic gels, these materials are investigated for use as solid sorbents.74 The role of the solvent as gelator was recently clarified by Li et al. who propose a general synthetic route for Al(III) MOF aerogels.75 They state that the formation of MOF gels evolves from two stages. The first stage is the nucleation step where the metal-clusters form, which polymerize or aggregate. In the second stage two directions can be taken, one of which leads to ordinary crystallization, but in the second stage, it is assumed that coordination equilibria are perturbed by competing reactions, and non-crystallographic branching may occur, thus resulting in mismatched growth or cross-linking and providing the probability for gelation. The theory is summarized in Fig. 5.
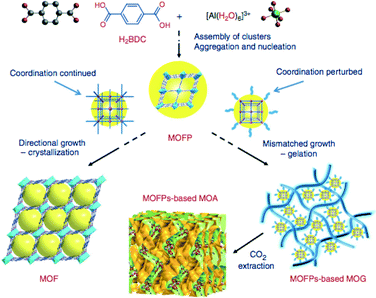 | ||
| Fig. 5 MOF gelation mechanism as proposed by Li et al. Reproduced with permission from ref. 75. | ||
We thus conclude that the role of the solvent in MOF synthesis is ambiguous and unpredictable. It might act as coordinating agent, (hydrogen-binding) non-coordinating template and even as homogeneous catalyst and gelator.
4. Final remarks
Reticular design is a tremendously attractive concept as it essentially presents a crystallographic analogue of retrosynthesis, which we know from organic chemistry, but without the tedious step-by-step assembly towards the final structure. It would make use of self-assembly instead. As we know by now, the concept of reticular design is a highly controversial one.76 A must-read 2006 article by Schön and Jansen slams the concept of solid-state chemical design, calling it an illusion, and states about the field of coordination polymers that one should critically note that typically, in those cases in which a synthesis is claimed to have been successfully designed, the design and actual synthesis are published in the same paper. This does not strike us as very convincing. They show that in our field the number of possible structures, which correspond to local minima in energy landscapes, is so large that one is effectively unable to predict anything.77 This is certainly seen in laboratory practice as the stunning amount of polynuclear tectons that form in near-identical synthetic environments for the aluminium-based frameworks treated in the text above is a testimony towards coordination chemistry unpredictability as described by Ribas Gispert.24As we see that our cognizance within the chemistry field is insufficient to truly design novel crystalline materials that self-assemble from newly designed tectons, we can but conclude that we are left to study the chemistry around pre-existing clusters, and assemble novel materials correspondingly, using a priori information about the connectivity of the pre-studied metal cluster. We can take soothe from the fact that this approach has indeed been successful in several cases, and various functionalities have been implemented in MOFs with predictable topology. In this paper we have seen that we are learning, and in some cases the full multiscale pathway – from molecular to crystal scale – has been revealed (always a posteriori). This obtained knowledge opens the door to a new step in crystal engineering. Yet again we underline that this does not mean the obscure prediction and assembly of new materials, but manipulation and adaptation of studied ones, in order to design structured sorbents, catalysts, magnetic materials, luminescent materials etc. One particular field that is opening up as knowledge around the coordination chemistry of MOFs is expanding is the case where we add structure-directing moieties, which are not necessities (as ions and solvents usually are). Adding structure-directing agents is common in zeolite synthesis, as many topologies actually require the use of an auxiliary template, yet the mechanistic effect of these structure-directing agents had for long been poorly understood and therefore called for specialist studies on the subject.78,79 As resulting knowledge of the mechanism of zeolite templation increased over the years, chemists have been able to tune their templates so they are capable of more than just stabilizing a topology. This resulted and is still resulting into some spectacular results, such the hierarchically structured zeolite catalysts presented by Ryong Ryoo in 2009.80 In the field of MOFs, the first reports on structure-directed synthesis, attempts at obtaining mesoporosity through the use of surfactants are starting to appear.81–83 True templation and corresponding structure stabilization has been observed for several high-profile MOFs such as the phosphotungstic acid enhanced synthesis of MIL-100(Cr) and Cu BTC.84,85
It is needless to say that templation chemistry in MOFs is much less developed than it is in zeolite synthesis, and it is considerably more complex due to the much larger diversity and complexity of tectonic units. This makes the development of tailored templates in MOF synthesis look like a daunting challenge, but the field is highly researched, and innovative breakthroughs in shaping MOFs, such as development of ultrathin films, aerogels, and even energy-harnessing “motors”, appear by year-to-year basis, so we are positive.75,86,87 We finish by stating that in order to apply metal–organic frameworks and see them ‘make it’ to industry, we must keep investigating their synthetic pathways. The unpredictability and pervasiveness described in the review above are perhaps beautiful for the coordination chemist, they are a setback for the crystallographer believing in reticular design, and a drawback for the industrialist.
Notes and references
- M. Simard, D. Su and J. D. Wuest, J. Am. Chem. Soc., 1991, 113, 4696–4698 CrossRef CAS.
- J. V. Smith, Chem. Rev., 1988, 88, 149–182 CrossRef CAS.
- X. Wang, M. Simard and J. D. Wuest, J. Am. Chem. Soc., 1994, 116, 12119–12120 CrossRef CAS.
- A. I. Cooper, Angew. Chem., Int. Ed., 2012, 51, 7892–7894 CrossRef CAS PubMed.
- B. F. Hoskins and R. Robson, J. Am. Chem. Soc., 1989, 111, 5962–5964 CrossRef CAS.
- N. W. Ockwig, O. Delgado-Friedrichs, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2005, 38, 176–182 CrossRef CAS PubMed.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef CAS PubMed.
- A. R. Millward and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 17998–17999 CrossRef CAS PubMed.
- O. K. Farha, I. Eryazici, N. C. Jeong, B. G. Hauser, C. E. Wilmer, A. A. Sarjeant, R. Q. Snurr, S. T. Nguyen, A. Ö. Yazaydın and J. T. Hupp, J. Am. Chem. Soc., 2013, 134, 15016–15021 CrossRef PubMed.
- D. Liu, H. Wu, S. Wang, Z. Xie, J. Li and W. Lin, Chem. Sci., 2012, 3, 3032–3037 RSC.
- B. Moulton, J. Lu, R. Hajndl, S. Hariharan and M. J. Zaworotko, Angew. Chem., Int. Ed., 2002, 41, 2821–2824 CrossRef CAS.
- T. Loiseau, L. Lecroq, C. Volkringer, J. Marrot, G. Férey, M. Haouas, F. Taulelle, S. Bourrelly, P. L. Llewellyn and M. Latroche, J. Am. Chem. Soc., 2013, 128, 10223–10230 CrossRef PubMed.
- M. C. Escher, Metamorphosis, http://www.mcescher.com/ Search PubMed.
- S. R. Batten, J. Solid State Chem., 2005, 178, 2475–2479 CrossRef CAS PubMed.
- M. O'Keeffe and O. M. Yaghi, Chem. Rev., 2012, 112, 675–702 CrossRef CAS PubMed.
- K. S. Park, Z. Ni, A. P. Côté, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186–10191 CrossRef CAS PubMed.
- J. Cravillon, C. A. Schröder, R. Nayuk, J. Gummel, K. Huber and M. Wiebcke, Angew. Chem., Int. Ed., 2011, 50, 8067–8071 CrossRef CAS PubMed.
- J. Juan-Alcañiz, R. A. M. Gielisse, A. B. Lago, E. V. Ramos-Fernandez, P. Serra-Crespo, T. Devic, N. Guillou, C. Serre, F. Kapteijn and J. Gascon, Catal. Sci. Technol., 2013, 3, 2311 Search PubMed.
- M. G. Goesten, J. Juan-Alcañiz, E. V. Ramos-Fernandez, K. B. Sai Sankar Gupta, E. Stavitski, H. Van Bekkum, J. Gascon and F. Kapteijn, J. Catal., 2011, 281, 177–187 CrossRef CAS PubMed.
- M. G. Goesten, K. B. Sai Sankar Gupta, E. V. Ramos-Fernandez, H. Khajavi, J. Gascon and F. Kapteijn, CrystEngComm, 2012, 14, 4109 RSC.
- K. M. L. Taylor-Pashow, J. Della Rocca, Z. Xie, S. Tran and W. Lin, J. Am. Chem. Soc., 2009, 131, 14261–14263 CrossRef CAS PubMed.
- Z. Wang and S. M. Cohen, Chem. Soc. Rev., 2009, 38, 1315–1329 RSC.
- J. Ribas Gispert, Coordination Chemistry, Wiley-VCH Verlag, 1st edn, Weinheim, 2008 Search PubMed.
- M. Kondo, T. Yoshitomi, K. Seki, H. Matsuzaka and S. Kitagawa, Angew. Chem., Int. Ed. Engl., 1997, 36, 1725–1727 CrossRef CAS.
- P. Ayyappan, O. R. Evans and W. Lin, Inorg. Chem., 2002, 41, 3328–3330 CrossRef CAS PubMed.
- R. Vaidhyanathan, S. S. Iremonger, K. W. Dawson and G. K. H. Shimizu, Chem. Commun., 2009, 5230–5232 RSC.
- H. Li, M. Eddaoudi, T. L. Groy and O. M. Yaghi, J. Am. Chem. Soc., 1998, 120, 8571–8572 CrossRef CAS.
- C. Serre, N. Stock, T. Bein and G. Férey, Inorg. Chem., 2004, 43, 3159–3163 CrossRef CAS PubMed.
- B. Q. Ma, D. S. Zhang, S. Gao, T. Z. Jin, C. H. Yan and G. X. Xu, Angew. Chem., Int. Ed., 2000, 39, 3644–3646 CrossRef CAS.
- A. Violante and P. M. Huang, Clays Clay Miner., 1985, 33, 181–192 CAS.
- A. Violante and P. M. Huang, Clays Clay Miner., 1993, 41, 590–597 Search PubMed.
- M. Haouas, C. Volkringer, T. Loiseau, G. Férey and F. Taulelle, Chem. Mater., 2012, 24, 2462–2471 CrossRef CAS.
- G. Férey, C. Serre, C. Mellot-Draznieks, F. Millange, S. Surblé, J. Dutour and I. Margiolaki, Angew. Chem., Int. Ed., 2004, 43, 6296–6301 CrossRef PubMed.
- A. Martinez Joaristi, J. Juan-Alcañiz, P. Serra-Crespo, F. Kapteijn and J. Gascon, Cryst. Growth Des., 2012, 12, 3489–3498 CAS.
- R. Mondal, T. Basu, D. Sadhukhan, T. Chattopadhyay and M. K. Bhunia, Cryst. Growth Des., 2009, 9, 1095–1105 CAS.
- H. M. Park, I. H. Hwang, J. M. Bae, Y. D. Jo, C. Kim, H. Y. Kim, Y. Kim and S. J. Kim, Bull. Korean Chem. Soc., 2012, 33, 1517–1522 CrossRef CAS.
- B. Notash, N. Safari and H. R. Khavasi, CrystEngComm, 2012, 14, 6788–6796 RSC.
- W. Wei, H. Yu, F. Jiang, B. Liu, J. Ma and M. Hong, CrystEngComm, 2012, 14, 1693–1700 RSC.
- S. H. Zhang, L. F. Ma, H. H. Zou, Y. G. Wang, H. Liang and M. H. Zeng, Dalton Trans., 2011, 40, 11402–11409 RSC.
- H. M. Hao, F. P. Huang, H. D. Bian, Q. Yu, X. L. Sun and H. Liang, Polyhedron, 2011, 30, 2099–2105 CrossRef CAS PubMed.
- T. Loiseau and G. Férey, J. Fluorine Chem., 2007, 128, 413–422 CrossRef CAS PubMed.
- K. S. Min and M. P. Suh, J. Am. Chem. Soc., 2000, 122, 6834–6840 CrossRef CAS.
- Y.-S. Li, H. Bux, A. Feldhoff, G.-L. Li, W.-S. Yang and J. Caro, Adv. Mater., 2010, 22, 3322–3326 CrossRef CAS PubMed.
- M. Goesten, E. Stavitski, E. A. Pidko, C. Gücüyener, B. Boshuizen, S. N. Ehrlich, E. J. M. Hensen, F. Kapteijn and J. Gascon, Chem.–Eur. J., 2013, 19, 7809–7816 CrossRef CAS PubMed.
- L. Carlucci, G. Ciani and D. M. Proserpio, J. Chem. Soc., Dalton Trans., 1999, 1799–1804 RSC.
- M. Kondo, T. Yoshitomi, K. Seki, H. Matsuzaka and S. Kitagawa, Angew. Chem., Int. Ed. Engl., 1997, 36, 1725–1727 CrossRef CAS.
- H.-P. Wu, C. Janiak, L. Uehlin, P. Klüfers and P. Mayer, Chem. Commun., 1998, 2637–2638 RSC.
- Y. Zhang and P. S. Cremer, Curr. Opin. Chem. Biol., 2006, 10, 658–663 CrossRef CAS PubMed.
- R. Custelcean and B. A. Moyer, Eur. J. Inorg. Chem., 2007, 1321–1340 CrossRef CAS.
- V. Trukil, L. Fábián, D. G. Reid, M. J. Duer, G. J. Jackson, M. Eckert-Maksić and T. Friić, Chem. Commun., 2010, 46, 9191–9193 RSC.
- B. C. Tzeng, H. T. Yeh, T. Y. Chang and G. H. Lee, Cryst. Growth Des., 2009, 9, 2552–2555 CAS.
- C. P. Li and M. Du, Chem. Commun., 2011, 47, 5958–5972 RSC.
- I. Senkovska and S. Kaskel, Eur. J. Inorg. Chem., 2006, 4564–4569 CrossRef CAS.
- M. Dincǎ and J. R. Long, J. Am. Chem. Soc., 2005, 127, 9376–9377 CrossRef PubMed.
- C. A. Williams, A. J. Blake, C. Wilson, P. Hubberstey and M. Schroder, Cryst. Growth Des., 2008, 8, 911–922 CAS.
- S.-I. Noro, S. Horike, D. Tanaka, S. Kitagawa, T. Akutagawa and T. Nakamura, Inorg. Chem., 2006, 45, 9290–9300 CrossRef CAS PubMed.
- N. N. Adarsh, D. K. Kumar and P. Dastidar, CrystEngComm, 2008, 10, 1565–1573 RSC.
- S. Couck, J. F. M. Denayer, G. V. Baron, T. Remy, J. Gascon and F. Kapteijn, J. Am. Chem. Soc., 2009, 131, 6326–6327 CrossRef CAS PubMed.
- S. Couck, T. Remy, G. V. Baron, J. Gascon, F. Kapteijn and J. F. M. Denayer, Phys. Chem. Chem. Phys., 2010, 12, 9413–9418 RSC.
- A. Boutin, S. Couck, F. X. Coudert, P. Serra-Crespo, J. Gascon, F. Kapteijn, A. H. Fuchs and J. F. M. Denayer, Microporous Mesoporous Mater., 2011, 140, 108–113 CrossRef CAS PubMed.
- P. Serra-Crespo, M. A. Van Der Veen, E. Gobechiya, K. Houthoofd, Y. Filinchuk, C. E. A. Kirschhock, J. A. Martens, B. F. Sels, D. E. De Vos, F. Kapteijn and J. Gascon, J. Am. Chem. Soc., 2012, 134, 8314–8317 CrossRef CAS PubMed.
- P. Serra-Crespo, E. V. Ramos-Fernandez, J. Gascon and F. Kapteijn, Chem. Mater., 2011, 23, 2565–2572 CrossRef CAS.
- M. Schubert, U. Mueller and C. Kiener, Patent WO 2008061958 A1, 2008 Search PubMed.
- E. Stavitski, M. Goesten, J. Juan-Alcañiz, A. Martinez-Joaristi, P. Serra-Crespo, A. V. Petukhov, J. Gascon and F. Kapteijn, Angew. Chem., Int. Ed., 2011, 50, 9624–9628 CrossRef CAS PubMed.
- A. F. Gualtieri, Phys. Chem. Miner., 2001, 28, 719–728 CrossRef CAS.
- F. Millange, R. El Osta, M. E. Medina and R. I. Walton, CrystEngComm, 2011, 13, 103–108 RSC.
- F. Millange, R. El Osta, M. E. Medina and R. I. Walton, CrystEngComm, 2011, 13, 7318 RSC.
- A. C. Sudik, A. P. Côté and O. M. Yaghi, Inorg. Chem., 2005, 44, 2998–3000 CrossRef CAS PubMed.
- M. G. Goesten, J. Juan-Alcañiz, E. Stavitski, A. Martinez-Joaristi, A. V. Petukhov, F. Kapteijn and J. Gascon, Catal. Today, 2013, 205, 120–127 CrossRef CAS PubMed.
- M. G. Goesten, P. Magusin, B. Mezari, E. A. Pidko, E. J. M. Hensen, F. Kapteijn and J. Gascon, submitted.
- J. Juan-Alcañiz, M. G. Goesten, E. V. Ramos-Fernandez, J. Gascon and F. Kapteijn, New J. Chem., 2012, 36, 977–987 RSC.
- M. R. Lohe, M. Rose and S. Kaskel, Chem. Commun., 2009, 6056–6058 RSC.
- S. K. Nune, P. K. Thallapally and B. P. McGrail, J. Mater. Chem., 2010, 20, 7623–7625 RSC.
- L. Li, S. Xiang, S. Cao, J. Zhang, G. Ouyang, L. Chen and C. Y. Su, Nat. Commun., 2013, 4 Search PubMed.
- G. Férey, Chem. Soc. Rev., 2008, 37, 191–214 RSC.
- M. Jansen and J. C. Schön, Angew. Chem., Int. Ed., 2006, 45, 3406–3412 CrossRef CAS PubMed.
- R. F. Lobo, S. I. Zones and M. E. Davis, J. Inclusion Phenom. Mol. Recognit. Chem., 1995, 21, 47–78 CAS.
- S. L. Burkett and M. E. Davis, J. Phys. Chem., 1994, 98, 4647–4653 CrossRef CAS.
- M. Choi, K. Na, J. Kim, Y. Sakamoto, O. Terasaki and R. Ryoo, Nature, 2009, 461, 246–249 CrossRef CAS PubMed.
- X. D. Do, V. T. Hoang and S. Kaliaguine, Microporous Mesoporous Mater., 2011, 141, 135–139 CrossRef CAS PubMed.
- L. G. Qiu, T. Xu, Z. Q. Li, W. Wang, Y. Wu, X. Jiang, X. Y. Tian and L. D. Zhang, Angew. Chem., Int. Ed., 2008, 47, 9487–9491 CrossRef CAS PubMed.
- L. Peng, J. Zhang, J. Li, B. Han, Z. Xue and G. Yang, Chem. Commun., 2012, 48, 8688–8690 RSC.
- S. R. Bajpe, C. E. A. Kirschhock, A. Aerts, E. Breynaert, G. Absillis, T. N. Parac-Vogt, L. Giebeler and J. A. Martens, Chem.–Eur. J., 2010, 16, 3926–3932 CrossRef CAS PubMed.
- J. Juan-Alcañiz, M. Goesten, A. Martinez-Joaristi, E. Stavitski, A. V. Petukhov, J. Gascon and F. Kapteijn, Chem. Commun., 2011, 47, 8578–8580 RSC.
- S. Hermes, F. Schröder, R. Chelmowski, C. Wöll and R. A. Fischer, J. Am. Chem. Soc., 2005, 127, 13744–13745 CrossRef CAS PubMed.
- Y. Ikezoe, G. Washino, T. Uemura, S. Kitagawa and H. Matsui, Nat. Mater., 2012, 11, 1081–1085 CAS.
Footnotes |
| † Covalent–organic frameworks (COFs) are sometimes referred to as organic polymers, but may factually represent a case of ‘metalloid–organic frameworks’, since very often these polymers contain boron and its strong directional binding. |
| ‡ MIL-101(Al) only forms when using the aminated version of the original terephthalic linker, 2-aminoterephthalic acid. The resulting framework is named as NH2-MIL-101(Al). |
| § MIL-96 and MIL-100 have also been synthesized using Fe and Cr. |
| ¶ Exceptions may always exist, and a few syntheses make use of metallic precursor, such as the synthesis of MIL-100(Cr) as first reported by Ferey34 and MOFs synthesized via electrochemical synthesis.35 Anions are however present in both cases, as mineralizing agent and electrolyte species, respectively. |
| || ZIF-7 is based on zinc and benzimidazole.17 |
| ** Both anions and cations are taken into account, but anions appear to have a much bigger effect. The ‘macromolecules’ initially investigated were proteins, and the anionic effect on protein folding and ‘salting in/out’ behaviour delivered the Hofmeister series. |
| †† Like it is technically possible to avoid the presence of anions, MOF synthesis without solvent, mechanosynthesis, is possible, but rarely applied.51 |
| ‡‡ 1,3,5-tricarboxylic acid which makes up MIL-100(Al) is a highly deactivated (electron poor) organic strut. 2-aminoterephthalato is much more electron-rich and was post-synthetically decorated with chloromethylene groups in our laboratories. |
| §§ Their industrial significance is reflected in a BASF patent which effectively claims NH2–MIL-53(Al) and NH2–MIL-101(Al).64 |
| This journal is © The Royal Society of Chemistry 2013 |