Recent advances in high mobility donor–acceptor semiconducting polymers
Laure
Biniek
*,
Bob C.
Schroeder
,
Christian B.
Nielsen
and
Iain
McCulloch
Department of Chemistry, Imperial College London, London SW7 2AZ, UK. E-mail: l.biniek@imperial.ac.uk
First published on 31st May 2012
Abstract
A combination of improved understanding of molecular design criteria and polymer purification techniques, as well as optimised fabrication techniques and device surface treatments, have driven recent advances in the performance of semiconducting polymers for transistor applications. This development has allowed polymer-based devices to reach parity with the best values obtained for small molecule evaporated devices. Herein, we present the most recent work on solution processable high mobility donor–acceptor type polymers. We discuss the approaches that have been taken to improve both hole (and electron) mobility further. We will not only focus on chemical design criteria, but also describe certain processing approaches which have led to impressive hole mobilities of up to 5.5 cm2 V−1 s−1.
 Laure Biniek | Laure Biniek is a research associate in the Chemistry Department at Imperial College London. She obtained her PhD in Chemistry from the University of Strasbourg in 2010. Her current research focuses on the design and synthesis of low band-gap light absorbing polymers with improved solar cell stability. |
 Bob C. Schroeder | Bob C. Schroeder is currently a doctoral student in the Chemistry Department at Imperial College London. He received both his B.Sc. (2008) and M.Sc. (2010) in Chemistry from the Free University of Brussels (ULB). His current research focuses on the design and synthesis of novel organic semiconducting materials for applications in organic field effect transistors and photovoltaic devices. Bob is the recipient of a Fellowship from the National Research Fund of Luxembourg (FNR). |
 Christian B. Nielsen | Christian B. Nielsen is a research associate in the Department of Chemistry at Imperial College London. He received his PhD in 2004 from the University of Copenhagen. His current research is focused on the design and synthesis of novel π-conjugated materials aimed at the continued development of organic electronic applications such as field-effect transistors and solar cells. |
 Iain McCulloch | Iain McCulloch is Professor of Polymer Materials in the Department of Chemistry at Imperial College London, with research interests in organic semiconducting materials synthesis, characterization, and devices. He was previously the Organic Electronics Research Manager at Merck Chemicals. His achievements have been recognized with several awards including the Royal Society of Chemistry Creativity in Industry Prize. |
Introduction
Early research in the field revealed that in the case of polythiophenes, when a π stacked, lamellar microstructure was obtained through optimal processing and regioregularity within the molecular structure, charge carrier mobilities of the order of 0.1 cm2 V−1 s−1 could be achieved.1 In this case, the plane of the polymer backbone was preferentially oriented orthogonal to the transport direction (i.e. the lamellae oriented in the plane). This characteristic morphology was shown to be further enhanced through liquid crystalline self-assembly promoting larger and more ordered domains2,3 with the result that mobilities could be increased to values approaching unity.4 For this kind of structure, the transport occurs within the two dimensional aromatic lamella. Subsequently, the improvement of the device air stability has been addressed by increasing the ionisation potential of the semiconducting polymer through the incorporation into the conjugated backbone of an electron deficient unit. For example, a bithiazole based copolymer has illustrated the potential of donor–acceptor alternating polymers for high charge carrier mobility materials by providing an air stable device with a hole mobility of 0.14 cm2 V−1 s−1.5 It has recently been demonstrated that both highly polar diketopyrrolopyrrole (DPP) and benzothiadiazole (BT) containing donor–acceptor polymers are able to afford even higher carrier mobilities,6–8 despite having apparently less ordered microstructures, and functional groups that have previously been expected to potentially act as charge traps. These results suggest that one dimensional charge transport along the polymer chains may dominate in these low ordered systems. This short highlight article attempts to collect and describe the literature reports since beginning of 2011.Discussion
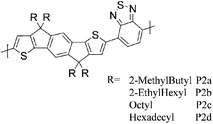
A series of results related to donor–acceptor type copolymers and their application in field-effect transistor (FET) devices have recently been published by Müllen et al. The alternating copolymer of cyclopentadithiophene (CPDT) and 2,1,3-benzothiadiazole (BT) (P1, Table 1) initially achieved a hole mobility of 0.17 cm2 V−1 s−1 in FET devices,9 but through optimisation of the polymer's molecular weight and the device fabrication conditions, a higher hole mobility of 1.4 cm2 V−1 s−1 was reported in 2009.10 Additional optimisation work from the same group afforded a thin film FET hole mobility of 3.3 cm2 V−1 s−1, and subsequently an impressive value of 5.5 cm2 V−1 s−1 was reported for a single fibre of P1.12,13P2 with the large fused indacenodithiophene (IDT) motif was designed to have a planar, stiff backbone with low energetic disorder, thus promoting good transport properties.14 A series of different alkyl chain analogues of P2 were evaluated, and the best transistor performance was found with P2d (where R = hexadecyl) giving a hole mobility of 1.2 cm2 V−1 s−1. The silicon analogue of IDT (Si-IDT) was reported in FET applications,25 with a similar approach adopted in the design of P3 and P4.15 The longer C–Si bond is expected to move the out-of-plane alkyl groups further away from the backbone and therefore promote stronger intermolecular interactions and more efficient π-stacking.11P3 afforded a hole mobility of 0.28 cm2 V−1 s−1 after thermal annealing at 180 °C. With the introduction of fluorine atoms incorporated on the BT units in P4, favourable intramolecular sulphur–fluorine interactions were expected to help planarize the polymer backbone, but although a slightly higher degree of crystallinity was observed for P4 compared to P3, a slightly inferior hole mobility of 0.19 cm2 V−1 s−1 was measured under identical processing conditions. Further elongation of the fused donor unit as in the case of P5 did not afford improved properties; the material was found to be amorphous and a lower hole mobility of 0.11 cm2 V−1 s−1 was reported.16| Polymer structure | Ref. | Max μh (cm2 V−1 s−1) as spun/after annealing (T) | M n/Mw (kDa) | HOMO (eV) | Crystallinity information |
|---|---|---|---|---|---|
| a The HOMO level has been estimated by cyclic voltammetry. b The HOMO level has been estimated by photoelectron spectroscopy in air (PESA). | |||||

|
12 | —/3.3 (200 °C) | 35/— | Highly crystalline, lamellar like structure. | |
| 13 | —/5.5 (200 °C) | 50/— | Single fibre obtained by growing method. | ||
|
14 P2a | 0.003 | 9/13 | −5.3b | |
| 14 P2b | 0.57 | 40/87 | −5.3b | ||
| 14 P2c | 0.15 | 33/283 | −5.3b | ||
| 14 P2d | 1.2 | 38/108 | −5.4b | ||

|
15 P3 | —/0.28 (180 °C) | 22/56 | −5.2a | Slightly crystalline. |
| −5.0b | |||||
| 15 P4 | —/0.19 (180 °C) | 26/71 | −5.2a | ||
| −5.1b | |||||
|
16 | —/0.11 (120 °C) | 38/61 | −5.31a | Amorphous. |

|
17 | —/0.2 (200 °C) | 35/57 | −5.18a | Fibres. |
|
18 | —/0.35 (200 °C) for HMDS modified surface | 52.6/127 | −5.54a | Semi-crystalline with a predominant edge-on orientation. |
| —/0.55 (200 °C) for FDTS modified surface | |||||

|
19 | —/0.13 (240 °C) | 17/47.6 | −4.8a | Lack of order. |
|
8 P9 | 0.6 (90 °C) | 104/310 | −5.2b | Mixture of face-on and edge-on orientation after annealing. |
| 8 P10 | 0.2 (90 °C) | 55/175 | −5.25b | ||
| 20 P10 | 0.89/0.97 (100 °C) | 106/365 in THF at 40 C | −5.2a | Layer by layer lamellar structure with an edge-on orientation. | |
| 25.4/60.6 in TCB at 160 C | |||||
| 8 P11 | 0.38 (90 °C) | 15/39 | −5.4b | Semi-crystalline. | |
| 21 P11 | 0.47/1.23 (200 °C) | 50/1935 | −5.33b | Edge-on orientation, crystallinity improved with annealing. | |

|
22 | 0.68/0.75 (250 °C) | 101/525 | −5.19a | Highly crystalline with an edge-on orientation. |

|
7 | 1.95 | 14/75 | −5.06b | Semi-crystalline. |
|
23 | —/0.20 (150 °C) | 89/206 | −5.14a | Lamellar crystalline packing. |
|
24 | 0.47/0.65 (120 °C) | 64/91 | −5.29a | Slightly crystalline. |
|
25 | 0.2/0.65 (200 °C) | 29/80 | −5.1b | |

|
26 P17 | —/0.1 (200 °C) | 70/210 | −5.2b | Semi-crystalline. |
|
26 P18 | —/1.1 (250 °C) | 100/250 | −5.1b | Semi-crystalline. |
|
27 | 0.67/1.62 (150 °C) from TCB solution | 97/204 | −5.09a | Highly crystalline, (nanofibres observed by TEM). |

|
28 | 0.13/1.54 (200 °C) | 130.5/296 | −5.32a | Amorphous before annealing, slightly crystalline after with a “layer by lamellar packing manner”. |
|
29 | 0.11 | 54/86 | −5.34a | Slightly crystalline. |
| −5.50b | |||||

|
30 | 0.53 | 42/60 | −5.2a | Highly ordered “layer by layer lamellar packing motif with an edge-on orientation”. |
|
31 | 0.06 | 136/578 | −5.1a | Amorphous. |
|
32 | —/0.46 (200 °C) | 30/69 | −5.16b | Semi-crystalline with a bimodal distribution of face-on and edge-on crystallites. |
|
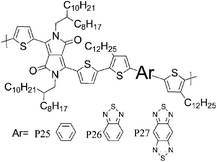
33 P25 | —/0.27 (160 °C) | 21/44 | −4.7a | Lack of crystallinity, face-on orientation. |
| 33 P26 | —/0.23 (160 °C) | 9.4/15 | −4.75a | Edge on orientation, more crystalline than P25. | |
| 33 P27 | —/1.17 (240 °C) | 8.7/13 | −4.55a | Edge-on orientation, poorly crystalline. | |
|
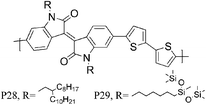
34 P28 | 0.2/0.79 (170 °C) | 135/742 | −5.38b | Lamellar packing both in plane and out of plane orientation. |
| 34 P29 | 0.45/2.48 (170 °C) | 138/454 | −5.20b | ||
|
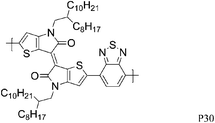
35 | 0.16/0.15 (250 °C) | 40/90 | −4.8b | Slightly crystalline. |

|
36 | —/0.27 (150 °C) | 12/38 | −5.20a | Highly crystalline. |
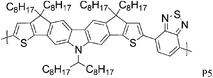
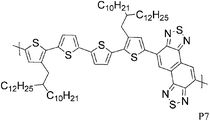
Quaterthiophenes with solubilising alkyl chains have also been used recently in donor–acceptor type copolymers as illustrated by P6 and P7. With BT as the acceptor unit, Ong and co-workers achieved a FET hole mobility of 0.2 cm2 V−1 s−1,17 while Osaka and colleagues used the naphthobisthiadiazole acceptor and a slightly different quaterthiophene unit to fabricate a FET device with a maximum hole mobility of 0.55 cm2 V−1 s−1.18 Interestingly, Osaka and co-workers found that the corresponding BT copolymer performed much worse (0.074 cm2 V−1 s−1), which demonstrates that the substitution pattern on the quaterthiophene unit is of great importance for the molecular packing and consequently for the device properties. Another variation of BT, namely the benzobisthiadiazole, was used by Yuen and Wudl to afford polymer P8, which showed a hole mobility of 0.13 cm2 V−1 s−1 after thermal annealing at 240 °C.19P8 was moreover found to have ambipolar properties as reflected in a maximum electron mobility of 0.10 cm2 V−1 s−1, which closely matches the hole mobility. Yuen and Wudl also compared the CPDT unit to other donor moieties such as fluorene, dithienopyrrole and dithienosilole, all of which afforded polymers with much lower charge carrier mobilities.
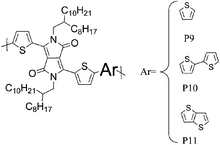
Besides the various benzothiadiazole based donor–acceptor polymers, several polymers with repeat units based on classical dye designs, have shown exceptionally high mobilities recently. One class of these dye based polymers showing high mobilities is composed of two families, the well characterized diketopyrrolopyrrole (DPP) polymers and the more recently introduced indigo based polymers. Long branched alkyl side chains, which improve polymer solubility, were for a long time considered to be counterproductive to high mobility and to efficient π–π stacking, for both steric and insulating reasons, but this hypothesis has been challenged with the introduction of DPP into the polymer backbone. In order to make DPP a viable unit in donor–acceptor polymers, long, branched and bulky 2-octyldodecyl side chains were attached to the otherwise insoluble DPP core, and high charge carrier mobilities were obtained despite the alkyl chain branching. Zhang et al. investigated a series of DPP polymers with various alkyl chain densities along the polymer backbone and they concluded that the organization of the polymer backbone is not necessarily interrupted by the presence of bulky and disordered side chains.8 Despite the sterically hindered side chains, hole mobilities of 0.6 cm2 V−1 s−1 were obtained with P9 and the authors speculate that mostly local coplanar intermolecular associations and short contacts are responsible for these high mobility values while the overall orientation and crystallinity of the polymer is less crucial for charge transport. Li and co-workers synthesized a DPP–quaterthiophene copolymer (P10), which forms highly crystalline thin films despite the long bulky alkyl chains and hole mobilities of 0.97 cm2 V−1 s−1 were achieved after mild annealing at 100 °C.20 The introduction of thieno[3,2-b]thiophene further stiffens the polymer backbone and lowers the HOMO energy level. Chen et al. demonstrated balanced hole and electron mobilities exceeding 1 cm2 V−1 s−1 for P11 by thoroughly optimizing the device architecture and material processing conditions.21 Jung et al. stiffened the polymer backbone further by introducing dithieno[3,2-b:2′,3′-d]thiophene, leading to a highly crystalline polymer P12.22 A high hole mobility of 0.68 cm2 V−1 s−1 could be measured for P12 and it is noteworthy that this high mobility could be obtained with as-spun films. Bronstein and co-workers designed and prepared thieno[3,2-b]thiophene flanking groups for the DPP unit rather than the ubiquitous thiophene groups, thereby further extending the distribution of the HOMO energy level along the polymer backbone, and also further stiffening the backbone.7 The resulting polymer P13 achieved an impressive hole mobility of 1.95 cm2 V−1 s−1 in top-gate/bottom-contact transistors without the need for high temperature annealing. It is interesting to note there was not a very high level of crystallinity observed.
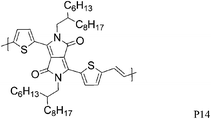
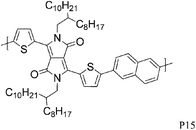
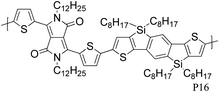
Varying the neighbouring group of the 1,4-diketo-3,6-bis(thien-2-yl)pyrrolo[3,4-c]pyrrole (T-DPP-T) core was demonstrated to have an impact on the polymer solubility, planarity, self-assembly properties and electronic structure. Wu et al. investigated the effect of an arylvinylene linkage (P14) on DPP based polymers.23 Although the crystallinity of the T-DPP-T based polymer was improved due to the rigidity of the arylenevinylene linkage, this did not change the hole mobility (measured at 0.2 cm2 V−1 s−1). In order to increase the overall planarity of the conjugated backbone, two fused aromatic systems, naphthalene (P15)24 and Si-IDT (P16)25 were chosen as co-repeat units to be copolymerised with T-DPP-T. Due to strong intermolecular interactions and favourable π–π stacking, both polymers achieved a high hole mobility of 0.65 cm2 V−1 s−1 after annealing at 120 and 200 °C respectively. It is interesting to note that by changing the device architecture to a dual gate structure, the mobility of P15 could be further increased to 0.98 cm2 V−1 s−1.
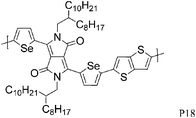
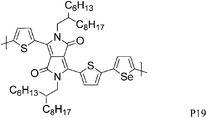
The introduction of selenophene into donor–acceptor type polymer backbones has recently attracted some interest including incorporation into DPP polymers. The substitution of thiophene with selenophene results in lower band-gap polymers, mainly due to the stabilisation of the polymer LUMO with little to no effect on the HOMO energy level. Shahid et al. reported the synthesis of selenophene analogues of polymer P9 and P11.26 Initial charge carrier mobilities of P18 were rather low, but thermal annealing at 250 °C and a change from bottom-contact to top-contact device configuration resulted in high ambipolar charge carrier mobilities (values of 1.1 and 0.15 cm2 V−1 s−1 for hole and electron mobility, respectively), reported to be due to improved thin film organisation and reduced contact resistance. The lower hole mobility observed for P17 (0.1 cm2 V−1 s−1) may arise from poorer polymer backbone alignment normal to the substrate, as evidenced by X-ray diffraction studies. In addition to thermal treatment, Lin et al. tuned the thin film morphology of P19 by using high boiling solvents.27 By using high boiling 1,2,4-trichlorobenzene as solvent, the initial hole mobility of 0.12 cm2 V−1 s−1, when spun from chloroform, could be increased to 1.62 cm2 V−1 s−1. The authors explain this dramatic increase in mobility by the fact that the high boiling solvent evaporates slower from the polymer film, thus giving the polymer more time to self-assemble and form a dense network of organized polymer chains.
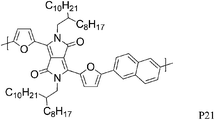
The substitution of thiophene by a furan ring in the polymer backbone led to a less crystalline polymer P20, even after thermal annealing at high temperature.28 Nevertheless, P20 showed a higher hole mobility (1.54 cm2V−1 s−1 after annealing at 200 °C) than the more crystalline thiophene polymer P10. With the introduction of naphthalene into the polymer backbone, reported by Sonar et al., a hole mobility of 0.11 cm2 V−1 s−1 was achieved in a bottom gate/top contact FET for P21,29 lower than for the previously described thiophene analogue P15.24
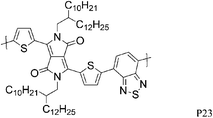
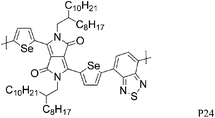
Most high mobility DPP polymers discussed so far have been obtained by co-polymerisation with thiophene-based donor units (or O and Se analogues). Having a thiophene based co-monomer however is no necessity for high charge carrier mobility in FETs and a couple of high-performing BT-containing DPP polymers are described in the literature. P22 and P23 differ only by the length of the solubilising alkyl chain and yet there are significant differences between the physical properties of the two polymers. Whereas P22, with 2-octyldodecyl side chains, adopts an ordered lamellar structure in the solid state and a hole mobility of 0.53 cm2 V−1 s−1 was obtained,30P23 with the slightly longer 2-decyltetradecyl chains is rather amorphous, which translates into a lower hole mobility of 0.1 cm2 V−1 s−1.31 The selenophene analogue P24 has been reported very recently and exhibited high hole and electron mobilities of 0.46 and 0.84 cm2 V−1 s−1 after thermal annealing at 200 °C of the spin coated polymer film.32 The authors of the study suggest that inclusion of the more polarisable selenium atoms into the polymer backbone is responsible for the improved charge carrier mobilities of P24 compared to P22, rather than structural differences between the two polymers. Wudl and co-workers reported the co-polymerisation of DPP with benzobisthiadiazole (BBT) to afford P27.33 Due to the incorporation of the strong BBT acceptor, the resulting polymer shows ambipolar FET properties and impressive charge carrier mobilities reaching unity could be achieved after annealing at 240 °C for both holes and electrons (1.17 and 1.32 cm2 V−1 s−1 respectively).
Over the last year, isoindigo has been introduced as a building block for organic semi-conducting materials. Similar to the well-known DPP, isoindigo is a pigment with an intramolecular dipole. Bao and co-workers were amongst the first to report a high mobility isoindigo based polymer P28.34 A hole mobility of 0.79 cm2 V−1 s−1, after annealing at 170 °C was measured for P28, but the mobility could be dramatically increased by substituting the branched alkyl chains of P28 with siloxane terminated alkyl chains. Due to the presence of the siloxane groups the π–π stacking distance of P29 is reduced to 3.58 Å, compared to 3.76 Å for P28, and a maximum hole mobility of 2.48 cm2 V−1 s−1 was achieved. Ashraf et al. further fine tuned the isoindigo unit by substituting the flanking benzene rings with thiophenes, thus minimizing backbone twisting when polymerized with BT.35P30 exhibits excellent ambipolar characteristics with hole and electron mobilities exceeding 0.1 cm2 V−1 s−1. Osaka et al. investigated another pigment, the quinacridone based polymer for FET applications and hole mobilities of 0.27 cm2 V−1 s−1 were measured for P31.36 Interestingly, the mobility seemed to be independent of polymer molecular weight and of orientational order, which the authors ascribe to limited electron delocalization along the polymer backbone.
Conclusion
It has become increasingly clear that the development of new semiconducting polymers is expanding from “conventional” nanocrystalline polymers with lamella morphologies, exemplified by polymers such as P3HT and pBTTT, to now also include donor–acceptor type polymers, with dipolar functionalities along the backbone. These high-performing polymers often incorporate strongly electron deficient units such as benzothiadiazole, or pyrrolidone with a high alkyl chain density. The alkyl chains are often oriented out of the plane of the polymer backbone, being grafted onto sp3-hybridised bridging atoms. It is remarkable that long branched alkyl chains do not seem to suppress efficient charge transport as seen especially for the DPP-containing polymers such as P13, P19 and P20. The most recent developments show that these alternating co-polymers can be excellent charge carriers and mobilities comparable to evaporated small molecules have already been achieved. Nevertheless, many are lacking long range order and crystallinity, which over many years have been the well-established criteria for high charge carrier mobilities. One of the possible explanations of this high mobility is that the intramolecular dipoles play a role in generating short contacts between chains. In the case of the polymers including extended π-planes motif, local coplanar intermolecular associations can also take place due to the local planarization and lead to these high mobility values. As outlined in this short highlight article, the field of high mobility polymers is progressing quickly with the continual development of new backbone designs and more advanced processing techniques. We believe that this trend will continue and that higher mobilities and more robust and commercially viable semiconducting polymers will be conceived in the near future.References
- (a) H. Sirringhaus, P. J. Brown, R. H. Friend, M. M. Nielsen, K. Bechgaard, B. M. W. Langeveld-Voss, A. J. H. Spiering, R. A. J. Janssen, E. W. Meijer, P. Herwig and D. M. de Leeuw, Nature, 1999, 401, 685–688 CrossRef CAS; (b) D. M. DeLongchamp, B. M. Vogel, Y. Jung, M. C. Gurau, C. A. Richter, O. A. Kirillov, J. Obrzut, D. A. Fischer, S. Sambasivan, L. J. Richter and E. K. Lin, Chem. Mater., 2005, 17, 5610–5612 CrossRef CAS.
- I. McCulloch, M. Heeney, C. Bailey, K. Genevicius, I. MacDonald, M. Shkunov, D. Sparrowe, S. Tierney, R. Wagner, W. Zhang, M. L. Chabinyc, R. J. Kline, M. D. McGehee and M. F. Toney, Nat. Mater., 2006, 5, 328–333 CrossRef CAS.
- B. S. Ong, Y. Wu, P. Liu and S. Gardner, J. Am. Chem. Soc., 2004, 126, 3378–3379 CrossRef CAS.
- B. H. Hamadani, D. J. Gundlach, I. McCulloch and M. Heeney, Appl. Phys. Lett., 2007, 91, 243512 CrossRef.
- J. Liu, R. Zhang, I. Osaka, S. Mishra, A. E. Javier, D.-M. Smilgies, T. Kowalewski and R. D. McCullough, Adv. Funct. Mater., 2009, 19, 3427–3434 CrossRef CAS.
- Z. Chen, M. J. Lee, R. S. Ashraf, Y. Gu, S. Albert-Seifried, M. Meedom Nielsen, B. Schroeder, T. D. Anthopoulos, M. Heeney, I. McCulloch and H. Sirringhaus, Adv. Mater., 2012, 24, 647–652 CrossRef CAS.
- H. Bronstein, Z. Chen, R. S. Ashraf, W. Zhang, J. Du, J. R. Durrant, P. Shakya Tuladhar, K. Song, S. E. Watkins, Y. Geerts, M. M. Wienk, R. A. J. Janssen, T. Anthopoulos, H. Sirringhaus, M. Heeney and I. McCulloch, J. Am. Chem. Soc., 2011, 133, 3272–3275 CrossRef CAS.
- X. Zhang, L. J. Richter, D. M. DeLongchamp, R. J. Kline, M. R. Hammond, I. McCulloch, M. Heeney, R. S. Ashraf, J. N. Smith, T. D. Anthopoulos, B. Schroeder, Y. H. Geerts, D. A. Fischer and M. F. Toney, J. Am. Chem. Soc., 2011, 133, 15073–15084 CAS.
- M. Zhang, H. N. Tsao, W. Pisula, C. Yang, A. K. Mishra and K. Müllen, J. Am. Chem. Soc., 2007, 129, 3472 CrossRef CAS.
- H. N. Tsao, D. Cho, J. W. Andreasen, A. Rouhanipour, D. W. Breiby, W. Pisula and K. Müllen, Adv. Mater., 2009, 21, 209 CrossRef CAS.
- (a) M. C. Scharber, M. Koppe, J. Gao, F. Cordella, M. A. Loi, P. Denk, M. Morana, H.-J. Egelhaaf, K. Forberich, G. Dennler, R. Gaudiana, D. Waller, Z. Zhu, X. Shi and C. J. Brabec, Adv. Mater., 2010, 22, 367–370 CrossRef CAS; (b) H.-Y. Chen, J. Hou, A. E. Hayden, H. Yang, K. N. Houk and Y. Yang, Adv. Mater., 2010, 22, 371–375 CrossRef CAS.
- H. N. Tsao, Do. M. Cho, I. Park, M. R. Hansen, A. Mavrinskiy, D. Y. Yoon, R. Graf, W. Pisula, H. W. Spiess and K. Müllen, J. Am. Chem. Soc., 2011, 133(8), 2605–2612 CrossRef CAS.
- S. Wang, M. Kappl, I. Liebewirth, M. Müller, K. Kirchhoff, W. Pisula and K. Müllen, Adv. Mater., 2012, 24, 417–420 CrossRef CAS.
- H. Bronstein, D. S. Leem, R. Hamilton, P. Woebkenberg, S. King, W. Zhang, R. Shahid Ashraf, M. Heeney, T. D. Anthopoulos, J. de Mello and I. McCulloch, Macromolecules, 2011, 44, 6649–6652 CrossRef CAS.
- B. C. Schroeder, Z. Huang, R. S. Ashraf, J. Smith, P. D'Angelo, S. E. Watkins, T. D. Anthopoulos, J. R. Durrant and I. McCulloch, Adv. Funct. Mater., 2012, 22, 1663–1670 CrossRef CAS.
- J.-S. Wu, Y.-J. Cheng, T.-Y. Lin, C.-Y. Chang, Pe.-I. Shih and C.-S. Hsu, Adv. Funct. Mater., 2012, 22, 1711–1722 CrossRef CAS.
- K.-H. Ong, S.-L. Lim, H.-S. Tan, H.-K. Wong, J. Li, Z. Ma, L. C. H. Moh, S.-H. Lim, J. C. de Mello and Z.-K. Chen, Adv. Mater., 2011, 23, 1409–1413 CrossRef CAS.
- I. Osaka, M. Shimawaki, H. Mori, I. Doi, E. Miyazaki, T. Koganezawa and K. Takimiya, J. Am. Chem. Soc., 2012, 134, 3498–3507 CrossRef CAS.
- J. D. Yuen, R. Kumar, D. Zakhidov, J. Seifter, B. Lim, A. J. Heeger and F. Wudl, Adv. Mater., 2011, 23, 3780–3785 CAS.
- Y. Li, P. Sonar, S. P. Singh, M. S. Soh, M. van Meurs and J. Tan, J. Am. Chem. Soc., 2011, 133, 2198–2204 CrossRef CAS.
- Z. Chen, M. J. Lee, R. S. Ashraf, Y. Gu, S. Albert-Seifried, M. Meedom Nielsen, B. Schroeder, T. D. Anthopoulos, M. Heeney, I. McCulloch and H. Sirringhaus, Adv. Mater., 2012, 24, 647–652 CrossRef CAS.
- J. W. Jung, F. Liu, T. P. Russell and W. H. Jo, Energy Environ. Sci., 2012, 5, 6857–6881 CAS.
- P.-T. Wu, F. S. Kim and S. A. Jenekhe, Chem. Mater., 2011, 23, 4618–4624 CrossRef CAS.
- P. Sonar, S. P. Singh, Y. Li, Z.-E. Ooi, T.-J. Ha, I. Wong, M. S. Soh and A. Dodabalapur, Energy Environ. Sci., 2011, 4, 2288–2296 CAS.
- R. S. Ashraf, Z. Chen, D. S. Leem, H. Bronstein, W. Zhang, B. Schroeder, Y. Geerts, J. Smith, S. Watkins, T. D. Anthopoulos, H. Sirringhaus, J. C. de Mello, M. Heeney and I. McCulloch, Chem. Mater., 2011, 23, 768–770 CrossRef CAS.
- M. Shahid, T. McCarthy-Ward, J. Labram, S. Rossbauer, E. B. Domingo, S. E. Watkins, N. Stingelin, T. D. Anthopoulos and M. Heeney, Chem. Sci., 2012, 3, 181–185 RSC.
- H.-W. Lin, W.-Y. Lee and W.-C. Chen, J. Mater. Chem., 2012, 22, 2120–2128 RSC.
- Y. Li, P. Sonar, S. P. Singh, W. Zeng and M. Si. Soh, J. Mater. Chem., 2011, 21, 10829–10835 RSC.
- Pr. Sonar, S. P. Singh, E. L. Williams, Y. Li, M. S. Soh and A. Dodabalapur, J. Mater. Chem., 2012, 22, 4425–4435 RSC.
- (a) P. Sonar, S. P. Singh, Y. Li, M. S. Soh and A. Dodabalapur, Adv. Mater., 2010, 22, 5409–5413 CrossRef CAS; (b) T.-J. Ha, P. Sonar, B. Cobb and A. Dodabalapur, Org. Electron., 2012, 13, 136–141 CrossRef CAS.
- S. Cho, J. Lee, M. Tong, J. H. Seo and C. Yang, Adv. Funct. Mater., 2011, 21, 1910–1916 CrossRef CAS.
- A. J. Kronemeijer, E. Gili, M. Shahid, J. Rivnay, A. Salleo, M. Heeney and H. Sirringhaus, Adv. Mater., 2012, 24, 1558–1565 CrossRef CAS.
- J. D. Yuen, J. Fan, J. Seifter, B. Lim, R. Hufschmid, A. J. Heeger and F. Wudl, J. Am. Chem. Soc., 2011, 133, 20799–20807 CrossRef CAS.
- J. Mei, D. H. Kim, A. L. Ayzner, M. F. Toney and Z. Bao, J. Am. Chem. Soc., 2011, 133, 20130–20133 CrossRef CAS.
- R. S. Ashraf, A. J. Kronemeijer, D. James, H. Sirringhaus and I. McCulloch, Chem. Commun., 2012, 48, 3939–3941 RSC.
- I. Osaka, M. Akita, T. Koganezawa and K. Takimiya, Chem. Mater., 2012, 24, 1235–1243 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |