(1R,4S,5R)-3-Fluoro-1,4,5-trihydroxy-2-cyclohexene-1-carboxylic acid: the fluoro analogue of the enolate intermediate in the reaction catalyzed by type II dehydroquinases
Martyn
Frederickson
a,
Aleksander W.
Roszak
b,
John R.
Coggins
c,
Adrian J.
Lapthorn
b and
Chris
Abell
*a
aUniversity Chemical Laboratory, Lensfield Road, Cambridge, UK CB2 1EW. E-mail: ca26@cam.ac.uk; Fax: +44 (0)1223 336362; Tel: +44 (0)1223 336405
bDepartment of Chemistry, University of Glasgow, Glasgow, UK G12 8QQ
cDivision of Biochemistry and Molecular Biology, Institute of Biomedical and Life Sciences, University of Glasgow, Glasgow, UK G12 8QQ
First published on 6th May 2004
Abstract
The fluoro analogue of the enolate intermediate in the reaction catalyzed by type II dehydroquinases has been prepared from naturally occuring (−)-quinic acid over seven steps and has been shown to be the most potent inhibitor reported to date of the type II enzyme from Mycobacterium tuberculosis.
Introduction
Dehydroquinase [3-dehydroquinate dehydratase; E.C. 4.2.1.10] is common to both the shikimate1,2 and quinate3 pathways. It catalyses the reversible interconversion of 3-dehydroquinate (DHQ) 1 and 3-dehydroshikimate (DHS) 2. The enzyme occurs in two chemically and biochemically distinct forms4 (type I and type II) that catalyse the same overall chemical transformation via different mechanisms. Type I enzymes are heat labile protein dimers5 that catalyse a syn elimination6 of water through Schiff's base intermediates7 involving a conserved lysine. Type II dehydroquinases are more thermally robust dodecameric species8 that effect an anti dehydration9via an enzyme-stabilized enolate anion 3 (Scheme 1).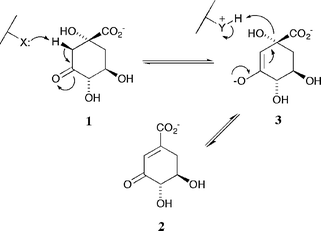 | ||
| Scheme 1 | ||
The mechanistic differences of the two enzyme types led us to design inhibitors that were selective for the type II proteins which are found in a number of clinically important bacterial strains including Mycobacterium tuberculosis and Helicobacter pylori. Based upon the idea that compounds which mimicked the enolate intermediate 3 were likely to show such selectivity we sought derivatives which, when compared to DHQ 1, possessed either increased sp2 character at C-2 and C-3 or had superior hydrogen bonding potential at C-3; accordingly we prepared alkene 4 and oxime 5 (Fig. 1). We were thus able to demonstrate that 4 and 5 were potent and highly selective (up to 1000 fold) competitive reversible inhibitors of type II dehydroquinases.10 X-ray crystallographic studies11 on the complexes formed between alkene 4 and oxime 5 and the type II dehydroquinases from Streptomyces coelicolor and Mycobacterium tuberculosis (PDB accession codes for 4: 1GU1 and 1H0R; for 5: 1H0S) allowed us to fully determine their modes of action.
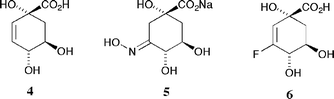 | ||
| Fig. 1 Selective inhibitors of type II dehydroquinases. | ||
Our search for even more effective inhibitors focused on derivatives in which the true geometric and electronic features of enolate 3 were more accurately mimicked, particularly the electron distribution and high electron density generated at C-3 during the course of the reaction. We thus chose to replace the enolate oxyanion with its closest chemical equivalent, the isosteric and isoelectronic fluorine atom and sought to prepare vinyl fluoride 6. In a recent communication12 we briefly outlined the synthesis of 6 (together with the 3,3-difluoro acid 7). Herein we report more fully on our studies towards the synthesis of 6 and describe in detail the inhibitory profiles of both 6 and 7 against dehydroquinases (both types I and II) from a variety of bacterial sources. In addition we comment briefly on the X-ray crystallographic structure of the co-complex of 6 with the type II dehydroquinase from Streptomyces coelicolor.
Results and discussion
Acyclic vinyl fluorides have gained popularity in recent years as isosteric and isoelectronic replacements for scissile amide bonds in non-hydrolysable protease inhibitors.13,14 In contrast, functionalized endocyclic vinyl fluorides remain far less well exemplified.15,16 We sought a reliable and relatively mild method for fluorine incorporation as we were wary of the fact that the desired polyfunctionalized acid 6 (and closely related derivatives) might prove to be prone to aromatization if subjected to more forcing reaction conditions; we thus looked to the very well developed chemistry of the closely related semi-aromatic shikimate ring system for inspiration.Methods for the introduction of fluorine into the shikimate nucleus have been investigated by several research groups with 2-fluoro,15,17 3-fluoro,18,19 3,5-difluoro,20 6-fluoro,21–24 6,6-difluoro25 and 6-trifluoromethyl26 derivatives having been described. Amongst these endeavours is work by the group of Bartlett15 describing the synthesis of 2-fluoroshikimate 11 (Scheme 2). In the key synthetic step incorporation of fluorine was effected upon treatment of cyclohexanone 8 with diethylaminosulfur trifluoride (DAST) in hot 1,2-dimethoxyethane (DME). The reaction afforded a mixture of gem-difluoride 9 (28%) and the desired vinyl fluoride 10 (40%) which was transformed into the fluoro acid 11 over four steps; conversion of the undesired product 9 to 11 was achieved via aldehyde 12 which readily eliminated hydrogen fluoride under mildly basic conditions. We thus envisaged a synthesis of fluoro acid 6 involving reaction between DAST and an appropriately protected cyclohexanone derivative of naturally occuring (−)-quinic acid 13.
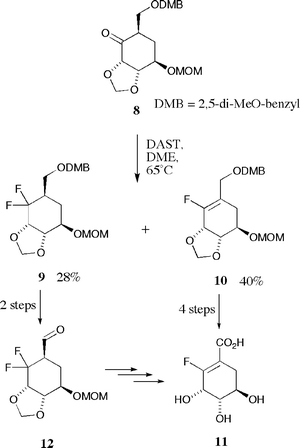 | ||
| Scheme 2 | ||
(−)-Quinic acid 13 was smoothly protected as the cyclic bis-ketal 14 with concomitant protection of the C-1 carboxylate functionality as the methyl ester upon treatment with 2,3-butanedione ketal27 in an acidified mixture of trimethyl orthoformate and methanol at reflux (Scheme 3). The reaction afforded essentially a single product in high yield (79%), the ketal protecting group being selective for the vicinal trans-diequatorial diol with a double anomeric effect controlling both ketal stereogenic centres. Oxidation of the C-3 secondary alcohol of 14 was readily achieved upon treatment with RuCl3 and KIO4 under biphasic conditions in a mixture of chloroform and aqueous potassium carbonate. Cyclohexanone 1528 (obtained as a colourless solid in 80% yield after column chromatography) is an exceptionally crystalline compound with a propensity to crystallize directly from concentrated solutions in diethyl ether obtained upon purification on silica. Protection of the remaining C-1 tertiary alcohol as the methoxymethyl (MOM) ether was effected with dimethoxymethane and excess phosphorus pentoxide in anhydrous chloroform. The resulting fully protected product 16 was obtained in essentially quantitative yield.
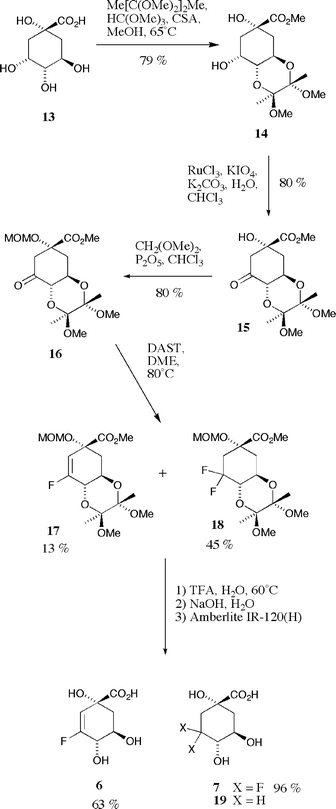 | ||
| Scheme 3 | ||
Initial attempts to introduce fluorine into the cyclohexanone ring of 16 (DAST, DME, 80 °C, nitrogen atmosphere) proved to be frustratingly poor in terms of both the yields and ratios of products 17 and 18 so obtained; in addition reaction reproducibility was found to be extremely capricious with some reactions failing altogether with rapid decomposition of starting material. DAST has been observed29,30 to decompose rapidly and with great exotherm at elevated temperatures resulting in violent decomposition of the contents of reaction, however, it is far more stable at moderate reaction temperatures (below 90 °C) in dilute solution and we thus suspected other factors to be the cause of such poor yields and reproducibility.
Factors that we considered included purity of starting material, purity and moisture content of the solvent and the age of DAST samples; to ensure purity of starting material ketone 16 was recrystallized from a mixture of hexane and diethyl ether as fine colourless needles (yields of 16 from 15 after crystallization ∼80%). Reactions were performed using fresh samples of DAST dissolved in fresh samples of anhydrous DME under an argon atmosphere to maintain anhydrous conditions. A solution of ketone 16 in boiling DME was treated with 3 portions of DAST (1.2 equivalents each) at 2 hourly intervals. After 6 h the reaction was cooled to room temperature and quenched by the addition of a saturated aqueous solution of sodium bicarbonate. Under these conditions we were successfully able to control the yield of fluorinated products; moreover the reaction was shown to be very reproducible.
Extensive analysis (1H, 13C and 19F NMR spectra recorded in CDCl3 and mass spectra) of the two reaction products obtained (3 : 1 ratio) after column chromatography on silica (with particular reference to H–F, C–F and F–F coupling constants) allowed for elucidation of their structures. The more chromatographically mobile isomer (colourless oil) contained a vicinal fluorine coupled olefinic hydrogen (δH 5.62, JH–F 15 Hz) and one fluorine atom (δF −115.9) coupled to an olefinic carbon (δC 159.6, JC–F 277 Hz). Mass spectral data (m/z 333.1349, M − OMe+) confirmed the product to be the desired vinyl fluoride 17. The less mobile isomer on silica (colourless solid) lacked olefinic hydrogens, contained a pair of coupled fluorine atoms (δF −103.3, JF–F 245 Hz; −113.3, JF–F 245 Hz) that were both coupled to the same sp3 carbon (δC 119.7, JC–F 251 and 245 Hz) and gave mass spectral data (m/z 385.1692, M+H+) indicative of a difluoride; this product was subsequently unequivocally shown to be the C-3 gem-difluoride 18 by single crystal X-ray analysis.12,31
Somewhat disappointingly, under these conditions the desired vinyl fluoride 17 proved to be the minor product (13%) with the difluoride 18 (45%) being the major component of the mixture, an isomeric reversal compared to the two products formed in the key fluorination step of the 2-fluoroshikimate synthesis (9 and 10, 28% and 40% respectively). Despite numerous attempts we were unable to discover conditions to increase the yield and proportion of 17 relative to 18. Attempts to effect conversion of 18 to 17 were similarly unsuccessful. A plethora of basic conditions were tested in an attempt to induce elimination of hydrogen fluoride from 18; in all cases 18 was recovered unchanged. Addition of soluble silver salts to these reactions (e.g. AgPF6, AgBF4 and AgOTf) to encourage precipitation of fluoride anion (as insoluble AgF) also proved unsuccessful. In spite of these results the conditions developed for incorporation of fluorine at C-3 of the quinate ring were robust and reproducible enough to consistently afford acceptable quantities of material (up to 3 mmol scale) and we were thus routinely able to prepare hundreds of milligrams of pure samples of both 17 and 18.
Both fluorides 17 and 18 were readily deprotected over three steps to afford the desired vinyl fluoride 6 and the difluoro acid 7 in good yields. Treatment of 17 and 18 with 50% aqueous trifluoroacetic acid at 60 °C resulted in the removal of both the MOM group protecting the C-1 tertiary alcohol and the 2,3-butanedione derived bis-ketal protecting the 4,5-diol functionality. The colour of the solutions changed dramatically during the course of the reactions from colourless to an intense yellow due to the liberation of 2,3-butanedione. Upon cooling of the reaction mixtures and removal of solvent under reduced pressure, purification was effected simply by partitioning between ethyl acetate and water followed by lyophilization of the resulting colourless aqueous phase. The intermediate methyl esters were readily saponified upon treatment with aqueous sodium hydroxide and the free acids 6 and 7 obtained after cation exchange [Amberlite IR-120(H)] and lyophilization of the resulting aqueous solutions (63% and 96% yields respectively over the three steps).
Acids 6 and 7 were assayed against purified samples of both type I and type II dehydroquinases from a variety of bacterial sources (Type I: from Salmonella typhi; Type II: from Mycobacterium tuberculosis and Streptomyces coelicolor). As expected from a compound specifically designed to closely mimic the reaction intermediate, the kinetics observed for 6 were indicative of it being a competitive reversible inhibitor of each of the enzymes. Difluoride 7, the 3,3-difluoro analogue of 3-deoxyquinic acid 1910 (a weak competitive reversible inhibitor), also showed the expected competitive reversible inhibitory behaviour. Ki values for 6 and 7 against each of the three dehydroquinases (Table 1) were determined directly from Dixon plots (Fig. 2).
![Dixon plot of reciprocal of rate versus inhibitor concentration [I] for four differing concentrations of 1 showing inhibition of the type II dehydroquinase from Mycobacterium tuberculosis by 6.](/image/article/2004/OB/b404535a/b404535a-f2.gif) | ||
| Fig. 2 Dixon plot of reciprocal of rate versus inhibitor concentration [I] for four differing concentrations of 1 showing inhibition of the type II dehydroquinase from Mycobacterium tuberculosis by 6. | ||
The two fluoro acids 6 and 7 showed similar inhibitory profiles. Both compounds were selective for the type II enzymes over the type I but in each case showed essentially no selectivity between the two type II proteins. As expected the vinyl fluoride 6 was significantly more potent than the difluoride 7 (particularly so against the type II enzymes) consistent with our previous findings10 that inhibitors with sp2 character at C-2 and C-3 had significantly greater potency.
The inhibitory behaviour of vinyl fluoride 6 towards the type II proteins contrasts sharply with those of the low micromolar inhibitors 4 and 5,10 both of which show a marked selectivity (up to 25 fold) amongst type II dehydroquinases from a variety of sources; the ‘ring flattened’ alkene inhibitor 4 is more potent against the enzyme from S. coelicolor whereas the ‘enhanced hydrogen bonding’ mimicking oxime 5 shows selectivity for the M. tuberculosis protein. Vinyl fluoride 6 is the first effective pan-type II dehydroquinase inhibitor reported to date with, in all cases, a Ki significantly lower than the corresponding Km but with a molecular mass and degree of molecular complexity essentially identical to the natural substrate DHQ 1. As such, fluoride 6 would appear to effectively mimic structural features that are critical to turnover for all type II dehydroquinases.
Similar subtleties in the kinetic profiles of type II dehydroquinases determined in the presence of simple polydentate anions (phosphate and sulfate) have also been observed35 and have been rationalized in terms of their binding characteristics with particular reference to data obtained from X-ray crystallographic studies. The results of these structural studies, when taken in conjunction with the findings outlined herein, strongly suggest that the binding characteristics of type II dehydroquinases are acutely sensitive to both the precise geometric features as well as the chemical functionalities present within the ligand.
Vinyl fluoride 6 is the most potent inhibitor of the type II dehydroquinase from Mycobacterium tuberculosis reported to date and the second most potent36 for the enzyme from Streptomyces coelicolor. The increase in potency shown by 6 over both 4 and 5 is only relatively modest (approximately twofold) for the enzymes against which 4 and 5 are most potent, however fluoride 6 offers a much more significant increase in potency (20–30 fold) against the enzymes for which 4 and 5 are less effective.
Vinyl fluoride 6 was co-crystallized with the type II dehydroquinase from Streptomyces coelicolor (Fig. 3). The structure was collected at room temperature as the ligand could not be detected in data collected from frozen crystals (at 100 K). In accord with both the previously reported apo structure11 (PDB: 1GU0) and co-complexes with phosphate anion36 (PDB: 1D0I), 2 (PDB: 1GTZ) and 4 (PDB: 1GU1) the co-complex (2.2 Å resolution) is a homo-dodecamer and contains an equivalent copy of 6 bound at each of the twelve separate active sites within the structure. The orientation of the vinyl fluoride 6 is essentially identical to that of the alkene 4 with the additional fluorine atom involved in a long hydrogen bond (3.2 Å) to a conserved water in the active site. The absence of any tartrate or glycerol in the active site (both of which are present in the alkene 4 inhibitor structure 1GU1) results in a shift in the lid domain and brings the Arg23 residue into the active site. Although the positively charged guanidinium sidechain of R23 is still relatively distant from the vinyl fluoride 6 (over 4.5 Å away from the fluorine atom) this observation may have implications in the binding of the natural substrate 1 and its subsequent turnover to 2.
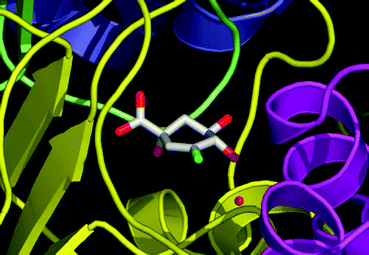 | ||
| Fig. 3 X-ray crystal structure (PDB: 1V1J) of the co-complex between vinyl fluoride 6 and the type II dehydroquinase from Streptomyces coelicolor. The conserved water molecule described in the text is shown as a red sphere. The figure was prepared using PyMol.37 | ||
Dehydroquinases are utilized by bacteria and fungi for the synthesis of essential aromatics and, in common with the other enzymes of the shikimate and quinate pathways, are foreign to all mammalian systems. Type II dehydroquinases are present in a number of bacterial strains that are responsible for a variety of disease states; they have therefore been seen as potential targets for novel therapeutics, particularly given the recently reported structural data for type II dehydroquinases complexed to small molecule inhibitors. The availability of this wealth of data opens up the possibility of rational drug design38 using a structure-aided approach against clinically important pathogens responsible for a number of increasingly prevalent diseases that include tuberculosis, ulceration and stomach cancer.
Experimental
All organic solvents were freshly distilled prior to use and Milli-Q deionized water was used for all biochemical work. Amberlite IR-120(H) was washed alternately with water, 5 M sodium hydroxide, water, 6 M HCl and water prior to use. Flash column chromatography was carried out using silica gel 60 (Merck 9385). Rf values are quoted with respect to the solvent used for elution. Melting points were recorded on a Kofler hot-stage and are uncorrected. Specific rotations were recorded in the solvent specified at ambient temperature on a Perkin Elmer 241 instrument and are quoted in 10−1 deg cm2 g−1. Microanalyses were determined by the Departmental Microanalytical Service, University Chemical Laboratory, Cambridge using an Exeter Analytical 440 elemental analyzer. Infrared spectra were recorded on an ATI Mattson Genesis Series FTIR spectrometer as a thin film or Nujol mull as stated. NMR spectra were recorded on a Bruker AC-250 spectrometer operating at 250.1 MHz (for 1H), 62.9 MHz (for 13C) or 234.5 MHz (for 19F) in the deuterated solvents stated. High resolution mass spectra were recorded on a Kratos MS890 or Kratos Concept 1H instrument under positive electron impact (+EI) conditions or under positive fast atom bombardment (+FAB) conditions (using xenon as the ionizing source) as stated. Compounds were assayed for inhibitory activity against dehydroquinases in the presence of 1 by monitoring changes in the rate of increase in absorbance at 234 nm in UV spectra due to formation of 2 (ε 12000 M−1cm−1). Kinetic assays were performed on a Varian Carey spectrophotometer in 50 mM pH 7 buffer (potassium phosphate and Tris-HCl for type I and type II dehydroquinases respectively) at 25 °C using black walled quartz cuvettes. The assay mixture was prepared in situ and the reaction initiated by addition of the enzyme solution to the mixture. Concentrations of solutions of 1 were accurately determined by equilibration with type I dehydroquinase and measurement of the change in UV absorbance at 234 nm. To measure inhibition the concentration of inhibitor (I) was varied at a constant concentration of substrate and enzyme and assays were repeated for a number of different concentrations of substrate. Ki values were obtained through Dixon plots (1/v versus [I]).Acknowledgements
We thank the BBSRC and the Wellcome Trust for postdoctoral support (to M.F. and A.W.R. respectively).References
- E. Haslam, Shikimic Acid: Metabolism and Metabolites, John Wiley & Sons, Chichester, 1993 Search PubMed.
- C. Abell, in Comprehensive Natural Products Chemistry; ed. U. Sankawa, Elsevier, Amsterdam, 1999, vol. 1, p. 573 Search PubMed.
- N. H. Giles, M. E. Case, J. A. Baum, R. F. Geever, L. Huiet, V. B. Patelt and B. M Tyler, Microbiol. Rev., 1985, 49, 338 Search PubMed.
- D. G. Gourley, A. K. Shrive, I. Polikarpov, T. Krell, J. R. Coggins, A. R. Hawkins, N. W. Isaacs and L. Sawyer, Nat. Struct. Biol., 1999, 6, 521 CrossRef CAS.
- S. Chaudhuri, K. Duncan, L. D. Graham and J. R. Coggins, Biochem. J., 1991, 275, 1 CAS.
- M. J. Turner, B. W. Smith and E. Haslam, J. Chem. Soc., Perkin Trans. 1, 1975, 52 RSC.
- A. Schneier, C. Kleanthous, R. Deka, J. R. Coggins and C. Abell, J. Am. Chem. Soc., 1991, 113, 9416 CrossRef.
- A. R. Hawkins, W. R. Reinhert and N. H. Giles, Biochem. J., 1982, 203, 769 CAS.
- J. M. Harris, C. González-Bello, C. Kleanthous, A. R. Hawkins, J. R. Coggins and C. Abell, Biochem. J., 1996, 319, 333 CAS.
- M. Frederickson, E. J. Parker, A. R. Hawkins, J. R. Coggins and C. Abell, J. Org. Chem., 1999, 64, 2612 CrossRef CAS.
- A. W. Roszak, D. A. Robinson, T. Krell, I. S. Hunter, M. Frederickson, C. Abell, J. R. Coggins and A. J. Lapthorn, Structure, 2002, 10, 493 CrossRef.
- M. Frederickson, J. R. Coggins and C. Abell, Chem. Commun., 2002, 1886 RSC.
- R. J. Abraham, S. L. R. Ellison, P. Schonholzer and W. A. Thomas, Tetrahedron, 1986, 42, 2101 CrossRef CAS.
- P. A. Bartlett and A. Otake, J. Org. Chem., 1995, 60, 3107 CrossRef CAS.
- R. H. Rich and P. A. Bartlett, J. Org. Chem., 1996, 61, 3916 CrossRef CAS.
- G. A. Boswell, Jr., U.S. Patent 4 212 815, 1980.
- C. González-Bello, M. K. Manthey, J. M. Harris, A. R. Hawkins, J. R. Coggins and C. Abell, J. Org. Chem., 1998, 63, 1591 CrossRef.
- R. Brettle, R. Cross, M. Frederickson, E. Haslam, F. S. MacBeath and G. M. Davies, Bioorg. Med. Chem. Lett., 1996, 6, 1275 CrossRef CAS.
- R. Brettle, R. Cross, M. Frederickson, E. Haslam, F. S. MacBeath and G. M. Davies, Tetrahedron, 1996, 52, 10547 CrossRef CAS.
- F. J. Weigert and A. Shenvi, J. Fluorine Chem., 1994, 66, 19 CrossRef CAS.
- J. K. Sutherland, W. J. Watkins, J. P. Bailey, A. K. Chapman and G. M. Davies, J. Chem. Soc., Chem. Commun., 1989, 1386 RSC.
- S. A. Bowles, M. M. Campbell, M. Sainsbury and G. M. Davies, Tetrahedron, 1990, 46, 3981 CrossRef CAS.
- J. K. Sutherland, R. C. Whitehead and G. M. Davies, J. Chem. Soc., Chem. Commun., 1993, 464 RSC.
- P. J. Duggan, E. Parker, J. R. Coggins and C. Abell, Bioorg. Med. Chem. Lett., 1995, 5, 2347 CrossRef CAS.
- J. L. Humphreys, D. J. Lowes, K. A. Wesson and R. C. Whitehead, Tetrahedron Lett., 2004, 45, 3429 CrossRef CAS.
- J. Leroy, N. Fischer and C. Wakselman, J. Chem. Soc., Perkin Trans. 1, 1990, 1281 RSC.
- J.-L. Montchamp, F. Tian, M. E. Hart and J. W. Frost, J. Org. Chem., 1996, 61, 3897 CrossRef CAS.
- F. Tian, J.-L. Montchamp and J. W. Frost, J. Org. Chem., 1996, 61, 7373 CrossRef CAS.
- P. A. Messina, K. C. Mange and W. J. Middleton, J. Fluorine Chem., 1989, 42, 137 CrossRef CAS.
- G. S. Lal, G. P. Pez, R. J. Pesaresi, F. M. Prozonic and H. Cheng, J. Org. Chem., 1999, 64, 7048 CrossRef CAS.
- X-ray crystallographic data for 18 have been deposited with the Cambridge Crystallographic Data Centre and are available upon request. CCDC reference number 184932. See http://www.rsc.org/suppdata/ob/b4/b404535a/ for crystallographic data in .cif or other electronic format.
- J. D. Moore, A. R. Hawkins, I. G. Charles, R. Deka, J. R. Coggins, A. Cooper, S. M. Kelly and N. C. Price, Biochem. J., 1993, 295, 277 CAS.
- T. Garbe, S. Servos, A. Hawkins, G. Dimitriadis, D. Young, G. Dougan and I. Charles, Mol. Gen. Genet., 1991, 228, 385 Search PubMed.
- P. J. White, J. Young, I. S. Hunter, H. G. Nimmo and J. R. Coggins, Biochem. J., 1990, 265, 735 CAS.
- L. Evans, A. Roszak, L. Noble, D. Robinson, P. Chalk, J. Matthews, J. Coggins, N. Price and A. Lapthorn, FEBS Lett., 2002, 530, 24 CrossRef CAS.
- C. González-Bello, E. Lence, M. D. Toscano, L. Castedo, J. R. Coggins and C. Abell, J. Med. Chem., 2003, 46, 5735 CrossRef CAS.
- W. L. DeLano, The PyMol Molecular Graphics System (2002), DeLano Scientific, San Carlos, California, USAhttp://www.pymol.org.
- J. R. Coggins, C. Abell, L. B. Evans, M. Frederickson, D. A. Robinson, A. W. Roszak and A. J. Lapthorn, Biochem. Soc. Trans., 2003, 31, 548 CAS.
| This journal is © The Royal Society of Chemistry 2004 |