
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
π-Extended dihydrophenazine based redox responsive polymers of intrinsic microporosity†
Grazia C. Bezzu a,
Beatrice Bartolomeibc,
Yue Wua,
Martina Vaccarod,
Mariagiulia Longod,
Maria Penelope De Santoe,
Alessio Fuoco*d,
Maurizio Prato*bfg,
Mariolino Carta*a and
Jacopo Dosso*b
a,
Beatrice Bartolomeibc,
Yue Wua,
Martina Vaccarod,
Mariagiulia Longod,
Maria Penelope De Santoe,
Alessio Fuoco*d,
Maurizio Prato*bfg,
Mariolino Carta*a and
Jacopo Dosso*b
aDepartment of Chemistry, Faculty of Science and Engineering, Swansea University, Grove Building, Singleton Park, Swansea, SA2 8PP, UK. E-mail: mariolino.carta@swansea.ac.uk
bDepartment of Chemical and Pharmaceutical Sciences, CENMAT, Centre of Excellence for Nanostructured Materials, INSTM UdR Trieste, University of Trieste, Via Licio Giorgieri 1, Trieste, 34127, Italy. E-mail: prato@units.it; jacopo.dosso@units.it
cDepartment of Chemistry, Northwestern University, Evanston, Illinois 60208, USA
dInstitute on Membrane Technology, National Research Council of Italy (CNR-ITM), Via P. Bucci 17/C, Rende, CS 87036, Italy. E-mail: alessio.fuoco@cnr.it
eDepartment of Physics and CNR-Nanotec, University of Calabria, Rende, 87036, Italy
fCentre for Cooperative Research in Biomaterials (CIC BiomaGUNE), Basque Research and Technology Alliance (BRTA), Paseo de Miramón 194, Donostia San Sebastián, 20014, Spain
gBasque Fdn Sci, Ikerbasque, Bilbao, 48013, Spain
First published on 3rd June 2025
Abstract
Redox-switchable Polymers of Intrinsic Microporosity (PIMs) are a promising yet underexplored class of materials. Here, we introduce π-extended dihydrophenazine-based PIMs for gas separation. The tert-butyl substituted Phen-PIM-1 stands out as a rare example of a redox-active switchable polymer with a high surface area (BET >600 m2 g−1) and excellent balance of porosity, pore size, and gas selectivity. Phen-PIM-1 is soluble in N-methyl pyrrolidone (NMP), enabling membrane fabrication, while the methyl-substituted Phen-PIM-2 is insoluble, highlighting the role of bulky tert-butyl groups (tBu) in solubility and film formation. Gas separation studies, performed on powder (IAST), demonstrate outstanding performance, with CO2/N2 selectivity up to 49. As a membrane material, Phen-PIM-1 shows competitive separation within the Robeson upper bound for several commercially important gas pairs, proving its potential for carbon capture and molecular sieving. Furthermore, these materials exhibit efficient and reversible redox switching upon chemical stimuli, leading to marked differences in properties and enhanced selectivity. This study establishes dihydrophenazine-based PIMs as a versatile platform for developing tunable, high-performance membranes for energy and environmental applications.
1. Introduction
The development of novel porous polymeric materials is currently one of the most investigated areas of chemistry.1,2 This interest arises from the great impact of such materials for applications in gas adsorption/separation,1,3,4 energy storage,5–8 and catalysis,9–12 which are of paramount importance to tackle the climate crisis that we are currently witnessing. As such, the number of works revolving around the development of innovative Metal organic frameworks (MOFs), Covalent organic frameworks (COFs) and in general, polymeric porous materials, is constantly rising. From this point of view, polymers of intrinsic microporosity (PIMs) have been extensively studied due to their highly promising surface areas and tunable functionalization, especially considering that many PIMs can be solution cast into membranes, and thus they can be efficiently exploited for gas separation applications. From the structural point of view, PIMs are a class of highly porous materials defined by their rigid and contorted monomeric structures, which prevent efficient chain packing and create interconnected micropores (<2 nm). Their microporosity arises from a combination of intrinsic backbone rigidity and sites of contortion, such as spiro-centers or ladder-like linkages, that disrupt dense packing.13 This results in high free volume and permanent porosity without the need for post-synthetic processing. PIMs' high surface area and tunable chemistry make them ideal for gas separation, CO2 capture, and membranes.14,15 Post-polymerization changes and further functionalization of the PIMs' backbones are extensively used strategies to improve their potential in gas separation and adsorption applications. For example, Weng16 and co-workers exploited the nitrile groups present in pre-formed PIMs to generate triazine cross-links in situ, leading to improved separation performance despite a reduction in porosity. Similarly, Rizzuto et al.17 demonstrated that reducing nitrile groups in PIM-1 to primary amines significantly enhanced CO2 selectivity, attributing this effect to competitive sorption interactions confirmed via mixed-gas studies, solid-state NMR, and molecular dynamics, which showed chemical bonds between CO2 and the amino groups. Beyond gas separation, Song's18 group engineered spirobifluorene-based PIMs to incorporate sulfonic acid in the repeat units, resulting in a significant improvement in redox flow battery performance. Building on this, stimuli-responsive structural modifications could further enhance PIM selectivity and transport properties. In fact, despite a growing number of works reporting new types of PIMs with continuously improved properties, the concept of introducing responsive units in such materials is still underexplored, with most examples focusing on using light as stimulus.4,19 Indeed, building PIMs functionalized with redox responsive molecular switches could produce materials that can change their properties “on demand”. By employing chemical or electrical stimuli these changes could lead to the tuning of their porosity, their polarity and charge, and their gas selectivity, all parameters that may greatly affect the final properties of a material.14,15 Building on this idea, we recently reported the development and reversible molecular switching of symmetrically π-extended dihydrophenazines.20,21 These molecules can be easily oxidized to their aromatic dicationic form, which is permanent once induced but can be reduced back to the neutral form by treatment with soft nucleophiles (e.g., Triphenylphosphine, Fig. 1A). Moreover, the oxidation to the dicationic form is also associated with a dramatic electrochromic and conformational switch, which can have important effects when introduced in porous networks.21 Given the rigidity and contortion of these molecules, which are typical features of monomers that produce high performing PIMs,22 and the relative ease of functionalization with catechol units (Fig. 1B),9 π-extended dihydrophenazines can be ideal candidates for the synthesis of redox switchable PIMs, especially exploiting nucleophilic aromatic substitutions as the polymerization reaction, which is typical of polybenzodioxin-based PIMs22 (Fig. 1C).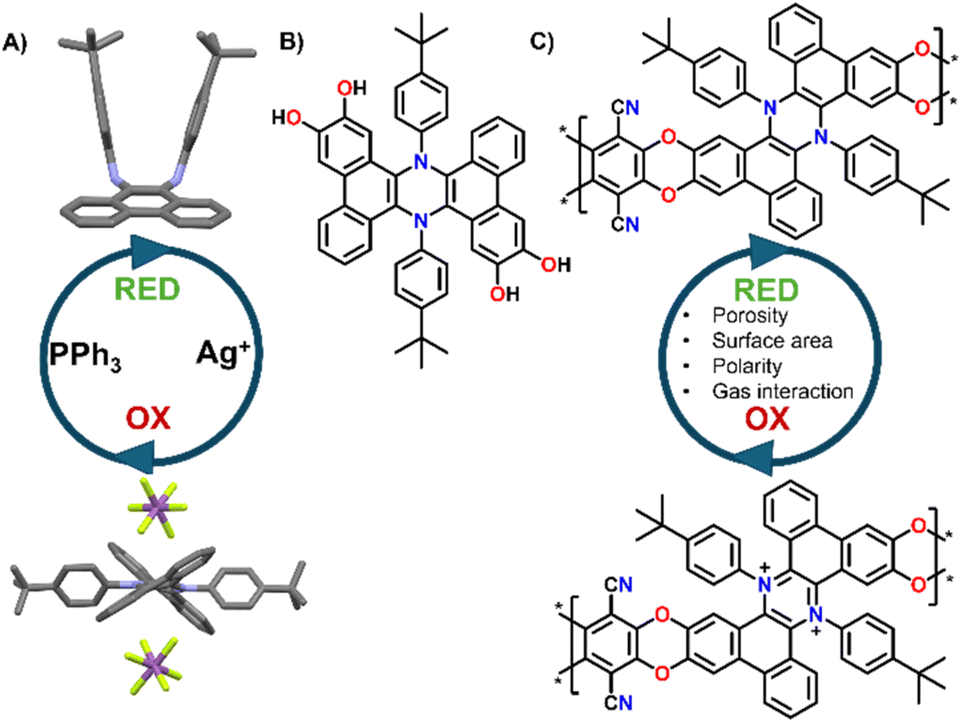 | ||
| Fig. 1 (A) Parent π-extended dihydrophenazine chemical oxidation/reduction cycle; (B) catechol functionalized derivatives; (C) proposed structure of the switchable π-extended dihydrophenazine based PIMs. | ||
As such, in this work we report the first synthesis and complete characterization of π-extended dihydrophenazine based PIMs. These materials present promising properties and can be reversibly oxidized and reduced, resulting in an increased selectivity between CO2 and N2 upon oxidation.
2. Results and discussion
2.1 Synthesis of polymeric materials
Building on our previous work on π-extended dihydrophenazine systems,20,21 we sought to utilize the presence of catechol moieties9 and the rigid contorted nature of these backbones to synthesize polybenzodioxin-based polymers of intrinsic microporosity. We deem this investigation to be particularly significant because, as demonstrated in prior studies,21 the dihydrophenazine backbone can be reversibly oxidized to its aromatic dicationic form, which leads to a conformational change that could potentially modify the microporosity of the material. In fact, based on a qualitative assessment of previously published X-Ray diffraction (XRD) crystal structures (Fig. 1A),21 we believe this change results in a more rigid and contorted arrangement, characteristic of highly porous PIMs,13 which may impact gas uptake, diffusion and release when used as a gas separation material. Herein we exploit this reversibility but within the polymeric chains, and not just on discreet molecules, to assess whether the expected changes also produce an alteration of the textural properties of the materials, such as porosity and pore size distribution (PSD). More specifically, we characterized the polymers in their initial pristine state (before oxidation, Fig. 2A), after oxidation of the dihydrophenazine system (Fig. 2B), and following its subsequent reduction to go back to the original form. In this way we aimed to confirm the polymers' ability to fully recover its initial properties, effectively demonstrating a chemo-switchable material. Building on previous findings about the irreversible structural changes in oxidized dihydrophenazine when exposed to nucleophilic molecules such as H2O, we also investigated how this degradation occurs within the polymer (Fig. 2C), aiming for a deeper understanding of the associated properties changes. This study is particularly focused on evaluating the potential of this chemo-switchable material for applications such as carbon capture and gas separation. In particular, any alterations in porosity and pore size distribution during oxidation–reduction cycles could significantly impact the permeability and selectivity of the polymers for commercially important gas pairs, especially CO2/N2. These insights are crucial for assessing their suitability in separation technologies.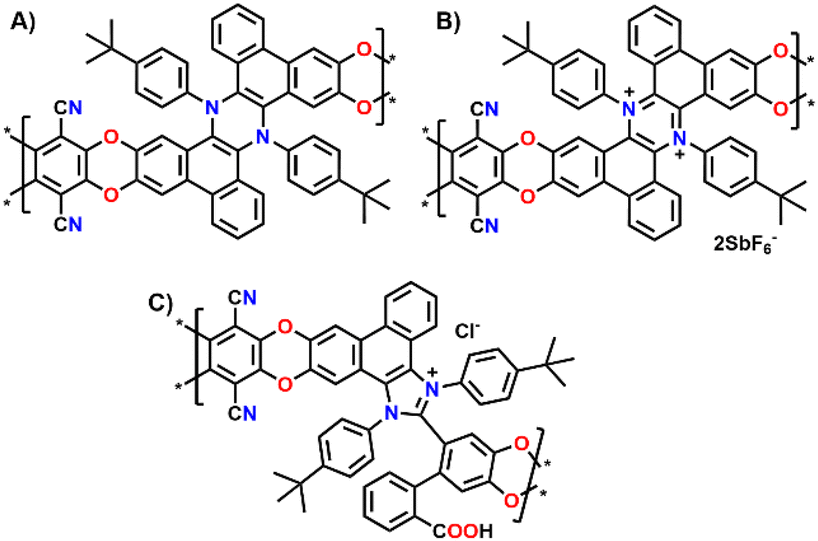 | ||
| Fig. 2 Structures of the three different versions of (A) Phen-PIM-1; (B) Phen-PIM-1ox; and (C) Phen-PIM-1deg. | ||
We started the synthesis by preparing dihydrophenazine biscatechol 1 (Scheme 1) following a procedure reported in our previous contribution.9 Since these substrates were never used in combination with tetrafluoroterephthalonitrile 5, which is one of the most common fluorinated monomers used for the synthesis of PIMs, the reaction conditions were initially tested on 1 using an excess of 5 in the presence of K2CO3 in anhydrous DMF at 60 °C. As a result, the fluorinated monomeric unit 3 was obtained in a quantitative yield. Derivative 3 proved to be soluble enough in chlorinated solvents to be completely characterized using nuclear magnetic resonance spectroscopy (1D and 2D NMR). Moreover, the monomeric units presented a marked visible absorption in the 350–450 nm region of the spectra as typically associated with the introduction of oxygen atoms on the phthalonitrile system (Fig. S31†). The efficient preparation of model compound 3 confirmed that the same conditions would work in the polymerization and that the dihydrophenazine would not hinder it. Thus, 1 was treated with an equimolar amount of 5 in the presence of K2CO3 in anhydrous and degassed DMF at 65 °C for 70 h (Scheme 1). The long reaction time was used to ensure complete reaction of the starting materials, and the formation of long polymeric chains.23 As a result, a dense bright yellow solid was formed, which was extensively purified by washing with several solvents (Section 2.6 in the ESI†) to ensure removal of short oligomers and salts and affording Phen-PIM-1 as a dark yellow solid in almost quantitative yield.
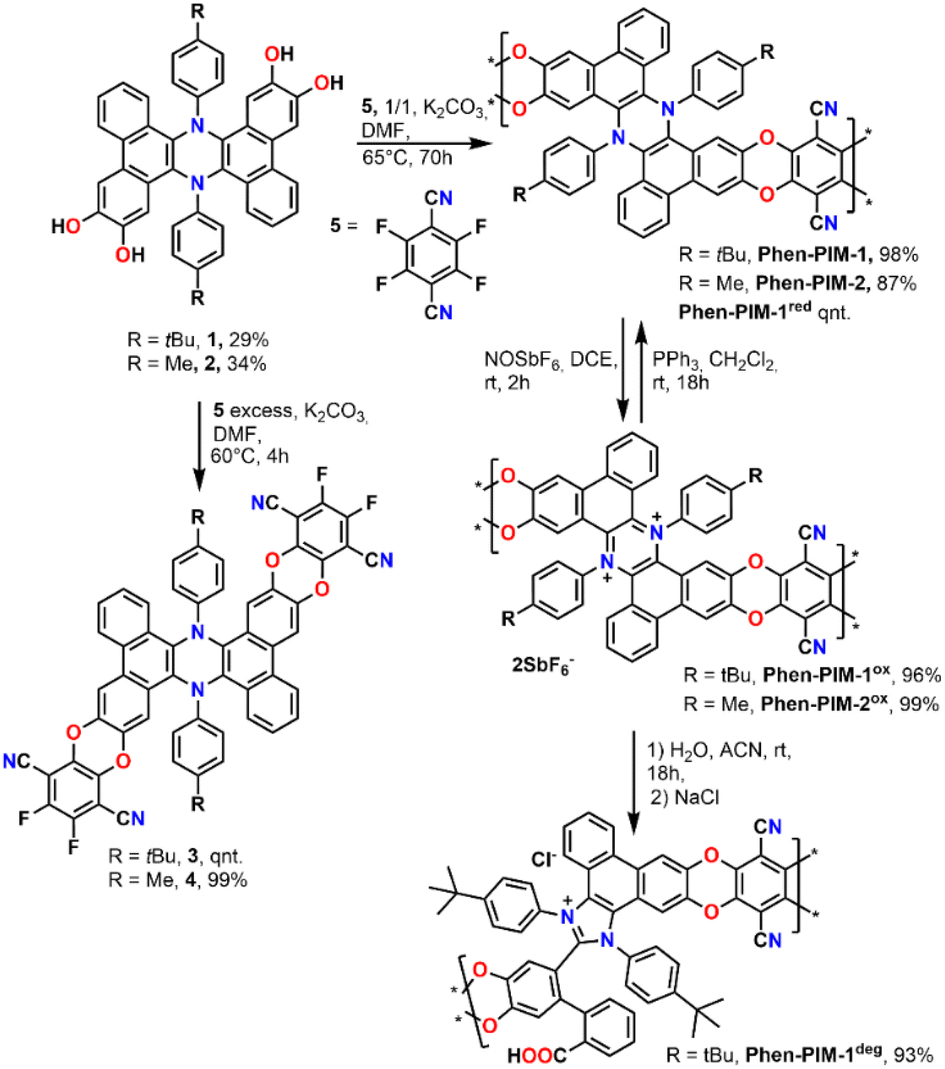 | ||
| Scheme 1 Synthetic procedure for the preparation of dihydrophenazine models (3 and 4), Phen-PIM-1 and Phen-PIM-2 and their oxidation, reduction, and degradation. | ||
To assess the effects on the material properties of the tert-butyl group (tBu), the same synthetic strategy was repeated with methyl functionalized dihydrophenazine 2, resulting in the formation of Phen-PIM-2. We anticipated that the presence of the tBu groups may slightly reduce the porosity compared to the methyl version, by a pore filling effect.24 However, the larger aliphatic tBu groups are also expected to enhance the solubility in organic solvents.25 To test the feasibility of the oxidation reaction on the system, monomer 3 was treated with an excess of NOSbF6 as an oxidant, resulting in the formation of a deep green solution (Section 2.3 in the ESI†). NMR analysis of the green solid highlighted the presence of a closed shell system fully consistent with the quantitative formation of dicationic 32+ (Fig. S15–S17†). Moreover, a UV-vis investigation highlighted a profile also compatible with the formation of the dicationic system (Fig. S31†) in line with previously reported examples. Based on this, the same reaction was then carried out on the two polymers to generate their oxidized form. As such, Phen-PIM-1 and Phen-PIM-2 were suspended in dichloroethane (DCE) and treated with a slight excess of NOSbF6 (Sections 2.7 and 2.8 in the ESI†). As a result, an immediate color change of the solids was visible and after purification Phen-PIM-1ox and Phen-PIM-2ox could be isolated in almost quantitative yield. To assess the reversible nature of the transformation, Phen-PIM-1ox was then also treated with PPh3 which is known to reversibly reduce the dicationic form to the initial neutral one, resulting in quantitative formation of Phen-PIM-1red. Finally, a batch of Phen-PIM-1ox was also treated with H2O in acetonitrile (ACN) solution, which, as expected based on previous reports by our group,21 resulted in the irreversible degradation of the material generating Phen-PIM-1deg.
2.2 Characterization of PIMs
Due to the lack of solubility in common deuterated solvents, typically used for solution NMR, the Phen-PIMs structures were characterized by FT-IR (Fig. S32†) and 13C solid-state-NMR (ssNMR, Fig. S22–S27†). In Fig. 3, we display the ssNMR spectra of the two forms of Phen-PIM-1: the original (black trace) and the oxidized (purple), and we observe that all expected peaks are present and consistent with previously reported structures of similar phenazines. Although the two spectra appear rather similar, subtle differences are also evident, suggesting that the transformation of the original Phen-PIM-1 into its oxidized form has occurred.21 Specifically, the peaks at ∼30–33 ppm, which can be readily attributed to the tBu substituents, in the oxidized form (purple) are slightly shifted to a lower field compared to the original (black). The signal at ∼94 ppm, likely assigned to the carbon linked to the nitrile group, remains in the same position as it is distant from the nitrogen.26 The aromatic region peaks are overlapped and difficult to assign precisely, but they are consistent with the reported structures, and the peaks at ∼139–142 ppm show a slight shift and change in shape. Also, comparing the 13C ssNMR of Phen-PIM-1 and Phen-PIM-1ox with the solution 13C NMR of the monomeric units 3 and 32+ highlights an excellent correspondence between signals, suggesting the presence of very similar units in the materials. Finally, the signal centered at 167 ppm in Phen-PIM-1deg, which may be related to the COOH groups generated by the degradation (as confirmed by the degradation of 32+, see the ESI†), is not observed in Phen-PIM-1ox, which rules out the occurrence of extensive degradation in the latter and confirms the expected nature of the different polymers.
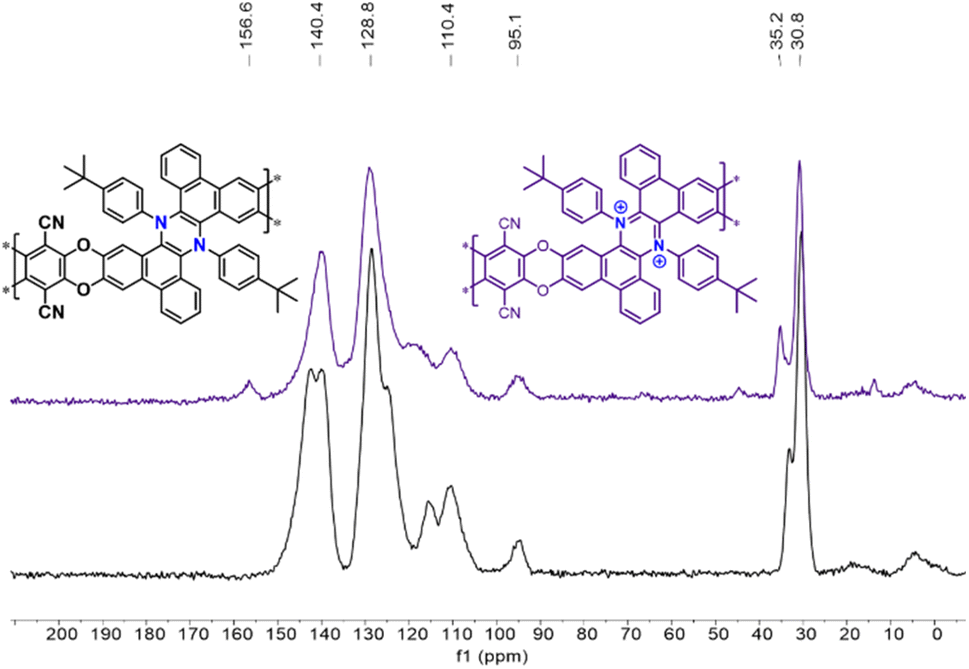 | ||
| Fig. 3 13C ssNMR of Phen-PIM-1 (black trace) and Phen-PIM-1ox (purple trace). | ||
 | ||
| Fig. 4 SEM images of the Phen-PIMs: (A) Phen-PIM-1 pristine; (B) Phen-PIM-1ox-red; (C) Phen-PIM-1ox; (D) Phen-PIM-1deg. | ||
As anticipated from the contorted structure of the monomers, both Phen-PIM-1 (Fig. 2A) and Phen-PIM-2 exhibited the high porosity characteristic of high-performing PIMs.22,23 The methyl functionalized polymer (Phen-PIM-2) proved to be only slightly more porous, with a SABET of 633 m2 g−1, compared to 618 m2 g−1 for the tBu version (Phen-PIM-1), as determined by nitrogen adsorption at 77 K. This was expected due to the pore filling effect of the tBu group,24 as already mentioned. Nevertheless, the difference is not particularly significant, which confirms that also Phen-PIM-1 is highly microporous. Upon oxidation, both materials demonstrated a substantial reduction in porosity, likely due to the incorporation of the bulky SbF6− counter-anion, which is expected to occupy the pores and significantly reduce the initial free volume associated with the inefficient packing typical of PIMs, by generating an even more pronounced pore filling.27 Because of that, the kinetics of nitrogen adsorption at 77 K proved to be significantly slow, resulting in negligible surface area measurements (Table 1). The pronounced hysteresis observed in the adsorption isotherms (see Fig. S36 and S37†), however, indicated that the materials retained some degree of porosity, and suggests the presence of inaccessible or partially buried pores that are difficult for the relatively large N2 probe gas to penetrate. To address this limitation, as reported in previous studies,11 and to more properly assess the intrinsic porosity of these samples, carbon dioxide adsorption isothermal measurements at 273 K were performed. In fact, the smaller molecular size of CO2 and the higher temperature of adsorption enhance diffusion and grant access to smaller pores.28 Indeed, these measurements confirmed that the oxidized PIMs remained porous, with surface areas of 244 m2 g−1 for Phen-PIM-2ox and 321 m2 g−1 for the tBu-functionalized polymer (Phen-PIM-1ox, Fig. 2B), calculated via the Grand Canonical Monte Carlo (GCMC) method.28 Interestingly, in contrast to Phen-PIM-2ox, in this case the tBu-functionalized variant exhibited slightly higher porosity. As previously anticipated, Phen-PIM-1 was also subjected to irreversible degradation, a process initially observed in the phenazine-backbone in the original study.21 The resulting Phen-PIM-1deg (Fig. 2C) showed intermediate porosity, with a surface area of 246 m2 g−1 calculated from the N2 isotherm measured at 77 K, and 377 m2 g−1 when evaluated with CO2 at 273 K. This result aligns with expectations, as the chain rearrangements hinder efficient packing, and the counter-anion can be exchanged from SbF6− to the less bulky chloride counter-anion. Pore volume calculations, determined using both N2 and CO2 adsorption, confirmed that all polymers possessed substantial free volume, ranging from 7.460 × 10−2 to 5.620 × 10−1 cc g−1 (Table 1). A high free volume is obviously crucial for applications such as gas separation, as will be discussed later. A key aspect of this study is represented by the reversibility of the porosity of the samples when subjected to oxidation and subsequent reduction, and this represents one of the most intriguing and distinctive properties of the new Phen-PIMs. As discussed, and shown in Fig. S37,† the oxidation of Phen-PIM-1 leads to the expected decrease in adsorption and, consequently, in surface area. However, upon reduction, adsorption measurements revealed nearly identical isothermal curves and surface area calculations to those observed prior to oxidation (Fig. 5A). This demonstrates that the porosity can be effectively restored, highlighting the material's ability to undergo cycles of oxidation and reduction while retaining its original performance.
| Polymer | BET (m2 g−1) | Pore volume a (cc g−1) | CO2 adsorption | Selectivityb | Qstc (kJ mol−1) | |
|---|---|---|---|---|---|---|
| 273 K (1 bar) (cc g−1) (mmolg−1) | 298 K (1 bar) (cc g−1) (mmolg−1) | CO2/N2 | ||||
| a BET values from adsorption of N2 at 77 K. In parentheses the surface areas calculated from CO2 @273 K using the GCMC method.28b Selectivity calculated according to IAST at 298 K and 1 bar.10,29c Isosteric heat of adsorption (Qst in kJ mol−1) of the corresponding gas at zero coverage calculated from isotherms collected at 273 and 298 K and fitted with the Langmuir-Freundlich equation and calculated via the Clausius Clapeyron equation. | ||||||
| This work | ||||||
| Phen-PIM-2 | 633 (509)a | 4.473 × 10−1 | 51 (2.28) | 32 (1.41) | 38 | 30.9 |
| Phen-PIM-1 | 618 (501)a | 5.620 × 10−1 | 55 (2.45) | 38 (1.70) | 49 | 29.1 |
| Phen-PIM-2ox | 1.7 (244)a | (7.460 × 10−2)a | 25 (1.12) | 19 (0.85) | 61 | 29.9 |
| Phen-PIM-1ox | 1.05 (321)a | (1.127 × 10−1)a | 32 (1.43) | 21 (0.94) | 64 | 32.2 |
| Phen-PIM-1deg | 246 (377)a | 1.887 × 10−1 | 43 (1.92) | 26 (1.16) | 42 | 32.9 |
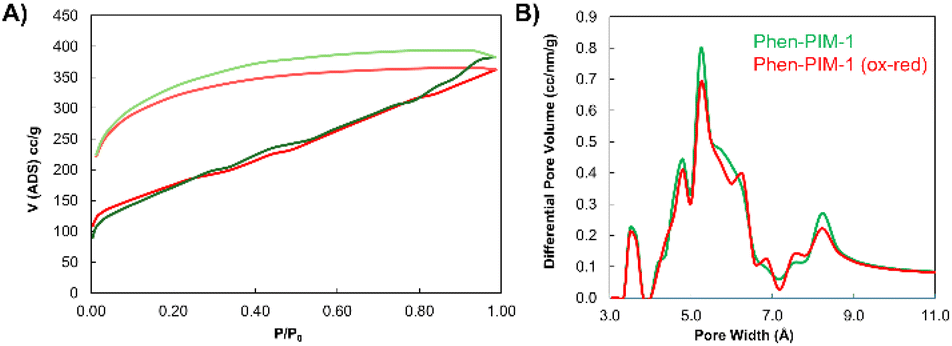 | ||
| Fig. 5 (A) N2 isotherms for Phen-PIM-1 original (green) and after oxidation/reduction (red). The lighter curves represent the desorption pathway; (B) pore size distribution calculated from CO2 adsorption at 273 K and via NLDFT. Color code original (green) vs. oxidation/reduction (red). | ||
The PSD of Phen-PIMs was assessed via CO2 adsorption at 273 K, with calculations performed via the Non-Local Density Functional Theory (NLDFT) method. The graph presented in Fig. 5B reveals a PSD typical of PIMs,30 featuring an initial set of peaks centered at 3–4 Å, corresponding to the ultra-microporous region, a broader range between 4.5 and 7 Å, and a final tail centered around 7–10 Å. The reduction in surface area following the oxidation of the Phen-PIM backbone is also reflected in the changes to the pore size distribution. Specifically, the peaks of the oxidized versions are significantly diminished (see Fig. S39† for a full comparison), whereas the irreversibly modified polymer exhibits an intermediate PSD. Importantly, Fig. 5B also demonstrates the complete reversibility of the PSD upon oxidation and subsequent reduction, as the two traces overlap almost perfectly.
As anticipated, the presence of the tBu groups had the effect of increasing polymer solubility. In fact, while Phen-PIM-2 was poorly soluble in all solvents, Phen-PIM-1 was soluble in N-methyl pyrrolidone (NMP), which allowed the casting of a robust film, suitable for the evaluation of its properties for potential applications as membranes for gas separation.
Moreover, it is worth mentioning that the limited solubility of the oxidized form, Phen-PIM-1ox, prevented the formation of a stable film. As a result, the selectivity comparison between the two forms is based on the powder form using IAST calculations, as shown in Table 1. The pristine Phen-PIM-1 film was insoluble in methanol, which could therefore be used for the standard soaking and drying treatment typically employed to remove traces of residual casting solvent from PIM films and to reverse the effect of physical aging.34,35 Unfortunately, the scarce solubility of the methyl counterpart (Phen-PIM-2) prevented its gas permeability evaluation and, so, a full comparison of the two new PIMs. The order of gas permeability for Phen-PIM-1 is CO2 > H2 >He > O2 > CH4 > N2 (Table 2); this is half way between the trend of the ultra-permeable PIMs, in which CO2 has a higher permeability than H2,25,36,37 and the typical trend of PIMs with very high diffusivity selectivity, in which He has a higher permeability than O2.38–40 The size-sieving character of Phen-PIM-1 is highlighted by the steep correlation between the diffusion coefficient and the squared effective diameter of the gas with a slope increase for the aged film (Fig. S43†). The slope increase is usually associated with an increase of the Young's moduli of the film due to aging.41 AFM force spectroscopy measurements were performed on both freshly methanol treated and aged samples in different positions, and the sampling scans show that the freshly treated sample has a narrow distribution of the Young's modulus with an average of 1.79 ± 0.12 GPa (Fig. S44A†). Thus, this polymer proved to be stiffer with respect to PIM-1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 42 but with similar Young's moduli or slightly more flexible than the ultrapermeable PIMs (Fig. S44B†).37,41 The aged film shows a broader distribution, with two main peaks centered at 3.01 ± 0.16 GPa and 5.58 ± 0.32 GPa (Fig. S44A†). The aging trend is similar to what has been previously observed for other PIMs and the bimodal distribution can be associated with the fact that aging occurs predominantly at the surface of the film and this creates a concentration gradient of fractional free volume near the surface.41,43
42 but with similar Young's moduli or slightly more flexible than the ultrapermeable PIMs (Fig. S44B†).37,41 The aged film shows a broader distribution, with two main peaks centered at 3.01 ± 0.16 GPa and 5.58 ± 0.32 GPa (Fig. S44A†). The aging trend is similar to what has been previously observed for other PIMs and the bimodal distribution can be associated with the fact that aging occurs predominantly at the surface of the film and this creates a concentration gradient of fractional free volume near the surface.41,43
| Transport properties | Permeability, diffusivity and solubility | |||||
|---|---|---|---|---|---|---|
| N2 | O2 | CO2 | CH4 | H2 | He | |
| a Da and Sa values for H2 and He are not reported due to the fast membrane time-lag. | ||||||
| Pa [Barrer] | 688.6 | 2628 | 12492 | 933.8 | 8254 | 3332 |
| α (i/N2) | (–) | (3.82) | (18.1) | (1.35) | (11.9) | (4.84) |
| Aged | 356.8 | 1713 | 7994 | 470.6 | 6851 | 2868 |
| α (i/N2) | (–) | (4.80) | (22.4) | (1.31) | (19.2) | (8.04) |
| Da [10−12 m2 s−1] | 105.6 | 383.5 | 136.0 | 38.8 | –a | –a |
| α (i/N2) | (–) | (3.63) | (1.29) | (0.36) | ||
| Aged | 50.3 | 203 | 66.7 | 15.3 | –a | –a |
| α (i/N2) | (–) | (4.03) | (1.32) | (0.30) | ||
| Sa [cm3STP cm−3 bar−1] | 4.89 | 5.14 | 68.9 | 18.0 | –a | –a |
| α (i/N2) | (–) | (1.05) | (14.1) | (3.68) | ||
| Aged | 5.32 | 6.31 | 89.9 | 23.0 | –a | –a |
| α (i/N2) | (–) | (1.19) | (16.9) | (4.32) | ||
The relative position of permeability and selectivity for selected gas pairs, which is displayed in the Robeson plot (Fig. 6), serves as a critical tool for researchers and engineers to evaluate and compare the efficiency of new membrane materials in gas separation applications, such as carbon capture and natural gas purification. Developed by L. M. Robeson in 1991, revised in 2008 for most of the commercially important gas pairs and then updated by other groups for specific separations,44–47 the plot depicts the performance of various polymeric membranes for gas separation, with permeability on the x-axis and selectivity (often represented as the ratio of the permeabilities of two gases) on the y-axis. In our case, the CO2 permeability of Phen-PIM-1 proved to be similar to or higher than that of other tBu containing PIMs (i.e., PIM-SBF-5,40 PIM-TOT-SBF-5,38 and DPt-TMPD48). Its higher selectivity, however, places this π-extended dihydrophenazine-based PIM above the 2008 upper bound in a more interesting region of the Robeson plots (Fig. 6A and B), close to the requirements for high permeance/low-energy membranes for power plant post-combustion carbon dioxide capture foreseen by Merkel et al.49 The high O2/N2 and He/N2 selectivity (Fig. 6C and D) is a further indication that the separation is strongly dependent on a molecular sieving mechanism, as both O2 and especially He have a smaller kinetic diameter with respect to N2 although their solubilities are not so different in polymers. In comparison with traditional low free volume polymers containing tBu groups,48,50–56 the Phen-PIM-1 shows 2–3 orders of magnitude higher permeability values, while maintaining a similar (CO2/CH4, O2/N2) or slightly lower (CO2/N2) selectivity (Fig. 6), thus showing encouraging performance for gas separation application for biogas upgrading, CO2 capture from flue gas and O2/N2 enrichment from air. In this work, we also investigated physical aging by repeating gas permeability experiments 211 days after the first measurements. Physical aging is a common phenomenon observed in highly microporous materials that are out of their thermodynamic equilibrium state, which in highly microporous polymers is characterized by the relaxation of the polymer's molecular chains, which leads to a reduction in internal free volume and, consequently, a decrease in permeability.57 As shown in Fig. 6, the expected loss of free volume resulted in a relative decrease in permeability for all gas pairs, accompanied by an increase in selectivity. Notably, the reduction in permeability is effectively counterbalanced by enhanced selectivity, ensuring that the overall performance remains relatively unchanged on the Robeson plot. This further demonstrates the enhanced molecular sieving effect achieved through the use of the dihydrophenazine monomer.
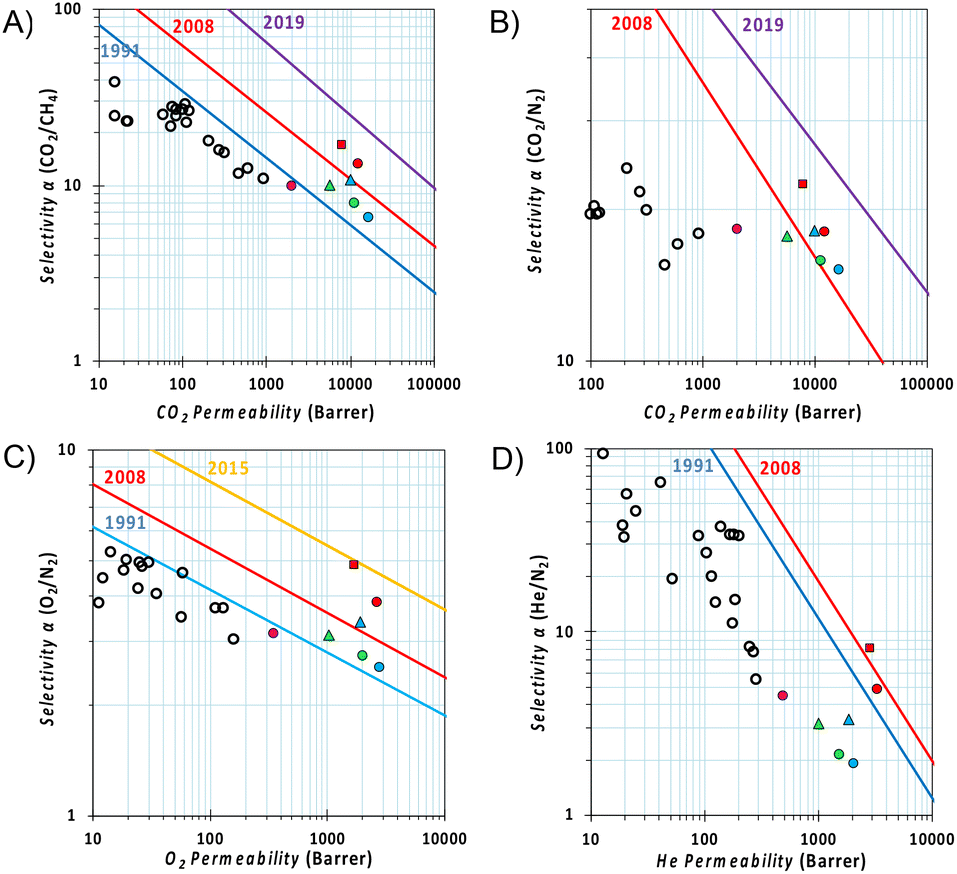 | ||
Fig. 6 Robeson diagrams for the (A) CO2/CH4; (B) CO2/N2; (C) O2/N2; and (D) He/N2 gas pairs with the upper bounds represented by blue lines for 1991,44 red lines for 2008,45 yellow lines for 2015,47 and purple lines for 2019.46 The gas permeabilities are reported for Phen-PIM-1 in red (circle  fresh and square fresh and square  211 day aged), for PIM-SBF-5 in blue (circle 211 day aged), for PIM-SBF-5 in blue (circle  fresh and triangle fresh and triangle  1439 day aged),40 for PIM-TOT-SBF-5 in green (circle 1439 day aged),40 for PIM-TOT-SBF-5 in green (circle  fresh and triangle fresh and triangle  543 day aged),38 for DPt-TMPD in pink (circle 543 day aged),38 for DPt-TMPD in pink (circle  fresh),48 and empty black circles are data for low free volume polymers with pendant tBu groups.48,50–56 fresh),48 and empty black circles are data for low free volume polymers with pendant tBu groups.48,50–56 | ||
3. Conclusions
In this work, we have successfully demonstrated the potential of π-extended dihydrophenazine scaffolds as versatile building blocks for the development of novel microporous polymers. This study represents one of the few examples of structural redox-switchable PIMs reported in the literature, further expanding the scope of functional materials in this class. Comprehensive characterization was carried out to assess the textural properties (surface area and pore size distribution), morphology (SEM), and chemical composition of the synthesized materials.Our findings highlight a remarkable balance between porosity, pore size, and gas selectivity, making these materials highly promising for gas separation applications. The solubility of Phen-PIM-1 in NMP enabled its processing into a self-standing membrane with mechanical properties in the range of other thin film forming materials, a crucial feature for practical implementation for gas separation. In contrast, the methyl-substituted counterpart Phen-PIM-2 exhibited poor solubility that prevented direct comparison of their membrane gas separation performance. These findings, although unfortunate, emphasize the critical role of large aliphatic tBu groups in enhancing solubility and facilitating the formation of robust films. Gas separation studies, conducted both in powder form (via IAST) and in membrane form (via time-lag measurements), revealed excellent separation performance. Notably, Phen-PIM-1 showed gas transport properties that place it in a highly competitive region of the Robeson upper bound for key gas pair separations such as CO2/CH4 and CO2/N2, demonstrating its strong potential for carbon capture applications. Additionally, its performance in O2/N2 and He/N2 separations suggests an enhanced molecular sieving effect, further broadening its applicability in gas purification processes.
Beyond its gas separation capabilities, this work marks an important milestone in the integration of dihydrophenazine-based compounds into the PIM family. The reversible redox behavior of these materials offers a rare example of switchable PIMs, opening new avenues for the design of stimuli-responsive porous materials. Given their highly promising properties, these materials lay a strong foundation for future optimization and functionalization, paving the way for their practical implementation in energy and environmental applications.
Data availability
The data that support the findings of this study are available in the ESI† of this article.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
J. D. kindly acknowledges FRA2024-2025 funded by the University of Trieste and Microgrants 2024 funded by Regione FVG (LR 2/2011, ART. 4). M. P. is the AXA-Chair for Bionanotechnology (2016–2026). This work was supported by the University of Trieste, INSTM, the Italian Ministry of Education MIUR (cofin Prot. 20228YFRNL), and la Agencia Estatal de Investigaciones through grant PID2022-140419OB-I00 funded by MCIN/AEI/10.13039/501100011033. M. V. and A. F. received funding from the European Union's Horizon Europe research and innovation program under grant agreement no. 101115488, within the EIC pathfinder project “Double-Active Membranes for a sustainable CO2 – DAM4CO2” (HORIZON-EIC-2022-PATHFINDERCHALLENGES-01). C.G.B and M.C. gratefully acknowledge UK Research and Innovation (UKRI) under the UK government's Horizon Europe funding guarantee [grant number 10083164] associated with DAM4CO2. The authors gratefully acknowledge Daniel M. Dawson and the University of St Andrews for the 13C ssNMR service.Notes and references
- A. G. Slater and A. I. Cooper, Science, 2015, 348, aaa8075 CrossRef PubMed.
- T. D. Bennett, F. X. Coudert, S. L. James and A. I. Cooper, Nat. Mater., 2021, 20, 1179–1187 CrossRef CAS PubMed.
- Y. Li, J. Brückel, M. Jereb, A. Zupanc, S. P. Hirvonen, S. Hietala, M. Kemell, Y. Wu, O. Fuhr, R. D. Jansen-van Vuuren, M. Carta and S. Bräse, Adv. Funct. Mater., 2024, 34, 2401957 CrossRef CAS.
- A. Gafiullina, B. P. Ladewig and J. Zhang, ACS Appl. Polym. Mater., 2023, 5, 1–30 CrossRef CAS.
- J. Zhou and B. Wang, Chem. Soc. Rev., 2017, 46, 6927–6945 RSC.
- X. Liu, C. F. Liu, W. Y. Lai and W. Huang, Adv. Mater. Technol., 2020, 5, 1–20 Search PubMed.
- J. Wu, F. Xu, S. Li, P. Ma, X. Zhang, Q. Liu, R. Fu and D. Wu, Adv. Mater., 2019, 31, 1–45 Search PubMed.
- J. Dosso, H. Oubaha, F. Fasano, S. Melinte, J.-F. Gohy, C. E. Hughes, K. D. M. Harris, N. Demitri, M. Abrami, M. Grassi and D. Bonifazi, Chem. Mater., 2022, 34, 10670–10680 CrossRef CAS PubMed.
- G. Gentile, B. Bartolomei, J. Dosso, N. Demitri, G. Filippini and M. Prato, Chem. Commun., 2023, 60, 602–605 RSC.
- A. L. Myers and J. M. Prausnitz, AlChE, 1964, 11, 121–127 CrossRef.
- A. R. Antonangelo, N. Hawkins, E. Tocci, C. Muzzi, A. Fuoco and M. Carta, J. Am. Chem. Soc., 2022, 144, 15581–15594 CrossRef CAS PubMed.
- S. Mondal, A. Hassan, S. A. Wahed, A. Kumar and N. Das, ACS Appl. Polym. Mater., 2025, 1503–1512 CrossRef CAS.
- N. B. McKeown, Polymer, 2020, 202, 122736 CrossRef CAS.
- H. S. Lau, A. Eugenia, Y. Weng and W. F. Yong, Prog. Mater. Sci., 2024, 145, 101297 CrossRef CAS.
- H. W. H. Lai, F. M. Benedetti, J. M. Ahn, A. M. Robinson, Y. Wang, I. Pinnau, Z. P. Smith and Y. Xia, Science, 2022, 375, 1390–1392 CrossRef CAS PubMed.
- W. Han, C. Zhang, M. Zhao, F. Yang, Y. Yang and Y. Weng, J. Membr. Sci., 2021, 636, 119544 CrossRef CAS.
- C. Rizzuto, F. Nardelli, M. Monteleone, L. Calucci, C. G. Bezzu, M. Carta, E. Tocci, E. Esposito, G. De Luca, B. Comesaña-Gándara, N. B. McKeown, B. Sayginer, P. M. Budd, J. C. Jansen and A. Fuoco, J. Mater. Chem. A, 2025, 13, 17865–17876 RSC.
- C. Ye, A. Wang, C. Breakwell, R. Tan, C. Grazia Bezzu, E. Hunter-Sellars, D. R. Williams, N. P. Brandon, P. A. A. Klusener, A. R. Kucernak, K. E. Jelfs, N. B. McKeown and Q. Song, Nat. Commun., 2022, 13, 3184 CrossRef CAS PubMed.
- D. Becker, N. Konnertz, M. Böhning, J. Schmidt and A. Thomas, Chem. Mater., 2016, 28, 8523–8529 CrossRef CAS.
- J. Dosso and M. Prato, Chem.–Eur. J., 2023, 29, e202203637 CrossRef CAS PubMed.
- J. Dosso, B. Bartolomei, N. Demitri, F. P. Cossío and M. Prato, J. Am. Chem. Soc., 2022, 144, 7295–7301 CrossRef CAS PubMed.
- N. B. McKeown, Curr. Opin. Chem. Eng., 2022, 36, 100785 CrossRef.
- C. Pathak, A. Gogoi, A. Devi and S. Seth, Chem.–Eur. J., 2023, 29, e202301512 CrossRef CAS PubMed.
- J. Jeromenok and J. Weber, Langmuir, 2013, 29, 12982–12989 CrossRef CAS PubMed.
- I. Rose, C. G. Bezzu, M. Carta, B. Comesanã-Gándara, E. Lasseuguette, M. C. Ferrari, P. Bernardo, G. Clarizia, A. Fuoco, J. C. Jansen, K. E. Hart, T. P. Liyana-Arachchi, C. M. Colina and N. B. McKeown, Nat. Mater., 2017, 16, 932–937 Search PubMed.
- B. Satilmis, M. Lanč, A. Fuoco, C. Rizzuto, E. Tocci, P. Bernardo, G. Clarizia, E. Esposito, M. Monteleone, M. Dendisová, K. Friess, P. M. Budd and J. C. Jansen, J. Membr. Sci., 2018, 555, 483–496 CrossRef CAS.
- A. Wang, C. Breakwell, F. Foglia, R. Tan, L. Lovell, X. Wei, T. Wong, N. Meng, H. Li, A. Seel, M. Sarter, K. Smith, A. Alvarez-Fernandez, M. Furedi, S. Guldin, M. M. Britton, N. B. McKeown, K. E. Jelfs and Q. Song, Nature, 2024, 635, 353–371 Search PubMed.
- D. Lozano-Castelló, D. Cazorla-Amorós and A. Linares-Solano, Carbon, 2004, 42, 1233–1242 Search PubMed.
- S. Lee, J. H. Lee and J. Kim, Korean J. Chem. Eng., 2018, 35, 214–221 CrossRef CAS.
- M. Tian, S. Rochat, H. Fawcett, A. D. Burrows, C. R. Bowen and T. J. Mays, Adsorption, 2020, 26, 1083–1091 CrossRef CAS.
- K. T. Leperi, R. Q. Snurr and F. You, Ind. Eng. Chem. Res., 2016, 55, 3338–3350 CrossRef CAS.
- R. Mobili, Y. Wu, C. X. Bezuidenhout, S. La Cognata, S. Bracco, M. Carta and V. Amendola, RSC Sustainability, 2024, 3345–3352 RSC.
- H. Zhou, C. Rayer, A. R. Antonangelo, N. Hawkins and M. Carta, ACS Appl. Mater. Interfaces, 2022, 14, 20997–21006 CrossRef CAS PubMed.
- P. M. Budd, N. B. McKeown, B. S. Ghanem, K. J. Msayib, D. Fritsch, L. Starannikova, N. Belov, O. Sanfirova, Y. Yampolskii and V. Shantarovich, J. Membr. Sci., 2008, 325, 851–860 Search PubMed.
- K. Nagai, A. Higuchi and T. Nakagawa, J. Polym. Sci., Part B Polym. Phys., 1995, 33, 289–298 CrossRef CAS.
- J. Chen, M. Longo, A. Fuoco, E. Esposito, M. Monteleone, B. Comesaña Gándara, J. Carolus Jansen and N. B. McKeown, Angew. Chem., Int. Ed., 2023, 62, 1–9 Search PubMed.
- C. G. Bezzu, A. Fuoco, E. Esposito, M. Monteleone, M. Longo, J. C. Jansen, G. S. Nichol and N. B. McKeown, Adv. Funct. Mater., 2021, 31, 1–10 CrossRef.
- S. A. Felemban, C. G. Bezzu, B. Comesaña-Gándara, J. C. Jansen, A. Fuoco, E. Esposito, M. Carta and N. B. McKeown, J. Mater. Chem. A, 2021, 9, 2840–2849 RSC.
- R. Malpass-Evans, I. Rose, A. Fuoco, P. Bernardo, G. Clarizia, N. B. McKeown, J. C. Jansen and M. Carta, Membranes, 2020, 10, 62–74 CrossRef CAS PubMed.
- C. G. Bezzu, M. Carta, M. C. Ferrari, J. C. Jansen, M. Monteleone, E. Esposito, A. Fuoco, K. Hart, T. P. Liyana-Arachchi, C. M. Colina and N. B. McKeown, J. Mater. Chem. A, 2018, 6, 10507–10514 RSC.
- M. Longo, M. P. De Santo, E. Esposito, A. Fuoco, M. Monteleone, L. Giorno, B. Comesaña-Gándara, J. Chen, C. G. Bezzu, M. Carta, I. Rose, N. B. McKeown and J. C. Jansen, Ind. Eng. Chem. Res., 2020, 59, 5381–5391 CrossRef CAS.
- C. R. Mason, L. Maynard-Atem, N. M. Al-Harbi, P. M. Budd, P. Bernardo, F. Bazzarelli, G. Clarizia and J. C. Jansen, Macromolecules, 2011, 44, 6471–6479 CrossRef CAS.
- B. W. Rowe, S. J. Pas, A. J. Hill, R. Suzuki, B. D. Freeman and D. R. Paul, Polymer, 2009, 50, 6149–6156 CrossRef CAS.
- L. M. Robeson, J. Membr. Sci., 1991, 62, 165–185 Search PubMed.
- L. M. Robeson, J. Membr. Sci., 2008, 320, 390–400 CrossRef CAS.
- B. Comesaña-Gándara, J. Chen, C. G. Bezzu, M. Carta, I. Rose, M. C. Ferrari, E. Esposito, A. Fuoco, J. C. Jansen and N. B. McKeown, Energy Environ. Sci., 2019, 12, 2733–2740 RSC.
- R. Swaidan, B. Ghanem and I. Pinnau, ACS Macro Lett., 2015, 4, 947–951 CrossRef CAS PubMed.
- R. Sulub-Sulub, M. I. Loría-Bastarrachea, H. Vázquez-Torres, J. L. Santiago-García and M. Aguilar-Vega, J. Membr. Sci., 2018, 563, 134–141 CrossRef CAS.
- T. C. Merkel, H. Lin, X. Wei and R. Baker, J. Membr. Sci., 2010, 359, 126–139 CrossRef CAS.
- Z. Qiu, G. Chen, Q. Zhang and S. Zhang, Eur. Polym. J., 2007, 43, 194–204 CrossRef CAS.
- N. Belov, R. Chatterjee, R. Nikiforov, V. Ryzhikh, S. Bisoi, A. G. Kumar, S. Banerjee and Y. Yampolskii, Sep. Purif. Technol., 2019, 217, 183–194 CrossRef CAS.
- S. Sánchez-García, F. A. Ruiz-Treviño, M. J. Aguilar-Vega and M. G. Zolotukhin, Ind. Eng. Chem. Res., 2016, 55, 7012–7020 CrossRef.
- D. Ayala, A. E. Lozano, J. De Abajo, C. García-Perez, J. G. De La Campa, K. V. Peinemann, B. D. Freeman and R. Prabhakar, J. Membr. Sci., 2003, 215, 61–73 CrossRef CAS.
- N. Esteban, M. Juan-y-Seva, C. Aguilar-Lugo, J. A. Miguel, C. Staudt, J. G. de la Campa, C. Álvarez and Á. E. Lozano, Polymers, 2022, 14, 5517–5538 CrossRef CAS PubMed.
- M. Calle, Á. E. Lozano, J. G. De La Campa and J. De Abajo, Macromolecules, 2010, 43, 2268–2275 CrossRef CAS.
- M. Calle, C. García, A. E. Lozano, J. G. De la Campa, J. De Abajo and C. Álvarez, J. Membr. Sci., 2013, 434, 121–129 CrossRef CAS.
- Z. X. Low, P. M. Budd, N. B. McKeown and D. A. Patterson, Chem. Rev., 2018, 118, 5871–5911 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ta02477c |
| This journal is © The Royal Society of Chemistry 2025 |