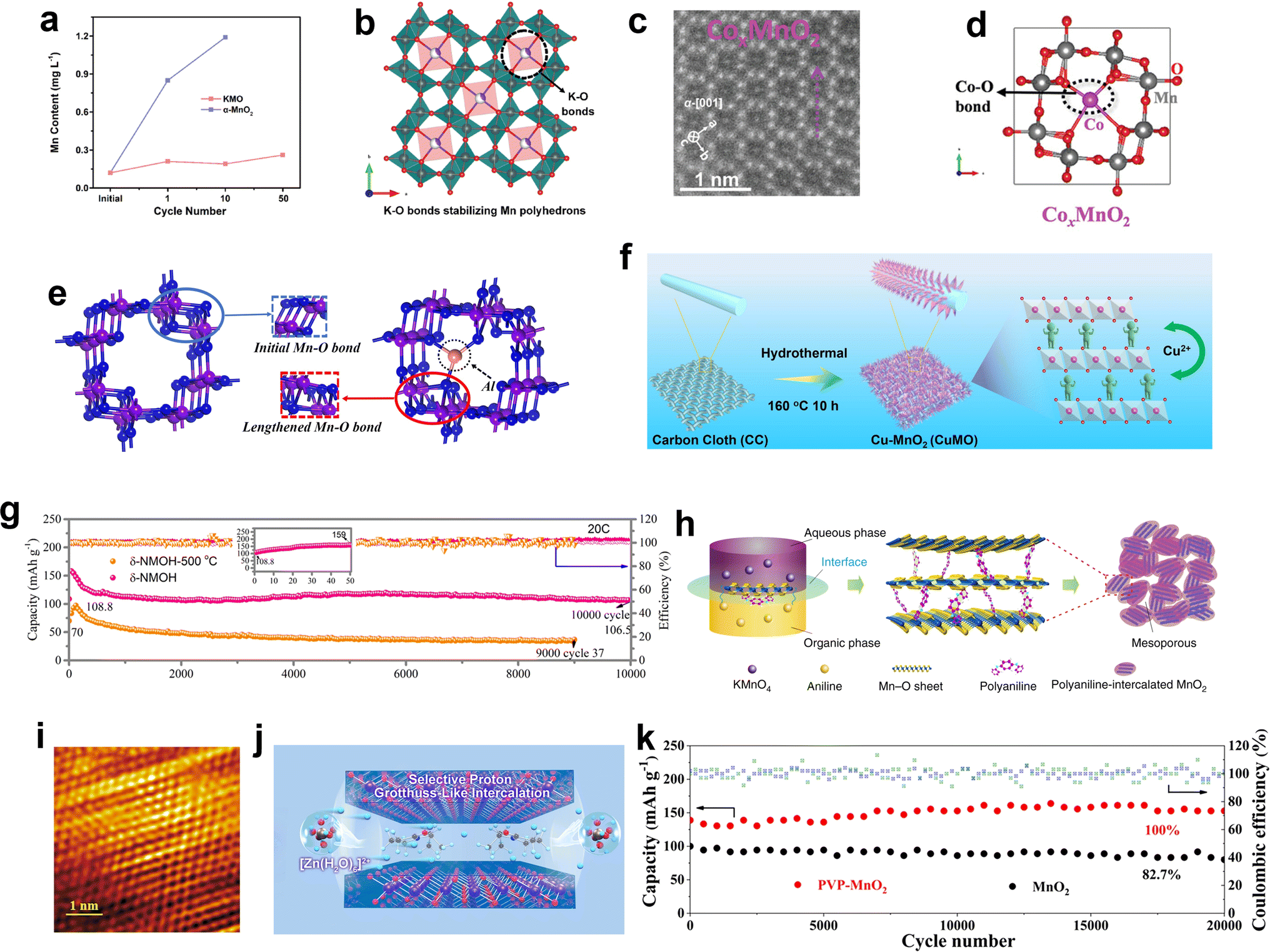
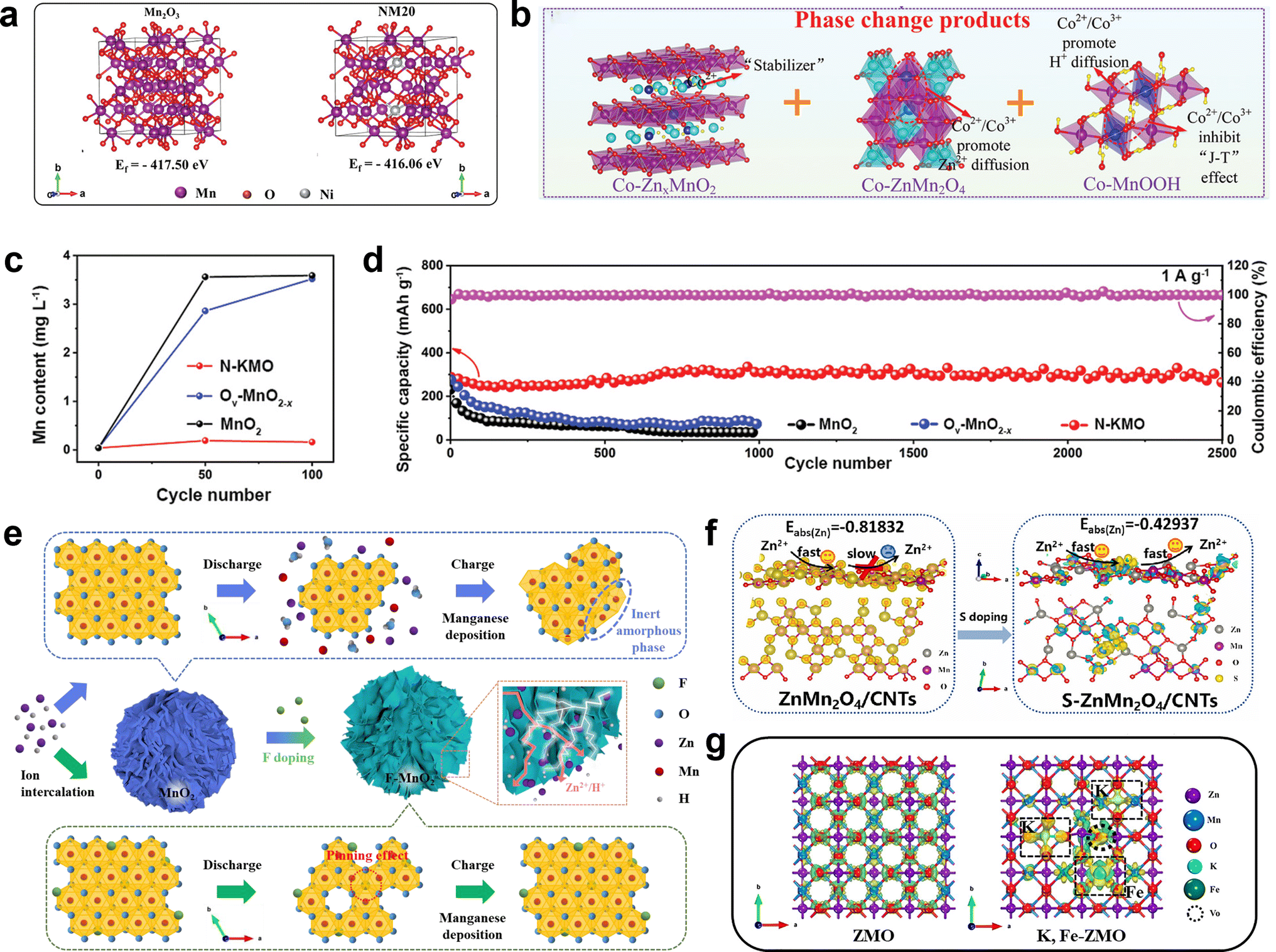
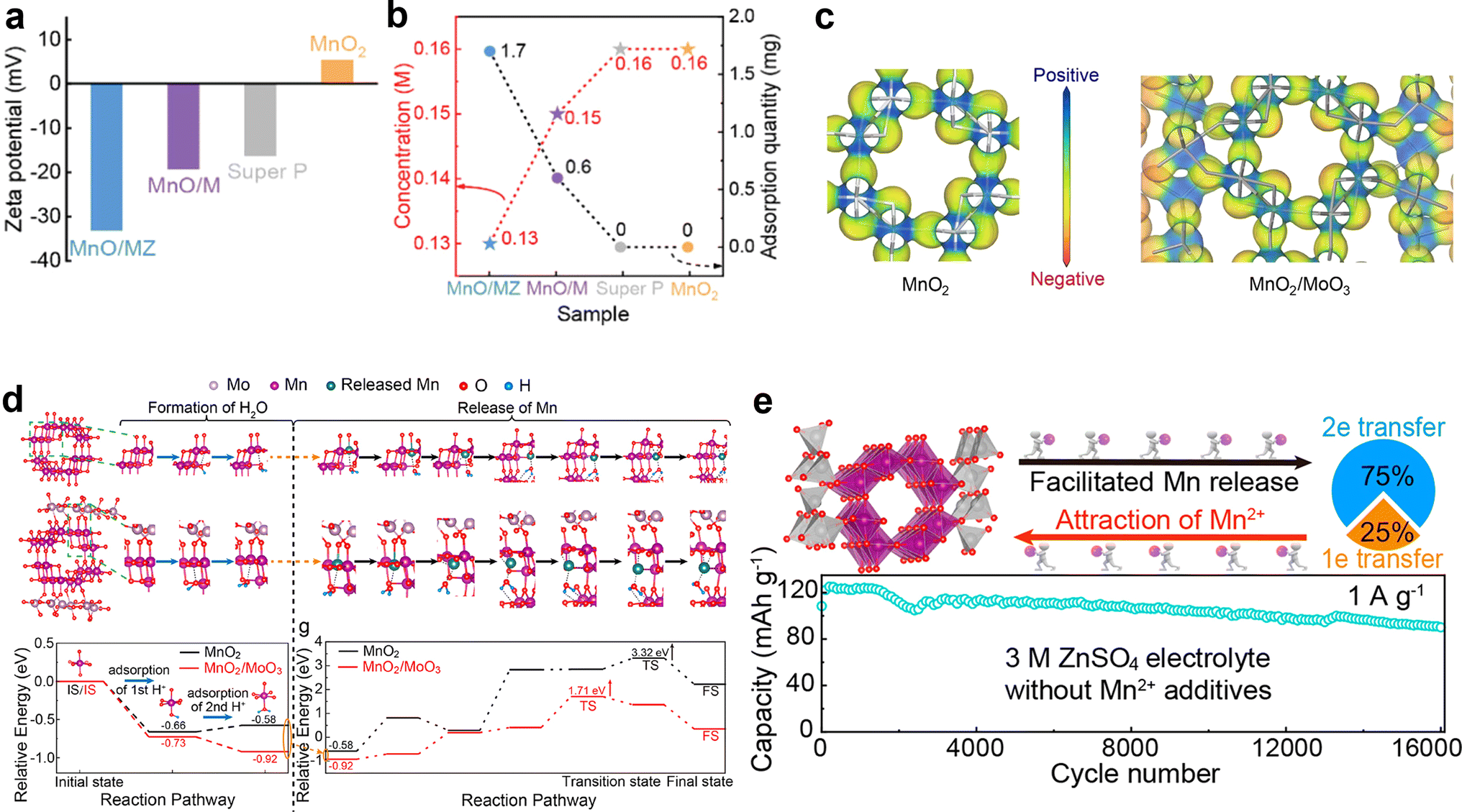
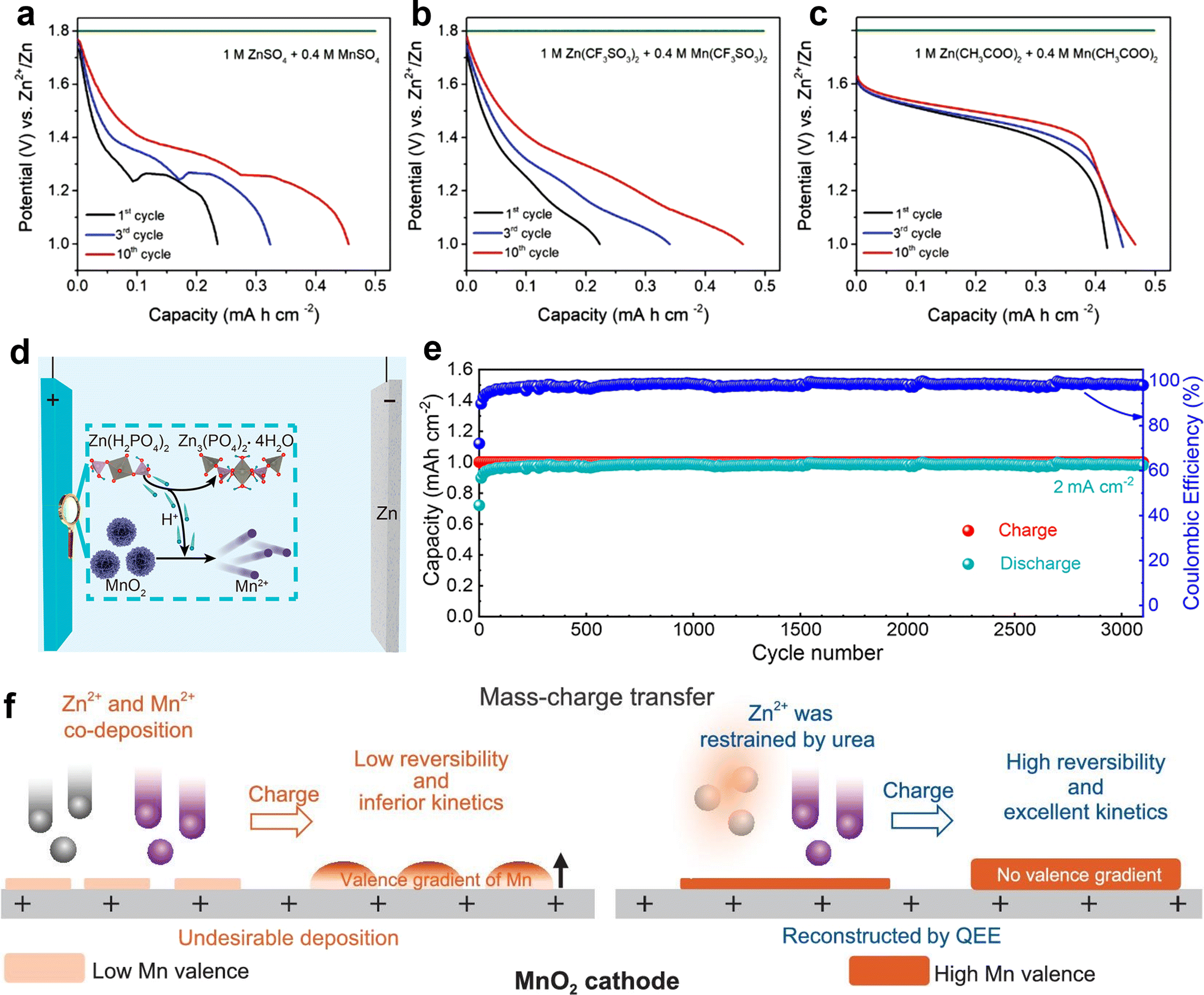
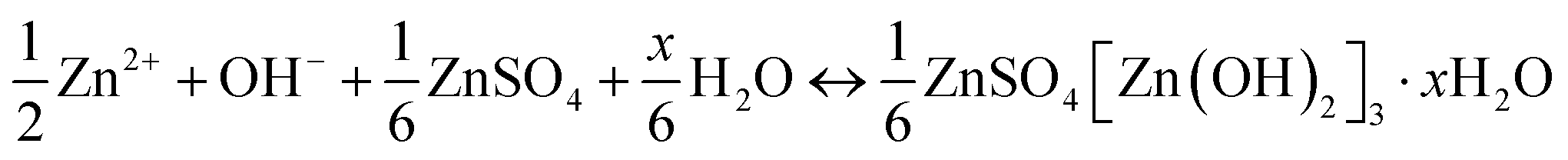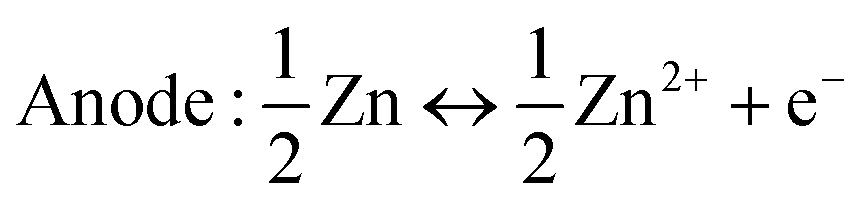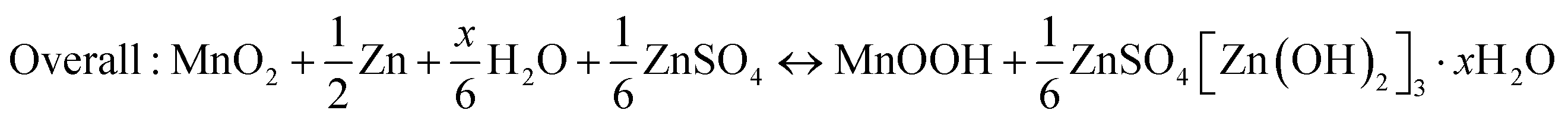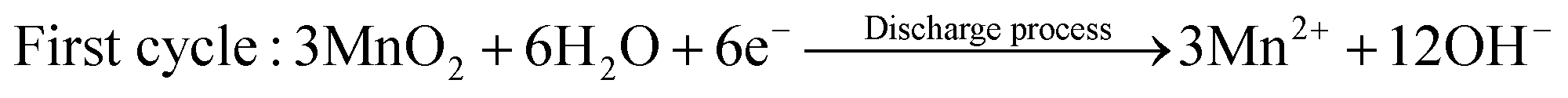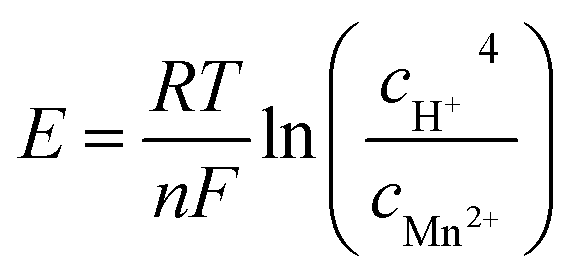DOI:
10.1039/D4SC00510D
(Review Article)
Chem. Sci., 2024,
15, 7441-7473
Insights into the cycling stability of manganese-based zinc-ion batteries: from energy storage mechanisms to capacity fluctuation and optimization strategies
Received
22nd January 2024
, Accepted 18th March 2024
First published on 11th April 2024
Abstract
Manganese-based materials are considered as one of the most promising cathodes in zinc-ion batteries (ZIBs) for large-scale energy storage applications owing to their cost-effectiveness, natural availability, low toxicity, multivalent states, high operation voltage, and satisfactory capacity. However, their intricate energy storage mechanisms coupled with unsatisfactory cycling stability hinder their commercial applications. Previous reviews have primarily focused on optimization strategies for achieving high capacity and fast reaction kinetics, while overlooking capacity fluctuation and lacking a systematic discussion on strategies to enhance the cycling stability of these materials. Thus, in this review, the energy storage mechanisms of manganese-based ZIBs with different structures are systematically elucidated and summarized. Next, the capacity fluctuation in manganese-based ZIBs, including capacity activation, degradation, and dynamic evolution in the whole cycle calendar are comprehensively analyzed. Finally, the constructive optimization strategies based on the reaction chemistry of one-electron and two-electron transfers for achieving durable cycling performance in manganese-based ZIBs are proposed.
1 Introduction
The rapidly increasing energy consumption and environmental issues make it urgent to utilize large-scale electrical energy storage (EES) systems to store intermittent but renewable energy, such as solar energy, wind, and tidal energy.1–3 Among the various EES systems, lithium-ion batteries (LIBs) have been widely used for dozens of years owing to their high gravimetric energy density and excellent cycling performance.4,5 However, the high price of lithium and its limited resources coupled with safety and environmental concerns for the use of flammable organic electrolytes hinder the application of LIBs as large-scale EES systems.6,7
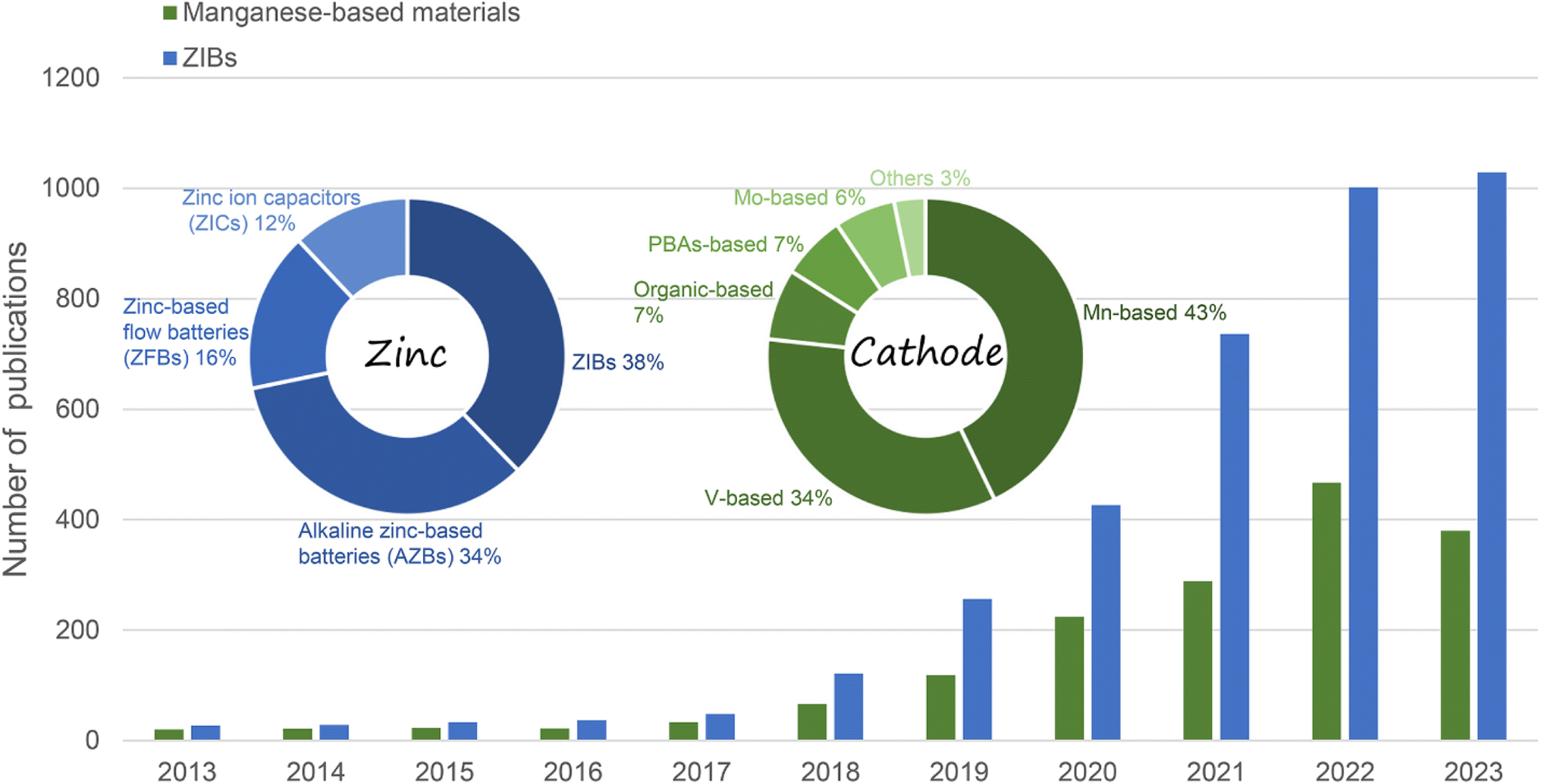
Fortunately, the emerging aqueous rechargeable batteries, particularly aqueous zinc-ion batteries (ZIBs) hold great promise for large-scale EES because of the low cost, natural abundance, high stability, low electrochemical potential (−0.763 V vs. the NHE) and high theoretical capacity (5855 mA h cm−3 and 820 mA h g−1) of Zn metal coupled with the high safety and high ionic conductivity of aqueous electrolytes.8–12 In general, the energy storage performance of ZIBs is mainly related to the cathode material, Zn anode, electrolyte, and separator.13 Recently, although studies in the literature have focused on the issues associated with the Zn anode,14–17 the nature of cathode materials essentially dominates the electrochemical performance of ZIBs, such as capacity, working potential, cycling stability, and rate performance. However, owing to the much larger radius of hydrated Zn2+ (4.3 Å) than Li+ (0.76 Å), many cathode materials with excellent electrochemical performance in LIBs are not suitable for ZIBs.18 Consequently, the limited performance of cathode materials for ZIBs greatly impede the development of high-performance EES systems. Thus far, manganese-based materials,19,20 vanadium-based materials,21–23 Prussian blue and its analogues,24–26 Chevrel phase compounds,27,28 and organic compounds have been exploited as cathode materials for ZIBs.29,30 Amongst them, manganese-based materials are considered the most attractive cathodes for ZIBs and have been widely and intensively investigated owing to their low cost, natural abundance, low toxicity, multivalent states, high operation voltage, and satisfactory capacity (Fig. 1).31–33
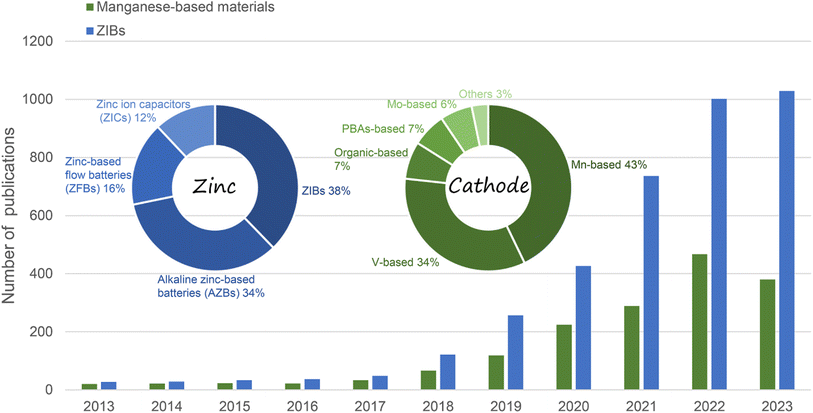 |
| | Fig. 1 Number of publications in recent years on manganese-based ZIBs and ZIBs (data from Web of Science, December 2023). | |
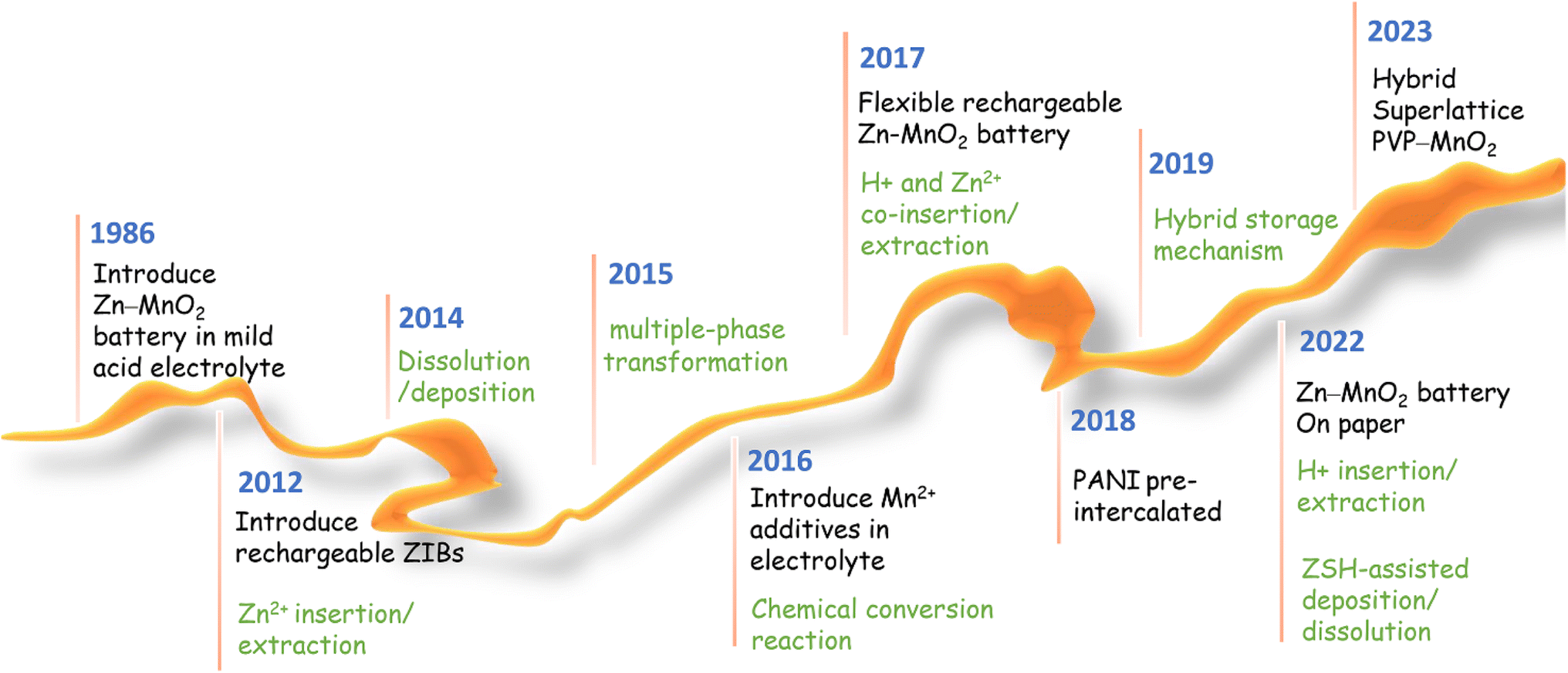
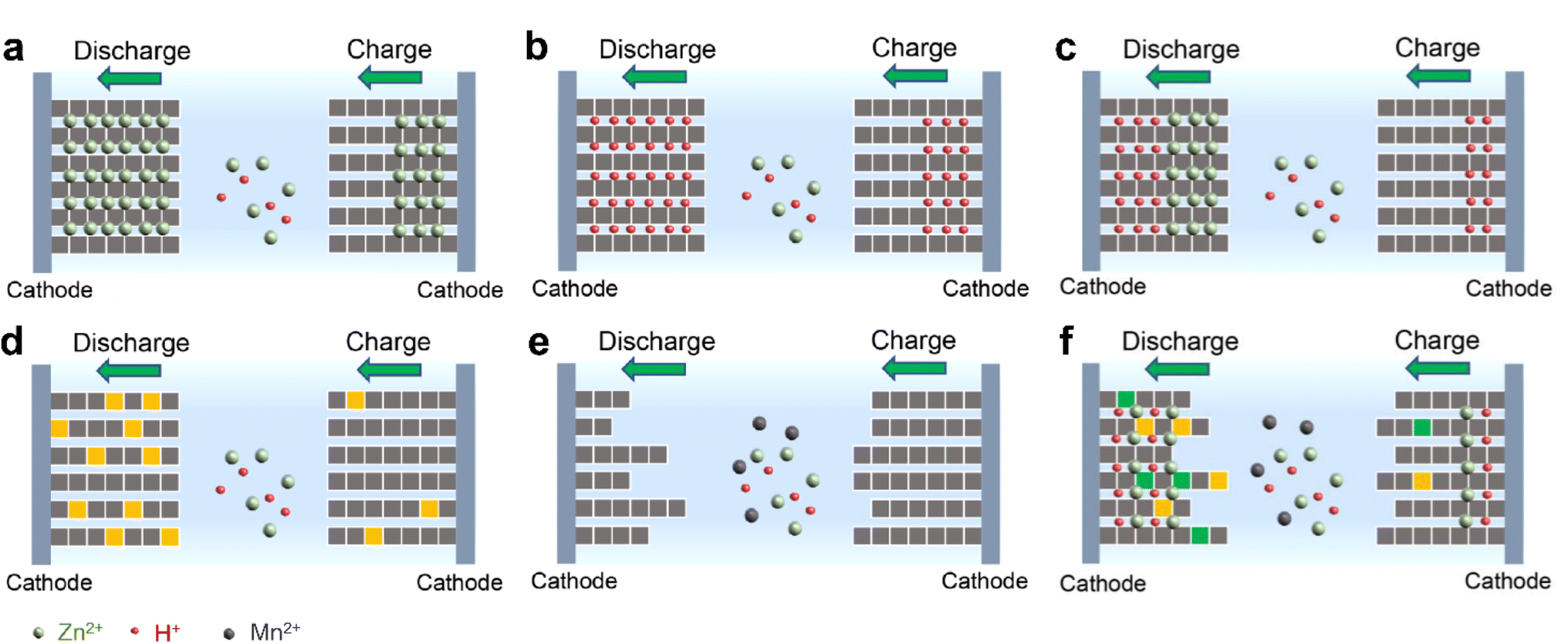
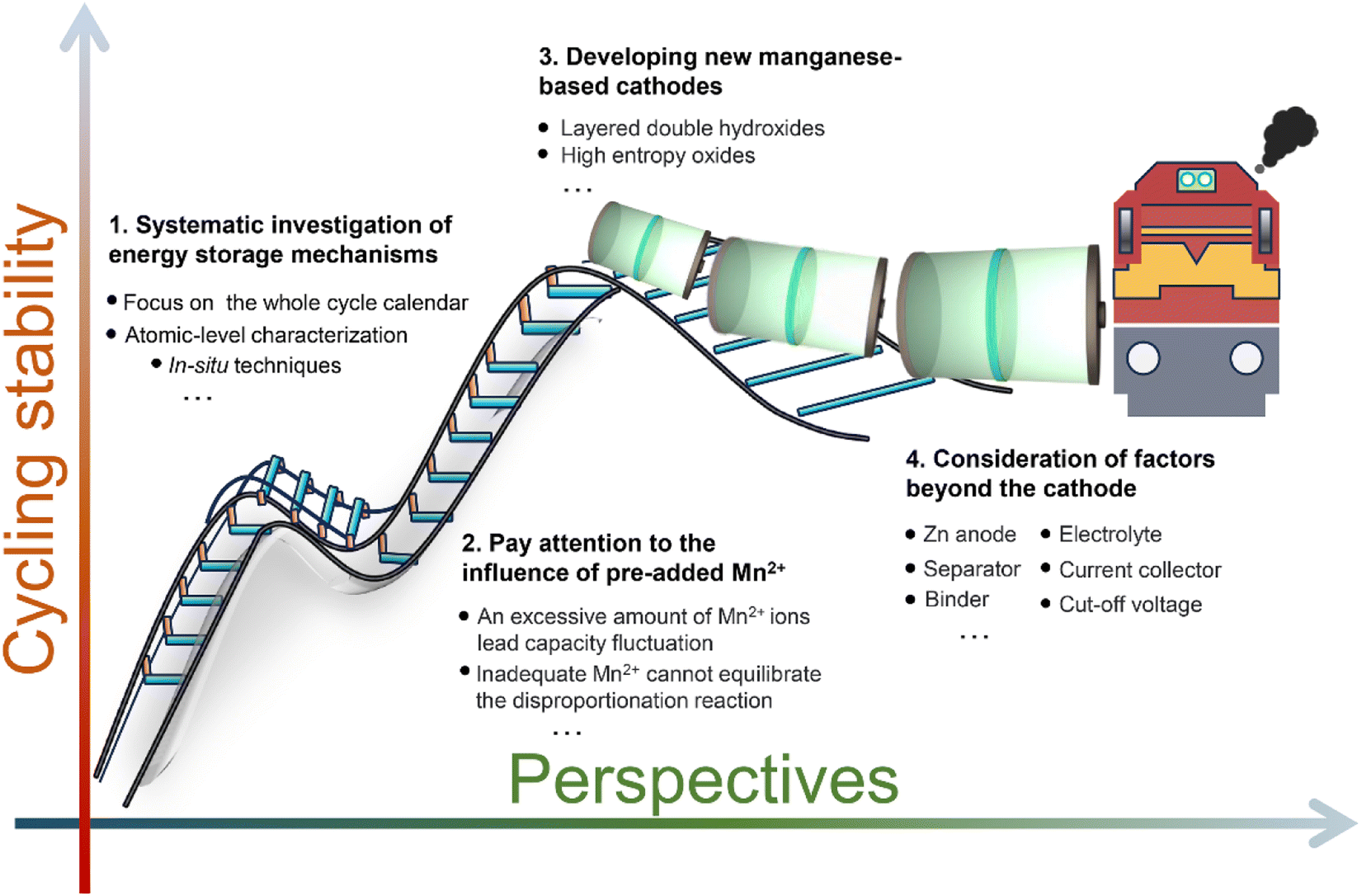
Significant progress has been made in manganese-based ZIBs over the past decade, as depicted in Fig. 2. Nevertheless, manganese-based cathodes in ZIBs involve various and controversial energy storage mechanisms, and six major energy storage mechanisms have been successively discovered in the past ten years: (1) Zn2+ insertion/extraction, (2) H+ insertion/extraction, (3) H+ and Zn2+ co-insertion/extraction, (4) chemical conversion, (5) dissolution–deposition, and (6) hybrid storage mechanisms (Fig. 3). These complex mechanisms invariably confuse readers, particularly for recent students in this field, and thus need to be comprehensively and clearly summarized. Moreover, the unsatisfactory long-term cycling stability (particularly at a low current) of manganese-based ZIBs has severely hampered their practical application. Meanwhile, a fluctuating capacity has been common observed (including capacity activation process and capacity degradation process) during long-term cycling and its mechanism has not been clearly understood, leading to a lack of robust methods to improve their cycling stability. Although some good reviews have been published on manganese-based ZIBs to guide researchers,34–43 these reviews mainly focused on optimization strategies for high capacity and fast reaction kinetics, seldomly mentioning the dynamic fluctuation of capacity and lacking a deep comprehension of essential reasons for capacity activation and degradation. Consequently, a comprehensive understanding of the capacity fluctuation is urgently needed to guide researchers to design manganese-based ZIBs with stable cycling performances.
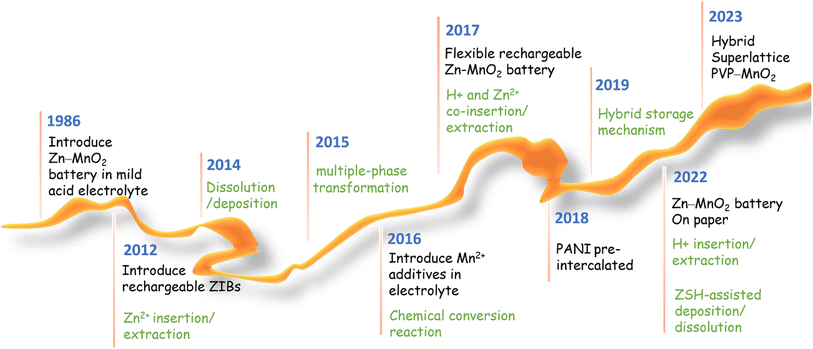 |
| | Fig. 2 Progress on manganese-based ZIBs. | |
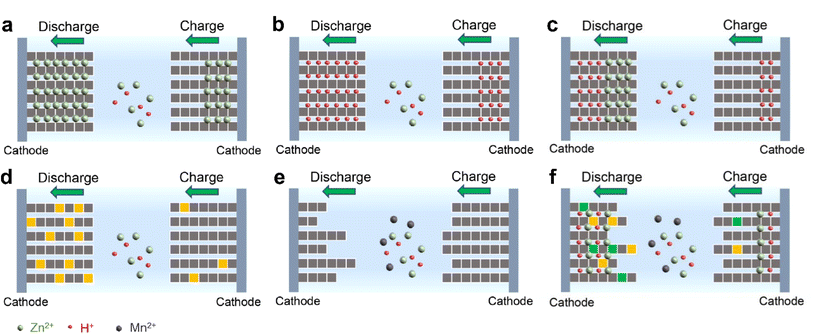 |
| | Fig. 3 Illustration of energy storage mechanisms for manganese-based ZIBs. (a) Zn2+ insertion/extraction, (b) H+ insertion/extraction, (c) H+ and Zn2+ co-insertion/extraction, (d) chemical conversion reaction, (e) dissolution–deposition, and (f) hybrid storage mechanism. | |
In this review, the energy storage mechanisms of manganese-based ZIBs with different structures are systematically clarified and summarized. More importantly, the capacity fluctuation of manganese-based ZIBs is comprehensively analyzed for the first time. We not only summarize the immediate causes of capacity activation and degradation, but also analyze the inherent dynamic evolutions (such as pH, reaction mechanisms, and active materials) that result in capacity fluctuations. Furthermore, the constructive optimization strategies based on the reaction chemistry of one-electron and two-electron transfer for achieving durable cycling performance manganese-based ZIBs are proposed, respectively. Finally, the conclusions and outlooks on the present reaction mechanisms and optimization strategies for achieving durable cycling performances based on our personal perspectives are also provided. This review provides a comprehensive overview and the research directions focusing on the recent reaction chemistry and cycling stability, which will provide a guide for future studies on manganese-based ZIBs.
2 Crystal structure of manganese-based materials
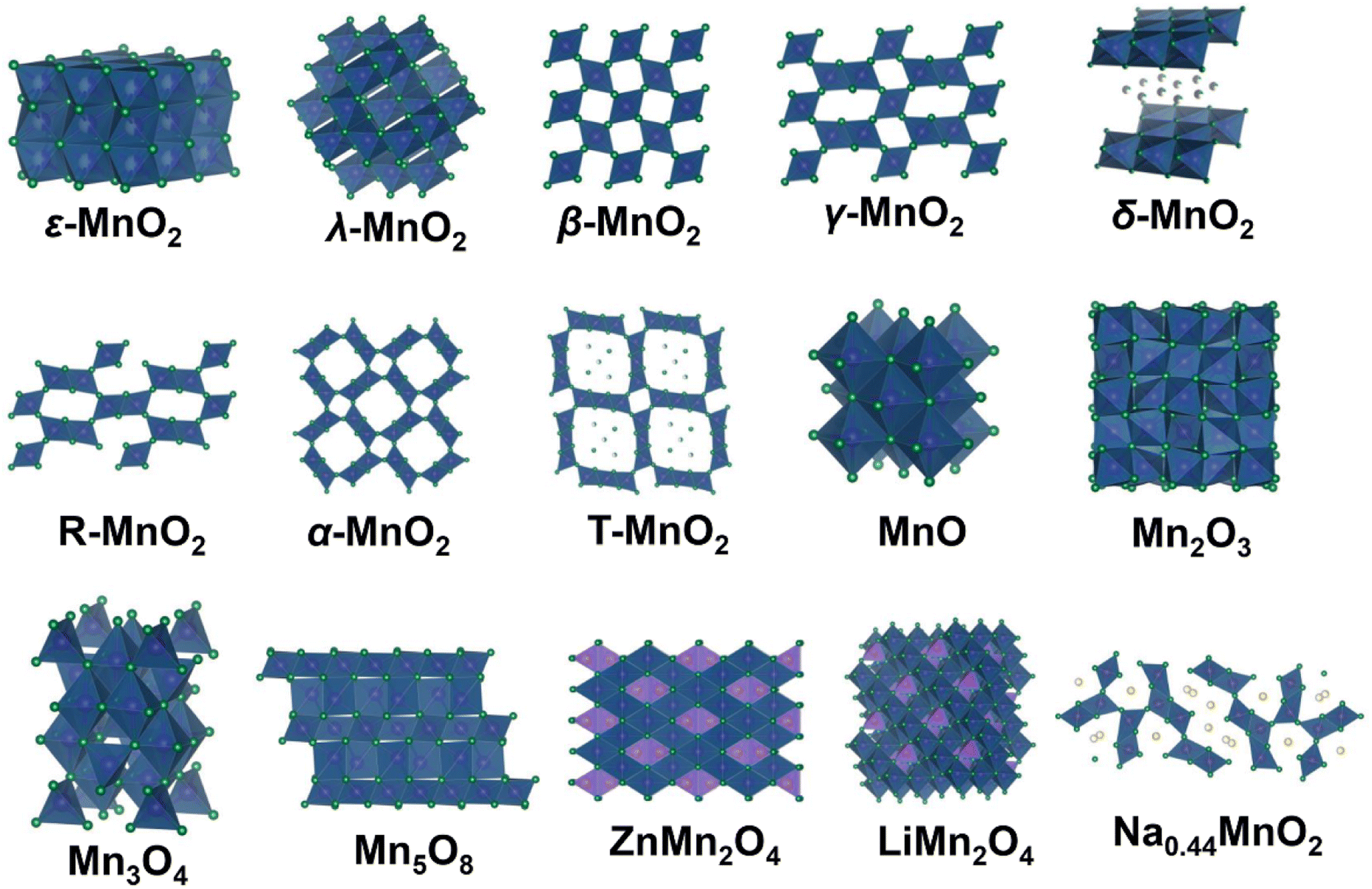
In this review, we divide manganese-based materials into four categories, including MnO2 polymorphs, low-valence manganese oxides, metal manganate (AMnxOy, A = Li, Na, Zn, Mg, Ca, etc.) and other manganese-based compounds. Various atomic structures and chemical constituents result in a variety of multivalent phases (+2, +3, +4, +5, and +7) and crystal structures for manganese-based materials (Fig. 4). The crystal structures of manganese-based materials are closely related to the energy storage performance of the corresponding ZIBs and dominate the reaction chemistry of cathodes. Therefore, it is essential to first comprehend the crystal structures of manganese-based materials.
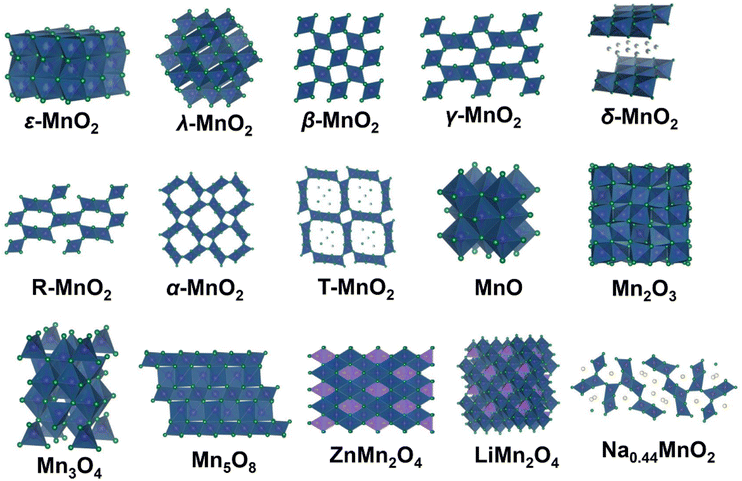 |
| | Fig. 4 Structural drawings of various manganese-based materials. | |
2.1 MnO2 polymorphs
MnO2 is one of the most popular cathodes for ZIBs, which possesses a high theoretical capacity of 308 mA h g−1 or 616 mA h g−1 based on one-electron or two-electron transfer, respectively. Impressively, MnO2 has diverse polymorphs with distinct tunneled or layered structures composed of basic octahedral units (MnO6) connected in different ways. These diverse polymorphs can be summarized based on their tunnel size, as shown in Fig. 4. ε-MnO2 (akhtenskite) is a special polymorph, which is densely packed without any tunnel structure. Its dense packing structure is not conducive to rapid cation insertion/extraction, resulting in low electrochemical activity for ZIBs.44 Regulating structures such as introducing defects is an efficient strategy to increase the number of active sites to improve the electrochemical performance of ε-MnO2.45 λ-MnO2 (spinel) possesses a three-dimensional (3D) spinel structure with [1 × 1] tunnels, which is a metastable form of MnO2 and can be easily obtained through acid treatment of LiMn2O4 at ambient temperature.46 It has been reported that λ-MnO2 only can deliver a poor capacity of 33.8 mA h g−1,47 showing the lowest electrochemical activity among the MnO2 polymorphs. Its unsatisfactory capacity may result from its limited number of 3D tunnels, restricting the migration of Zn2+.48 β-MnO2 (pyrolusite) is one of the most thermodynamically stable structure, which also has narrow [1 × 1] tunnels. However, the numerous open channels of β-MnO2 can accommodate more cation insertion/extraction than λ-MnO2. Recent studies have demonstrated the excellent electrochemical performance of β-MnO2,49–52 for example, Chen et al.53 reported that β-MnO2 can deliver a remarkable reversible capacity of 225 mA h g−1 and outstanding long-term cycling stability with 94% capacity retention over 2000 cycles. γ-MnO2 (nsutite) possesses a hybrid tunnel structure with [1 × 1] and [1 × 2] tunnels, which has been successfully applied in alkaline Zn–MnO2 batteries and is also promising in ZIBs.54,55 δ-MnO2 (birnessite) features a typical two-dimensional (2D) layered structure with a wide interlayer spacing of 7.0 Å, which is favorable for cation intercalation/extraction. In addition, the layered structure of δ-MnO2 originating from its source avoids a phase transition from tunneled MnO2 to a layered phase, which greatly reduces severe volume changes. Notably, the interlayer space of δ-MnO2 usually contains many cations (such as K+ and Na+) and structural water, which are beneficial to stabilize the MnO6 layers. R-MnO2 (ramsdellite) has a moderate tunnel size of [1 × 2] and is larger than β-MnO2 but narrower than α-MnO2, which can be regarded as a part of γ-MnO2. Chen et al.56 found that R-MnO2 uniquely shows a low manganese dissolution property compared to other MnO2 polymorphs due to the thermodynamic feasibility and moderate volume changes of its [1 × 2] tunnels. α-MnO2 (hollandite) possesses large [2 × 2] tunnels, which is considered an ideal material for cation intercalation/extraction. To date, α-MnO2 exhibits one of the most comprehensive electrochemical performances among manganese-based materials and has been extensively studied by researchers.38,44 T-MnO2 (todorokite) has large [3 × 3] tunnels with many intercalated metal cations and water molecules, and these guest species can effectively stabilize the tunnel framework and prevent structural collapse. However, although T-MnO2 possesses a much larger tunnel structure than α-MnO2, its practical capacity is much lower than α-MnO2, which can be ascribed to the pre-intercalation of too many metal cations and water molecules, reducing the number of potential Zn2+ insertion sites.57
2.2 Low-valence manganese oxides
Low-valence manganese oxides, including MnO, Mn2O3, Mn3O4 and Mn5O8, are also important cathode materials for ZIBs. In the case of MnO, with Mn2+ ions possessing a coordination number of six, it displays tetrahedral or octahedral coordination, together with specific magnetic anisotropy.58 MnO is intrinsically inactive without any tunnels for Zn2+ and H+ insertion/extraction. However, it exhibits an excellent electrochemical performance comparable to MnO2 after the initial activation cycles,59,60 and its activation mechanisms will be discussed in detail in Section 4.2. In the structure of Mn2O3, the Mn3+ ions are arranged in an octahedral coordination with each O ion surrounded by four Mn ions. Similar to MnO, it lacks migration channels, and thus it has been found that pristine Mn2O3 can only deliver a limited capacity of 78 mA h g−1 in the first discharge.61 In the case of spinel Mn3O4 (chemical formula of Mn2+Mn23+O4), Mn2+ and Mn3+ occupy the tetrahedral (4a) and octahedral (8d) sites in the intermediate distorted cubic close-pack array of oxygen anions (16h), respectively. Mn3O4 demonstrates thermodynamic stability, making it typically irreversible as the final discharge product.43 Mn5O8 (chemical formula of Mn22+Mn34+O8) is another layered structure manganese oxide composed of 2D octahedral sheets of [Mn34+O8] with Mn2+ located above and below the vacant Mn4+ sites.62 The large interlayer space of Mn5O8 offers abundant active sites and allows facile ion diffusion, and it has been reported that Mn5O8/rGO can deliver a high reversible capacity of 260 mA h g−1 at 100 mA g−1 with a stable cycling performance over 1000 cycles.63 However, there are few reports on the application of Mn5O8 as a cathode material for ZIBs, and thus more efforts should be devoted to the study of Mn5O8.
2.3 Metal manganate AMnxOy
Metal manganate AMnxOy (A refers to Zn, Li, Na, Mg, etc.) can also be utilized as a cathode material for ZIBs, including ZnMn2O4, LiMn2O4, MgMn2O4, Na0.44MnO2, and CoMn2O4. In the structure of spinel-type ZnMn2O4, oxygen atoms are arranged in a hexagonal close-packed configuration, forming an octahedral site. In a perfect spinel lacking cation deficiency, the Zn2+ ions migrate from one tetrahedral site (4a) to another by passing through an unoccupied octahedral site (8c), encountering significant electrostatic repulsion from the Mn cations in the neighboring octahedral site (8d), thereby strongly impeding Zn2+ diffusion.64 Meanwhile, due to the thermodynamic stability of ZnMn2O4, which is commonly the final product of manganese-based ZIBs in the capacity attenuation period, the capacity degradation caused by ZnMn2O4 will be discussed in detail in Section 4.1.3. Defect engineering is an effective strategy to reduce the electrostatic barrier, leading to easier Zn2+ diffusion.39 The spinel MgMn2O4 possesses an analogous tetragonal symmetry structure to ZnMn2O4. Soundharrajan et al.65 fabricated MgMn2O4 as a cathode with ZnSO4, MgSO4, and MnSO4 as the electrolyte, showing the reversible insertion/extraction of both Mg2+ and Zn2+. In the structure of spinel-type LiMn2O4, the Mn3+/Mn4+ ions occupy the octahedral sites (16d) within a cubic close-packed array of oxygen atoms, while the Li+ ions occupy the tetrahedral sites (8a) and migrate through a three-dimensional channel.66 LiMn2O4 has been widely investigated in LIBs due to the inherent advantages of the manganese element and its high specific power.67 Interestingly, LiMn2O4 also shows an excellent performance in ZIBs. For instance, Zhang et al.68 reported the fabrication of a Zn/LiMn2O4 battery containing ZnSO4, Li2SO4, and MnSO4 electrolyte, which exhibited a high capacity of more than 300 mA h g−1 with long-term cycling stability over 2000 cycles. In addition, tunnel-type Na0.44MnO2 with interconnected 3D S-shaped channels allows facile cation diffusion. Similar to LiMn2O4 and MgMn2O4, Na0.44MnO2 demonstrates highly reversible insertion/extraction of Na+ and Zn2+ in hybrid electrolytes composed of ZnSO4 and Na2SO4.69,70
2.4 Other manganese-based compounds
Compared with crystalline manganese oxides, amorphous manganese oxides with short-range ordered atomic arrangements are ignored in ZIBs due to their lack of a high manganese valency and cation host structure. However, it was demonstrated that amorphous manganese oxides with unique superiority of structural plasticity are beneficial to accommodate the volume changes caused by Zn2+/H+ intercalation/extraction. In addition, the abundant defects in amorphous manganese oxides can serve as reversible cation storage sites, giving rise to fast ion diffusion kinetics and high specific capacity.71,72 Besides, MnS,73 Mn2SiO4,74,75 LaMnO3,76 MnSe,77etc. have been developed as available cathode materials for ZIBs. However, most of these cathode materials exhibit unsatisfactory electrochemical performances compared to manganese oxides owing to their intrinsically poor electrochemical activity and structural stability. Therefore, more efforts are needed to improve the inherent shortcomings of these newly developed manganese-based compounds.
In summary, the crystal structure of MnO2 polymorphs, low-valence manganese oxides, metal manganate, and other manganese-based compounds was discussed. It has been found that the electrochemical performance of these materials in ZIBs is predominantly influenced by their crystal structure. Materials with large tunnel or layered structures generally exhibit superior insertion/extraction capabilities for Zn2+/H+, whereas those with dense and spinel-type structures, which offer fewer reaction sites for Zn2+/H+ ions, typically demonstrate inferior electrochemical performances. Table 1 provides a compilation of the previously reported electrochemical performances of manganese-based materials in ZIBs.
Table 1 Summary of the electrochemical performance of manganese-based materials in ZIBs
| Cathodes |
Electrolyte |
Voltage window |
Specific capacity (current density) |
Cycling performance |
Ref. |
| α-MnO2 |
2 M ZnSO4 + 0.1 M MnSO4 |
1–1.8 V |
285 mA h g−1 (1/3C) |
92% capacity retention over 5000 cycles at 5C |
78
|
| β-MnO2 |
3 M Zn(CF3SO3)2 + 0.1 M Mn(CF3SO3)2 |
0.8–1.9 V |
225 mA h g−1 (0.65C) |
94% capacity retention over 2000 cycles at 6.5C |
53
|
| γ-MnO2 |
1 M ZnSO4 |
1–1.8 V |
285 mA h g−1 (0.05 mA cm−2) |
63% capacity retention over 40 cycles at 0.5 mA cm−2 |
54
|
| δ-MnO2 |
1 M Zn(TFSI)2 + 0.1 M Mn(TFSI)2 |
1–1.8 V |
238.8 mA h g−1 (0.2C) |
93% capacity retention over 4000 cycles at 20C |
79
|
| ε-MnO2 |
3 M ZnSO4 + 0.1 M MnSO4 |
0.8–1.8 V |
312 mA h g−1 (0.1 A g−1) |
80.1% capacity retention over 1000 cycles at 1 A g−1 |
45
|
| λ-MnO2 |
0.2 M ZnSO4 + 0.1 M MnSO4 |
0.8–1.9 V |
250 mA h g−1 (0.02 A g−1) |
115 mA h g−1 over 500 cycles at 1 A g−1 |
80
|
| R-MnO2 |
2 M ZnSO4 + 0.1 M MnSO4 |
0.8–1.85 V |
168 mA h g−1 (0.1 A g−1) |
118.3 mA h g−1 over 500 cycles at 0.3 A g−1 |
56
|
| T-MnO2 |
1 M ZnSO4 |
0.7–2.0 V |
108 mA h g−1 (1/2C) |
83% capacity retention over 50 cycles at 50 mA g−1 |
57
|
| MnO |
2 M ZnSO4 |
1–1.9 V |
330 mA h g−1 (0.1 A g−1) |
80.7% capacity retention over 300 cycles at 0.3 A g−1 |
81
|
| Mn2O3 |
2 M ZnSO4 + 0.2 M MnSO4 |
1.0–1.8 V |
292 mA h g−1 (0.2C) |
89% capacity retention over 3000 cycles at 3.08 A g−1 |
82
|
| Mn3O4 |
2 M ZnSO4 |
0.8–1.9 V |
85.6 mA h g−1 (0.2 A g−1) |
106.1 mA h g−1 over 300 cycles at 0.5 A g−1 |
83
|
| Mn8O5/rGO |
2 M ZnSO4 + 0.1 M MnSO4 |
0.8–1.9 V |
260 mA h g−1 (0.1 A g−1) |
98.8% capacity retention over 1000 cycles at 0.5 A g−1 |
63
|
| ZnMn2O4 |
1 M ZnSO4 + 0.05 M MnSO4 |
0.8–1.9 V |
70.2 mA h g−1 (3.2 A g−1) |
106.5 mA h g−1 over 300 cycles at 0.1 A g−1 |
84
|
| MgMn2O4 |
1 M ZnSO4 + 1 M MgSO4 + 0.1 M MnSO4 |
0.5–1.9 V |
247 mA h g−1 (0.05 A g−1) |
80% capacity retention over 500 cycles at 0.5 A g−1 |
65
|
| LiMn2O4 |
1 M ZnSO4 + 1 M Li2SO4 + 0.1 M MnSO4 |
0.8–1.85 V |
300 mA h g−1 (0.1 A g−1) |
93.5% capacity retention over 500 cycles at 1 A g−1 |
68
|
| CoMn2O4 |
2 M ZnSO4 + 0.2 M MnSO4 |
0.8–1.9 V |
232.3 mA h g−1 (0.1 A g−1) |
— |
85
|
| MnCo2O4 |
2 M ZnSO4 + 0.5 M MnSO4 |
0.8–1.8 V |
595.3 mA h g−1 (0.05 A g−1) |
85% capacity retention over 250 cycles at 0.2 A g−1 |
86
|
| Na0.44MnO2 |
3 M ZnSO4 + 0.1 M MnSO4 |
1.0–1.8 V |
301.3 mA h g−1 (0.1 A g−1) |
69.3% capacity retention over 800 cycles at 1 A g−1 |
87
|
| MnS |
2 M ZnSO4 + 0.1 M MnSO4 |
0.8–2 V |
335.7 mA h g−1 (0.3 A g−1) |
104 mA h g−1 over 4000 cycles at 3 A g−1 |
73
|
| Mn2SiO4 |
1 M ZnSO4 + 0.1 M MnSO4 |
1.0–1.8 V |
154 mA h g−1 (0.1 A g−1) |
55% capacity retention over 40 cycles at 0.1 A g−1 |
74
|
| LaMnO3 |
2 M ZnSO4 + 0.2 M MnSO4 |
1.0–1.8 V |
226 mA h g−1 (0.1 A g−1) |
113 mA h g−1 over 1000 cycles at 0.5 A g−1 |
76
|
3 Energy storage mechanisms
The energy storage mechanisms of manganese-based ZIBs are very complicated and controversial due to the various crystal structures of manganese-based materials and the intricate environment of the aqueous electrolyte in comparison to LIBs with insertion–alloying–conversion reactions.88 In addition, the reaction of Zn2+ deposition to generate large flake-like Zn4SO4(OH)6·xH2O (ZHS) on the cathode surface during discharge makes the determination of the real reactions more difficult. Undoubtedly, numerous researchers have devoted significant efforts to studying the real reaction mechanisms for a long time. Accordingly, to date, the storage mechanisms of manganese-based ZIBs can be divided into six categories, as presented here.
3.1 Zn2+ insertion/extraction mechanism
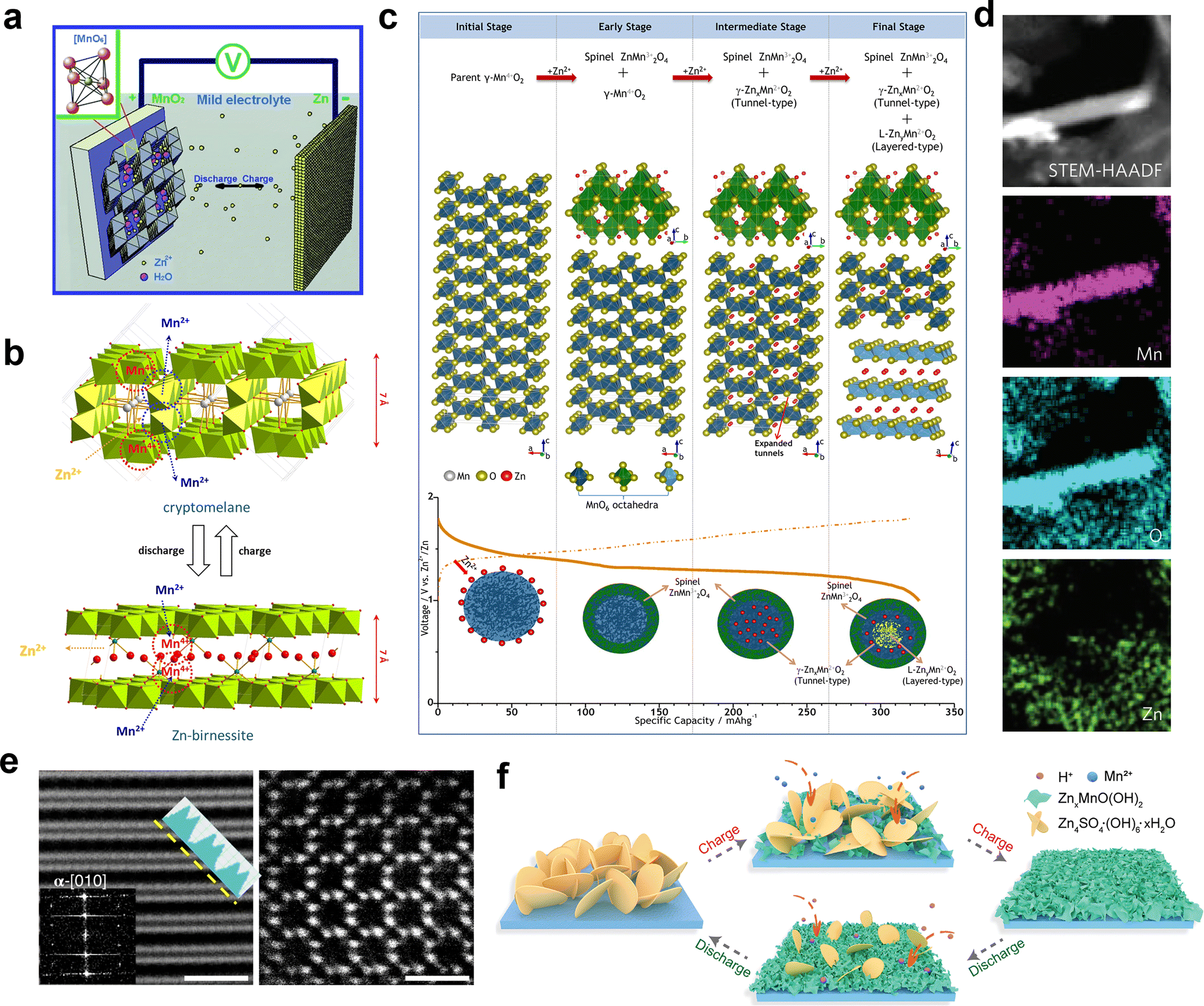
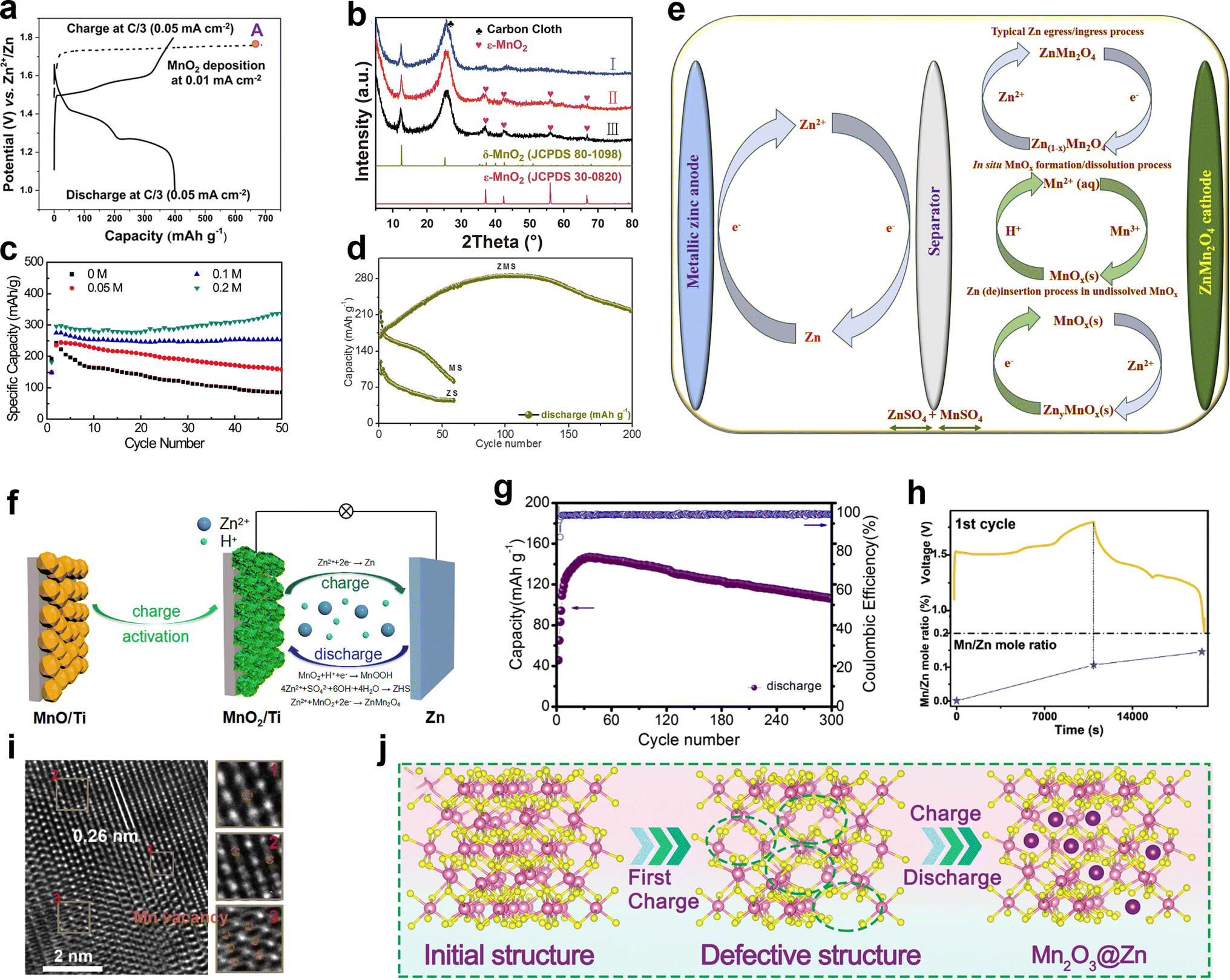
The Zn2+ insertion/extraction mechanism is the earliest accepted mechanism, which was first systematically proven by Kang et al.89 They concluded that the Zn2+ from the ZnSO4 or Zn(NO3)2 aqueous electrolyte can be reversibly intercalated into the tunnels of α-MnO2, and the anodic zinc can reversibly undergo dissolution/deposition upon discharging and charging (Fig. 5a). The reactions are summarized as below:| | | Cathode: Zn2+ + 2e− + 2α-MnO2 ↔ ZnMn2O4 | (1) |
| | | Anode: Zn ↔ Zn2+ + 2e− | (2) |
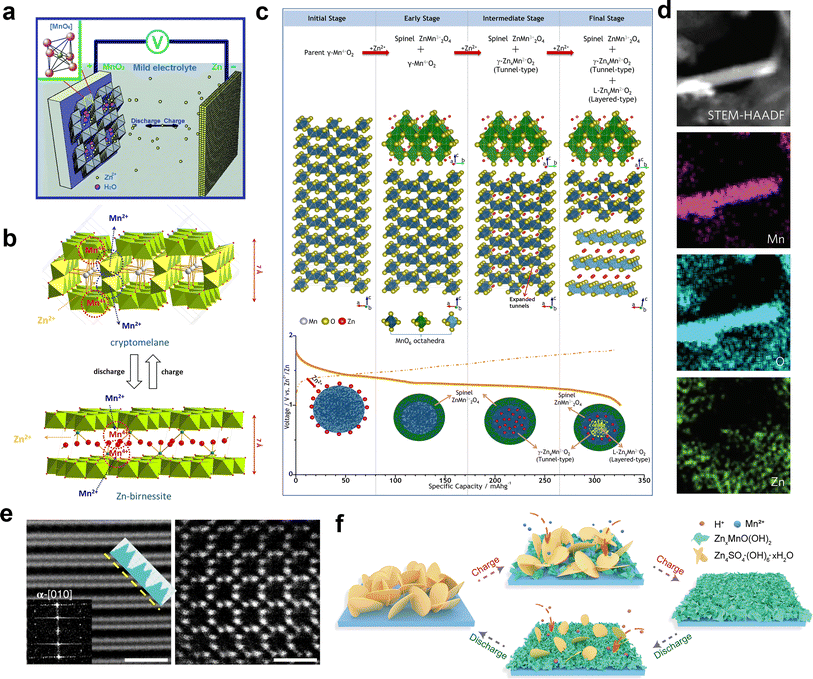 |
| | Fig. 5 Various energy storage mechanisms of manganese-based ZIBs. (a) Schematic of the chemistry of a zinc-ion battery. Reproduced with permission.89 Copyright 2012, Wiley-VCH. (b) Schematic illustrating the mechanism of zinc intercalation in α-MnO2. Reproduced with permission.90 Copyright 2014, Springer Nature. (c) Schematic illustration of the reaction pathway of Zn-insertion in a γ-MnO2 cathode. Reproduced with permission.54 Copyright 2015, the American Chemical Society. (d) STEM-EDS mappings of the elemental distributions of Mn, O and Zn in the MnO2 electrode in the discharged state during the first cycle. Reproduced with permission.78 Copyright 2016, Springer Nature. (e) Atomic scale [001] and projections of MnO2 demonstrating essentially ‘empty’ tunnels without presence of heavy cations. Reproduced with permission.94 Copyright 2022, Springer Nature. (f) Schematic of the ZSH-assisted deposition–dissolution reaction model. Reproduced with permission.95 Copyright 2022, Wiley-VCH. | |
Oh et al.90 demonstrated the intercalation mechanism of Zn2+ in α-MnO2 during discharge, involving an electrochemical-induced reversible phase transition of α-MnO2 from a tunneled to layered structure. This transition is induced by the dissolution of manganese from α-MnO2 during the discharge process, resulting in the formation of layered Zn-birnessite (11 Å). The original tunneled structure is restored through the incorporation of Mn2+ ions back into the layers of Zn-birnessite during charging (Fig. 5b). The reaction can be depicted as follows:
| | | Mn4+(s) + e− → Mn3+(s) | (3) |
| | | 2Mn3+(s) → Mn4+(s) + Mn2+(aq) | (4) |
| | | Mn2+(aq) → Mn4+(s) + 2e− | (5) |
Later, the intercalation of Zn2+ ions into α-MnO2 was rectified, resulting in the formation of buserite (11 Å) and the previously observed Zn-birnessite is attributed to the dehydration of the original buserite structure.91 Owing to the narrow [1 × 1] tunnels in β-MnO2, it has been suggested to be unfavorable for Zn intercalation. Interestingly, Chen et al.53 revealed that tunnel-type β-MnO2 undergoes a phase transition to layer-type Zn-buserite during the initial discharge process, followed by the reversible insertion/extraction of Zn2+ ions in the layered structure. The phase transition of MnO2 during charge/discharge was deeply investigated by Alfaruqi et al.,54 revealing a complex multiple-phase transformation process for γ-MnO2. It was proposed that the original tunnel-type γ-MnO2 undergoes a structural transformation to form a spinel-type Mn(III) phase (ZnMn2O4), as well as two new intermediary Mn(II) phases, namely tunnel-type γ-ZnxMnO2 and layered L-ZnyMnO2. These phases with multi-oxidation states coexist after complete electrochemical Zn-insertion (Fig. 5c). In addition, the presence of cation-defects in ZnMn2O4 spinel permits reversible Zn2+ insertion/extraction reactions, and the Mn deficiency in ZnMn2O4 can effectively reduce the electrostatic barrier for Zn2+ diffusion, resulting in the enhanced mobility of Zn2+ ions, and consequently faster electrode reaction kinetics.64 However, it is worth noting that while the mechanism of Zn2+ insertion/extraction was initially demonstrated by researchers, some of the early analyses may have been misleading due to the absence of advanced characterization techniques and limited understanding of ZIBs.
3.2 Chemical conversion reaction mechanism
Different from Zn2+ insertion in the MnO2 host, which is responsible for energy storage, the chemical conversion reaction mechanism is a new discovery, which was first proposed by Pan et al.78 During the discharge process, MnO2 undergoes a reaction with H+ derived from H2O to form MnOOH. To achieve charge neutrality in the system, the remaining OH− ions react with ZnSO4 and H2O in the aqueous electrolyte, resulting in the formation of large flake-like ZnSO4[Zn(OH)2]3·xH2O. The XRD pattern of the MnOOH phase and STEM-EDS mappings of the elemental distributions of Mn, O, and Zn in the discharged state strongly demonstrate the reaction chemistry between H+ and MnO2 in the cathode (Fig. 5d). In the absence of H+, organic Zn2+-based electrolytes exhibit limited capacity; however, upon the addition of H2O, the Zn/α-MnO2 battery demonstrates electrochemical behavior similar to that observed in aqueous electrolyte, thereby providing further evidence for the occurrence of a chemical conversion reaction. The reaction chemistry can be formulated as follows:| | | Cathode: H2O ↔ H+ + OH− | (6) |
| | | MnO2 + H+ + e− ↔ MnOOH | (7) |
| | 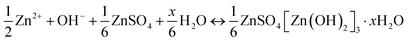 | (8) |
| | 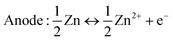 | (9) |
| | 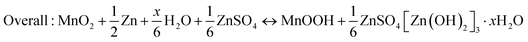 | (10) |
Although ample evidence has indicated the occurrence of chemical conversion reactions leading to the transformation of MnO2 into MnOOH, the operando XRD study unequivocally confirmed the absence of a crystalline MnOOH phase. It is worth noting that ex situ analysis methods, such as highly crystalline ZHS, may introduce external interferences and occasionally mistake the formation of MnOOH.92,93 Therefore, more advanced in situ characterization methods are required to further validate the mechanism of the chemical conversion reaction.
3.3 H+ insertion/extraction mechanism
Different from the chemical conversion between MnO2 and MnOOH revealed by Pan et al., a view of H+ intercalation in α-MnO2 tunnels was proved by combining atomic-scale electron microscopy, electrochemistry and atomistic simulations.94 The in situ XRD analysis revealed the absence of new phases except for the reversible generation and disappearance of ZHS. However, during discharge, the slight shifts in the peaks of α-MnO2 towards lower angles indicate cation insertion. Down-tunnel STEM imaging demonstrated severe distortion of the tunnels near the surface region of MnO2 nanowires, strongly supporting cation intercalation. Nevertheless, EDS mapping showed the absence of Zn element in MnO2 nanowires, suggesting that Zn2+ insertion in the tunnels is unlikely. Furthermore, atomic-scale imaging clearly indicated the presence of unoccupied tunnels without any heavy cations, providing crucial evidence against Zn2+ insertion in the MnO2 lattice (Fig. 5e). The computational results further support the favorability of H+ insertion over Zn2+. The reaction chemistry can be denoted as follows:| | | α-MnO2 + H+ + e− ↔ α-H1MnO2 | (11) |
3.4 H+ and Zn2+ co-insertion/extraction mechanism
The H+ and Zn2+ insertion/extraction mechanism is widely accepted and has been reported in numerous studies. Considering the existence of both H+ and Zn2+ cations in the electrolyte, Wang et al.96 were the first to demonstrate that MnO2 undergoes successive H+ and Zn2+ insertion processes. The reaction kinetics at different depths of discharge, as observed through GITT and EIS, indicated that the reaction on the first plateau occurs much faster than on the second plateau due to the smaller size of H+ compared to Zn2+. Furthermore, even without ZnSO4 in the electrolyte, there was still evidence of H+ insertion reactions occurring, with the first sloping plateau being attributed solely to H+ insertion. Additionally, discharge down to 1.3 V and 1.0 V resulted in the formation of MnOOH and ZnMn2O4 respectively, further confirming the subsequent insertion of H+ and Zn2+. Subsequently, the study conducted by Jin et al.79 also provided compelling evidence to substantiate the co-insertion mechanism. However, this investigation revealed that the intercalation of Zn2+ in MnO2 is governed by a non-diffusion-controlled process, followed by H+ conversion reaction, rather than the consequent insertion of H+ and Zn2+. These two distinct conclusions confuse researchers in terms of which cation reacts with MnO2 firstly and should be disclosed by further evidence.
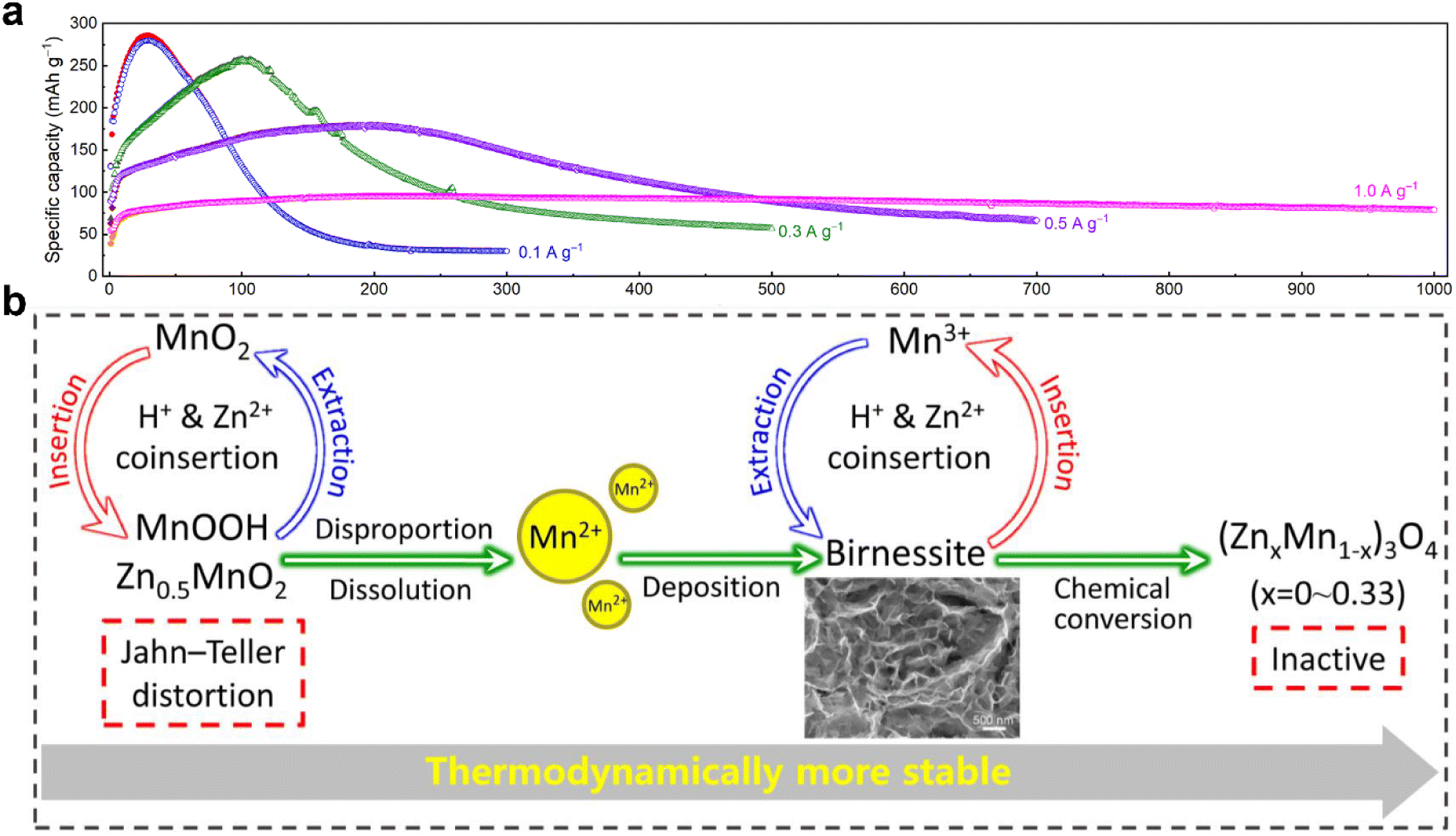
3.5 Dissolution/deposition mechanism
The dissolution of Mn2+ caused by Jahn–Teller distortion is commonly considered the reason for capacity attenuation.42,44,97 Thus, to alleviate the issue of Mn dissolution, Mn2+ additives have been introduced to shift the disproportionation reaction 2Mn3+ ↔ Mn2+ + Mn4+.78 However, Oh et al.98 discovered that Mn2+ dissolved in the discharge state can be electro-oxidized back to MnO2 through a reversible dissolution/deposition process of ZHS on the electrode surface, which is triggered by pH changes in the electrolyte. This phenomenon has been observed in MnO2-based flow batteries with acidic electrolytes.99| | | Cathode: 3MnO2 + 8Zn2+ + 2SO42− + 16H2O + 6e− → 3Mn2+ + 2Zn4(OH)6(SO4)·5H2O↓ | (12) |
| | | Anode: 3Zn → 3Zn2+ + 6e− | (13) |
| | | Overall: 3MnO2 + 3Zn + 5Zn2+ + 2SO42− + 16H2O ↔ 3Mn2+ + 2Zn4(OH)6(SO4)·5H2O↓ | (14) |
The dissolution/deposition mechanism was also confirmed by Liang and coworkers.100 They concluded that the dissolution and deposition of MnO2 contribute major capacity in the energy storage process. During the initial discharge, the dissolution of the original MnO2 turns it to Mn2+ in the electrolyte, and in the subsequent charging process, Mn2+ is deposited on the electrode surface as birnessite-MnO2 rather than the original MnO2. In the subsequent cycles, birnessite-MnO2 and Mn2+ are reversibly dissolved/deposited during charge/discharge.
| | 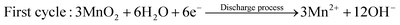 | (15) |
| |  | (16) |
| | | Subsequent cycles: 3 birnessite-MnO2 + 6H2O + 6e− ↔ 3Mn2+ + 12OH− | (17) |
| | | Side reaction: 12OH− + 2SO42− + 8Zn2+ + 8H2O ↔ 2Zn4(OH)6SO4·4H2O | (18) |
Wu et al.101 further verified the Mn dissolution/deposition faradaic mechanism that governs the electrochemistry by providing direct evidence through operando, spatiotemporal-resolved synchrotron X-ray fluorescence mapping measurements on a custom aqueous Zn/α-MnO2 battery. By simultaneously visualizing and quantifying the distribution of Mn species in the electrolyte, they observed the formation of aqueous Mn species during discharge and depletion upon charge.
Different from the MnO2/Mn2+ dissolution/deposition reaction, Chen et al.95 reported a zinc sulfate hydroxide-assisted dissolution/deposition reaction for aqueous Zn–Mn batteries. They found that the role of the α-MnO2 cathode is to consume H+ driving the generation of ZHS and the release of Mn2+ in the electrolyte provides an Mn source for the next charge deposition, with α-MnO2 contributing little capacity in the subsequent cycles. The energy conversion occurs by the reversible dissolution/deposition between the layered ZnxMnO(OH)2 and ZHS (Fig. 5f). According to this energy storage mechanism, it is suggested that ZSH plays an essential role as an active material rather than being solely a byproduct affecting the cycling performance, contrary to previous literature reports.43,100,102 Based on this understanding of ZSH-assisted deposition/dissolution reactions, successful designs of Zn–ZnO, Zn–MgO, and Zn–CaO batteries have been achieved. The energy storage mechanism is summarized as follows:
| | | Charge: Zn4(OH)6SO4·4H2O + Mn2+ → ZnxMnO(OH)2 + 4H+ + SO42− + 2e− | (19) |
| | | Discharge: ZnxMnO(OH)2 + 4H+ + 2e− → Zn2+ + Mn2+ + 3H2O | (20) |
| | | 6OH− + SO42− + 4Zn2+ + 4H2O → Zn4(OH)6SO4·4H2O | (21) |
3.6 Hybrid storage mechanism
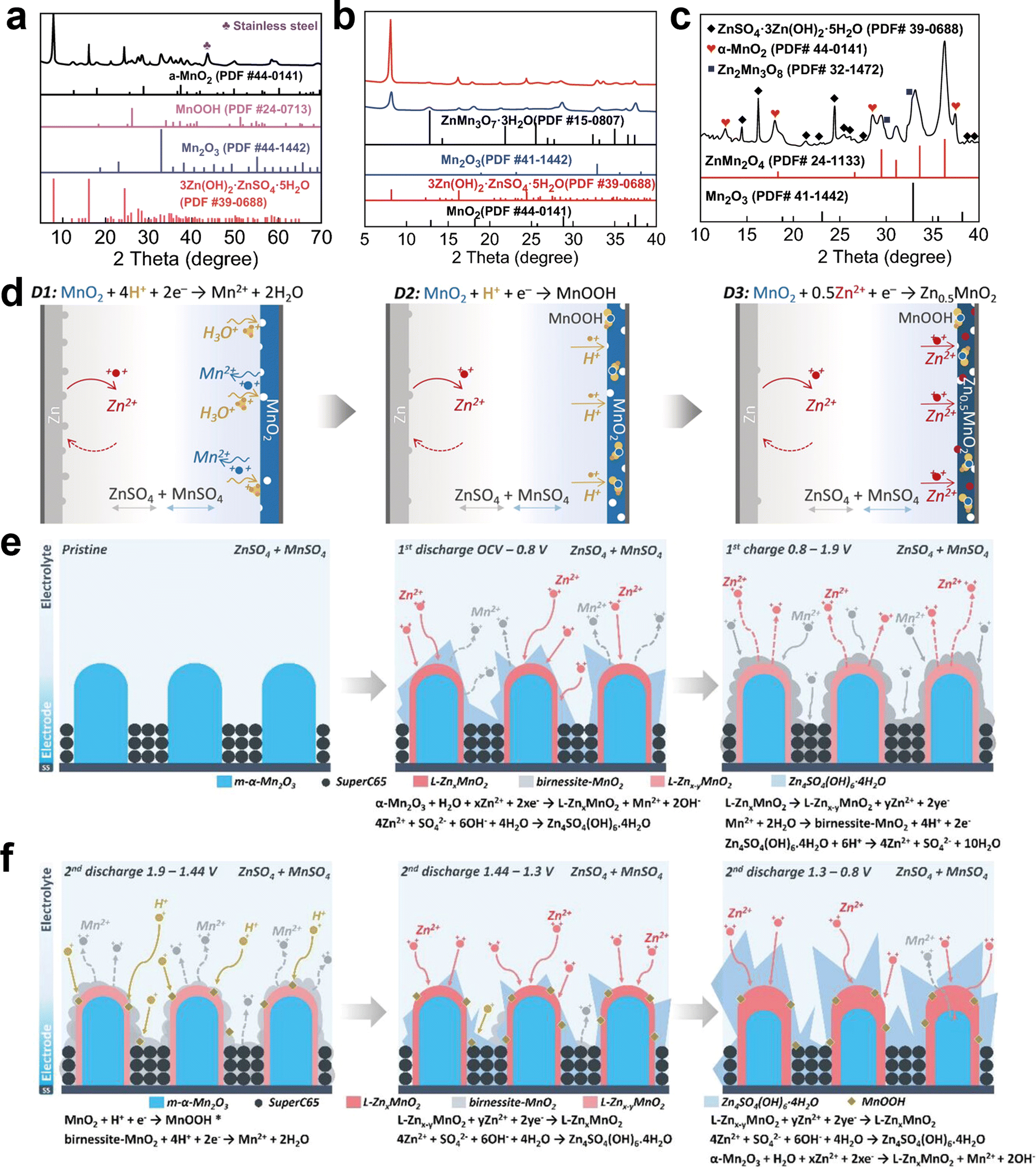
In recent years, an increasing number of reports has demonstrated that the energy storage mechanisms in Zn–Mn cells exhibit a hybrid storage mechanism rather than a singular mechanism. For example, in contrast to the previous simple phase transition, Xu et al.103 revealed a complex phase evolution involving both intercalation and conversion reactions. During the initial discharge process, α-MnO2 undergoes Zn2+ intercalation at around 1.4 V, resulting in the formation of α-ZnxMnO2. Subsequently, an H+ conversion reaction occurs between 1.0 and 1.3 V, leading to the generation of MnOOH and Mn2O3 in the cathode, thereby increasing the electrolyte pH value and producing a byproduct known as ZnSO4·3Zn(OH)2·5H2O (BZSP) (Fig. 6a). The reaction can be described as follows:| | | MnO2 + xZn2+ + 2xe− → ZnxMnO2 | (22) |
| | | 4Zn2+ + SO42− + 5H2O + 6OH− → ZnSO4·3Zn(OH)2·5H2O | (23) |
| | | MnO2 + H+ + e− → MnOOH | (24) |
| | | 2MnO2 + 2H+ + 2e− → Mn2O3 | (25) |
 |
| | Fig. 6 Hybrid storage mechanism. XRD patterns of the α-MnO2 cathode at different states: (a) discharged to 1.4 V in the first cycle, (b) fully charged state in the first cycle, and (c) after 100 cycles. (a)–(c) Reproduced with permission.103 Copyright 2019, Springer Nature. (d) Schematic representation and charge storage mechanism of an MnO2–Zn ion battery in 1 M ZnSO4 and 1 M MnSO4 electrolyte. Reproduced with permission.104 Copyright 2019, Wiley-VCH. Schematic illustration of the different contributions to the charge storage mechanism of m-α-Mn2O3 during (e) first discharge and charge process and (f) second discharge process. (e) and (f) Reproduced with permission.105 Copyright 2021, Wiley-VCH. | |
During the initial charging process, α-ZnxMnO2, MnOOH, and Mn2O3 revert to α-MnO2 by the release of Zn2+ and H+, while BZSP reacts with Mn2+ to form ZnMn3O7·3H2O (Fig. 6b). This reaction can be described as follows:
| | | 3(ZnSO4·3Zn(OH)2·5H2O) + 3Mn2+ + 8e− → ZnMn3O7 + 3ZnSO4 + 18OH− + 8Zn2+ + 12H2O | (26) |
In the subsequent cycles, Zn2+ undergoes reversible insertion/extraction in/from the α-MnO2, α-ZnxMnO2, and ZnMn3O7·3H2O hosts. The participation of Mn2+ leads to the formation of new phases including Zn2Mn3O8 and ZnMn2O4 through reversible conversion (Fig. 6c). The reaction can be described as follows:
| | | ZnxMnO2 + (0.5 − x)Zn2+ + (1 − 2x)e− → 0.5ZnMn2O4 | (27) |
| | | Zn2Mn3O8 + Mn2+ → 2ZnMn2O4 | (28) |
Qiao et al.104 reported the occurrence of multi-redox reactions during the discharge process, with three distinct discharge regions corresponding to three different reaction mechanisms. The region between 2–1.7 V results from the dissolution of MnO2, while the region between 1.7–1.4 V is responsible for H+ insertion into the residual MnO2, and the region between 1.4–0.8 V is dominated by Zn2+ insertion in the residual MnO2 (Fig. 6d), as confirmed by ex situ XRD and XPS measurements. Furthermore, the optimized addition 0.1 M H2SO4 significantly enhanced the energy density and maximized the electrolysis efficiency for two-electron reactions. The reaction can be written as follows:
| | | Cathode: MnO2 + 4H+ + 2e− ↔ Mn2+ + 2H2O | (29) |
| | | MnO2 + H+ + e− ↔ MnOOH | (30) |
| | | MnO2 + 0.5Zn2+ + e− ↔ Zn0.5MnO2 | (31) |
| | | Anode: Zn2+ + 2e− ↔ Zn | (32) |
The H+ conversion and Mn2+ dissolution/deposition mechanism of β-MnO2 was demonstrated by Liu et al.50 In the initial discharge cycle, β-MnO2 undergoes a transformation to MnOOH and Mn2+, while during charge, Mn2+ deposition and MnOOH conversion lead to the formation of ε-MnO2. In subsequent cycles, the deposited ε-MnO2 reacts with H+ to generate MnOOH, which partially dissolves to form Mn2+, accompanied by the formation of ZHS during the discharge process. Upon charging, Mn2+ is redeposited into the previously deposited ε-MnO2, resulting in the disappearance of ZHS. The chemical equation can be written as follows:
| | | Cathode: MnO2 + H+ + e− ↔ MnOOH | (33) |
| | | MnOOH + 3H+ + 3e− ↔ Mn2+ + H2O | (34) |
| | | 4Zn2+ + SO42− + 8H2O ↔ Zn4(OH)6SO4·5H2O + 6H+ | (35) |
| | | Anode: Zn ↔ Zn2+ + 2e− | (36) |
The energy storage mechanism of α-Mn2O3 was systematically and comprehensively investigated by Ma et al.105 They demonstrated that α-Mn2O3 is unsuitable for H+ insertion. In fact, α-Mn2O3 undergoes an irreversible phase transition to layer-type L-ZnxMnO2 by electrochemical reaction with H2O and Zn2+, accompanied by the dissolution of Mn2+ in the electrolyte. This process leads to the formation of OH−, ultimately generating ZHS in the first discharge process. During the first charge process, Zn2+ extraction from L-ZnxMnO2 forms L-Znx–yMnO2, while Mn2+ is oxidized to Mn(IV), resulting in the deposition of birnessite-MnO2 on the outer surface of the electrode (Fig. 6e). The ZHS fades due to the formation of H+ by deposition of Mn2+. In the second discharge process between 1.9–1.44 V, H+ insertion in MnO2 (formed from L-ZnxMnO2 and Mn2+) leads to the generation of MnOOH. In the range of 1.44–0.8 V, Zn2+ inserts in L-Znx–yMnO2, together with formation of ZSH, and it should be noted that the residual α-Mn2O3 will continue undergoing an irreversible phase transition in the range of 1.3–0.8 V until it completely transforms into L-ZnxMnO2 and Mn2+, as shown in Fig. 6f.
In summary, the storage mechanisms of manganese-based ZIBs including Zn2+ insertion/extraction, chemical conversion reaction, H+ insertion/extraction, H+ and Zn2+ co-insertion/extraction, dissolution/deposition and hybrid storage mechanisms have been extensively investigated by researchers. However, although significant advancements have been made in determining the storage mechanisms of manganese-based ZIBs over the past decade, their intricate reaction chemistry is still under debate, hindering a consensus. Moreover, the underlying causes of different storage mechanisms and some abnormal phenomena such as the unusual “turning point” of the GCD curve have not been reasonably explained, resulting in obscure optimization strategies based on the storage mechanism and further impeding their practical application. Therefore, more efforts are urgently needed to determine the real reaction chemistry of manganese-based ZIBs during charge/discharge.
4 The mechanisms of capacity fluctuation
Although numerous studies have demonstrated the long-term cycling performance of manganese-based ZIBs at high C-rates (≥5C),56,79,102 it should be noted that this excellent performance is primarily attributed to capacitive processes dominating the capacity contribution rather than the typical diffusion-controlled charge storage mechanisms for batteries.18 Given the ongoing controversy regarding the primary causes of battery failure, there is a lack of robust methods to enhance the cycling stability of manganese-based ZIBs, particularly at a low current. Besides the crucial challenges encountered by Zn anodes such as dendrite growth, passivation, corrosion and hydrogen evolution,106–108 on the cathode side, issues such as Mn dissolution, dramatic volume changes, and the irreversible consumption of Mn2+ lead to the generation of electrochemically inactive materials, which can result in severe performance degradation and even battery failure.109–111 Furthermore, the phenomenon of capacity activation, which commonly occurs during the initial cycles, has received limited attention from researchers. In general, the long-term cycling behavior of manganese-based ZIBs consists of an activation period, a stabilization period, and ultimately a decay period. However, previous review articles failed to address the underlying reasons for the dynamic capacity fluctuation from capacity activation to capacity attenuation, resulting in inadequate guidance and references for enhancing the cycling stability of manganese-based ZIBs. Thus, the mechanisms of capacity degradation, capacity activation, and dynamic capacity fluctuation are discussed in the following sections.
4.1 Capacity degradation mechanisms
4.1.1 Jahn–Teller distortion.
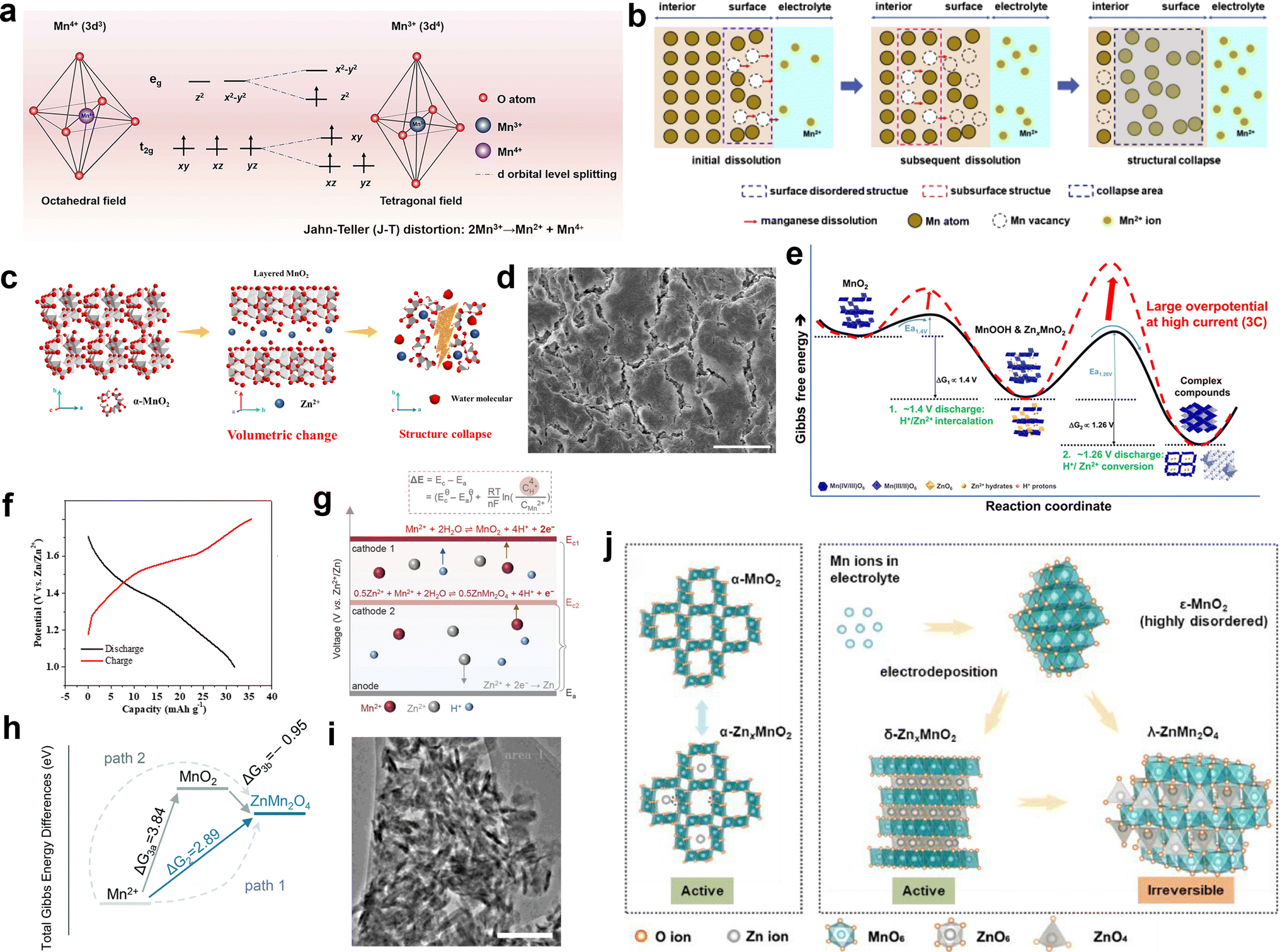
Most manganese-based materials consist of basic octahedral units (MnO6) interconnected through corner and/or edge sharing, with the Mn cation located at the center of each MnO6 octahedron adopting a six-oxide coordination. Under the influence of an electric field, the 3d orbitals of the Mn cation in the MnO6 octahedra split into two degenerate orbitals, namely eg and t2g. Due to the lower formation energy of the triply degenerate t2g orbitals (dxy, dxz and dyz) compared to the doubly degenerate eg orbitals (dz2 and dx2–y2), the three electrons in the 3d orbital in Mn4+ will occupy the dxy, dxz and dyz orbitals, respectively, forming a stable structure. However, once Mn4+ gains an electron through the reduction process to form Mn3+, in addition to occupying the dxy, dxz and dyz orbitals, the remaining electron in the 3d orbital of Mn3+ will occupy the dz2 orbital in eg. The asymmetry of the electron distribution between the dz2 and dx2–y2 orbitals in Mn3+ leads to the distortion of the MnO6 geometry from octahedral to tetragonal symmetry by lowering the overall energy.34,112 Consequently, this distortion induces elongation of two axial Mn–O bonds and contraction of four equatorial Mn–O bonds (Fig. 7a). These changes can be observed through in situ Raman spectroscopy as a shift towards higher wavenumbers for the peak corresponding to the Mn–O stretching vibrations during the discharge processes.112
 |
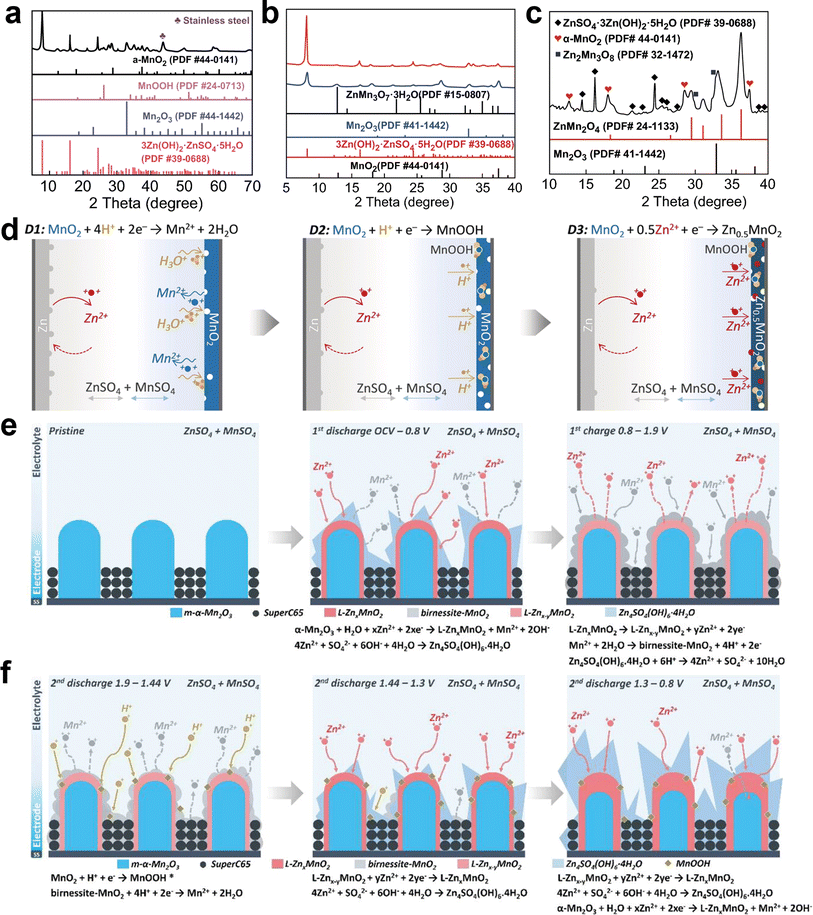
| | Fig. 7 Mechanisms of capacity degradation. (a) Mechanism of Jahn–Teller distortion. Reproduced with permission.34 Copyright 2023, the Royal Society of Chemistry. (b) Manganese dissolution caused by the unstable or disordered surface. Reproduced with permission.113 Copyright 2020, Elsevier. (c) Large volumetric change induced by phase transition. Reproduced with permission.116 Copyright 2020, Elsevier. (d) Electrode pulverization induced by large volumetric change. Reproduced with permission.53 Copyright 2017, Springer Nature. (e) Kinetic properties of the redox reactions in Zn/MnO2 cells. Reproduced with permission.117 Copyright 2019, the American Chemical Society. (f) Galvanostatic charge/discharge curve of ZnMn2O4. Reproduced with permission.49 Copyright 2019, Elsevier. (g) Relationship between pH values and the potentials of Mn2+ deposition based on the Nernst equation. Reproduced with permission.119 Copyright 2023, Wiley-VCH. (h) Total Gibbs energy differences for two possible reaction paths. Reproduced with permission.120 Copyright 2022, the Royal Society of Chemistry. (i) TEM image of the MnO2 electrode with 100 cycles. Reproduced with permission.94 Copyright 2022, Springer Nature. (j) Schematic illustration of the crystal structural transformation. Reproduced with permission.102 Copyright 2021, Elsevier. | |
It has been widely acknowledged that the Jahn–Teller distortion during discharge leads to the disproportionation of Mn3+ (2Mn3+ → Mn2+ + Mn4+), resulting in the release of Mn2+ and causing severe dissolution issues. A consensus has been reached regarding the detrimental impact of inevitable Mn dissolution on the rapid capacity decay observed in manganese-based ZIBs, which is attributed to the loss of active cathode materials for reactions. Also, it has been unveiled that approximately 1/3 of the total manganese on the electrode dissolves in the electrolyte after the full discharge.90 Once the outermost manganese atoms dissolve in the electrolyte, the subsequent dissolution of subsurface atoms occurs through a chain reaction, leading to structural collapse (Fig. 7b).113 Moreover, the irreversible phase transformations, considerable strain, and unstable interface triggered by the Jahn–Teller distortion lead to structural degradation, eventually resulting in capacity degradation. In short, during the cycling process of manganese-based ZIBs, the presence of Jahn–Teller distortion poses a significant challenge given that it leads to crystal structure deterioration and the gradual loss of active materials, resulting in a rapid performance degradation.
4.1.2 Dramatic volume changes.
Previous studies have demonstrated that manganese-based LIBs undergo large volume changes due to the shuttle of Li+ caused by lattice expansion or contraction.114 Similarly, repetitive insertion/extraction of hydrated H+/Zn2+ ions will cause significant volume changes in manganese-based ZIBs. In comparison with Li+ ions (0.76 Å), the larger hydrated radius of Zn2+ ions (4.3 Å) will destabilize and weaken the crystal framework, resulting in irreversible volumetric alterations and structural damage. In addition, the strong electrostatic force between Zn2+ ions and the host materials results in aggravated volume expansion and sluggish Zn2+ diffusion.115 Furthermore, the irreversible phase transformation from a tunnel to layer or spinel structure during the charge/discharge process can lead to drastic volume changes, causing pulverization, cracking, and collapse of the active materials (Fig. 7c and d).34,53,116 Once structural collapse occurs, some of the active materials will lose electrical contact with the current collector, resulting in the formation of dead Mn. The dramatic volume changes not only compromise the structure stability and lead to the loss of active materials, but also reduce the electrochemical activity of manganese-based cathode materials, resulting in capacity fading and poor rate performance. Yang et al.117 conducted a comprehensive investigation combining experimental and theoretical approaches to elucidate the rapid capacity degradation observed in Zn/MnO2 batteries, corresponding to the irreversible phase transformations occurring at the discharge plateaus of around 1.26 V, which are considered kinetically limiting reactions (Fig. 7e). The conversion reactions induce a large volume change and lead to the formation of electrochemically inactive ZHS. Thus, by increasing the lower cutoff voltage from 1.0 to 1.3 V, the kinetically limited irreversible reactions can be directly eradicated. As a result, when charged/discharged in the potential range of 1.3–1.8 V at a low current density of 1C, stable capacity retention over 150 cycles was achieved.
4.1.3 Irreversible consumption of Mn2+ to generate inactive manganese compounds.
The essential reason for the capacity fading of manganese-based cathodes upon cycling is the loss of active materials. During the period of capacity decay, it is commonly observed that the original active cathode materials exhibit reduced crystallinity with the formation of electrochemically inactive manganese compounds on the cathode, such as ZnMn2O4, Mn3O4, and ZnMn3O7. Consequently, many reports suggested that the formation of an inert manganese compound phase is responsible for the ultimate performance attenuation.43,49,93
Taking hetaerolite ZnMn2O4 as a representative example, Zn2+ migration in the perfect spinel ZnMn2O4 is difficult due to their strong electrostatic repulsion. Additionally, ZnMn2O4 exhibits high resistivity, which is six orders of magnitude higher than that of MnO2,118 resulting in poor electrode conductivity. Consequently, when ZnMn2O4 was used as a cathode material, only an ultralow discharge capacity of ∼32.6 mA h g−1 could be delivered at 0.05C (Fig. 7f), manifesting its strong electrochemical inactivity.49 Moreover, once a few of irreversible ZnMn2O4 nanoparticles are accumulated on the cathode surface, a mass of nucleation sites will be produced, inducing the formation of more ZnMn2O4 nanoparticles. The coverage of abundant ZnMn2O4 nanoparticles on the electrode would impede cation diffusion and the charge transport of the inner active materials. Unfortunately, these ZnMn2O4 nanoparticles cannot be electrochemically decomposed during the charging process as long as they are formed and attached to the electrode, which can be a crucial reason accounting for the capacity degradation of manganese-based cathodes after long-term cycling.
There are two primary mechanisms responsible for the formation of inactive ZnMn2O4 nanoparticles, where one is attributed to the conversion of tunnel or layered-ZnxMn2O4 due to the higher thermodynamic stability of spinel ZnMn2O4, as shown in the following process:
| | | MnO2 + xZn2+ + 2xe− → ΖnxMnO2 | (37) |
| | | ΖnxMnO2 → λ-ΖnMnO2 + 2(x − 1)Zn2+ + (4x − 2)e− | (38) |
The other involves an electrodeposition reaction from an Mn2+-containing electrolyte, which can be denoted as:
| | | 2Mn2+ + Zn2+ + 8OH− → ZnMn2O4 + 4H2O + 2e−, E = 1.320 V vs. Zn/Zn2+ | (39) |
The electrodeposition potential of Mn2+ to form MnO2 is known to be 1.869 V (vs. Zn/Zn2+),121 which is higher than that of ZnMn2O4 (Fig. 7g). In addition, the Gibbs energy of path 1 is lower than that of path 2 (Fig. 7h). Moreover, theoretical calculation indicates a lower formation energy compared to MnO2.120 These pieces of evidences indicate that the generation of ZnMn2O4 is favored by electrodeposition. Based on the aforementioned analyses and equation, it is evident that the concentration of Mn2+ will eventually decrease significantly due to the continuous formation of an irreversible ZnMn2O4 phase, leading to loss of active materials based on the Mn2+ electrodeposition reaction.
The capacity decay resulting from H+ insertion/extraction was elucidated by Yuan et al.94 With an increase in the cycle number, MnO2 nanowires gradually dissolved, and some tiny needle-like nanograins were discovered after 50 cycles, which were identified as the Zn–Mn–O phase. Notably, the number of Zn–Mn–O nanograins kept increasing with an increase in the number of cycles, and almost all the nanowires were converted to Zn–Mn–O after 100 cycles, as shown in Fig. 7i. Meanwhile, the concentration of Mn2+ kept increasing within 50 cycles due to the Mn3+ disproportionation reaction, which is consistent with the TEM results. However, the Mn3+ disproportionation reaction was largely but not totally reversible, leading to the gradual loss of active MnO2, and the accumulation of dissolved Mn2+ was thermodynamically favored to form the irreversible Zn–Mn–O phase due to the reprecipitation Mn2+, resulting in a rapid decrease in Mn2+ concentration and capacity. In addition, by disassembling an in situ cell after 5000 cycles, Wei et al.122 found that the capacity fading during long-term cycling was attributed to the irreversible consumption of Mn2+ in the electrolyte, where Mn2+ is irreversible converted to the woodruffite phase (ZnMn3O7·2H2O) with poor electrochemical activity, leading to a performance decay based on the capacity contribution from the dissolution/deposition of Zn-birnessite. Liang et al.102 concluded that the in situ electrodeposition of MnO2 from pre-added Mn2+ during the charging process should be a side reaction, where Mn2+ in the electrolyte is consumed to form ε-MnO2. However, ε-MnO2 will irreversibly convert to the inactive λ-ZnMn2O4, leading to capacity decay (Fig. 7j), which was also confirmed by Xia and coworkers.123 In addition, the depletion of the Mn2+ electrolyte to generate irreversible λ-ZnMn2O4 will accelerate the disproportionation reaction, thereby triggering the dissolution of more original active MnO2. Moreover, it was found that pre-adding a large amount of Mn2+ did not improve the cycle stability; in contrast, excess Mn2+ in the electrolyte promoted the conversion of Mn2+ to the ZnMn2O4 phase, leading to severe capacity fading and even battery failure.124
4.2 Capacity activation mechanisms
4.2.1 Electrodeposition of Mn2+.
It is recognized that the Mn2+ additive in the electrolyte plays an important role in achieving a sustainable cycling performance for manganese-based ZIBs. Early studies have found that the pre-added Mn2+ can effectively suppress the dissolution of the cathode by alleviating the disproportionation of Mn3+ (2Mn3+ → Mn2+ + Mn4+) caused by Jahn–Teller distortion.78 Nevertheless, according to the Nernst eqn (40), a high pH value can reduce the oxidation and deposition potential of Mn2+, and recent investigations have concluded that Mn2+ in a high pH environment created by ZSH can be oxidized at a low charging potential.95 It has even been reported that active MnO2 can be electrodeposited on the substrate from the MnSO4-based electrolyte, with an electrodeposition plateau at around 1.73 V. Also, the in situ formed MnO2 can be directly used for subsequent discharge/charge with the typical GCD curve (Fig. 8a).117 In electrolyte containing 2 M ZnSO4 and 0.1 M MnSO4, when the charge cut-off voltage was 1.7 V, no new diffraction peaks could be detected in the XRD pattern in the fully charged state; meanwhile, the cycling performance showed fast capacity fading. However, when the charge cut-off voltage increased to 1.8 or 1.9 V, a new set of peaks emerged in the fully charged state, which can be well indexed to ε-MnO2 (Fig. 8b), accompanied by an increase in capacity with an increase in the number of cycles.123 These findings suggest that the Mn2+ ions in the electrolyte can be electro-oxidized at a high potential, and the increased capacity is attributed to the newly formed MnO2.| | 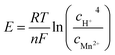 | (40) |
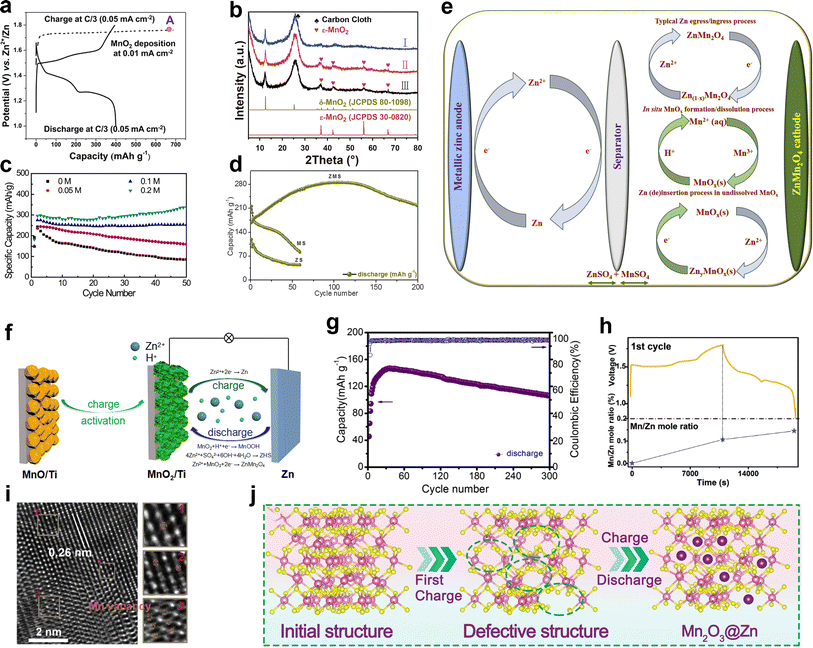 |
| | Fig. 8 Mechanisms of capacity activation. (a) Electrodeposition of MnO2. Reproduced with permission.117 Copyright 2019, the American Chemical Society. (b) XRD patterns of the MnO2/CC electrodes in the first charge state with different charge cut-off voltages. Reproduced with permission.123 Copyright 2020, Elsevier. (c) Effect of the Mn2+ additive concentration in the electrolyte on the cycling performance. Reproduced with permission.125 Copyright 2019, Springer Nature. (d). Capacity activation of the ZnMn2O4/Zn cell. (e) Electrochemical reactions inside the ZnMn2O4/Zn cell. (d and e) Reproduced with permission.126 Copyright 2020, Elsevier. (f) Diagram of energy storage mechanism for the MnO cathode of ZIBs. Reproduced with permission.81 Copyright 2019, Elsevier. (g) Cycling performance of the Zn/Mn3O4 battery. Reproduced with permission.83 Copyright 2018, Elsevier. (h) Elemental analysis of dissolved Mn2+ ions in 2 M ZnSO4 electrolyte during cycling. (i) HRTEM of 1st fully charged state. (h and i) Reproduced with permission.60 Copyright 2020, Elsevier. (j) Schematic diagram of Zn2+ storage mechanism of Mn2O3. Reproduced with permission.127 Copyright 2023, Wiley-VCH. | |
The identification of electro-oxidized compounds with a nanosheet morphology is challenging due to their low crystallinity and complex components in the bulk cathode. In the literature, ε-MnO2,102,123 birnessite,56,122 vernadite,95,128 and their Zn compounds have been reported for electro-oxidized manganese oxides. But the newly formed MnOx on the electrode surface caused by re-oxidation of Mn2+ compensating the capacity decay has reached a consensus. It has been reported that about 18.9% of the capacity is attributed to the contribution of pre-added Mn2+.129 However, the concentration of the Mn2+ additive has a significant influence on the electrodeposited MnOx. In an electrolyte with no Mn2+ additive, the capacity of MnO2 decayed rapidly; however, with an increasing concentration of Mn2+ additive, the cycling stability gradually improves (Fig. 8c). Nevertheless, when the additive concentration reached 0.2 M, an increase in capacity was observed with cycle number due to the deposition of a large amount of aggregated nanosheets on the electrode surface.125 These aggregated nanosheets augmented the active mass at the cathode, and consequently enhanced the cycling performance. Furthermore, Kim et al.126 provided a deep comprehension of the role played by pre-added Mn2+ in the exceptional performance of a ZnMn2O4 cathode (Fig. 8d). They proposed that the additional capacity comes from two parts, where one is based on the quasi-reversible in situ electro-deposition/dissolution of MnOx from pre-added Mn2+, and the other is based on the reversible insertion/extraction of Zn2+ from undissolved MnOx (Fig. 8e), which may be generated from the incomplete dissolution process due to the gradual pH changes during the electrochemical cycling process.
Besides the contribution of pre-added Mn2+ to the increased capacity, the dissolved Mn2+ from the original cathode material also leads to performance activation. For instance, it has been observed that MnO, which was previously considered inactive for Zn2+ storage due to its lack of tunnels, exhibits a competitive electrochemical performance comparable to MnO2 after undergoing an activation process. Chen et al.130 attributed the activation of MnO to its spontaneous dissolution in ZnSO4 electrolyte, which can be denoted as eqn (41). The spontaneous dissolution behavior of MnO provides adequate Mn2+ for the ZSH-assisted deposition/dissolution reaction and results in performance activation, where the energy storage mechanism can be described by eqn (19)–(21). Besides, it was found that there is a positive correlation between capacity and rest time of MnO, manifesting that the dissolution of Mn plays a key role in the performance activation.
| | | MnO + 2H+ → Mn2+ + H2O | (41) |
4.2.2 Electrochemical oxidation in the charging process.
Additionally, it has been reported that the inert MnO can be activated by electrochemical oxidation in the charging process, leading to the formation of active MnOx (x > 2), which serves as the active phase for the reversible insertion/extraction in the following cycles. Kang et al.81 claimed that the electrochemical oxidation of MnO into porous layered-type MnO2 nanosheets in the initial charging process is responsible for MnO activation. Combined with the phase, morphology evolution and increased Mn valence (3.9), it is evident that the surface of the MnO particles is partially oxidized into layered-type MnO2 nanosheets, serving as electro-active sites for subsequent energy storage (Fig. 8f). Sun et al.131 further confirmed this activation transition process, wherein MnO undergoes sequential oxidization to form layered-type MnO2 in the initial few cycles, resulting in performance activation. In addition, Mn3O4, which is electrochemically inactive in alkaline Zn–MnO2 batteries, it has also been reported to exhibit a good electrochemical performance in ZIBs.83 An ex situ XRD analysis revealed the formation of Mn5O8 and birnessite after the first charging process. As cycling proceeded, the peak intensity of Mn3O4 weakened, while the peaks corresponding to birnessite became much stronger, indicating a transformation from Mn3O4 to birnessite. It should be noted that Mn5O8 is an intermediate product resulting from the incomplete electrochemical oxidation reaction and will finally transfer to birnessite after further electrochemical oxidation. The gradual generation of both the Mn5O8 intermediate and birnessite through the progressive electrochemical oxidation of Mn3O4 contributes to a gradual increase in capacity during the initial tens of cycles (Fig. 8g).
4.2.3 Mn-defects.
Besides spontaneous dissolution and electrochemical oxidation, which contribute to the performance activation of MnO, in situ-generated Mn-defects also play a significant role in comprehending its performance activation. Zhou et al.60 first reported that the induction of Mn defects through an in situ electrochemical process can achieve the activation of MnO (Fig. 8h and i). The electrochemical extraction of Mn2+ from the MnO host during charging provides more accessible channels for Zn2+ insertion/extraction. DFT calculations further confirmed the electrochemical activation of MnO caused by Mn defects, given that the differential charge density implies that electrons easily accumulate around the Mn defects to form a strong electrostatic field, which is highly favorable for Zn2+ adsorption. However, the uniform charge distribution of pristine MnO may cause severe destruction to the MnO framework upon Zn2+ insertion. The activation of MnO through charge-induced Mn dissolution was also verified by Zhao et al.59 They concluded that the cycling performance of MnO is strongly related to the concentration of pre-added Mn2+, where insufficient amounts of Mn2+ in the electrolyte can lead to severe structural deterioration due to the greater dissolution of Mn, while an excess amount may over-suppress its dissolution. In addition, the Mn2O3/CoO heterointerface with poor initial discharge capacity was found to be activated by the Mn-defects in the charging process.127 The distortion of Mn2O3 due to Zn2+ insertion in the initial cycles will lead to the easy release of Mn atoms from the framework during charging, resulting in the formation of Mn vacancies (Fig. 8j). Furthermore, a strong internal electric field is generated at the interface of the heterostructure due to the homogeneous charge distribution of Mn2O3/CoO heterointerface, which will induce strong electrostatic fields at the Mn species for electron rearrangement. Owing to the similar ionic radius of Zn2+ and Mn2+, the Mn dissolved from the host structure acts as a sacrificial site, providing more insertion sites and channels for Zn2+ insertion, leading to performance activation.
4.3 Dynamic capacity fluctuation for long-term cycling
4.3.1 The evolution of active materials.
In conjunction with the analysis of cycling performance, ex situ XRD results, and morphology evolutions, Chen and colleges concluded that the evolution of the active materials is responsible for capacity fluctuation.56 As shown in Fig. 9a, it is noticeable that the cycling performance of MnO2 shows an obviously capacity fluctuation with an arched trend. The capacity increased rapidly during the initial cycles but subsequently decayed rapidly after reaching the peak value. The ex situ XRD patterns show that the diffraction peak intensity of MnO2 gradually decreased in the initial cycles, manifesting the continuous dissolution of MnO2 with an increase in the number of cycles. Moreover, numerous newly deposited birnessite nanosheets with low crystallinity were found on the electrode surface, indicating that the dissolved Mn2+ from the original MnO2 was converted to birnessite during charging. Birnessite with specific nanosheet structures is electrochemical active, which can provide more reaction sites than the original MnO2. Additionally, the deposition of birnessite compensates for the capacity loss caused by MnO2 dissolution, resulting in performance activation. A similar activation mechanism of MnO was investigated simultaneously by Sun et al.132 However, with a further increase in the number of cycles, the birnessite nanosheets faded and the ZnMn2O4/Mn3O4 phase emerged. Unfortunately, both ZnMn2O4 and Mn3O4 are electrochemically inactive due to their inaccessible pathways for Zn2+/H+ insertion/extraction, resulting in limited capacity delivery. As the intensity of the signals for the ZnMn2O4/Mn3O4 phase continued to increase, a noticeable capacity degradation was observed during the subsequent cycling. In addition, owing to the competition between activation towards birnessite and degradation towards ZnMn2O4/Mn3O4, a transitional period with a relatively stable capacity existed between the activation and decay processes. However, once most of the original MnO2 was converted to birnessite, severe capacity attenuation occurred (Fig. 9b). Moreover, it should be noted that the fluctuation tendency is closely associated with the current density, where with an increase in current density, MnO2 exhibited enhanced cycling stability. This can be attributed to the mild dissolution issue of the original MnO2, followed by the slow deposition and conversion reaction rate under high currents.
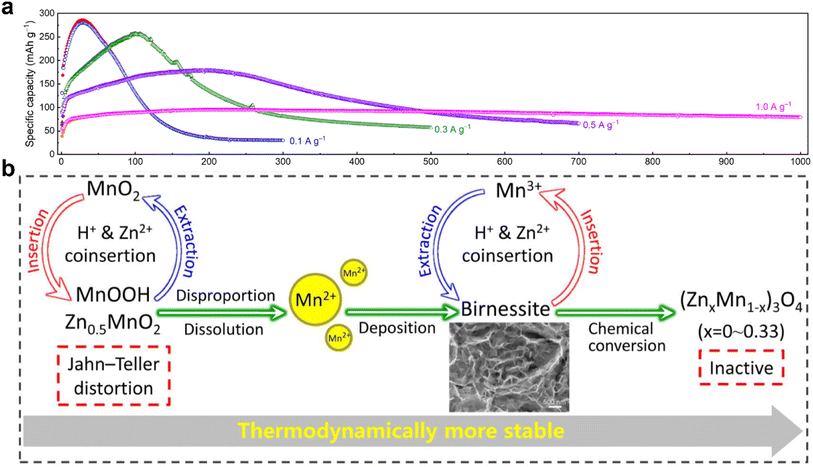 |
| | Fig. 9 Evolution of the active materials during long-term cycling. (a) Cycling performance at different specific currents of β-MnO2 electrode. (b) Schematic illustration of the transformation process and reaction mechanism in each step of MnO2 during long-term cycling. (a) and (b) Reproduced with permission.56 Copyright 2022, Elsevier. | |
4.3.2 The evolution of capacity contribution ratio from Mn2+.
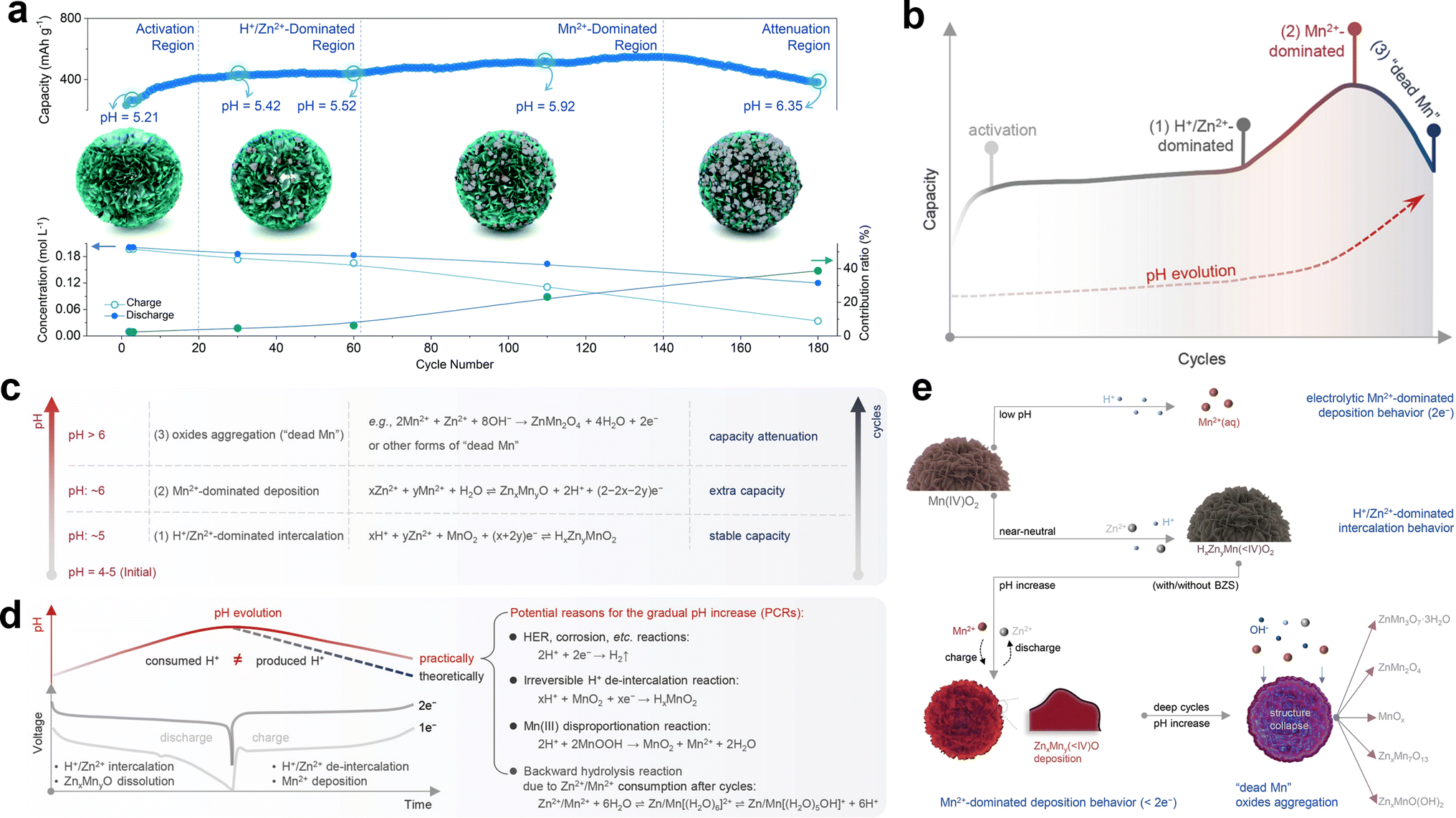
Chao et al.120 proposed a competitive capacity evolution (Mn-CCE) protocol to elucidate the capacity fluctuation behavior of manganese-based ZIBs. Based on the fluctuation characteristics and capacity contribution ratio from Mn2+ (CfM), the cycle curve can be divided into four distinct regions, as illustrated in Fig. 10a. Firstly, region I exhibits a rapid capacity increase at the beginning and ends with a slower growth rate; next, region II presents a mild ascending trend of capacity after region I; then, region III displays an evident rising trend of capacity, which ends with the peak value; finally, region IV demonstrates a quick downward trend in capacity. The magnitude of the variation in Zn2+ within region I was found to be significantly higher than that of Mn2+ during charge/discharge, with a calculated CfM value of less than 5%. This suggests that Zn2+ insertion/extraction plays a dominant role in the capacity contribution within region I. Therefore, the observed increase in capacity in this region can be attributed to electrode wetting. In region II, CfM slightly increases and the deposition of ZnMn2O4 is quasi-reversible due to the low pH environment. The capacity contribution mainly originates from the reversible intercalation/deintercalation of H+/Zn2+, resulting in a relatively flat cycle curve. Instead, CfM significantly increases with a rapidly decrease in the concentration of Mn2+ in region III, manifesting that Mn2+ dominates the reaction chemistry rather than H+/Zn2+ insertion/extraction, leading to an obvious increase in capacity. Besides, Qiu et al.123 found that properly increasing the pre-added Mn2+ can expand the trend of capacity increase and achieve a higher maximum capacity value. The irreversible deposition of ZnMn2O4 nanoparticles provides nucleation sites for subsequent Mn2+ deposition, thereby inducing the accumulation of more ZnMn2O4 nanoparticles on the electrode surface, giving rise to a fast increase in CfM. However, the abundant accumulation of ZnMn2O4 nanoparticles on the cathode surface significantly increases the resistance and restricts the ion/charge transfer between the electrolyte and inner MnO2, causing rapid capacity fading in region IV. In brief, the different capacity contributions originating from cation insertion/extraction and Mn2+ conversion during long-term cycling result in a dynamic performance evolution.
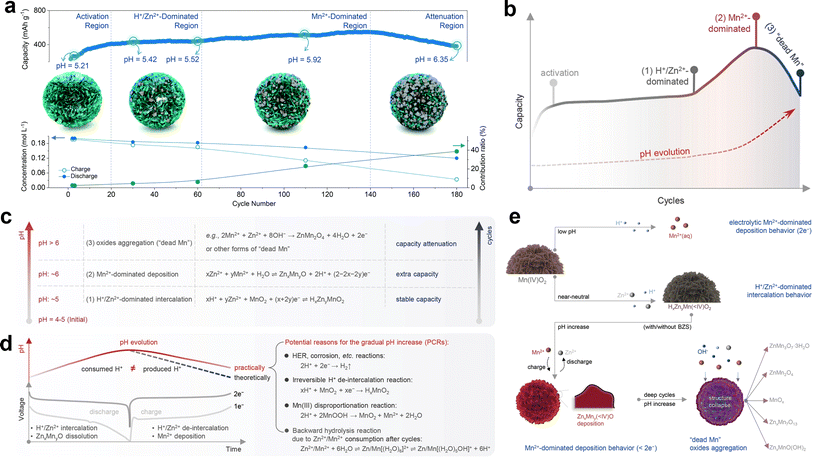 |
| | Fig. 10 Evolution of CfM and pH during long-term cycling. (a) Mn-based competitive capacity evolution protocol. Reproduced with permission.120 Copyright 2022, The Royal Society of Chemistry. (b) Evolution of capacity and charge storage mechanisms during cycles associated with pH change. (c) Dominant reactions determined by the pH environment. (d) Summary of potential reasons for gradual pH increase. (e) Illustration of the H+/Zn2+/Mn2+-dominated electrochemical process at different pH environments. (b)–(e) Reproduced with permission.119 Copyright 2023, Wiley-VCH. | |
4.3.3 The evolution of pH.
Moreover, new insight into the pH evolution in near-neutral pH electrolytes rationally makes the underlying cause for capacity fluctuation clear (Fig. 10b and c).119 Owing to the unavoidable occurrence of the hydrogen evolution reaction,133 irreversible H+ extraction reaction, Mn3+ disproportionation reaction and backward hydrolysis reaction,134,135 complete restoration of the initial pH state within a single cycle becomes unattainable, giving rise to a continuous increase in pH in the whole cycle calendar (Fig. 10d). The deposition potential of Mn2+ is high in the initial cycles due to the relatively low pH, wherein cation intercalation reactions dominate this period and result in a relatively stable evolution curve of capacity. As the pH continues to increase, the formation of deposited manganese oxide becomes more easily triggered with the dominant process changing to Mn deposition, leading to a rapid increase in capacity. As the cycle proceeds, the dissolution of manganese oxide becomes harder due to the high pH value and the formation of abundant irreversible manganese oxide consumes a large amount of H+, which causes a significant increase in pH, ultimately depleting the Mn2+ electrolyte and resulting in capacity attenuation (Fig. 10e). Consequently, the capacity fluctuation of manganese-based ZIBs with rapid increase and decay can be attributed to pH evolution in near-neutral pH electrolytes.
In summary, the mechanisms of capacity attenuation, capacity activation and dynamic capacity fluctuation in manganese-based ZIBs were comprehensively discussed. The inevitable dissolution of Mn, dramatic volume changes, and irreversible consumption of Mn2+ lead to capacity attenuation. Capacity activation is attributed to electrolyte Mn2+ electrodeposition, electrochemical oxidation and induced Mn-defects of low-valence manganese oxides during the charging process. Also, the evolution of the active materials, capacity contribution ratio from Mn2+, and pH results in dynamic capacity fluctuation.
5 Strategies for improving cycling stability
Undoubtedly, the capacity fluctuation poses a significant challenge for the commercial application of manganese-based ZIBs. Although converting Mn2+ to manganese oxides can provide additional capacity, resulting in an increase in capacity, this “fake” high-performance is transitory and the capacity will rapidly decay due to the generation of “dead” Mn. Consequently, enhanced cycling stability can be achieved by mitigating Mn dissolution and Mn2+ deposition, thereby enabling the dominant capacity contribution from Zn2+/H+ insertion/extraction. Additionally, improving the reversibility of the dissolution/deposition reaction represents an alternative approach to enhance the cycling stability. Thus, strategies aimed at optimizing the cycling stability should focus on either promoting one-electron transfer in the Mn4+ ↔ Mn3+ reaction or facilitating two-electron transfer in the Mn4+ ↔ Mn2+ reaction.
5.1 Strategies based on one-electron transfer reaction
5.1.1 Rational composite design.
Rationally compositing conductive materials or transition metal oxides not only significantly enhances the electronic conductivity and ionic diffusion efficiency of semi-conductive manganese-based materials, but importantly improves their structural stability and mitigates side reactions during cycling. The enhanced structural stability primarily originates from the synergistic effects generated by two stable interfaces (the interface between the outer component and electrolyte, as well as the interface between components) through chemical bonding or physical isolation.
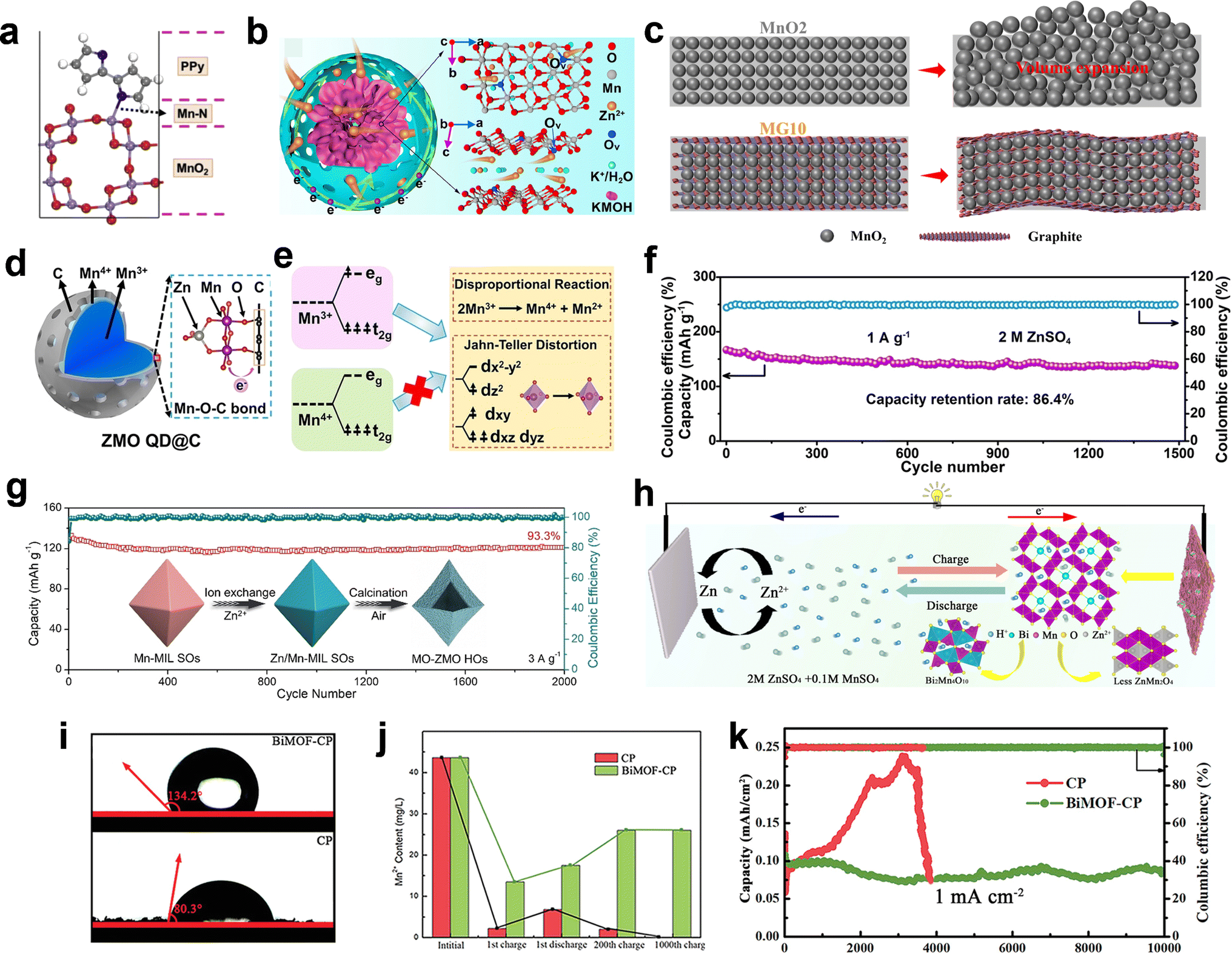
The multilayered rGO coated on MnO2 nanowires with a thickness of 5 nm, as reported by Mai et al.,136 was found to effectively alleviate direct contact between the internal active MnO2 and the electrolyte. In pure ZnSO4 electrolyte, it was observed that the concentration of Mn2+ significantly decreased during the discharge process after rGO coating, indicating the effective inhibition of the Mn dissolution issue. Additionally, the coating of polypyrrole (PPy) on MnO2 also proved to suppress Mn dissolution, where PPy not only serves as a protective layer but also forms an Mn–N bond at the interface between MnO2 and PPy (Fig. 11a), resulting in cycle performance of 500 cycles without capacity fading.113 Recently, Zhu et al. successfully fabricated a hydrophobic and uniform amino-propyl phosphonic acid (AEPA) coating layer with a few nanometer thickness on the surface of MnO2. It was found that AEPA can form strong chemical bonds with MnO2, effectively inhibiting the dissolution of Mn by creating a barrier between the electrolyte and cathode.137 Yolk–shell structured K-birnessite@mesoporous carbon nanospheres (KMOH@C) with abundant void space between the KMOH core and carbon shell were synthesized by Zhai et al.138 The mass of internal voids between the KMOH core and the carbon skeleton could efficiently restrain the volume changes, aggregation, and further pulverization of the KMOH nanosheets during the charging/discharging (Fig. 11b).116 As a result, KMOH@C showed excellent long-term cycle stability over 6000 cycles at 3 A g−1. Also, graphite nanosheets, which possess superior mechanical properties, were demonstrated to enhance the stress of MnO2 and suppress the further expansion and extrusion during the phase change process (Fig. 11c).
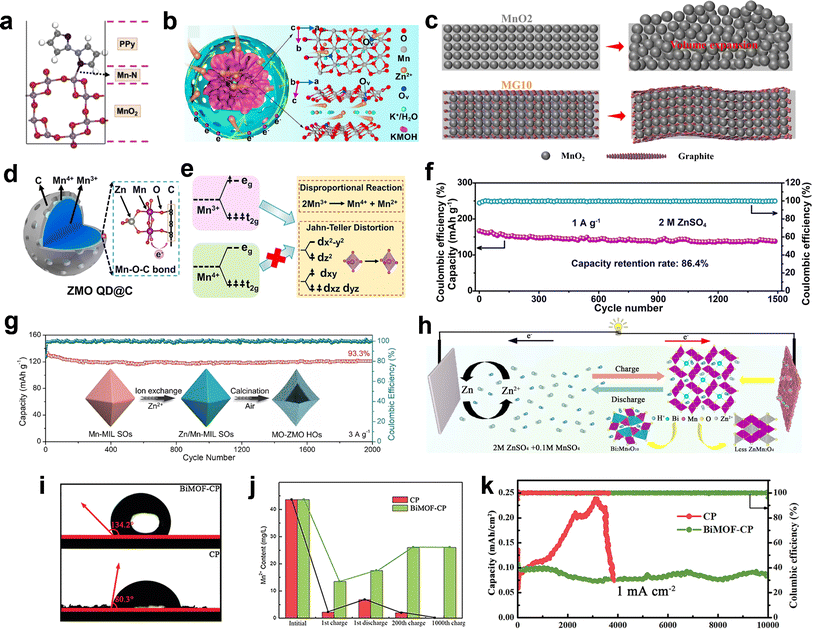 |
| | Fig. 11 Strategy for rational composite design based on one-electron transfer reaction. (a) Optimal configuration of the disordered MnO2 surface covered by PPy. Reproduced with permission.113 Copyright 2020, Elsevier. (b) Schematic illustration of the Zn-ion/electron transport in KMOH@C with oxygen vacancies. Reproduced with permission.138 Copyright 2021, Elsevier. (c) Schematic illustration of the volume change in MnO2 and MnO2/graphite. Reproduced with permission.116 Copyright 2020, Elsevier. (d) Illustration of the Mn valence state distribution in ZMO QD@C. (e) Mechanism for the structural stability of ZMO QD@C. (f) Long-term cycle test. (d)–(f) Reproduced with permission.139 Copyright 2022, Wiley-VCH. (g) Cycling performance of hetero-structured Mn2O3–ZnMn2O4 hollow octahedrons. Reproduced with permission.146 Copyright 2021, Wiley-VCH. (h) Schematic diagram showing the effects of bismuth in manganese-based ZIBs. Reproduced with permission.147 Copyright 2021, the American Chemical Society. (i) Contact angle tests of BiMOF-CP and CP samples. (j) Element analysis of deposited Mn2+ in 2 M ZnSO4/0.2 M MnSO4 aqueous electrolyte during cycling of BiMOF-CP and CP. (k) Cycling performance of BiMOF-CP and CP. (i)–(k) Reproduced with permission.148 Copyright 2023, Wiley-VCH. | |
ZnMn2O4 quantum dots (ZMO QD@C) were synthesized within a porous carbon framework through in situ electrochemically inducing Mn-MIL-100-derived Mn3O4 quantum dots and carbon composite, as reported by Niu et al.139 In comparison to ZMO-bulk, the presence of a new peak at 1025 cm−1 for ZMO QD@C is attributed to the formation of Mn–O–C bonds. The XPS spectra further confirmed the existence of Mn–O–C bonds, with new peaks observed at 531.9 eV for O 1s and 286.3 eV for C 1s in ZMO QD@C, respectively. Moreover, the analysis of the Mn valence states revealed that Mn predominantly exists as Mn(IV) at the interface between ZMO QDs and the carbon matrix, while being present as Mn(III) within the interior of ZMO QDs (Fig. 11d). The formation of stable Mn–O–C bonds at the interface effectively mitigates the disproportionation reactions and dissolution processes occurring on the surface of ZMO QDs (Fig. 11e). In addition, the establishment of a stable interface can also prevent direct contact between the electrolyte and the interior ZMO, thereby suppressing the dissolution of Mn3+ within the interior of ZMO QDs during discharge. Consequently, even in pure ZnSO4 electrolyte, a super stable cycling performance can be realized (Fig. 11f). Additionally, it has been reported that the formation of an Mn–O–Ce bond in the MnO2@CeO2 composite can also ameliorate the disproportionation reaction and enhance the cycling stability.140 Compositing with 2D layered MXenes not only enhances the charge transfer kinetics, but also mitigates issues such as volume expansion/contraction, structure dissolution, and side reactions of MnO2.141–145 For example, Yan et al.141 demonstrated that the incorporation of 3D Ti3C2Tx@MnO2 microflowers with superior structural integrity resulted in good cycle stability with capacity retention of 90.6% over 2000 cycles at 0.5 A g−1, which is much better than pure MnO2 with a capacity retention only 21.4%. Hetero-structured Mn2O3–ZnMn2O4 hollow octahedrons with superior structural stability were designed via a two-step metal–organic framework template strategy.146 The synergistic effects of the distinct composition and hollow heterostructure induced very stable Zn2+ storage, and the unique hollow octahedral structure could be well maintained even after 2000 cycles (Fig. 11g).
Gou et al.147 demonstrated that the MnO2–Bi2O3 composite undergoes a competitive reaction during the cycling process, resulting in the formation of Bi2Mn4O10 instead of electrochemically inactive ZnMn2O4 due to its lower formation energy, as shown in eqn (42) and (43). This leads to improved cycling performance (Fig. 11h). Thus, to avoid Mn2+ electrodeposition and its further transformation to electrochemically inactive λ-ZnMn2O4, a kinetic inhibition strategy was employed by incorporating graphite nanosheets with limited surface defects in MnO2 electrodes. The hydrophobicity and smooth surface of the graphite nanosheets effectively suppressed the rate-determining Mn2+ adsorption during nucleation, thereby ensuring a long-term cycling performance even at a high depth of discharge.102
| | | Bi2O3 + 6H+ → 2Bi3+ + 3H2O | (42) |
| | | 2Bi3+ + 4MnO2 + 2H2O + 2e− → Bi2Mn4O10 + 4H+ | (43) |
Instead of blocking Mn2+ deposition, pyridine-3,5-dicarboxylate (Bi-PYDC) with a specific poorly hydrophilic and manganophilic nature (Fig. 11i), was reported to regulate the Mn2+ deposition reaction in cathode-free ZIBs.148 As depicted in Fig. 11j, the concentration of Mn2+ rapidly decreased after the first cycle in the Zn/CP system. The fast deposition rate of CP induced the rapid generation of MnO2 and inert phase, resulting in an unstable cycling performance. In contrast, the Zn/Bi-PYDC system maintained a relatively high and steady Mn2+ concentration due the fewer nucleation sites for Mn2+ deposition. This indicates a moderate deposition rate of Mn2+ for Bi-PYDC, which effectively prevented the excessive depletion of Mn2+ in the electrolyte and maintained an equilibrium reaction between dissolution and deposition. Moreover, a certain amount of Mn2+ in the electrolyte could effectively suppress the disproportionation reaction of deposited MnO2. Consequently, the cathode-free ZIBs showed outstanding long-term cycling stability over 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles at 1 mA cm−2 (Fig. 11k).
000 cycles at 1 mA cm−2 (Fig. 11k).
5.1.2 Pre-intercalation.
The pre-intercalation of crystal water, ions, and organic species in manganese-based materials is an effective strategy for reinforcing the inherent structural stability. These intercalated guest species can form chemical bonds with basic polyhedron or act as pillars to stabilize the host framework. For example, Liang and coworkers claimed that K+ pre-intercalated in the [2 × 2] tunnels of α-MnO2 enables the formation of K–O bonds with MnO6 octahedra,149 which can stabilize the host framework and suppress the manganese dissolution to a great extent during cycling (Fig. 12a and b). Through atomic imaging, Yuan et al.150 directly observed that Co2+ was intercalated in the tunnel space of α-MnO2 [2 × 2] (Fig. 12c). The low binding energy (−6.62 eV) exhibited by Co2+ intercalated in tunnel MnO2 implies the possibility of the formation a Co–O bond between Co2+ and the surrounding O atoms, thereby enhancing the inherent stability of the Mn–O octahedron and effectively inhibiting the dissolution of Mn (Fig. 12d). The formation of metal–O bonds was further confirmed by pre-intercalated Al3+ in α-MnO2.151In situ Raman spectroscopy revealed a highly reversible phase transformation between α-MnO2 and layered birnessite, and the peak located at 280 cm−1 is attributed to Al–O bonds. Despite the phase transition inducing α-MnO2 to form layered birnessite during discharge, the signal of Al–O bonds persisted, indicating that the consistent presence of Al–O bonds not only stabilized the tunnel structure of α-MnO2 but also alleviated the structural deformation and dissolution of the layered birnessite (Fig. 12e).
 |
| | Fig. 12 Strategy for pre-intercalation based on one-electron transfer reaction. (a) Element analysis of dissolved Mn2+ in 2 M ZnSO4 aqueous electrolyte during cycling of KMO and α-MnO2. (b) Schematic of the incorporation of K+ ions stabilizing the Mn polyhedrons. (a) and (b) Reproduced with permission.149 Copyright 2019, Wiley-VCH. (c) [001] down-tunnel imaging showing the atomic structure of α-MnO2 with Co2+ and K+ occupying the tunnel center position. (d) Schematic of the incorporation of Co2+ ions stabilizing the Mn polyhedrons. (c) and (d) Reproduced with permission.150 Copyright 2023, Wiley-VCH. (e) Structure diagrams of pristine MnO2 and Al-intercalated MnO2. Reproduced with permission.151 Copyright 2021, Elsevier. (f) Schematic illustration of the synthetic process of Cu-intercalated MnO2 electrode. Reproduced with permission.152 Copyright 2022, Elsevier. (g) Long-term cycling stability of Zn-δ-NMOH battery and Zn-δ-NMOH-500 battery. Reproduced with permission.154 Copyright 2019, the American Chemical Society. (h) Schematic illustration of PANI-intercalated MnO2 nanolayers. Reproduced with permission.155 Copyright 2018, Springer Nature. (i) HAADF-STEM images of and PVP–MnO2. (j) Selective proton Grotthuss intercalation of interlayer configuration optimization. (k) Cycling stability of PVP–MnO2. (i)–(k) Reproduced with permission.156 Copyright 2023, Wiley-VCH. | |
The application of the pre-intercalation strategy in δ-MnO2 is considered promising due to its much larger interlamellar spacing (about 0.7 nm) compared to tunnel-type MnO2. Cu2+ intercalation in δ-MnO2 was reported by Zhang et al. (Fig. 12f).152 DFT calculations indicated that the pre-intercalation of Cu2+ can effectively enhance the structural stability of δ-MnO2, which is attributed to the strong ionic bonds with oxygen atoms. The pre-intercalation of alkali ions play an important role in reinforcing the structural stability of δ-MnO2, where Na+ or K+ acts as pillars stabilizing the layered structures. This led to a minimal loss capacity of 3% between the 20th and 100th cycles at 1/3C, and long cycling stability with 90% retention after 1000 cycles at 10C for the K0.27MnO2·0.54H2O cathode, as reported by Simon et al.153
The effect of crystal water on manganese-based materials cannot be neglected, where the critical role of crystal water in layered MnO2 was explored in SIBs previously.157,158 It was found that crystal water not only acts as interlayer pillars through hydrogen bonding with the MnO6 layers to suppresses Mn dissolution, but also exhibits a charge shielding effect that mitigates the strong electrostatic interactions between the cations and the host framework, thereby enhancing the ion diffusion kinetics. As reported by Zhi et al.,154 Na+ and crystal water pre-intercalated in the δ-MnO2 interlayer space (δ-NMOH), which acted as pillars to stabilize the layered structures. Benefitting from Na+ and crystal water pre-intercalation, δ-NMOH showed an ultra-high capacity retention of 98% and well-retained layered structures over 10000 cycles. However, when δ-NMOH was thermally treated at 500 °C to remove the crystal water, it showed a poor electrochemical performance compared with δ-NMOH due to the loss of a lubricant effect for cation insertion/extraction and the insufficient remaining Na+ ions to stabilize the host structure (Fig. 12g). Lou et al.159 devised a method to pre-intercalate ammonium ions in layered δ-MnO2 through an ion–exchange reaction between K+ ions and ammonium ions. These intercalated ammonium ions act as interlayer pillars, expanding the lattice spacing of δ-MnO2 and forming stable hydrogen-bond networks, thereby alleviating the Jahn–Teller effect and demonstrating superior cycling stability over 10000 cycles. Interestingly, anions were also reported to intercalate in MnO2 for enhanced cycle performance. For example, Zhang et al.160 successfully introduced PO43− in the MnO2 layer with rich oxygen defects via a simple phosphorization process.
Besides ions and crystal water, polymers have also been introduced as stabilizers for δ-MnO2. A one-step inorganic/organic interface reaction was utilized to synthesize polyaniline-intercalated δ-MnO2 (Fig. 12h), which was proven to be effective in preventing an irreversible phase transformation and structure collapse of δ-MnO2 during the repeated hydrated Zn2+/H+ intercalation/de-intercalation process, leading to a stable cycle performance even at a low current of 0.2 A g−1.155 In addition, it was reported that pre-intercalated poly(3,4-ethylenedioxythiophene) (PEDOT) with an enlarged interlayer spacing acts as a structural pillar, which is beneficial to stabilize the layered structure of MnO2.161 Recently, polyvinylpyrrolidone (PVP) pre-intercalation in MnO2 (PVP–MnO2) with a hybrid superlattice was reported by Bai et al. (Fig. 12i).156 PVP–MnO2 exhibited preferential H+ Grotthuss intercalation behavior and effectively mitigated structural collapse by impeding the transport of [Zn(H2O)6]2+ ions due to their larger radius and stronger coulombic interaction with the host material (Fig. 12j). Consequently, this resulted in outstanding cycle stability with nearly 100% capacity retention over 20000 cycles at 10 A g−1 (Fig. 12k).
5.1.3 Heteroatom doping.
Heteroatom doping is considered an effective strategy for regulating the chemical nature and geometrical configuration of manganese-based materials. On the one hand, the incorporation of heteroatoms not only facilitates ion transfer but also modifies the electronic structure to enhance the conductivity, ultimately improving the reaction kinetics and electrochemical performance. On the other hand, heteroatom doping can modulate the strength of Mn–O bonds or form chemical bonds with Mn atoms to enhance the crystal structure stability.162,163
Metal ion doping has been commonly employed to improve the cycling stability of manganese-based ZIBs. Also, various metal ions have been demonstrated as available dopants. For instance, it was reported that the substitution of Ti and the resulting oxygen vacancy can create a charge depletion zone, leading to the formation of a built-in electric field. The presence of oxygen vacancy can modify the internal electric field and improve the electrostatic stability by compensating for the nonzero dipole moment.170 As a result, this leads to a significantly reduced charge-transfer impedance and diffusion rate for both Zn2+ and H+, facilitating their intercalation within the MnO2 tunnel structure, and ultimately contributing to an outstanding long-term cycling performance.171 The incorporation of Ni2+ in Mn2O3 was demonstrated to reduce the formation energy of the Mn–O bond (Fig. 13a), which could effectively stabilize the Mn–O bond and alleviate the dissolution of manganese.164 This resulted in a remarkable capacity retention of 85.6% over 2500 cycles at 1 A g−1 for Ni-doped Mn2O3. Multivalent cobalt-doped Mn3O4 was successfully synthesized by Wang et al.,165 wherein the doped Co exhibited various valence states and played multiple roles. Specifically, Co2+ acts as a “structural pillar” to stabilize the framework of the phase change product δ-MnO2, while the Co4+ in the layer enhances the conductivity of Mn4+ and contributes to its high specific capacity. Importantly, doping with Co2+ and Co3+ can effectively suppress the Jahn–Teller distortion and facilitate ion diffusion (Fig. 13b). The doping of high-valent Mo6+ has been reported to result in an elevated oxidation state of Mn and a reduction in oxygen defects, thereby enhancing the structural stability and inhibiting the Jahn–Teller effect during discharge/charge processes.172 Analogously, the substitution of Fe3+ at the Mn site can alleviate the collapse of the MnO2 crystal structure caused by Jahn–Teller distortion due to the increase in the mean valence of manganese.173
 |
| | Fig. 13 Strategy for heteroatom doping based on one-electron transfer reaction. (a) Calculated formation energy (in eV) of Mn2O3 and Ni-doped Mn2O3. Reproduced with permission.164 Copyright 2021, Wiley-VCH. (b) Effect of various Co ions in phase change product. Reproduced with permission.165 Copyright 2020, Wiley-VCH. (c) Mass content of dissolved Mn element in 2 M ZnSO4 aqueous electrolyte during cycling. (d) Cycling performance of N-KMO at a current density of 1 A g−1. (c) and (d) Reproduced with permission.166 Copyright 2022, Wiley-VCH. (e) Schematic diagram of enhanced reaction feasibility of F-MnO2. Reproduced with permission.167 Copyright 2021, Elsevier. (f) Charge density difference distribution diagrams of ZMO and S-ZMO. Reproduced with permission.168 Copyright 2023, Elsevier. (g) Electron density difference of K, Fe-ZMO and ZMO. Reproduced with permission.169 Copyright 2022, Elsevier. | |
Nonmetal element doping is also important for enhancing the cycling stability of manganese-based ZIBs. For example, Lou et al.166 constructed N-doped KMn8O16 (N-KMO) with abundant oxygen vacancies and large specific surface area. It was found that N-doping effectively inhibited the dissolution of Mn2+ caused by the Jahn–Teller distortion, as confirmed by the ICP results (Fig. 13c), thereby resulting in an impressive capacity retention of 91% over 2500 cycles (Fig. 13d). Moreover, the introduction of N-doping facilitated the formation of Mn–N bonds, thereby promoting interfacial dynamics in manganese-based materials and effectively mitigating manganese dissolution to ensure structural integrity.174,175 In addition, N-doped ZnMn2O4 (N-ZMO) is conductive to the release of electrostatic forces caused by charge changes. The reduced electrostatic forces during Zn2+ insertion alleviate the volume expansion and structural collapse of N-ZMO.176 Owing to the extremely high electronegativity and electron absorption of fluorine, the fluorine doping can promote robust bonding with the surrounding atoms. The dispersed doped fluorine in F-MnO2 acts as a pin to anchor the lattice framework, preventing the excessive dissolution of the manganese-based framework in the discharge process. Although the partially dissolved manganese leads to Mn vacancy in the lattice, the “pinning effect” of fluorine protects against lattice collapse. During the charging process with manganese deposition, Mn2+ preferentially deposits into metastable coordination environments associated with Mn vacancy defects, thus facilitating the restoration of the original crystal structure (Fig. 13e).167 The sulfur-doping proposed by Zhao et al. aimed to enhance the structural stability of MnO2. This improvement was attributed to the substitution of the S anions, which have lower electronegativity than the O anions in the crystal structure. Consequently, this substitution reduced the diffusion barrier for Zn2+ ions across the cathode and prevented the formation of thermodynamically stable ZnMn2O4.177 Additionally, Yang et al.168 demonstrated that S-doping could effectively modulate the electrical structure of ZnMn2O4 by inducing the non-uniform distribution of charge distribution. The accumulation of electrons near the Mn atom signifies strong interactions between Mn and O (Mn–O) as well as between Mn and S (Mn–S) (Fig. 13f), thereby improving the structural stability of ZnMn2O4.
Dual heteroatom doping with distinct functionalities represents a novel approach to achieve a stable cycling performance in manganese-based ZIBs.169,178 For example, Shao et al.169 reported the preparation of K, Fe co-doped ZnMn2O4 (K, Fe-ZMO) as a cathode for stable ZIBs. In the structure of K, Fe-ZMO, K occupies partial Zn sites, while Fe occupies partial Mn sites. The dual doping strategy promotes strong interactions between the Fe atoms and O atoms as well as the Mn atoms and O atoms (Fig. 13g). Meanwhile, the lower formation energy of K, Fe-ZMO ensures structural stability and low manganese dissolution. However, it is important to note that the performance of batteries is intricately linked to both the doping sites and content of dopants. Not all forms of doping are beneficial; in fact, some can be detrimental. By employing controllable favorable heteroatom doping, significant enhancements in intrinsic reaction kinetics can be achieved, resulting in an increase in capacity and energy power density, as well as improved cycling life.179
5.2 Strategies based on two-electron transfer reaction
The two-electron transfer reaction can eliminate the shortcomings associated with the insertion mechanism, such as detrimental Mn dissolution, dramatic volume changes and complex phase transformation. Furthermore, batteries employing the deposition/dissolution mechanism exhibit enhanced capacity compared to those based on the insertion mechanism. However, achieving a stable cycling performance in manganese-based ZIBs using the deposition/dissolution mechanism still faces challenges due to the insufficient Mn2+ absorption sites and competing reactions of Mn2+/Mn3+ during charging, as well as the incomplete dissolution of MnO2 caused by its low electrical conductivity. The strategies for improving the reversibility of two-electron transfer reaction are discussed in the following sections.
5.2.1 Rational composite design.
A porous carbon matrix-encapsulated MnO (MnO@PC) nanocomposite demonstrated the enhanced dissolution of MnO. The hierarchical meso-microporous carbon matrix possesses a large specific surface area, facilitating electrolyte infiltration and providing a negatively charged surface, which promotes the adsorption of cations. This enables better utilization of Zn2+ and Mn2+ in the electrolyte for ZSH deposition and the reversible deposition/dissolution process of ZnxMnO(OH)2.130 The composite of negatively charged Zn–Mn MOF-derived porous carbon and MnO (MnO/MZ) exhibited strong adsorption of Mn2+ owing to the strong electrostatic interactions between the negatively charged MnO/MZ and positively charged Mn2+, which significantly promoted the dissolution/deposition reaction of Mn2+/MnO2. As depicted in Fig. 14a, compared to MnO2, Super P, and monometallic Mn-MOF-derived MnO/M, MnO/MZ demonstrated the highest negatively charged zeta potential in the same mild pH environment. Moreover, the similarly lower zeta potentials observed for Super P and MnO/M suggest that Zn plays a crucial role in generating negative charge, which is possibly attributed to the vaporization of positively charged Zn during calcination. The ICP results further confirmed the superior Mn2+ absorption ability of MnO/MZ (Fig. 14b), leading to a significant decrease in Mn2+ concentration after soaking in MnSO4 solution. Despite the continuous dissolution of MnO during cycling, the dissolved Mn2+ can effectively undergo electro-oxidation and back deposition to form MnO2, resulting in performance activation. Once all the MnO is completely dissolved, the highly reversible reaction between MnO2 and Mn2+ dominates the capacity contribution, thereby ensuring excellent cycling stability over 11000 cycles.132
 |
| | Fig. 14 Strategy for rational composite design based on two-electron transfer reaction. (a) Zeta potentials of MnO/MZ, MnO/M, Super P carbon, and MnO2. (b) Adsorption of Mn2+ by MnO/MZ, MnO/M, Super P carbon, and MnO2. (a) and (b) Reproduced with permission.132 Copyright 2022, Wiley-VCH. (c) MESP distributions of MnO2 and MnO2/MoO3. (d) DFT calculations on the formation of structural water (left) and release of Mn (right) and their relative energy diagrams. (e) Schematic illustration and long-term cycling of MnO2/MoO3. (c)–(e) Reproduced with permission.180 Copyright 2023, the American Chemical Society. | |
Liu and colleagues found that both the dissolution of MnO2 and back-deposition of Mn2+ can be facilitated by compositing with MoO3.180 After introducing MoO3, the Mn–O bond strength was weakened, leading to the formation of more oxygen vacancies by reacting with the oxygen atoms in MnO2. In addition, compared to pure MnO2, MnO2/MoO3 possessed more structural water during the whole discharge process, which resulted from the proton interactions with the lattice oxygen and the lower energy barrier for Mn release, as confirmed by the DFT results (Fig. 14d). Therefore, the inherent structural stability of MnO2 was weakened and the Mn release and dissolution process were promoted. Moreover, MnO2/MoO3 exhibited a negative charge potential, while MnO2 possessed a very high positive charge potential (Fig. 14c). This indicates that MnO2/MoO3 is beneficial to attract Mn2+ for deposition. Consequently, due to the high reversibility of the dissolution and deposition reaction, MnO2/MoO3 demonstrated a stable cycling performance with 92.6% capacity retention over 300 cycles at 0.1 A g−1 and 80.1% capacity retention over 16000 cycles at 1 A g−1 in pure ZnSO4 electrolyte (Fig. 14e). Besides, organic molecules were demonstrated to improve the reversibility of the dissolution/deposition reaction. For instance, poly(1,5-diaminoanthraquinone) (PDAAQ) plays a key role on the regulation of the Mn2+ concentration, which induced a long-term cycling life exceeding 2000 cycles.181
5.2.2 Electrolyte regulation.
A strongly acidic environment undoubtedly promotes the MnO2/Mn2+ reaction according to the Nernst equation; however, it severely corrodes the Zn metal anode. Zn(CH3COO)2 is a typical near-neutral electrolyte, which can alleviate the corrosion issues and enhance the coulombic efficiency for Zn plating/stripping. During the discharge process, the protonation of the O atoms on the cathode surface leads to the formation of OH* intermediates, and the continuous reactions between OH* and proton induce the formation and desorption of H2O, resulting in cathode dissolution. Due to the high polarizability and high electronegativity of the CH3COO− groups, they strongly adsorb on the surface Mn sites,182,183 effectively weakening the binding of H2O on MnO2 and reducing the barriers for MnO2 dissolution. The highly reversible Mn4+/Mn2+ redox reaction not only increased the working potential (Fig. 15a–c), but also ensured long-term cycling stability over 4000 cycles with a high capacity of 556 mA h g−1.184 The promotion of the dissolution/deposition reaction chemistry by acetate ions was also confirmed by Zhong et al.185 The co-deposition of Zn(H2PO4)2 with MnO2 at the cathode was demonstrated to function as a local proton reservoir, facilitating the dissolution of MnO2, as reported by Liu et al.186 During the discharge, Zn(H2PO4)2 deprotonates to form Zn3(PO4)2 and provides protons to promote the MnO2/Mn2+ dissolution reaction. During the charge process, Zn3(PO4)2 is reversibly protonated to Zn(H2PO4)2, restoring protons at the cathode (Fig. 15d), as indicated by eqn (44) and (45). Therefore, a high discharge voltage of 1.75 V was achieved with an enhanced reversible two-electron transfer reaction (Fig. 15e), as well as alleviating the corrosion of the Zn anode. In addition, pH buffer is ideal to achieve highly reversible MnO2/Mn2+ reaction in mild aqueous electrolyte, as demonstrated by Balland et al.187| | | MnO2 + 4H+ + 2e− ⇌ Mn2+ + 2H2O | (44) |
| | | Zn(H2PO4)2 + 2Zn2+ + 4H2O ⇌ Zn3(PO4)2·4H2O + 4H+ | (45) |
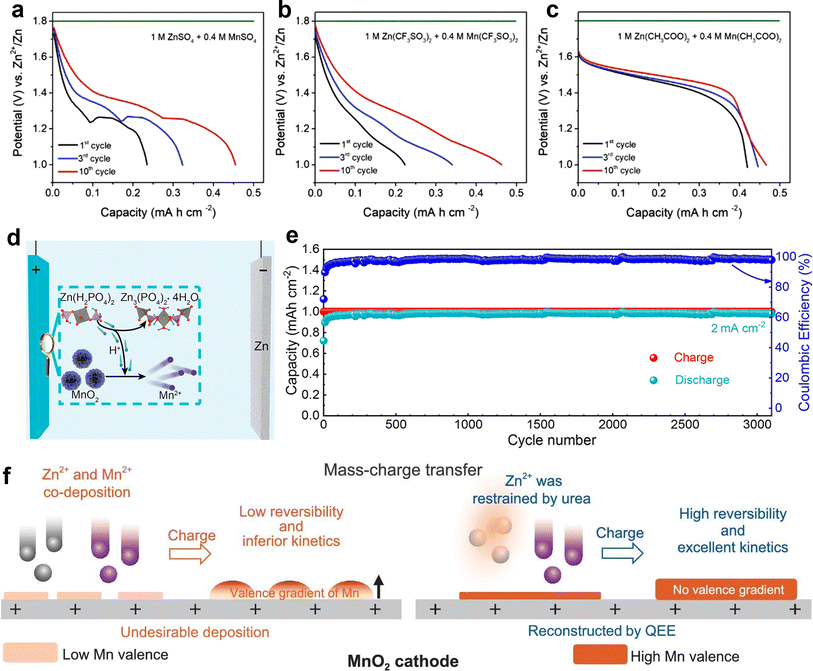 |
| | Fig. 15 Strategy for electrolyte regulation based on two-electron transfer reaction. (a) Galvanostatic discharge curves of a Zn/MnO2 battery in sulfate-based, (b) sulfonate-based, and (c) acetate-based aqueous electrolytes. (a)–(c) Reproduced with permission.184 Copyright 2020, Wiley-VCH. (d) Schematic illustration of Zn(H2PO4)2 for facilitating MnO2 dissolution. (e) Long-term cycling performance of a Zn–MnO2 cell with ZnCl2/Mn(H2PO4)2 electrolyte. (d) and (e) Reproduced with permission.186 Copyright 2022, the American Chemical Society. (f) Diagram of the electrochemical behavior of urea-based eutectic electrolyte at the cathode interface. Reproduced with permission.188 Copyright 2023, Oxford University Press. | |
The inevitable co-deposition of Zn2+ and Mn2+ to form an irreversible ZMO product will lead to poor cycling stability in manganese-based ZIBs. Through the modulation of the Zn2+-solvation shell to reduce the Zn2+ deposition at the interface, the reversibility of the dissolution/deposition reaction can be improved. For example, Hu et al.188 constructed a quasi-eutectic electrolyte composed of 2 M Zn(OTf)2 + 4 M urea + 0.25 M MnSO4. It was found that the binding energy of the Mn2+-solvation shell with urea was very weak, while the binding energy of the Zn2+-solvation shell was strong. The significant difference in the Mn2+/Zn2+ ionic solvation structure led to different mass transfer between Zn2+ and Mn2+ at the cathode interface (Fig. 15f). The high desolvation energy of Zn2+ makes Zn2+ co-deposition with Mn2+ at the cathode interface difficult, thereby restricting the mass transfer of Zn2+. When the electrolyte contains urea, ZMO cannot be discovered at the cathode during charging and discharging. In contrast, without urea additive, the inert ZnMn3O7 phase is obviously detected during charging and discharging. Moreover, with the addition of urea, the valence states of Mn in the deposition increased obviously, thus giving rise to a higher capacity and better reversibility. In addition, a cationic accelerator strategy (CA) by adding poly(vinylpyrrolidone) was proposed to regulate the cationic solvation structures, as reported by Chuai et al.189 Theoretical calculations and experimental results demonstrated the effective promotion of the rapid migration of Zn2+ and Mn2+ in the electrolyte by CA, facilitating sufficient charge transfer at electrode–electrolyte interface. This beneficially contributed to the reversible electrochemical deposition/dissolution chemistry of cathodic MnO2/Mn2+ and anodic Zn/Zn2+, leading long-term cycling stability for over 2000 cycles. The concentration of pre-added Mn2+ significantly influenced the long-term cycling performance. Shen et al.190 demonstrated that adjusting the Mn2+ concentration to a critical range can recycle Mn2+ from MnOOH disproportionation, enabling the reversible redox conversion between MnO2 and Mn2+. As a result, an ultra-long lifespan of 16000 cycles without noticeable capacity degradation was achieved.
In summary, a comprehensive discussion was presented on optimization strategies aimed at improving the cycling stability in manganese-based ZIBs, and the manganese-based materials for superior long-term cycling stability in ZIBs are summarized in Table 2. The formation of stable chemical bonds through compositing, pre-intercalation and heteroatom doping can mitigate the dissolution and structural collapse of manganese-based materials. Moreover, rational composite design can effectively reduce the deposition reaction of Mn2+ ions, preventing rapid electrolyte Mn2+ consumption and the formation of an inert ZMO phase. Meanwhile, reducing the dissolution barrier of manganese, promoting Mn2+ adsorption, and selectively alleviating Zn2+ deposition on the cathode by compositing or electrolyte regulation are beneficial for Mn dissolution and Mn2+ redeposition. Consequently, a stable cycling performance in manganese-based ZIBs can be achieved based on the reversible Mn3+ ↔ Mn2+ reaction or Mn4+ ↔ Mn2+ reaction, respectively.
Table 2 Summary of the stable cycling performance of manganese-based materials in ZIBs
| Reaction mechanism/electrodes |
Electrolyte |
Voltage window |
Capacity retention (cycles/current density) |
Strategies/causes |
Ref. |
|
One-electron transfer reaction
|
|
|
|
Rational composite design
|
|
| α-MnO2@graphite nanosheets |
2 M ZnSO4 + 0.1 M MnSO4 |
1–1.8 V |
95% (600/1C) |
Suppress Mn2+ electrodeposition |
102
|
| 98% (3000/6C) |
| MnO2@CeO2 |
2 M ZnSO4 + 0.1 M MnSO4 |
0.8–1.8 V |
89.68% (1000/1 A g−1) |
Suppress Jahn–Teller distortion |
140
|
| ZnMn2O4@C |
2 M ZnSO4 |
0.8–1.8 V |
86.4% (1500/1 A g−1) |
Suppress Jahn–Teller distortion |
139
|
| δ-MnO2-C NA |
2 M ZnSO4 + 0.1 M MnSO4 |
0.8–1.8 V |
90.4% (50000/4 A g−1) |
Buffer volume changes |
191
|
| N-doped carbon@δ-MnO2 |
2 M ZnSO4 + 0.3 M MnSO4 |
1–1.8 V |
87% (3500/2 A g−1) |
Buffer volume changes |
192
|
| α-MnO2/graphene scrolls |
2 M ZnSO4 + 0.2 M MnSO4 |
1–1.85 V |
94% (3000/3 A g−1) |
Suppress cathode dissolution |
136
|
| Mn2O3–ZnMn2O4 |
Zn(CF3SO3)2 + 0.1 M MnSO4 |
0.8–1.8 V |
93.3% (2000/3 A g−1) |
Superior structural stability |
146
|
| Ti3C2Tx@MnO2 |
2 M ZnSO4 + 0.2 M MnSO4 |
0.8–1.8 V |
90.6% (2000/0.5 A g−1) |
Suppress cathode dissolution |
141
|
| N-Mn3O4/MnO |
2 M ZnSO4 + 0.2 M MnSO4 |
0.8–1.8 V |
77.2% (2500/10 A g−1) |
Alleviate stress |
193
|
|
|
|
|
|
Pre-intercalation
|
|
| K0.27MnO2·0.54H2O |
2 M ZnSO4 + 0.3 M MnSO4 |
0.7–1.8 V |
91% (1000/10C) |
Suppress cathode dissolution |
153
|
| Al-intercalated MnO2 |
2 M ZnSO4 |
0.8–1.8 V |
94.5% (2000/2 A g−1) |
Suppress cathode dissolution |
151
|
| Na0.55Mn2O4·1.5H2O |
2 M ZnSO4 + 0.1 M MnSO4 + 0.5M Na2SO4 |
0.8–1.9 V |
70% (10000/6.5C) |
Suppress cathode dissolution |
194
|
| Na0.46Mn2O4·1.4H2O |
2 M ZnSO4 + 0.2 M MnSO4 |
0.95–1.85 V |
98% (10000/20C) |
Suppress cathode dissolution |
154
|
| Na+-intercalated MnO2 |
2 M ZnSO4 + 0.2 M MnSO4 |
0.9–1.8 V |
93% (1000/4 A g−1) |
Suppress cathode dissolution |
195
|
| Cu-MnO2 |
2 M ZnSO4 + 0.2 M MnSO4 |
0.8–1.9 V |
90.1% (700/5 A g−1) |
Suppress cathode dissolution |
152
|
| CoxMnO2 |
2 M ZnSO4 + 0.2 M MnSO4 |
0.8–1.8 V |
84.6 (500/0.5 A g−1) |
Suppress cathode dissolution |
150
|
| NH4+-intercalated MnO2 |
2 M ZnSO4 + 0.1 M MnSO4 |
0.8–1.8 V |
69% (10000/4 A g−1) |
Suppress cathode dissolution |
159
|
| PANI-intercalated MnO2 |
2 M ZnSO4 + 0.1 M MnSO4 |
1–1.8 V |
∼84% (5000/2 A g−1) |
Suppress cathode dissolution |
155
|
| PVP-MnO2 |
2 M ZnSO4 + 0.2 M MnSO4 |
0.8–1.8 V |
∼100% (20000/10 A g−1) |
Suppress cathode dissolution |
156
|
|
|
|
|
|
Heteroatom doping
|
|
| Al-doped MnO2 |
2 M ZnSO4 + 0.1 M MnSO4 |
0.8–1.8 V |
79% (15000/4 A g−1) |
Suppress cathode dissolution |
196
|
| Bi-doped MnO2 |
2 M ZnSO4 + 0.2 M MnSO4 |
0.8–1.8 V |
93% (10000/1 A g−1) |
Suppress cathode dissolution |
197
|
| Ni-doped MnO |
2 M ZnSO4 + 0.2 M MnSO4 |
0.8–1.8 V |
91% (6000/3 A g−1) |
Enhance structure stability |
198
|
| Co-doped Mn3O4 |
2 M ZnSO4 + 0.2 M MnSO4 |
0.2–2.2 V |
80% (1100/2 A g−1) |
Suppress Jahn–Teller distortion |
165
|
| N-doped KMn8O16 |
2 M ZnSO4 + 0.1 M MnSO4 |
0.8–1.8 V |
91% (2500/1 A g−1) |
Suppress Jahn–Teller distortion |
166
|
| N-doped Na2Mn3O7 |
2 M ZnSO4 + 0.2 M MnSO4 |
0.8–1.8 V |
78.9% (550/2 A g−1) |
Suppress cathode dissolution |
174
|
| K, Fe-doped ZnMn2O4 |
2 M ZnSO4 + 0.2 M MnSO4 |
0.8–1.8 V |
88.1% (500/1 A g−1) |
Suppress cathode dissolution |
169
|
|
Two-electron transfer reaction
|
|
|
|
Rational composite design
|
|
| MnO2/MoO3 |
3 M ZnSO4 |
0.2–2.2 V |
92.6% (300/0.1 A g−1) |
Facilitating dissolution/deposition |
180
|
| 80.1% (16000/1 A g−1) |
| MnO/MN |
3 M ZnSO4 |
0.6–1.9 V |
80.3% (200/0.1 A g−1) |
Facilitating deposition |
132
|
| −(11000/2 A g−1) |
| MnO/PC |
3 M ZnSO4 |
0.8–1.9 V |
93% (120/0.1 A g−1) |
Facilitating deposition/dissolution |
130
|
| −(2000/2 A g−1) |
| MnO2@PDAAQ |
2 M ZnSO4 + 0.2 M MnSO4 |
0.2–1.85 V |
−(2000/2 A g−1) |
Facilitating deposition/dissolution |
181
|
|
Electrolyte regulation
|
| MnO2/Zn(H2PO4)2 |
2 M ZnCl2 + 0.07 M Mn(H2PO4)2 |
0.5–2.1 V |
∼100% (3000/2 mA cm−2) |
Facilitating dissolution |
186
|
| MnO2 |
1 M ZnSO4 + 1 M MnSO4 + 0.1 M H2SO4 + 0.07 mM PVP |
1–2.2 V |
90% (2000/20C) |
Facilitating deposition/dissolution |
189
|
| MnO2 |
2 M Zn(OTf)2 + 4 M urea + 0.25 M MnSO4 |
0.8–1.8 V |
76.8% (350/0.5 A g−1) |
Restricting Zn2+ deposition |
188
|
| MnO2 |
2 M ZnSO4 + 0.4 M MnSO4 + 0.8 NaAc |
1–1.8 V |
−(350/0.5 A g−1) |
Facilitating deposition/dissolution |
185
|
| MnO2 |
1 M Zn(Ac)2 + 0.4 M Mn(Ac)2 |
1–1.8 V |
∼100% (4000/5 mA cm−2) |
Facilitating deposition/dissolution |
184
|
| MnO2@CNT |
2 M ZnSO4 + 0.005 M MnSO4 |
1–2.4 V |
∼100% (16000/10 mA cm−2) |
Facilitating deposition |
190
|
6 Summary and perspectives
Manganese-based ZIBs exhibit distinctive advantages and hold great promise for large-scale energy storage applications due to their cost-effectiveness, natural availability, low toxicity, multivalent states, high operation voltage, and satisfactory capacity. However, their unsatisfactory cycle stability coupled with intricate and contentious storage mechanisms hinder their commercial applications. In this review, the energy storage mechanisms of manganese-based ZIBs with different structures were systematically clarified and summarized. Subsequently, the capacity fluctuation of manganese-based ZIBs including capacity activation, degradation, and dynamic evolution in the whole cycle calendar were comprehensively analyzed. Lastly, the constructive optimization strategies based on the reaction chemistry of one-electron or two-electron transfer for achieving a durable cycling performance in manganese-based ZIBs were put forward, respectively. Nevertheless, challenges and disputes still persist in manganese-based ZIBs. Herein, we conclude some challenges and prospects in the reaction chemistry and optimization strategies for achieving a stable cycling performance in manganese-based ZIBs, as illustrated in Fig. 16.
 |
| | Fig. 16 Perspectives for achieving stable cycling performance in manganese-based ZIBs. | |
(i) Systematic investigation of energy storage mechanisms. Given that the storage mechanisms have been discovered successively and the quick development of advanced characterization techniques in recent years, early reports may give incorrect and incomplete analyses. For example, there have been reports suggesting the complete transformation of the tunnel phase in α-MnO2 during discharge, followed by subsequent recovery upon charging. However, the extent of this remarkable structural reversibility raises questions due to the high energy demand associated with charge ordering during tunnel formation.199 Moreover, conventional characterization of the reaction mechanisms heavily relies on interpretations based on bulk-level characterizations, which may overlook the crucial local changes occurring within cells and potentially lead to misinterpretation of the reaction mechanisms due to the presence of side products or residual electrolytes.200 For instance, routinely used XRD techniques may yield ambiguous data due to the formation of various phases of low symmetry, adding layers upon cycling. In addition, the specific roles of Zn2+ and H+ during charge/discharge remain ambiguous. However, employing crucial analysis methods, such as solid-state 67Zn and 1H (1D) NMR-mass spectroscopy with isotope labeling can potentially facilitate a more comprehensive evaluation of the contribution of Zn2+ and H+, although this task remains challenging.92 Furthermore, it is worth noting that in most reports, a single electrochemical reaction process is declared in the whole cycle calendar. However, the reaction chemistry usually consists of multiple processes that dominate at different stages with a dynamic change in the Mn valence and electrolyte pH. Therefore, investigations into energy storage mechanisms based solely on initial cycles may not fully account for the complexities of the entire cycle. Consequently, the development of more advanced characterization technologies such as atomic-level characterization and in situ techniques combined with the continuous monitoring of dynamic changes in the entire cycle, will facilitate a comprehensive understanding of the energy storage mechanisms inherent to manganese-based ZIBs.
(ii) Paying attention to the influence of pre-added Mn2+. On the one hand, the Mn2+ additives would stabilize the cycling performance to some extent by compensating for the capacity loss of Mn2+ electrodeposition or mitigating the Jahn–Teller effect. On the other hand, the pre-added Mn2+ increases the complexity of reaction chemistry in manganese-based ZIBs and may obscure the electrochemical behaviors of the original cathode due to its deposition. Although extra capacity can be obtained, the accumulation of by-products such as a ZMO phase resulting from the transformation of deposited Mn2+ can lead to rapid capacity degradation and even battery failure. In addition, the content of Mn2+ in the electrolyte not only impacts the cycle stability, but also dominates the reaction rate between insertion/extraction chemistry and dissolution/deposition chemistry. Excessive Mn2+ in the electrolyte may facilitate the dissolution/deposition reaction, while inadequate Mn2+ may compromise the cycling stability. Moreover, the presence of pre-added Mn2+ has an influence on pH fluctuations, thereby further impacting the energy storage mechanism. The excessive addition of Mn2+ can effectively suppress the formation of ZHS, but may also alter the reaction mechanism by reducing the electrolyte pH.92 Therefore, gaining a comprehensive understanding of pre-added Mn2+ is important for researchers to comprehend the underlying reaction mechanisms in manganese-based ZIBs and devise effective strategies to enhance their cycle stability.
(iii) Developing new manganese-based cathodes. Developing novel manganese-based cathodes is of paramount importance for the large-scale implementation of ZIBs. Despite the fact that traditional manganese oxides have been widely investigated as cathode materials for ZIBs, ZIBs still suffer from the lack of high-performance cathode materials. Ensuring the structural stability of manganese-based materials during Zn2+ storage is crucial for achieving cycling stability. The search for new stable manganese-based cathodes, such as manganese-based layered double hydroxide (LDH) and high entropy oxides (HEOs), presents a promising avenue.201 For example, HEOs (Co0.6Ni0.6Fe0.6Mn0.6Cu0.6O4) with long-range disorders and a fixed lattice structure can effectively alleviate the structural collapse induced by the strong electrostatic force between Zn2+, thereby achieving remarkable long-term stability.202 Additionally, a trinary LDH with hydrogen vacancies (Ni3Mn0.7Fe0.3-LDH) has been reported as a novel cathode material for ZIBs, exhibiting a reversible high capacity of up to 328 mA h g−1 and demonstrating excellent cycling stability over 500 cycles with a capacity retention of 85%.203 However, their stable cycling performance still relied on the presence of pre-added Mn2+, which may lead to a misinterpretation of their inherent structural stability. Therefore, to realize the stable cycling performance of manganese-based ZIBs, it is imperative to not only optimize traditional manganese oxides but also dedicate more efforts towards developing new manganese-based cathodes with intrinsic structural stability.
(iv) Consideration of factors beyond the cathode. Besides rational modifications to manganese-based cathodes and electrolytes, attention should also be given beyond the cathode. The Zn anode faces challenges such as dendrite formation, passivation, corrosion, and hydrogen evolution, which significantly impede the cycling performance stability. Effective strategies for achieving Zn anodes with a long lifespan include the deposition of artificial interface layers, electrode design, and electrolyte modifications.204 For example, the addition of DMSO to the electrolyte has been demonstrated to facilitate the formation of a solid electrolyte interphase on the anode, effectively preventing Zn dendrite growth and water decomposition.205 In addition, although glass fiber separators are commonly used in ZIBs, their inherent fragility and large pores make them susceptible to penetration by Zn dendrites, leading to battery failure after repetitive plating/stripping.206,207 Modifying traditional glass fiber separators or developing novel separators can help regulate the electric field distribution uniformly and suppress dendrite formation as well as parasitic side reactions. Besides, current collectors and binders cannot be neglected as well. Furthermore, optimizing the cut-off voltage from the perspective of electrochemical parameters is another way to enhance the cycling stability of manganese-based ZIBs, which could avoid side reactions or activate inactive materials.117,208,209 For instance, extending the voltage cut-off to 2.2 V enables the activation of previously inactive components, facilitating a reversible MnO2/Mn2+ reaction with two-electron transfer.210 However, the investigation of cycling stability beyond the cathode remains inadequate. Therefore, to achieve the maximum cycling stability, more attention should be paid to optimizing beyond the cathode.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
References
- Z. Yang, J. Zhang, M. Kintner-Meyer, X. Lu, D. Choi, J. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577–3613 CrossRef CAS PubMed.
- L. Yang, K. Yang, J. Zheng, K. Xu, K. Amine and F. Pan, Chem. Soc. Rev., 2020, 49, 4667–4680 RSC.
- B. Dunn, H. Kamath and J. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- J. W. Choi and D. Aurbach, Nat. Rev. Mater., 2016, 1, 16013 CrossRef CAS.
- M. Li, J. Lu, Z. Chen and K. Amine, Adv. Mater., 2018, 30, 1800561 CrossRef.
- B. Liu, Y. Jia, J. Li, S. Yin, C. Yuan, Z. Hu, L. Wang, Y. Li and J. Xu, J. Mater. Chem. A, 2018, 6, 21475–21484 RSC.
- S. Abada, G. Marlair, A. Lecocq, M. Petit, V. Sauvant-Moynot and F. Huet, J. Power Sources, 2016, 306, 178–192 CrossRef CAS.
- L. Blanc, D. Kundu and L. Nazar, Joule, 2020, 4, 771–799 CrossRef CAS.
- D. Selvakumaran, A. Pan, S. Liang and G. Cao, J. Mater. Chem. A, 2019, 7, 18209–18236 RSC.
- L. Chen, Q. An and L. Mai, Adv. Mater. Interfaces, 2019, 6, 1900387 CrossRef.
- J. Yan, J. Wang, H. Liu, Z. Bakenov, D. Gosselink and P. Chen, J. Power Sources, 2012, 216, 222–226 CrossRef CAS.
- Y. Zong, H. Chen, J. Wang, M. Wu, Y. Chen, L. Wang, X. Huang, H. He, X. Ning, Z. Bai, W. Wen, D. Zhu, X. Ren, N. Wang and S. Dou, Adv. Mater., 2023, 35, 2306269 CrossRef CAS PubMed.
- J. Yin, Y. Wang, Y. Zhu, J. Jin, C. Chen, Y. Yuan, Z. Bayhan, N. Salah, N. Alhebshi, W. Zhang, U. Schwingenschlögl and H. Alshareef, Nano Energy, 2022, 99, 107331 CrossRef CAS.
- Q. Zhang, J. Luan, Y. Tang, X. Ji and H. Wang, Angew. Chem., Int. Ed., 2020, 59, 13180–13191 CrossRef CAS.
- Y. Dai, C. Zhang, W. Zhang, L. Cui, C. Ye, X. Hong, J. Li, R. Chen, W. Zong, X. Gao, J. Zhu, P. Jiang, Q. An, D. J. L. Brett, I. P. Parkin, G. He and L. Mai, Angew. Chem., Int. Ed., 2023, 62, e202301192 CrossRef CAS PubMed.
- T. Wei, Y. Ren, Y. Wang, L. e. Mo, Z. Li, H. Zhang, L. Hu and G. Cao, ACS Nano, 2023, 17, 3765–3775 CrossRef CAS PubMed.
- Y. Lin, Z. Mai, H. Liang, Y. Li, G. Yang and C. Wang, Energy Environ. Sci., 2023, 16, 687–697 RSC.
- N. Chodankar, P. Shinde, S. Patil, G. Rama Raju, S. Hwang, S. Marje, H. Tyagaraj, E. Al Hajri, A. Al Ghaferi, Y. Huh and Y. Han, ChemSusChem, 2023, 16, e202300730 CrossRef CAS.
- J. Chen, W. Xu, H. Wang, X. Ren, F. Zhan, Q. He, H. Wang and L. Chen, J. Mater. Chem. A, 2022, 10, 21197–21250 RSC.
- F. Gao, B. Mei, X. Xu, J. Ren, D. Zhao, Z. Zhang, Z. Wang, Y. Wu, X. Liu and Y. Zhang, Chem. Eng. J., 2022, 448, 137742 CrossRef CAS.
- X. Zhao, L. Mao, Q. Cheng, F. Liao, G. Yang, X. Lu and L. Chen, Energy Storage Mater., 2021, 38, 397–437 CrossRef.
- M. Wu, J. Bai, M. Xue, X. Zhao, L. Mao and L. Chen, Chem. Commun., 2023, 59, 11668–11671 RSC.
- X. Zhao, L. Mao, Q. Cheng, F. Liao, G. Yang and L. Chen, Chem. Commun., 2022, 624, 482–493 Search PubMed.
- G. Zampardi and F. La Mantia, Curr. Opin. Electrochem., 2020, 21, 84–92 CrossRef CAS.
- H. Wang, Q. He, S. Liang, Y. Li, X. Zhao, L. Mao, F. Zhan and L. Chen, Energy Storage Mater., 2021, 43, 531–578 CrossRef.
- H. Yi, R. Qin, S. Ding, Y. Wang, S. Li, Q. Zhao and F. Pan, Adv. Funct. Mater., 2021, 31, 2006970 CrossRef CAS.
- H. Wang, X. Ren, J. Chen, W. Xu, Q. He, H. Wang, F. Zhan and L. Chen, J. Power Sources, 2023, 554, 232309 CrossRef CAS.
- Q. He, J. Bai, H. Wang and Y. Liao, Chem. Eng. J., 2024, 485, 149893 CrossRef CAS.
- T. Sun, J. Xie, W. Guo, D. Li and Q. Zhang, Adv. Energy Mater., 2020, 10, 1904199 CrossRef CAS.
- Z. Tie and Z. Niu, Angew. Chem., Int. Ed., 2020, 59, 21293–21303 CrossRef CAS.
- G. Fang, J. Zhou, A. Pan and S. Liang, ACS Energy Lett., 2018, 3, 2480–2501 CrossRef CAS.
- K. Nam, H. Kim, J. Choi and J. Choi, Energy Environ. Sci., 2019, 12, 1999–2009 RSC.
- T. Xiong, Z. Yu, H. Wu, Y. Du, Q. Xie, J. Chen, Y. Zhang, S. Pennycook, W. Lee and J. Xue, Adv. Energy Mater., 2019, 9, 1803815 CrossRef.
- A. Zhang, R. Zhao, Y. Wang, J. Yang, C. Wu and Y. Bai, Energy Environ. Sci., 2023, 16, 3240–3301 RSC.
- Q. Dai, L. Li, T. Hoang, T. Tu, B. Hu, Y. Jia, M. Zhang, L. Song and M. Trudeau, J. Energy Storage, 2022, 55, 105397 CrossRef.
- S. Xie, X. Li, Y. Li, Q. Liang and L. Dong, Chem. Rec., 2022, 22, e202200201 CrossRef CAS PubMed.
- N. Zhang, Y. Ji, J. Wang, P. Wang, Y. Zhu and T. Yi, J. Energy Chem., 2023, 82, 423–463 CrossRef CAS.
- X. Cui, Y. Zhang, J. Zhang, E. Xie and J. Fu, Adv. Mater. Technol., 2023, 8, 2300321 CrossRef CAS.
- T. Xiong, Y. Zhang, W. Lee and J. Xue, Adv. Energy Mater., 2020, 10, 2001769 CrossRef CAS.
- N. Zhang, J. Wang, Y. Guo, P. Wang, Y. Zhu and T. Yi, Coord. Chem. Rev., 2023, 479, 215009 CrossRef CAS.
- M. Han, L. Qin, Z. Liu, L. Zhang, X. Li, B. Lu, J. Huang, S. Liang and J. Zhou, Mater. Today Energy, 2021, 20, 100626 CrossRef CAS.
- W. Shi, W. Lee and J. Xue, ChemSusChem, 2021, 14, 1634–1658 CrossRef CAS PubMed.
- N. Zhang, J. Wang, X. Liu, P. Wang, Y. Liu, Y. Xie and T. Yi, Composites, Part B, 2023, 260, 110770 CrossRef CAS.
- X. Jia, C. Liu, Z. G. Neale, J. Yang and G. Cao, Chem. Rev., 2020, 120, 7795–7866 CrossRef CAS.
- Z. Zhang, H. Shang, X. Zhang, C. Liu, S. Li, Z. Wen, S. Ji and J. Sun, ACS Appl. Energy Mater., 2021, 4, 5113–5122 CrossRef CAS.
- X. Du, G. Guan, X. Li, A. D. Jagadale, X. Ma, Z. Wang, X. Hao and A. Abudula, J. Mater. Chem. A, 2016, 4, 13989–13996 RSC.
- C. Yuan, Y. Zhang, Y. Pan, X. Liu, G. Wang and D. Cao, Electrochim. Acta, 2014, 116, 404–412 CrossRef CAS.
- W. Chen, X. Zhan, B. Luo, Z. Ou, P. Shih, L. Yao, S. Pidaparthy, A. Patra, H. An, P. Braun, R. Stephens, H. Yang, J. Zuo and Q. Chen, Nano Lett., 2019, 19, 4712–4720 CrossRef CAS PubMed.
- M. Liu, Q. Zhao, H. Liu, J. Yang, X. Chen, L. Yang, Y. Cui, W. Huang, W. Zhao, A. Song, Y. Wang, S. Ding, Y. Song, G. Qian, H. Chen and F. Pan, Nano Energy, 2019, 64, 103942 CrossRef CAS.
- W. Liu, X. Zhang, Y. Huang, B. Jiang, Z. Chang, C. Xu and F. Kang, J. Energy Chem., 2021, 56, 365–373 CrossRef CAS.
- S. Islam, M. Alfaruqi, V. Mathew, J. Song, S. Kim, S. Kim, J. Jo, J. Baboo, D. Pham, D. Putro, Y. Sun and J. Kim, J. Mater. Chem. A, 2017, 5, 23299–23309 RSC.
- M. Han, J. Huang, S. Liang, L. Shan, X. Xie, Z. Yi, Y. Wang, S. Guo and J. Zhou, iScience, 2020, 23, 100797 CrossRef CAS PubMed.
- N. Zhang, F. Cheng, J. Liu, L. Wang, X. Long, X. Liu, F. Li and J. Chen, Nat. Commun., 2017, 8, 405 CrossRef.
- M. Alfaruqi, V. Mathew, J. Gim, S. Kim, J. Song, J. Baboo, S. Choi and J. Kim, Chem. Mater., 2015, 27, 3609–3620 CrossRef CAS.
- C. Wang, Y. Zeng, X. Xiao, S. Wu, G. Zhong, K. Xu, Z. Wei, W. Su and X. Lu, J. Energy Chem., 2020, 43, 182–187 CrossRef.
- Y. Liao, H. Chen, C. Yang, R. Liu, Z. Peng, H. Cao and K. Wang, Energy Storage Mater., 2022, 44, 508–516 CrossRef.
- J. Lee, J. Ju, W. Cho, B. Cho and S. Oh, Electrochim. Acta, 2013, 112, 138–143 CrossRef CAS.
- L. Chen, X. Guo, W. Lu, M. Chen, Q. Li, H. Xue and H. Pang, Coord. Chem. Rev., 2018, 368, 13–34 CrossRef CAS.
- W. Li, X. Gao, Z. Chen, R. Guo, G. Zou, H. Hou, W. Deng, X. Ji and J. Zhao, Chem. Eng. J., 2020, 402, 125509 CrossRef CAS.
- C. Zhu, G. Fang, S. Liang, Z. Chen, Z. Wang, J. Ma, H. Wang, B. Tang, X. Zheng and J. Zhou, Energy Storage Mater., 2020, 24, 394–401 CrossRef.
- B. Jiang, C. Xu, C. Wu, L. Dong, J. Li and F. Kang, Electrochim. Acta, 2017, 229, 422–428 CrossRef CAS.
- X. Shan, D. S. Charles, Y. Lei, R. Qiao, G. Wang, W. Yang, M. Feygenson, D. Su and X. Teng, Nat. Commun., 2016, 7, 13370 CrossRef CAS PubMed.
- J. Sun, D. Li, S. Wang, J. Xu, W. Liu, M. Ren, F. Kong, S. Wang and L. Yang, J. Alloys Compd., 2021, 867, 159034 CrossRef CAS.
- N. Zhang, F. Cheng, Y. Liu, Q. Zhao, K. Lei, C. Chen, X. Liu and J. Chen, J. Am. Chem. Soc., 2016, 138, 12894–12901 CrossRef CAS.
- V. Soundharrajan, B. Sambandam, S. Kim, V. Mathew, J. Jo, S. Kim, J. Lee, S. Islam, K. Kim, Y.-K. Sun and J. Kim, ACS Energy Lett., 2018, 3, 1998–2004 CrossRef CAS.
- Y. Gao, H. Yang, Y. Bai and C. Wu, J. Mater. Chem. A, 2021, 9, 11472–11500 RSC.
- X. Hou, X. Liu, H. Wang, X. Zhang, J. Zhou and M. Wang, Energy Storage Mater., 2023, 57, 577–606 CrossRef.
- T. Zhang, Y. Tang, G. Fang, C. Zhang, H. Zhang, X. Guo, X. Cao, J. Zhou, A. Pan and S. Liang, Adv. Funct. Mater., 2020, 30, 2002711 CrossRef CAS.
- H. Guo, Z. Shao, Y. Zhang, X. Cui, L. Mao, S. Cheng, M. Ma, W. Lan, Q. Su and E. Xie, J. Colloid Interface Sci., 2022, 608, 1481–1488 CrossRef CAS PubMed.
- X. Wu, Y. Li, Y. Xiang, Z. Liu, Z. He, X. Wu, Y. Li, L. Xiong, C. Li and J. Chen, J. Power Sources, 2016, 336, 35–39 CrossRef CAS.
- Y. Wu, J. Fee, Z. Tobin, A. Shirazi-Amin, P. Kerns, S. Dissanayake, A. Mirich and S. L. Suib, ACS Appl. Energy Mater., 2020, 3, 1627–1633 CrossRef CAS.
- Y. Cai, R. Chua, S. Huang, H. Ren and M. Srinivasan, Chem. Eng. J., 2020, 396, 125221 CrossRef CAS.
- X. Chen, W. Li, Y. Xu, Z. Zeng, H. Tian, M. Velayutham, W. Shi, W. Li, C. Wang, D. Reed, V. V. Khramtsov, X. Li and X. Liu, Nano Energy, 2020, 75, 104869 CrossRef CAS.
- M. Wang, C. Cao, F. Su, Y. Wang, W. Wang, C. Ding, J. Bai, T. Liu, X. Sun and J. Zhang, J. Power Sources, 2020, 477, 229013 CrossRef CAS.
- X. Dong, J. Sun, Y. Mu, Y. Yu, T. Hu, C. Miao, C. Huang, C. Meng and Y. Zhang, J. Colloid Interface Sci., 2022, 610, 805–817 CrossRef CAS.
- T. Zhu, K. Zheng, P. Wang, X. Cai, X. Wang, D. Gao, D. Yu, C. Chen and Y. Liu, J. Colloid Interface Sci., 2022, 610, 796–804 CrossRef CAS PubMed.
- S. Wang, G. Zeng, Q. Sun, Y. Feng, X. Wang, X. Ma, J. Li, H. Zhang, J. Wen, J. Feng, L. Ci, A. Cabot and Y. Tian, ACS Nano, 2023, 17, 13256–13268 CrossRef CAS PubMed.
- H. Pan, Y. Shao, P. Yan, Y. Cheng, K. S. Han, Z. Nie, C. Wang, J. Yang, X. Li, P. Bhattacharya, K. T. Mueller and J. Liu, Nat. Energy, 2016, 1, 16039 CrossRef CAS.
- Y. Jin, L. Zou, L. Liu, M. Engelhard, R. Patel, Z. Nie, K. Han, Y. Shao, C. Wang, J. Zhu, H. Pan and J. Liu, Adv. Mater., 2019, 31, 1900567 CrossRef PubMed.
- Z. Tang, W. Chen, Z. Lyu and Q. Chen, Energy Mater. Adv., 2022, 2022, 9765710 Search PubMed.
- J. Wang, J.-G. Wang, H. Liu, Z. You, C. Wei and F. Kang, J. Power Sources, 2019, 438, 226951 CrossRef CAS.
- D. Feng, T. Gao, L. Zhang, B. Guo, S. Song, Z. Qiao and S. Dai, Nano-Micro Lett., 2019, 12, 14 CrossRef.
- J. Hao, J. Mou, J. Zhang, L. Dong, W. Liu, C. Xu and F. Kang, Electrochim. Acta, 2018, 259, 170–178 CrossRef CAS.
- X. Wu, Y. Xiang, Q. Peng, X. Wu, Y. Li, F. Tang, R. Song, Z. Liu, Z. He and X. Wu, J. Mater. Chem. A, 2017, 5, 17990–17997 RSC.
- L. Jiang, Z. Wu, Y. Wang, W. Tian, Z. Yi, C. Cai, Y. Jiang and L. Hu, ACS Nano, 2019, 13, 10376–10385 CrossRef CAS PubMed.
- N. Yadav, S. Khamsanga, S. Kheawhom, J. Qin and P. Pattananuwat, J. Energy Storage, 2023, 64, 107148 CrossRef.
- J. Li, L. Li, H. Shi, Z. Zhong, X. Niu, P. Zeng, Z. Long, X. Chen, J. Peng, Z. Luo, X. Wang and S. Liang, ACS Sustainable Chem. Eng., 2020, 8, 10673–10681 CAS.
- M. R. Palacín, Chem. Soc. Rev., 2009, 38, 2565–2575 RSC.
- C. Xu, B. Li, H. Du and F. Kang, Angew. Chem., Int. Ed., 2012, 51, 933–935 CrossRef CAS PubMed.
- B. Lee, C. S. Yoon, H. R. Lee, K. Y. Chung, B. W. Cho and S. H. Oh, Sci. Rep., 2014, 4, 6066 CrossRef CAS PubMed.
- B. Lee, H. Lee, H. Kim, K. Chung, B. Cho and S. Oh, Chem. Commun., 2015, 51, 9265–9268 RSC.
- B. Sambandam, V. Mathew, S. Kim, S. Lee, S. Kim, J. Hwang, H. Fan and J. Kim, Chem, 2022, 8, 924–946 CAS.
- M. Alfaruqi, S. Islam, D. Putro, V. Mathew, S. Kim, J. Jo, S. Kim, Y. Sun, K. Kim and J. Kim, Electrochim. Acta, 2018, 276, 1–11 CrossRef CAS.
- Y. Yuan, R. Sharpe, K. He, C. Li, M. Saray, T. Liu, W. Yao, M. Cheng, H. Jin, S. Wang, K. Amine, R. Shahbazian-Yassar, M. Islam and J. Lu, Nat. Sustainability, 2022, 5, 890–898 CrossRef.
- H. Chen, C. Dai, F. Xiao, Q. Yang, S. Cai, M. Xu, H. J. Fan and S.-J. Bao, Adv. Mater., 2022, 34, 2109092 CrossRef CAS.
- W. Sun, F. Wang, S. Hou, C. Yang, X. Fan, Z. Ma, T. Gao, F. Han, R. Hu, M. Zhu and C. Wang, J. Am. Chem. Soc., 2017, 139, 9775–9778 CrossRef CAS.
- W. Du, E. Ang, Y. Yang, Y. Zhang, M. Ye and C. Li, Energy Environ. Sci., 2020, 13, 3330–3360 RSC.
- B. Lee, H. Seo, H. Lee, C. Yoon, J. Kim, K. Chung, B. Cho and S. Oh, ChemSusChem, 2016, 9, 2948–2956 CrossRef CAS PubMed.
- M. Wang, X. Zheng, X. Zhang, D. Chao, S. Qiao, H. Alshareef, Y. Cui and W. Chen, Adv. Energy Mater., 2021, 11, 2002904 CrossRef CAS.
- X. Guo, J. Zhou, C. Bai, X. Li, G. Fang and S. Liang, Mater. Today Energy, 2020, 16, 100396 CrossRef.
- D. Wu, L. Housel, S. Kim, N. Sadique, C. Quilty, L. Wu, R. Tappero, S. L. Nicholas, S. Ehrlich, Y. Zhu, A. Marschilok, E. Takeuchi, D. Bock and K. Takeuchi, Energy Environ. Sci., 2020, 13, 4322–4333 RSC.
- R. Liang, J. Fu, Y.-P. Deng, Y. Pei, M. Zhang, A. Yu and Z. Chen, Energy Storage Mater., 2021, 36, 478–484 CrossRef.
- Y. Huang, J. Mou, W. Liu, X. Wang, L. Dong, F. Kang and C. Xu, Nano-Micro Lett., 2019, 11, 49 CrossRef CAS.
- D. Chao, W. Zhou, C. Ye, Q. Zhang, Y. Chen, L. Gu, K. Davey and S. Qiao, Angew. Chem., Int. Ed., 2019, 58, 7823–7828 CrossRef CAS.
- Y. Ma, Y. Ma, T. Diemant, K. Cao, X. Liu, U. Kaiser, R. J. Behm, A. Varzi and S. Passerini, Adv. Energy Mater., 2021, 11, 2100962 CrossRef CAS.
- W. Xie, K. Zhu, H. Yang and W. Yang, Adv. Mater., 2023, 36, 2306154 CrossRef PubMed.
- R. He, F. Yu, K. Wu, H. Liu, Z. Li, H. Liu, S. Dou and C. Wu, Nano Lett., 2023, 23, 6050–6058 CrossRef CAS PubMed.
- L. Han, Q. Wang, R. Pang, D. Zhang, B. Zhao, W. Meng, M. Li, Y. Zhang, A. Cao and Y. Shang, Adv. Energy Mater., 2023, 13, 2302395 CrossRef CAS.
- J. Yue, S. Chen, J. Yang, S. Li, G. Tan, R. Zhao, C. Wu and Y. Bai, Adv. Mater., 2023, 36, 2304040 CrossRef PubMed.
- X. Li, L. Wang, Y. Fu, H. Dang, D. Wang and F. Ran, Nano Energy, 2023, 116, 108858 CrossRef CAS.
- M. Xue, J. Bai, M. Wu, Q. He, Q. Zhang and L. Chen, Energy Storage Mater., 2023, 62, 102940 CrossRef.
- T. Wu, Y. Lin, Z. Althouse and N. Liu, ACS Appl. Energy Mater., 2021, 4, 12267–12274 CrossRef CAS.
- J. Huang, X. Tang, K. Liu, G. Fang, Z. He and Z. Li, Mater. Today Energy, 2020, 17, 100475 CrossRef.
- L. Wang, Z. Wu, J. Zou, P. Gao, X. Niu, H. Li and L. Chen, Joule, 2019, 3, 2086–2102 CrossRef CAS.
- B. Tang, L. Shan, S. Liang and J. Zhou, Energy Environ. Sci., 2019, 12, 3288–3304 RSC.
- J. Cao, D. Zhang, X. Zhang, S. Wang, J. Han, Y. Zhao, Y. Huang and J. Qin, Appl. Surf. Sci., 2020, 534, 147630 CrossRef CAS.
- Y. Li, S. Wang, J. R. Salvador, J. Wu, B. Liu, W. Yang, J. Yang, W. Zhang, J. Liu and J. Yang, Chem. Mater., 2019, 31, 2036–2047 CrossRef CAS.
- J. Gallaway, M. Menard, B. Hertzberg, Z. Zhong, M. Croft, L. Sviridov, D. Turney, S. Banerjee, D. Steingart and C. Erdonmez, J. Electrochem. Soc., 2015, 162, A162 CrossRef CAS.
- H. Yang, T. Zhang, D. Chen, Y. Tan, W. Zhou, L. Li, W. Li, G. Li, W. Han, H. J. Fan and D. Chao, Adv. Mater., 2023, 35, 2300053 CrossRef CAS PubMed.
- H. Yang, W. Zhou, D. Chen, J. Liu, Z. Yuan, M. Lu, L. Shen, V. Shulga, W. Han and D. Chao, Energy Environ. Sci., 2022, 15, 1106–1118 RSC.
- L. Li, T. K. A. Hoang, J. Zhi, M. Han, S. Li and P. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 12834–12846 CrossRef CAS PubMed.
- S. Zhao, B. Han, D. Zhang, Q. Huang, L. Xiao, L. Chen, D. Ivey, Y. Deng and W. Wei, J. Mater. Chem. A, 2018, 6, 5733–5739 RSC.
- C. Qiu, X. Zhu, L. Xue, M. Ni, Y. Zhao, B. Liu and H. Xia, Electrochim. Acta, 2020, 351, 136445 CrossRef CAS.
- S. Cui, D. Zhang and Y. Gan, J. Power Sources, 2023, 579, 233293 CrossRef CAS.
- H. Ren, J. Zhao, L. Yang, Q. Liang, S. Madhavi and Q. Yan, Nano Res., 2019, 12, 1347–1353 CrossRef CAS.
- V. Soundharrajan, B. Sambandam, S. Kim, S. Islam, J. Jo, S. Kim, V. Mathew, Y. Sun and J. Kim, Energy Storage Mater., 2020, 28, 407–417 CrossRef.
- Q. Wang, G. Tian, C. Huang and D. Zhang, Small, 2023, 19, 2301189 CrossRef CAS PubMed.
- H. Chen, S. Cai, Y. Wu, W. Wang, M. Xu and S. J. Bao, Mater. Today Energy, 2021, 20, 100646 CrossRef CAS.
- N. Qiu, H. Chen, Z. Yang, S. Sun and Y. Wang, RSC Adv., 2018, 8, 15703–15708 RSC.
- Y. Liu, Y. Ma, W. Yang, S. Bao, H. Chen and M. Xu, Chem. Eng. J., 2023, 473, 145490 CrossRef CAS.
- B. Yang, X. Cao, S. Wang, N. Wang and C. Sun, Electrochim. Acta, 2021, 385, 138447 CrossRef CAS.
- Y. Liu, Z. Qin, X. Yang and X. Sun, Adv. Funct. Mater., 2022, 32, 2106994 CrossRef CAS.
- Y. Kim, Y. Park, M. Kim, J. Lee, K. Kim and J. Choi, Nat. Commun., 2022, 13, 2371 CrossRef CAS PubMed.
- P. Oberholzer, E. Tervoort, A. Bouzid, A. Pasquarello and D. Kundu, ACS Appl. Mater. Interfaces, 2019, 11, 674–682 CrossRef CAS PubMed.
- O. Fitz, C. Bischoff, M. Bauer, H. Gentischer, K. Birke, H. Henning and D. Biro, ChemElectroChem, 2021, 8, 3553–3566 CrossRef CAS.
- B. Wu, G. Zhang, M. Yan, T. Xiong, P. He, L. He, X. Xu and L. Mai, Small, 2018, 14, 1703850 CrossRef PubMed.
- X. Xiao, L. Zhang, W. Xin, M. Yang, Y. Geng, M. Niu, H. Zhang and Z. Zhu, Small, 2024, 2309271 CrossRef PubMed.
- X. Zhai, J. Qu, J. Wang, W. Chang, H. Liu, Y. Liu, H. Yuan, X. Li and Z. Yu, Energy Storage Mater., 2021, 42, 753–763 CrossRef.
- S. Deng, Z. Tie, F. Yue, H. Cao, M. Yao and Z. Niu, Angew. Chem., Int. Ed., 2022, 61, e202115877 CrossRef CAS PubMed.
- M. Xie, R. Wang, N. Wang, Q. Zhang, X. Zhang, C. Feng, L. Huang, Y. Xu, Y. Jiao and J. Chen, J. Mater. Chem. A, 2023, 11, 21927–21936 RSC.
- M. Shi, B. Wang, C. Chen, J. Lang, C. Yan and X. Yan, J. Mater. Chem. A, 2020, 8, 24635–24644 RSC.
- X. Zhu, Z. Cao, W. Wang, H. Li, J. Dong, S. Gao, D. Xu, L. Li, J. Shen and M. Ye, ACS Nano, 2021, 15, 2971–2983 CrossRef CAS PubMed.
- Y. Li, Z. Yuan, D. Li, J. Li, Y. Zhang, M. Wang, G. Li, L. Wang and W. Han, ACS Nano, 2024, 18, 4733–4745 CrossRef CAS PubMed.
- Z. Yuan, Q. Lin, Y. Li, W. Han and L. Wang, Adv. Mater., 2023, 35, 2211527 CrossRef CAS PubMed.
- B. Zhong, X. Qin, H. Xu, L. Liu, L. Li, Z. Li, L. Cao, Z. Lou, J. Jackman, N. Cho and L. Wang, Nat. Commun., 2024, 15, 624 CrossRef CAS PubMed.
- Y. Zeng, Y. Wang, Q. Jin, Z. Pei, D. Luan, X. Zhang and X. Lou, Angew. Chem., Int. Ed., 2021, 60, 25793–25798 CrossRef CAS PubMed.
- L. Gou, S. Zhao, W. Wang, L. Xu, W. Wang, J. Wu, Z. Ma, X. Fan and D. Li, ACS Appl. Energy Mater., 2021, 4, 7355–7364 CrossRef CAS.
- L. Gou, J. Li, K. Liang, S. Zhao, D. Li and X. Fan, Small, 2023, 19, 2208233 CrossRef CAS PubMed.
- G. Fang, C. Zhu, M. Chen, J. Zhou, B. Tang, X. Cao, X. Zheng, A. Pan and S. Liang, Adv. Funct. Mater., 2019, 29, 1808375 CrossRef.
- Q. Chen, X. Lou, Y. Yuan, K. You, C. Li, C. Jiang, Y. Zeng, S. Zhou, J. Zhang, G. Hou, J. Lu and Y. Tang, Adv. Mater., 2023, 35, 2306294 CrossRef CAS PubMed.
- C. Chen, M. Shi, Y. Zhao, C. Yang, L. Zhao and C. Yan, Chem. Eng. J., 2021, 422, 130375 CrossRef CAS.
- R. Zhang, P. Liang, H. Yang, H. Min, M. Niu, S. Jin, Y. Jiang, Z. Pan, J. Yan, X. Shen and J. Wang, Chem. Eng. J., 2022, 433, 133687 CrossRef CAS.
- L. Liu, Y. Wu, L. Huang, K. Liu, B. Duployer, P. Rozier, P. Taberna and P. Simon, Adv. Energy Mater., 2021, 11, 2101287 CrossRef CAS.
- D. Wang, L. Wang, G. Liang, H. Li, Z. Liu, Z. Tang, J. Liang and C. Zhi, ACS Nano, 2019, 13, 10643–10652 CrossRef CAS PubMed.
- J. Huang, Z. Wang, M. Hou, X. Dong, Y. Liu, Y. Wang and Y. Xia, Nat. Commun., 2018, 9, 2906 CrossRef PubMed.
- A. Zhang, R. Zhao, Y. Wang, J. Yue, J. Yang, X. Wang, C. Wu and Y. Bai, Angew. Chem., Int. Ed., 2023, 62, e202313163 CrossRef CAS PubMed.
- K. Nam, S. Kim, E. Yang, Y. Jung, E. Levi, D. Aurbach and J. Choi, Chem. Mater., 2015, 27, 3721–3725 CrossRef CAS.
- Y. Li, X. Feng, S. Cui, Q. Shi, L. Mi and W. Chen, CrystEngComm, 2016, 18, 3136–3141 RSC.
- H. Yao, H. Yu, Y. Zheng, N. Li, S. Li, D. Luan, X. Lou and L. Yu, Angew. Chem., Int. Ed., 2023, 62, e202315257 CrossRef CAS PubMed.
- Y. Zhang, S. Deng, G. Pan, H. Zhang, B. Liu, X. Wang, X. Zheng, Q. Liu, X. Wang, X. Xia and J. Tu, Small Methods, 2020, 4, 1900828 CrossRef CAS.
- H. Chen, W. Ma, J. Guo, J. Xiong, F. Hou, W. Si, Z. Sang and D. a. Yang, J. Alloys Compd., 2023, 932, 167688 CrossRef CAS.
- Y. Li, M. Chen, B. Liu, Y. Zhang, X. Liang and X. Xia, Adv. Energy Mater., 2020, 10, 2000927 CrossRef CAS.
- H. Qian, H. Ren, Y. Zhang, X. He, W. Li, J. Wang, J. Hu, H. Yang, H. Sari, Y. Chen and X. Li, Electrochem. Energy Rev., 2022, 5, 2 CrossRef CAS.
- D. Zhang, J. Cao, X. Zhang, N. Insin, S. Wang, J. Han, Y. Zhao, J. Qin and Y. Huang, Adv. Funct. Mater., 2021, 31, 2009412 CrossRef CAS.
- J. Ji, H. Wan, B. Zhang, C. Wang, Y. Gan, Q. Tan, N. Wang, J. Yao, Z. Zheng, P. Liang, J. Zhang, H. Wang, L. Tao, Y. Wang, D. Chao and H. Wang, Adv. Energy Mater., 2021, 11, 2003203 CrossRef CAS.
- G. Cui, Y. Zeng, J. Wu, Y. Guo, X. Gu and X. Lou, Adv. Sci., 2022, 9, 2106067 CrossRef CAS PubMed.
- Z. Liu, L. Qin, X. Chen, X. Xie, B. Zhu, Y. Gao, M. Zhou, G. Fang and S. Liang, Mater. Today Energy, 2021, 22, 100851 CrossRef CAS.
- Y. Yang, T. Shao, Y. Zhang, Y. Lu, M. Li, H. Liu, Q. Xu and Y. Xia, J. Power Sources, 2023, 564, 232863 CrossRef CAS.
- T. Shao, Y. Zhang, T. Cao, Y. Yang, Z. Li, H. Liu, Y. Wang and Y. Xia, Chem. Eng. J., 2022, 431, 133735 CrossRef CAS.
- N. Liu, X. Wu, Y. Yin, A. Chen, C. Zhao, Z. Guo, L. Fan and N. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 28199–28205 CrossRef CAS PubMed.
- S. Lian, C. Sun, W. Xu, W. Huo, Y. Luo, K. Zhao, G. Yao, W. Xu, Y. Zhang, Z. Li, K. Yu, H. Zhao, H. Cheng, J. Zhang and L. Mai, Nano Energy, 2019, 62, 79–84 CrossRef CAS.
- H. Wang, R. Guo, Y. Ma and F. Zhou, Adv. Funct. Mater., 2023, 33, 2301351 CrossRef CAS.
- H. Li, Z. Huang, B. Chen, Y. Jiang, C. Li, W. Xiao and X. Yan, J. Power Sources, 2022, 527, 231198 CrossRef CAS.
- X. Cheng, J. Xiao, M. Ye, Y. Zhang, Y. Yang and C. Li, ACS Appl. Mater. Interfaces, 2022, 14, 10489–10497 CrossRef CAS PubMed.
- Y. Zhang, Y. Liu, Z. Liu, X. Wu, Y. Wen, H. Chen, X. Ni, G. Liu, J. Huang and S. Peng, J. Energy Chem., 2022, 64, 23–32 CrossRef CAS.
- C. Huang, Q. Wang, D. Zhang and G. Shen, Nano Res., 2022, 15, 8118–8127 CrossRef CAS.
- Y. Zhao, P. Zhang, J. Liang, X. Xia, L. Ren, L. Song, W. Liu and X. Sun, Energy Storage Mater., 2022, 47, 424–433 CrossRef.
- G. Nam, C. Hwang, H. Jang, N. Kane, Y. Ahn, M. Kwak, Z. Luo, T. Li, M. Kim, N. Liu and M. Liu, Small, 2023, 2306919 CrossRef PubMed.
- Z. Li, C. Bommier, Z. Chong, Z. Jian, T. Surta, X. Wang, Z. Xing, J. Neuefeind, W. Stickle, M. Dolgos, P. Greaney and X. Ji, Adv. Energy Mater., 2017, 7, 1602894 CrossRef.
- Y. Liu, K. Wang, X. Yang, J. Liu, X. Liu and X. Sun, ACS Nano, 2023, 17, 14792–14799 CrossRef CAS PubMed.
- F. Ye, Q. Liu, C. Lu, F. Meng, T. Lin, H. Dong, L. Gu, Y. Wu, Z. Tang and L. Hu, Energy Storage Mater., 2022, 52, 675–684 CrossRef.
- M. Mautner, J. Am. Chem. Soc., 1988, 110, 3854–3858 CrossRef CAS.
- A. Gandhe, J. Rebello, J. Figueiredo and J. Fernandes, Appl. Catal., B, 2007, 72, 129–135 CrossRef CAS.
- X. Zeng, J. Liu, J. Mao, J. Hao, Z. Wang, S. Zhou, C. D. Ling and Z. Guo, Adv. Energy Mater., 2020, 10, 1904163 CrossRef CAS.
- Z. Zhong, J. Li, L. Li, X. Xi, Z. Luo, G. Fang, S. Liang and X. Wang, Energy Storage Mater., 2022, 46, 165–174 CrossRef.
- Y. Liu, Z. Qin, X. Yang, J. Liu, X. Liu and X. Sun, ACS Energy Lett., 2022, 7, 1814–1819 CrossRef CAS.
- M. Mateos, N. Makivic, Y. Kim, B. Limoges and V. Balland, Adv. Energy Mater., 2020, 10, 2000332 CrossRef CAS.
- Y. Hu, Z. Liu, L. Li, S. Guo, X. Xie, Z. Luo, G. Fang and S. Liang, Nat. Sci. Rev., 2023, 10, nwad220 CrossRef CAS PubMed.
- M. Chuai, J. Yang, R. Tan, Z. Liu, Y. Yuan, Y. Xu, J. Sun, M. Wang, X. Zheng, N. Chen and W. Chen, Adv. Mater., 2022, 34, 2203249 CrossRef CAS PubMed.
- X. Shen, X. Wang, Y. Zhou, Y. Shi, L. Zhao, H. Jin, J. Di and Q. Li, Adv. Funct. Mater., 2021, 31, 2101579 CrossRef CAS.
- X. Xu, Y. Chen, W. Li, R. Yin, D. Zheng, X. Niu, X. Dai, W. Shi, W. Liu, F. Wu, M. Wu, S. Lu and X. Cao, Small, 2023, 19, 2207517 CrossRef CAS PubMed.
- X. Zhao, L. Mao, Q. Cheng, F. Liao, G. Yang and L. Chen, Carbon, 2022, 186, 160–170 CrossRef CAS.
- T. Li, R. Guo, L. Meng, X. Sun, Y. Li, F. Li, X. Zhao, L. An, J. Peng and W. Wang, Chem. Eng. J., 2022, 446, 137290 CrossRef CAS.
- Y. Xu, J. Zhu, J. Feng, Y. Wang, X. Wu, P. Ma, X. Zhang, G. Wang and X. Yan, Energy Storage Mater., 2021, 38, 299–308 CrossRef.
- H. Peng, H. Fan, C. Yang, Y. Tian, C. Wang and J. Sui, RSC Adv., 2020, 10, 17702–17712 RSC.
- K. Wang, J. Wang, P. Chen, M. Qin, C. Yang, W. Zhang, Z. Zhang, Y. Zhen, F. Fu and B. Xu, Small, 2023, 19, 2300585 CrossRef CAS PubMed.
- Y. Ma, M. Xu, R. Liu, H. Xiao, Y. Liu, X. Wang, Y. Huang and G. Yuan, Energy Storage Mater., 2022, 48, 212–222 CrossRef.
- Y. Chen, X. Hu, X. Chen, J. Liu, Y. Huang and D. Cao, Chem. Eng. J., 2023, 478, 147411 CrossRef CAS.
- Y. Yuan, K. He, B. W. Byles, C. Liu, K. Amine, J. Lu, E. Pomerantseva and R. Shahbazian-Yassar, Chem, 2019, 5, 1793–1805 CAS.
- S. Kim, D. Wu, N. Sadique, C. Quilty, L. Wu, A. Marschilok, K. Takeuchi, E. Takeuchi and Y. Zhu, Small, 2020, 16, 2005406 CrossRef CAS PubMed.
- Q. Lin and L. Wang, J. Semicond., 2023, 44, 041601 CrossRef.
- K. Du, Y. Liu, Y. Yang, F. Cui, J. Wang, M. Han, J. Su, J. Wang, X. Han and Y. Hu, Adv. Mater., 2023, 35, 2301538 CrossRef CAS PubMed.
- Y. Zhao, P. Zhang, J. Liang, X. Xia, L. Ren, L. Song, W. Liu and X. Sun, Adv. Mater., 2022, 34, 2204320 CrossRef CAS PubMed.
- S. Yang, H. Du, Y. Li, X. Wu, B. Xiao, Z. He, Q. Zhang and X. Wu, Green Energy Environ., 2023, 8, 1531–1552 CrossRef CAS.
- L. Cao, D. Li, E. Hu, J. Xu, T. Deng, L. Ma, Y. Wang, X. Yang and C. Wang, J. Am. Chem. Soc., 2020, 142, 21404–21409 CrossRef CAS PubMed.
- Y. Zhang, X. Li, L. Fan, Y. Shuai and N. Zhang, Cell Rep. Phys. Sci., 2022, 3, 100824 CrossRef CAS.
- S. Xu, W. Nie, P. Sun, J. Sun, Z. Xu, C. Chu and L. Liu, Mater. Lett., 2023, 331, 133546 CrossRef CAS.
- H. Wang, T. Wang, G. Stevenson, M. Chamoun and R. W. Lindström, Energy Storage Mater., 2023, 63, 103008 CrossRef.
- P. Ruan, X. Xu, D. Zheng, X. Chen, X. Yin, S. Liang, X. Wu, W. Shi, X. Cao and J. Zhou, ChemSusChem, 2022, 15, e202201118 CrossRef CAS PubMed.
- Y. Liu, Z. Qin, X. Yang, J. Liu, X. Liu and X. Sun, Energy Storage Mater., 2023, 56, 524–531 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b,
Qichun
Zhang
*b,
Qichun
Zhang