
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExo- or endo-1H-pyrazole metal coordination modulated by the polyamine chain length in [1 + 1] condensation azamacrocycles. Binuclear complexes with remarkable SOD activity†
Irene
Bonastre-Sabater
a,
Alberto
Lopera
a,
Álvaro
Martínez-Camarena
 ab,
Salvador
Blasco
a,
Antonio
Doménech-Carbó
c,
Hermas R.
Jiménez
d,
Begoña
Verdejo
a,
Enrique
García-España
*a and
M. Paz
Clares
*a
ab,
Salvador
Blasco
a,
Antonio
Doménech-Carbó
c,
Hermas R.
Jiménez
d,
Begoña
Verdejo
a,
Enrique
García-España
*a and
M. Paz
Clares
*a
aDepartamento de Química Inorgánica, Instituto de Ciencia Molecular. Universidad de Valencia, Calle Catedrático José Beltrán 2, 46980 Paterna, Valencia, Spain. E-mail: m.paz.clares@uv.es; enrique.garcia-es@uv.es
bDepartamento de Química Inorgánica, Facultad de Ciencias Químicas, Universidad Complutense de Madrid, avda. Complutense s/n, 28040 Madrid, Spain
cDepartamento de Química Analítica, Universidad de Valencia, Calle Dr Moliner s/n, 46100 Burjassot, Valencia, Spain
dDepartamento de Química Inorgánica, Universidad de Valencia, Calle Doctor Moliner s/n, 46100 Burjasot, Valencia, Spain
First published on 2nd July 2024
Abstract
The Cu2+ complexes of three [1 + 1] azacyclophane macrocycles having the 1H-pyrazole ring as the spacer and the pentaamine 1,5,8,11,15-pentaazadecane (L1) or hexaamines 1,5,8,12,15,19-hexaazanonadecane (L2) and 1,5,9,13,17,21-hexaazaheneicosane (L3) as bridges show endo- coordination of the pyrazolate bridge giving rise to discrete monomeric species. Previously reported pyrazolacyclophanes evidenced, however, exo-coordination with the formation of dimeric species of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, 3:2 or even 4:2 Cu2+:L stoichiometry. The complexes have been characterized in solution using potentiometric studies, UV-Vis spectroscopy, paramagnetic NMR, cyclic voltammetry and mass spectrometry. The measurements show that all three ligands have as many protonation steps in water as secondary amines are in the bridge, while they are able to form both mono- and binuclear Cu2+ species. The crystal structures of the complexes [Cu(HL1)Br]Br(1+x)(ClO4)(1−x)·yH2O (1) and [Cu2(H−1L2)Cl(ClO4)](ClO4)·H2O·C2H5OH (2) have been solved by X-ray diffraction studies. In 1 the metal ion lies at one side of the macrocyclic cavity being coordinated by one nitrogen of the pyrazolate moiety and the three consecutive nitrogen atoms of the polyamine bridge. The other nitrogen of the pyrazole ring is hydrogen-bonded to an amine group. In 2 the two metal ions are interconnected by a pyrazolate bis(monodentate) moiety and complete their coordination spheres with three amines and either a bromide or a perchlorate anion, which occupy the axial positions of distorted square pyramid geometries. Paramagnetic NMR studies of the binuclear complexes confirm the coordination pattern observed in the crystal structures. Cyclic voltamperommetry data show potentials within the adequate range to exhibit superoxide dismutase (SOD) activity. The IC50 values calculated by McCord–Fridovich enzymatic assays show that the binuclear Cu2+ complexes of L2 and L3 have SOD activities that rank amongst the highest ones reported so far.
:2, 3:2 or even 4:2 Cu2+:L stoichiometry. The complexes have been characterized in solution using potentiometric studies, UV-Vis spectroscopy, paramagnetic NMR, cyclic voltammetry and mass spectrometry. The measurements show that all three ligands have as many protonation steps in water as secondary amines are in the bridge, while they are able to form both mono- and binuclear Cu2+ species. The crystal structures of the complexes [Cu(HL1)Br]Br(1+x)(ClO4)(1−x)·yH2O (1) and [Cu2(H−1L2)Cl(ClO4)](ClO4)·H2O·C2H5OH (2) have been solved by X-ray diffraction studies. In 1 the metal ion lies at one side of the macrocyclic cavity being coordinated by one nitrogen of the pyrazolate moiety and the three consecutive nitrogen atoms of the polyamine bridge. The other nitrogen of the pyrazole ring is hydrogen-bonded to an amine group. In 2 the two metal ions are interconnected by a pyrazolate bis(monodentate) moiety and complete their coordination spheres with three amines and either a bromide or a perchlorate anion, which occupy the axial positions of distorted square pyramid geometries. Paramagnetic NMR studies of the binuclear complexes confirm the coordination pattern observed in the crystal structures. Cyclic voltamperommetry data show potentials within the adequate range to exhibit superoxide dismutase (SOD) activity. The IC50 values calculated by McCord–Fridovich enzymatic assays show that the binuclear Cu2+ complexes of L2 and L3 have SOD activities that rank amongst the highest ones reported so far.
Introduction
1H-Pyrazole is an imidazole isomer that displays different hydrogen bond and coordination modes.1 Among the latter, the most relevant ones are monodentate coordination in its neutral form and bis(monodentate) or exo(bidentate) coordination in its deprotonated pyrazolate form. These coordination and hydrogen bond features have made pyrazole and its derivatives, particularly carboxylates, well-known building blocks in the preparation of metal–organic frameworks (MOFs).2,3 On the other hand, pyrazole and its derivatives have found interesting applications in pharmacology.4The bis(monodentate) coordination mode of the pyrazolate anion separates the metal ions from 3.7 to 4.0 Å, distances which are close to those found in type III copper centres of biomolecules such as hemocyanin or multinuclear copper enzymes.5 In this respect, several 1H-pyrazole open-chain ligands and complexes have been prepared to mimic enzymes or to analyse the electronic and magnetic properties of the interconnected metal ions.6
Previously, others and we have extensively worked on the synthesis and study of [2 + 2] condensation macrocycles in which two 1H-pyrazole units were bound through methylene groups to different polyamine chains.7 This work was also extended to [3 + 2] condensation cryptands in which three pyrazole spacers were linked to two tris(2-aminoethyl)amine (tren) moieties or analogue tripodal polyamines. These macrocycles showed interesting behaviours regarding their metal and anion coordination, biomedical chemistry and self-assembling properties. [2 + 2] Azamacrocycles proved to have strong binding to metal ions, amino acids and neurotransmitters.8 Interestingly, the interaction of a [2 + 2] macrocycle having cadaverine polyamines with Cu2+ led to the formation of a 4:2 Cu2+:ligand metallocage appropriately sized to host a water molecule that seemed not be hydrogen bonded to any other water molecule or to donor or acceptor groups within the cage.9
The cryptand containing tren units was able to encapsulate metal ions and different anions within its cavity as proved by a variety of techniques including single crystal X-ray diffraction.10 The combination of the pyrazole bis(monodentate)-binding motif and polyamine organization permitted interesting Cu2+-cryptand cages to be obtained in which six metal ions were shared by three cryptands through a right arrangement of the amines in axial positions.11 Moreover, we noticed that by regulating the pH we could reach a situation in which the protonated amino groups of the cryptand selectively hosted chloride anions and water molecules, while the pyrazole groups coordinated in an exo-monodentate fashion with Cu2+ ions linking cryptand moieties so that a 1D-helical coordination polymer behaving as a multi-anion receptor was formed.12
In spite of all this interest in [2 + 2] 1H-pyrazole macrocycles, the number of research works dealing with [1 + 1] condensation of 1H-pyrazole azamacrocycles is much more scarce. As far as we know, after a previous report showing the synthesis and characteristics of a series of 1H-pyrazole oxygen crown ethers as dopamine receptors,13 only three papers coming from our own laboratory have appeared describing [1 + 1] 1H-pyrazole azamacrocycles.14–16 These papers revealed that the binding of a single 1H-pyrazole to the ends of different open-chain tetra-amines to produce [1 + 1] azamacrocycles gave rise to Cu2+ complexes with exo-coordination of the 1H-pyrazole fragments so that complexes of 2:2, 3:2 and even 4:2 Cu2+:ligand stoichiometry were predominantly formed as assessed by solution studies and X-ray diffraction (Fig. 1). The ability of pyrazole to show an exo-binding mode was very nicely illustrated in the case of macrocycles formed by pyrazole and imidazole units whose Cu2+ complexes led to pillared structures.17 In none of these systems, discrete binuclear 2:1 Cu2+ complexes were detected. However, such binuclear metal complexes, in particular the Cu2+ ones, may have great relevance in biomimetic chemistry. As mentioned above, the positioning of the Cu2+ atoms at the ca. 3.7 Å distance dictated by the bis(monodentate) pyrazolate anion may have relevance for instance in the mimicking of enzymes involved in the protection against reactive oxygen species (ROS). The so far explored [2 + 2] 1H-pyrazole condensation macrocycles saturate the space between the metal ions making difficult their transient binding to exogenous ligands as the superoxide radical or hydrogen peroxide anions involved in these processes.
 | ||
| Fig. 1 Coordination modes in [1 + 1] condensation polyazapyrazolaphanes. | ||
Regarding this point, our group has been synthesizing and studying pyridinaphane macrocyclic receptors whose copper or manganese complexes had the capacity to scavenge superoxide radical anions promoting their disproportionation into hydrogen peroxide and dioxygen. In particular, mononuclear manganese complexes of tetraazapyridinaphane ligands having pending polyamine chains and binuclear copper complexes of hexaazapyridinaphane macrocycles have shown interesting superoxide dismutase (SOD) mimicking potentiality in vitro (see Fig. 2).18,19
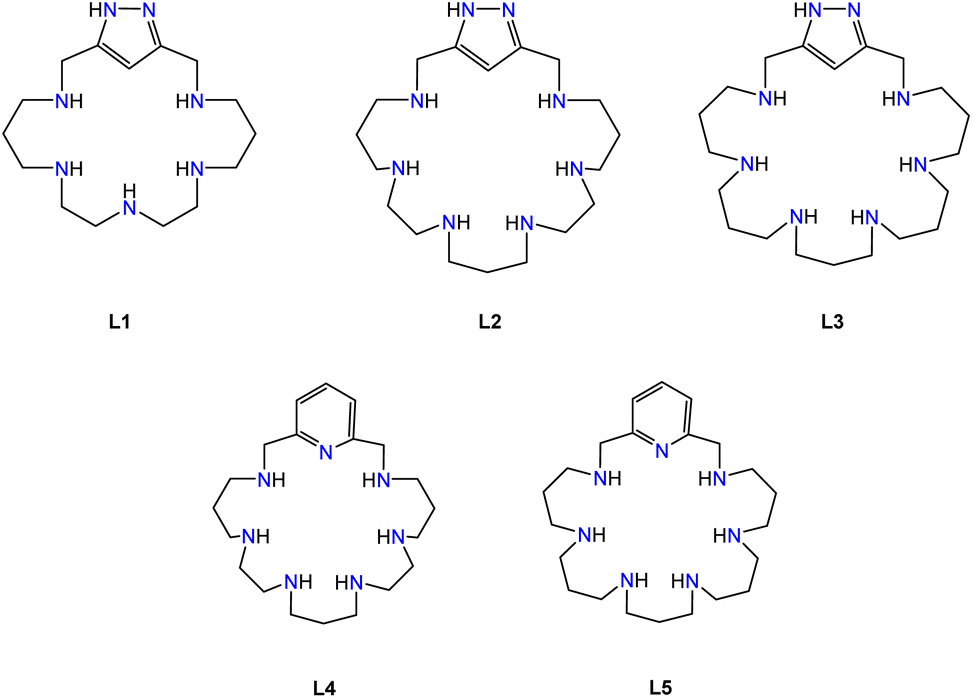 | ||
| Fig. 2 Ligand drawing. | ||
Moreover, we have reported that grafting of the active amines onto boehmite nanoparticles (γ-AlO(OH), BNPs) giving rise to amino-nanozyme systems led to a significant activity increase due to the positive charge of the BNPs and accumulation of the complexes on the surface.20,21 Literature reports show interesting properties in imaging and in therapeutic intervention against Alzheimer's disease of metal complexes of the derivatives of the molecule 3,6,9-triaza-1-(2,6)-pyridinacyclodecaphane (py22 or pyclen), which constitutes the macrocyclic core of our pending pyridinaphane ligands.22 In this line, we have recently shown that the Cu2+ complexes of a py22 derivative with a carboxylate group at the para position developed a striking SOD activity, while when grafted to BNPs amino-nanozymes were obtained displaying mitoROS scavenging properties and the ability to disaggregate mutant huntingtin deposits in cells.23
As mentioned above, in this work, we turn our attention towards macrocyclic polyamines and we want to learn how replacing the pyridine aromatic spacer by a 1H-pyrazole one influences the superoxide dismutase activity in 2:1 Cu2+:macrocycle binuclear systems. It has been shown that the way in which superoxide anions approach and bind to the active centre of the enzyme is a key factor in the catalytic cycle. Since the 1H-pyrazole spacer has the possibility to donate and accept hydrogen bonds, it may have some relevant effect on the catalysis of superoxide disproportionation.24 In this context, here we report on the Cu2+ complexes of the newly synthesized ligand 3,7,10,13,17-pentaaza-1-(3,5)-pyrazolacyclooctadecaphane (L1) and their potential capability to behave as superoxide dismutase mimics. Moreover, we have extended these studies to the 1H-pyrazole hexaaza-macrocycles 3,7,10,14,17,21-hexaaza-1-(3,5)-pyrazolacyclodocosaphane and 3,7,11,15,19,22-hexaaza-1-(3,5)-pyrazolacyclotetracosaphane, hereafter (L2) and (L3). We have used penta- and hexaamines in the bridges to avoid exo-binding to metal ions of the 1H-pyrazole unit and to permit endo coordination facilitating the formation of monomeric binuclear complexes involving just one macrocycle. We have used a handful of experimental techniques to establish the acid–base and Cu2+ coordination chemistry in solution of the three ligands and we describe the crystal structure of the mononuclear complex [Cu(HL1)Br]Br(1+x)(ClO4)(1−x)·yH2O (1) and the binuclear one [Cu2(H−1L2)Cl(ClO4)](ClO4)·H2O·C2H5OH (2).
Results and discussion
Synthetic procedures
The synthesis of L1 was carried out employing the modification of the Richman–Atkins procedure25 we had previously used for the synthesis of L2 and L3, and related tetraaza pyrazolacyclophanes.14–16 The procedure comprises the cyclisation reaction between the protected pyrazole moiety 3,5-bis(chloromethyl)-1-(tetrahydropyran-2-yl)-pyrazole and the corresponding tosylated pentaamine followed by the removal of the protecting groups in acid media using a hydrobromide/acetic acid mixture and phenol (Scheme 1). Protection of the pyrazole group is necessary to avoid side-reactions.26 | ||
| Scheme 1 Ligand synthesis. | ||
Acid–base behaviour
Table 1 shows the stepwise and cumulative protonation constants of L1, L2 and L3 determined by means of pH-metric titrations employing the HYPERQUAD software.27 Distribution diagrams calculated with the program HySS28 are collected in the ESI (Fig. S1–S3†). As shown in Table 1, the macrocycles display in the pH range available for potentiometric titrations (2.5–11.0) as many constants as the number of secondary amine groups present in their structures.| Reactiona | L1 | L2 | L3 |
|---|---|---|---|
|
a Charges omitted.
b Numbers in parenthesis are standard deviation in the last significant figure.
c Taken from ref. 16.
d Calculated as logβ = ΣjlogKHjL.
|
|||
| L + H ⇄ HL | 9.61(2)b | 10.06(1)c | 10.72(1)c |
| HL + H ⇄ H2L | 9.26(2) | 9.24(1) | 9.80(1) |
| H2L + H ⇄ H3L | 7.35(3) | 8.09(1) | 8.63(1) |
| H3L + H ⇄ H4L | 6.35(3) | 6.50(1) | 7.46(1) |
| H4L + H ⇄ H5L | 3.09(3) | 5.41(1) | 6.90(1) |
| H5L + H ⇄ H6L | — | 4.42(2) | 6.02(1) |
| logβd |
35.66(3) | 43.72(2) | 49.53(1) |
As observed in previous related systems neither protonation nor deprotonation of the 1H-pyrazole units occurs within the explored pH range.7,29,30 For the three macrocycles, the values of the logarithms of the protonation constants decrease as the number of positive charges in the receptor increases, following the expected trend for polyazamacrocycles, which can be ascribed to an increase of the electrostatic repulsions between the positively charged protonated amines.31L3 has, in general, higher basicity than L2 in all the protonation steps, while L1 has less basicity in all of them. Similar trends were obtained for pyridinaphanes with analogous polyaminic bridges.19 The greater number of carbon atoms between the secondary amino groups in L3 leads to larger inductive effects and to a greater minimization of electrostatic repulsions between the charged ammonium groups, resulting in higher stepwise protonation constants.31 As a matter of fact, L3 presents six orders of magnitude greater overall basicity than L2 (see the last entry in Table 1). The variations of the pyrazole UV band at ca. 205 nm with the pH value indicate that the amine groups closer to the pyrazole moieties are the first ones being protonated, since from pH 9 downwards no changes in absorptivity occur (see Fig. S1–S3 in the ESI†).
Cu2+ coordination
The stability constants of the Cu2+ complexes of the pyrazole cyclophanes L1, L2 and L3 have been obtained by potentiometric titration using the HYPERQUAD set of programs for the analysis of the data (Table 2).27 Distribution diagrams have been produced with the program HySS (Fig. 3, Fig. S4 and S5†).28 The titrations were carried with Cu2+:L molar ratios from 1:2 to 2:1 in the pH range 2.5–11.0. L1, L2 and L3 form mono- and binuclear monomeric complexes of [Cu(HxL)](2+x)+ or [Cu2(HyL)](4+y)+ stoichiometries. For L1 the x and y values range from 2 to 1 and from −1 to −3, respectively. For L2 and L3, x varied from 3 to −1 and from 4 to −1, respectively, while x changed from 0 to −2 for both systems.
 | ||
| Fig. 3 Distribution diagram for the system Cu2+–L1 overlapped with the absorbance at 259 nm [L1] = 1 × 10−3 M, (A) [Cu2+] = 1 × 10−3 M and (B) [Cu2+] = 2 × 10−3 M. | ||
| Reactiona | L1 | L2 | L3 |
|---|---|---|---|
| a Charges omitted. b Numbers in parenthesis are standard deviation in the last significant figure. | |||
| Cu + L + 4H ⇄ CuH4L | 43.71(3) | ||
| Cu + L + 3H ⇄ CuH3L | 38.04(4) | 38.97(4) | |
| Cu + L + 2H ⇄ CuH2L | 30.49(2)b | 34.82(2) | 34.42(1) |
| Cu + L + H ⇄ CuHL | 27.03(1) | 30.77(4) | 28.21(2) |
| Cu + L ⇄ CuL | 19.63(3) | 22.56(5) | 19.23(3) |
| Cu + L ⇄ CuH−1L + H | 9.21(4) | 11.15(6) | 8.62(3) |
| 2Cu + L ⇄ Cu2(H(H−1L)) | 30.44(6) | 28.00(4) | |
| 2Cu + L ⇄ Cu2(H−1L) + H | 20.50(2) | 26.68(2) | 23.20(1) |
| 2Cu + L ⇄ Cu2(H−1L)(OH) + 2H | 13.20(3) | 16.58(4) | 11.95(4) |
| 2Cu + L ⇄ Cu2(H−1L)(OH)2 + 3H | 2.84(4) | ||
| CuH3L + H ⇄ CuH4L | 4.74(3) | ||
| CuH2L + H ⇄ CuH3L | 3.21(3) | 4.55(4) | |
| Cu(H2(H−1L)) + H ⇄ CuH2L | 3.46(2) | 4.06(4) | 6.20(2) |
| Cu(H(H−1L)) + H ⇄ Cu(H2(H−1L)) | 7.40(3) | 8.20(4) | 8.98(2) |
| Cu(H−1L) + H ⇄ Cu(H(H−1L)) | 10.41(5) | 11.41(7) | 10.61(4) |
| Cu + Cu(H(H−1L)) ⇄ Cu2(H−1(HL)) | 7.87(7) | 8.77(5) | |
| Cu2(H(H−1L)) ⇄ Cu2(H−1L)+ H | −3.76(6) | −4.80(4) | |
| Cu2(H−1L) + H2O ⇄ Cu2(H−1L)(OH) + H | −7.30(2) | −10.10(4) | −11.25(4) |
| Cu2(H−1L)(OH) ⇄ Cu2(H−1L)(OH)2 + H | −10.36(5) | ||
HR-ESI-MS studies, performed at variable pH, permitted the identification of most of the species detected in the potentiometric studies, confirming their mono- and binuclear nature (Table 3). Fig. S6–S28† show the experimental and calculated spectra for the detected species.
| Ligand Species | L1 | L2 | L3 | |||
|---|---|---|---|---|---|---|
| Found | Calculated | Found | Calculated | Found | Calculated | |
| [CuL]2+ | 186.0950 | 186. 1018 | 214.6258 | 214.6252 | 228.6411 | 228.6401 |
| [CuL(Cl)]+ | 407.1608 | 407.1725 | 464.2193 | 462.2199 | 492.2513 | 492.2512 |
| [CuL(ClO4)]+ | 417.1433 | 417.1522 | 528.1984 | 528.1995 | 556.2309 | 556.2308 |
| [CuH−1L]+ | 371.1837 | 371.1959 | — | — | 456.2742 | 456.2745 |
| [Cu2H−1L(Cl)]2+ | — | — | 263.0712 | 263.0705 | 277.0871 | 277.0862 |
| [Cu2H−1L(ClO4)]2+ | — | — | 295.0610 | 295.0604 | 309.0775 | 309.0760 |
| [Cu2H−2L]2+ | 216.5522 | 216.5638 | 245.0833 | 245.0822 | 259.0992 | 259.0979 |
| [Cu2H−2L(Cl)]+ | 470.0763 | 470.0965 | 525.1331 | 525.1338 | 553.1654 | 553.1651 |
| [Cu2H−2L(ClO4)]+ | — | — | 589.1125 | 589.1135 | 617.1452 | 617.1448 |
In contrast to related shorter derivatives having tetraamine bridges,14,15 which showed the formation of dimeric bi-, tri or tetranuclear complexes (Cu2+:L stoichiometries 2:2. 3:2 or 4:2) (Fig. 1), for L1, L2 and L3 we have only been able to detect mono- and binuclear monomeric complexes. The larger size and number of amine groups, along with the greater flexibility of these ligands, permit an enough number of nitrogen donors to point inside their cavities, giving rise to an endo-coordination instead of the preferred exo-coordination mode observed for the parent tetraamine pyrazole macrocycles.14,15
The formation of discrete binuclear complexes for L1 and L3 has also been checked by paramagnetic NMR spectroscopy. We have recorded the 1H NMR spectra, measured the 1H transversal relaxation times, T2, and analysed the temperature dependence of the chemical shifts. The 1H NMR spectrum of the system Cu2+–L1 in a 2:1 molar ratio recorded in D2O at pH = 7 shows, in the downfield region, five well-resolved isotropically shifted signals (a–c, f and g) and three signals (d), (e) and (E). In addition, it displays four signals (h–k) shifted upfield (Fig. S29†). Chemical shift values, linewidths at half-height, transversal relaxation time values (T2) and assignments are reported in the ESI (Table S1†). The assignment of the isotropically shifted signals, as well as the description of the characteristic properties of the binuclear Cu2+ system have been performed taking into account previous reports.15,32
The isotropically shifted signals show linewidths, measured at half-height, of around ∼70 Hz, except for signals a–c and h–k with a linewidth of 2960 to 850 Hz, respectively. Transversal relaxation time values were below 1 ms, in the case of signals a–c and h–k, and 4 ms for signals f and g. The assignments of the isotropically shifted signals by means of the integration of signals and the transversal relaxation times of the paramagnetic signals are shown in Table S1.† Chemical shift values, transversal relaxation times and the broad linewidth at half-height of the Cu2+–L1 system in 2:1 molar ratio are characteristic of a spin-coupled binuclear Cu2+.15,19,32,33 The pattern of paramagnetic signals for the macrocyclic protons supports the formation of monomeric binuclear complexes with the nitrogen coordination pattern shown in Scheme 2.
 | ||
| Scheme 2 Nitrogen coordination pattern found in binuclear Cu2+ complexes of L1 and L3 suggested by paramagnetic 1H NMR spectra. | ||
The paramagnetic 1H NMR spectrum of the system Cu2+–L3 for 2:1 molar ratio in D2O at 298 K and pH = 6 is shown in Fig. S30.†
The hyperfine-shifted resonances, linewidth at half-height and T2 values are reported in Table S2.† The pattern of paramagnetic signals for this system also suggests in this case the formation of monomeric binuclear complexes in which the metal centres are bound to four nitrogen atoms as represented in Scheme 2.
Variable temperature 1H NMR spectra of both systems were recorded from 283 to 323 K. Isotropically shifted signals are temperature dependent following a Curie behaviour except for some signals belonging to β-CH2 or HmPz protons of the macrocyclic ligand that show an anti-Curie or temperature independent behaviour. In the Curie behaviour the paramagnetically shifted signals decrease with increasing temperature (see Tables S1 and S2†). The anti-Curie dependence results are indicative of spin-coupled dicopper(II) systems with antiferromagnetic coupling.
An interesting point concerns the actual protonation degree of the pyrazole moiety. As previously mentioned, 1H-pyrazole may either bind a proton behaving as a base or deprotonate to give the pyrazolate anionic form. In the absence of metal ions, these equilibria occur either at very acidic or very basic pH values falling outside the pH range of the technique.29,30 Formation of binuclear complexes induces the ready deprotonation of pyrazole to give rise to the preferred bis(monodentate) binding mode of this unit.10,11 However, a point that has not yet been fully addressed is the protonation state of pyrazole in the case of formation of mononuclear complexes, although the two nitrogen atoms of the pyrazole moiety can very hardly converge into a single metal ion since the formation of a three-membered chelate ring would be required. To shed some light on this aspect, we have recorded the variation with the pH of the UV spectra for all three systems both in 1:1 and 2:1 Cu2+:L molar ratios and the corresponding plots are given in Fig. 4A and B for L1 and in Fig. S4 and S5† for L2 and L3. Fig. 3A, which shows the distribution diagrams versus pH for the system Cu2+:L1 in a 1:1 molar ratio overlapped with the absorbance of pyrazole at 247 nm, shows that the formation of the mononuclear complexes at pH 3–4 produces an increase in the absorbance which reaches a plateau and remains thereon constant. The formation of binuclear complexes (Fig. 3B) produces a further increase of absorbance of about 20%. Since in binuclear complexes the pyrazole is deprotonated, one might think that the binding of the first metal ion already polarizes the N–H bond of pyrazole to a considerable extent, polarization that would be assisted by hydrogen bonding with an amine group. Interestingly, calculations about the location of the residual electron density around the pyrazole in the crystal structure of the mononuclear complex [Cu(HL1)Br]Br(1+x)(ClO4)(1−x)·yH2O (1) (vide infra) show that the residual electron density of the proton would be at about 80% located in the amine group and 20% in the pyrazole nitrogen (see below).
 | ||
| Fig. 4 Representation of the most stable structure for the monoprotonated Cu2+–L1 complex. (A) [Cu(H2(H−1L1)]3+ and (B) [Cu(HL1)]3+. Structure B is ca. 8.8 kJ mol−1 more stable than A. | ||
This indicates the formation of a relatively strong hydrogen bond with proton transfer from the pyrazole to the amine group. DFT calculation about the mono- and diprotonated species assuming that the proton is either in the polyamine chain or in the pyrazole ring, for the monoprotonated species (A) [Cu(H2(H−1L1)]3+ or (B) [Cu(HL1)]3+ (Fig. 4), and for the diprotonated species (A) [Cu(H3(H−1L1)]4+ and (B) [Cu(H2L1)]4+ (Fig. 5), reveals close energies between both structures, in both cases the structures with the proton located in the pyrazole ring being slightly favoured.
 | ||
| Fig. 5 Representation of the most stable structure for the diprotonated Cu2+–L1 complex. (A) [Cu(H3(H−1L1)]4+ and (B) [Cu(H2L1)]4+. Structure B is ca. 1.0 kJ mol−1 more stable than A. | ||
Similar calculations were performed for the diprotonated complex of L2, structures (A) [Cu(H2(H−1L2)]4+ and (B) [Cu(HL2)]4+ as shown in Fig. 6. The calculations denote a greater stabilisation of the structure in which the proton is placed at the pyrazole ring (structure B). In this case, the data do not suggest hydrogen bond formation between the pyrazole and the amine group in structure B, whereas hydrogen bonding is observed between protonated and non-protonated amines in the chain. In this structure hydrogen bonding is observed both within the chain and between the pyrazole N–H and an amine group of the chain.
 | ||
| Fig. 6 Representation of the most stable structure for the monoprotonated Cu2+–L2 complex. (A) [Cu(H2(H−1L2)]4+ and (B) [Cu(HL2)]4+. Structure B is ca. 25.3 kJ mol−1 more stable than A. | ||
Therefore, all the data seem to suggest that coordination of a single metal ion does not induce neat deprotonation of the pyrazole ring, even though hydrogen bonding with adjacent amine groups of the polyamine chain may contribute to the stabilisation of the structure.
Distribution diagrams in Fig. 3 for the system Cu2+–L1 and in Fig. S4 and S5† for the systems Cu2+–L2 and Cu2+–L3 show that the formation of mono- or binuclear complexes is clearly controlled by the Cu2+:L molar ratio used. In all three systems for 1:1 molar ratio only mononuclear complexes form, while for 2:1 molar ratio binuclear complexes prevail in a broad pH window. In the case of the systems Cu2+–L2 and Cu2+–L3 for 2:1 molar ratio the species [Cu2(H−1L)]2+ predominates from pH values of ca. 4 and 5 to 10 and 11, respectively. L2 due to its hydrocarbon sequence will provide alternate 5- and 6-membered chelate rings favouring a stronger interaction with the metal ions than the only 6-membered chelate ring sequence of L3.34 Consequently, in the Cu2+–L2 binuclear system the pyrazole is more strongly polarized and its deprotonation occurs at more acidic pH values. The structure of the [Cu2(H−1L)]3+ species of L2 and L3 should essentially correspond to that observed for crystals 2 discussed in the next section. Next deprotonation to give [Cu2(H−1L)(OH)]2+ would likely imply the loss of a proton by a coordinated water molecule. The values of the pKas of these processes of 10.10 and 11.25 log units found for L2 and L3 (Table 2), respectively, suggest that the OH− formed is not bridging both metal centres.
The system Cu2+–L1, even though related, has several particularities as there is the absence of a [Cu2(H(H−1L))]4+ species and the formation of both mono- and bis(hydroxylated) binuclear species. The pKa obtained for the formation of the first hydroxylated species [Cu2(H−1L1)(OH)]2+ (pKa = 7.30) may imply that the hydroxide formed is bridging both metal centres.35 However the pKa for the formation of the second one (pKa = 10.36) suggests again that this step corresponds to the deprotonation of a coordinated water molecule without giving rise to the formation of a bridging hydroxide ligand. Electronic spin resonance (esr) spectra, recorded at the pH values where the maximum formation of the mononuclear and binuclear complexes occur, somehow support the formation of hydroxide bridging ligands in the binuclear complexes of L1. While the formation of the different 1:1 complexes (Fig. S31†) provides a signal for high spin monomeric Cu2+ that practically does not change with pH, the signal corresponding to the [Cu2(H−1L1)]3+ species (pH = 6) (Fig. S32†) shows a significant decrease in intensity due to an antiferromagnetic coupling of the Cu2+ ions though the deprotonated pyrazolate bridge; the formation of a further hydroxo bridge between the metal ions (pH = 9) completely vanishes the signal. This is in agreement with previous reports in which Cu2+ ions were interconnected through 3,6-bis(2-pyridyl)pyridazine and hydroxo bridging ligands.36,37
The variations at pH = 6 in the visible spectra of L1 and L2 recorded upon addition of increasing amounts of Cu2+ denote some differential features for both systems (Fig. 7).
 | ||
| Fig. 7 Variation in the UV-Vis spectra of systems (A) Cu2+:L1, (B) Cu2+:L2 and (C) Cu2+–L3 upon increasing additions of Cu2+ at pH = 6, the initial concentration of the ligands 1 × 10−3 M. | ||
For L1, a d–d band centred at 560 nm is observed until a 1:1 molar ratio of Cu2+:L is reached (ε = 115 L mol−1 cm−1), then this band stops increasing in intensity and a new band bathochromically shifted at 716 nm appears that reaches its maximum intensity for 2:1 Cu2+:L1 molar ratio (ε = 140 L mol−1 cm−1) (Fig. 7A).
In the case of L2, however, for Cu2+:L2 ratios below 1 a continuous increase of the band at 570 nm is observed that progressively shifts bathochromically to 600 nm increasing its intensity until a 2:1 molar ratio is reached (ε = 240 L mol−1 cm−1) (Fig. 7B).
Interestingly, in the case of the largest ligand, L3, the addition of increasing amounts of Cu2+ at pH = 6 just leads to an increase in the absorbance of a band centred at 262 nm which reaches its maximum for 2:1 Cu2+–L3 without a significant shift in the wavelength of the band.
For a 1:1 Cu2+–L1 molar ratio, spectra recorded at pH values of 5, 8 and 11 where the mononuclear [CuHL1]3+, [CuL1]2+ and [CuL1(OH)]+ species predominate, respectively, are practically the same with a d–d band centred at around 560 nm (ε = 130 L mol−1 cm−1), only a small hypsochromic shift is observed for the last species (Fig. 8A).
 | ||
| Fig. 8 UV-Vis spectra for the system Cu2+–L1 at variable pH. (A) [Cu2+] = [L1] = 1 × 10−3 M. (B) [Cu2+]= 2 × 10–3 M, [L1] = 1 × 10−3 M. | ||
While for pH values below 5 the spectra reveal characteristics of the mononuclear species with an absorption band at 560 nm, at pH 6.6 the [Cu2(H−1L1)]3+ species predominates (Fig. 8B), in addition to the band at 560 nm, a new band centred at 710 nm (ε = 141 L mol−1 cm−1) is observed. The formation of the hydroxylated species [Cu2(H−1L1)(OH)]2+ and [Cu2(H−1L1)(OH)2]+, which predominate at pH values of 9 and 11 (Fig. 8B), leads to hypsochromic shifts of the less energetic band that now appears centred at ca. 650 nm (ε = 148 L mol−1 cm−1; 134 mol−1 cm−1). These spectral changes suggest a geometry change from essentially square planar for the mononuclear species as supported by the crystal structure 1 (vide infra) to a more pyramidal geometry38 once the binuclear complex is formed following the deprotonation of the pyrazole moiety.
Crystal structure of [Cu(HL1)Br]Br(1+x)(ClO4)(1−x)·yH2O (1)
Crystals of 1 evolved by slow diffusion of acetone into an aqueous solution containing L1·6HBr, Cu(ClO4)2·6H2O at pH 6. The crystals are composed of [Cu(H2(H−1L1))Br]2+ cations, bromide and perchlorate counter-anions and crystallization water molecules. Crystallographic data of the structures are provided in Table S3.†The Cu2+ ion is coordinated with an almost square pyramidal geometry (Addison parameter λ = 0.03)39 to one of the nitrogen donors of the pyrazole moiety, which acts as a monodentate ligand, and the three amine groups at the side of the coordinated pyrazole nitrogen donor that confirm the equatorial plane of the square pyramid (Fig. 9). As in previous cases, the shortest Cu2+–N distance is the one with the pyrazole moiety.
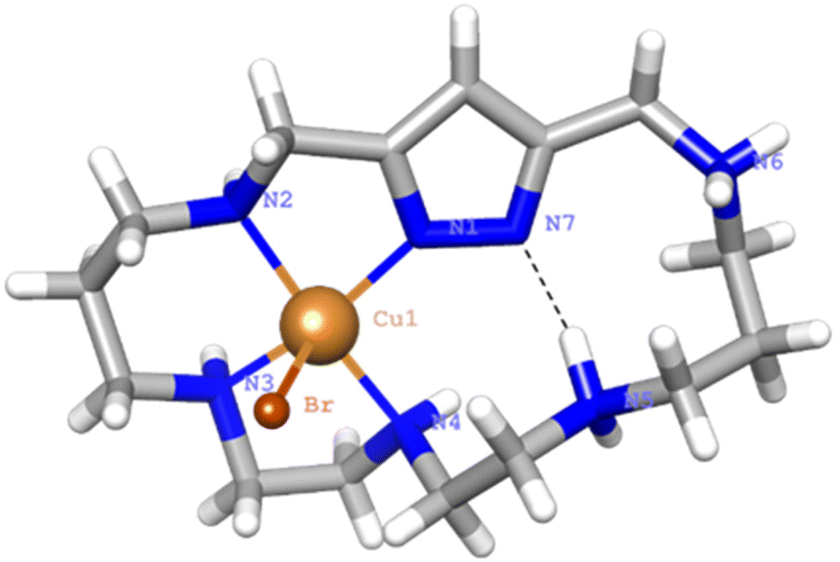 | ||
| Fig. 9 View of the X-ray crystal structure of the complex cation [Cu(H2(H−1L1))Br]2+. | ||
The elevation of the Cu2+ atom over the plane defined by the equatorial donor atoms is 0.29 Å. The molecule is rather flat with the angle between the mean planes defined by the coordinated and non-coordinated donor atoms of 22°. A list of bond distances and angles is shown in Table 4. As previously mentioned, one point of interest regards the nature of the pyrazole ligand in the mononuclear complexes. The location of the residual electron density indicates that only about 22(5)% would remain in the pyrazole nitrogen and the other 78(5)% would be shifted towards amine N5 that, thereby, would gain a significant ammonium group characteristic. Therefore, in this case coordination of only one copper to the pyrazole polarizes the N–H bond facilitating its partly transfer to an amine group. The situation might be interpreted as if a strong intramolecular hydrogen bond between the ammonium and the pyrazolate would be formed (N–H⋯N 1.75(4) Å, 160(4)°). The uncoordinated side of the molecule is of interest since different guests could be bound taking advantage of coulombic interactions, hydrogen bonding and the assistance of the coordinated metal ion as a Lewis acid centre.
| Distances (Å) | Angles (°) | ||
|---|---|---|---|
| Cu1–N1 | 1.985(2) | N1–Cu1–N2 | 81.9(1) |
| Cu1–N2 | 2.057(3) | N2–Cu1–N3 | 90.5(1) |
| Cu1–N3 | 2.021(3) | N3–Cu1–N4 | 84.9(1) |
| Cu1–N4 | 2.074(3) | N1–Cu1–N4 | 97.9(1) |
| Cu1–Br1 | 2.713(7) | N1–Cu1–Br1 | 102.69(7) |
| N2–Cu1–Br1 | 96.61(7) | ||
| N3–Cu1–Br1 | 94.21(8) | ||
| N4–Cu1–Br1 | 99.21(7) | ||
Also, it is interesting to remark that the coordination behaviour of this [1 + 1] pyrazole azacyclophane differs from that exhibited by related pyrazolaphanes with tetraamine bridges in which the formation of dimeric binuclear complexes was always observed. As commented before, the larger number of nitrogen atoms and greater flexibility of these ligands allow the convergence of an enough number of nitrogen atoms inwards facilitating the formation of monomeric complexes.
Crystal structure of [Cu2(H−1L2)Cl(ClO4)](ClO4)·H2O·C2H5OH (2)
Crystals of 2 were obtained by slow diffusion of ethanol into an aqueous solution of L2·6HCl and Cu(ClO4)2·6H2O at pH = 7. The asymmetric unit consists of [Cu2(H−1L2)Cl(ClO4)]+ cations, a perchlorate counter-anion and crystallisation water and ethanol molecules. The pyrazole moiety is deprotonated coordinating as a bis(monodentate) bridging ligand to the Cu2+ centres, which are placed at a distance of 4.213(3) Å (Fig. 10).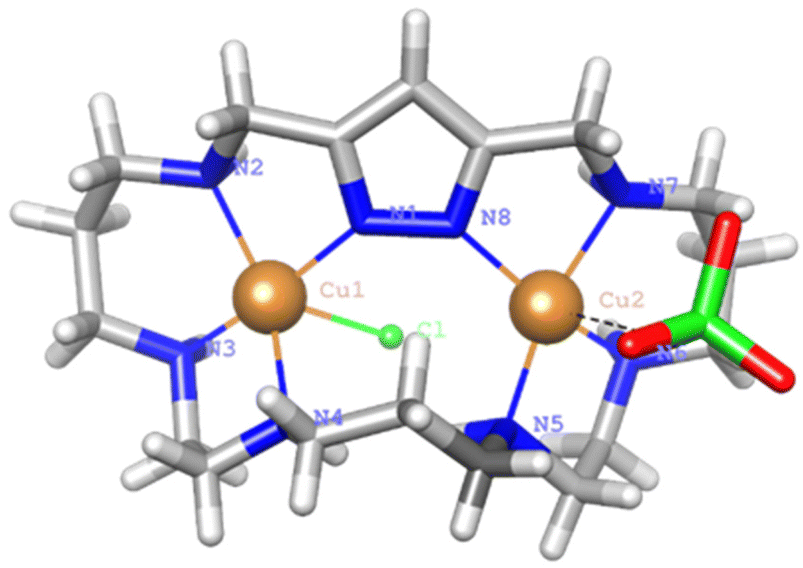 | ||
| Fig. 10 View of the crystal structure of the binuclear cation complex [Cu2(H−1L2)Cl(ClO4)]+. | ||
This distance is larger than those found in complexes where two pyrazolate moieties were simultaneously bridging the two Cu2+ metal ions. This was the case of monomeric complexes of either [2 + 2] pyrazole azamacrocycles7,8 or dimeric complexes of [1 + 1] azamacrocycles.14,15 In all these examples the Cu2+–Cu2+ distance was about 4 Å. The copper atoms complete their coordination spheres with three consecutive amine groups at each side of the bridge and with either a chloride or a perchlorate anion that points towards different sides of the macrocyclic cavity. The coordination geometry for Cu1 is slightly distorted square pyramidal, and essentially regular square pyramidal for Cu2, with the chloride or perchlorate anions occupying the strongly elongated axial positions. Addison parameters are λ = 0.23 and λ = 0.05, respectively.39 The shortest Cu–N distances are those established with the nitrogen atoms of the pyrazole rings. The distances and angles of the coordination sites are reported in Table 5.
| Distances (Å) | Angles (°) | ||||
|---|---|---|---|---|---|
| Cu1–N1 | 1.972(9) | N1–Cu1–N2 | 80.8(4) | N1–Cu1–Cl1 | 84.8(3) |
| Cu1–N2 | 2.081(9) | N2–Cu1–N3 | 92.9(4) | N2–Cu1–Cl1 | 117.0(3) |
| Cu1–N3 | 1.998(9) | N3–Cu1–N4 | 85.2(4) | N3–Cu1–Cl1 | 89.1(3) |
| Cu1–N4 | 2.11(1) | N1–Cu1–N4 | 104.9(4) | N4–Cu1–Cl1 | 89.1(3) |
| Cu1–Cl1 | 2.741(3) | ||||
| Cu2–N5 | 2.00(1) | N5–Cu2–N6 | 87.1(6) | N5–Cu2–O22 | 84.0(5) |
| Cu2–N6 | 2.02(1) | N6–Cu2–N7 | 88.9(7) | N6–Cu2–O22 | 90.8(6) |
| Cu2–N7 | 2.04(1) | N7–Cu2–N8 | 82.5(6) | N7–Cu2–O22 | 78.4(6) |
| Cu2–N8 | 1.97(1) | N5–Cu2–N8 | 107.3(5) | N8–Cu2–O22 | 108.3(5) |
| Cu2–O22 | 2.65(2) | ||||
The elevation of the metal ion with respect to the mean equatorial plane defined by the coordinated nitrogen atoms is 0.23 Å for Cu1 while Cu2 is completely embedded in the plane. The angle between the equatorial planes of both sites is 64°. In the tridimensional arrangement the binuclear complexes are organised in couples interconnected by a hydrogen bond network involving the chloride anion bound to Cu1, the ethanol molecules and two of the coordinated amine groups to Cu1 (N2–H2, N3–H3) (see Fig. 10).
Electrochemistry
The reduction potential is one of the most important chemical properties when designing superoxide dismutase mimetics, as at physiological pH it must be less negative than the reduction potential for the oxygen/superoxide anion couple (E0 O2/O2− = −0.33 V vs. NHE) but less positive than the superoxide anion/water couple (E0 O2−/H2O = +0.89 V vs. NHE).40 The voltammetric response of the studied complexes was highly homogeneous. As shown in Fig. 11 and Fig. S33,† the cyclic voltammograms consist of two more or less overlapped cathodic peaks at ca. −0.40 (C1) and −0.75 V (C2) vs. Ag/AgCl. In the subsequent anodic scan, a typical stripping peak was recorded at −0.10 V (A2) followed by a second weak oxidation wave at potentials between −0.05 and +0.18 V (A1) depending on the studied complex. Experiments performed in different potential ranges revealed that the process A2 is coupled with the previous signal C2. This denotes that this cathodic process leads to the formation of copper metal. In turn, the signal C1 can be attributed to the Cu2+ reduction to Cu+. This signal often appears as a separate peak (L1) or as a shoulder (L2, L3) preceding the cathodic peak C2. | ||
| Fig. 11 Cyclic voltammograms at the glassy carbon electrode for aqueous solution of the mononuclear (A) Cu–L1 and the binuclear (B) Cu2L1 systems, 1 × 10−4 M, in 0.15 M NaCl at pH 7.0. Scan rate 50 mV s−1. | ||
This suggests that there is no direct CuIIL → CuIL → Cu0 + L reduction via two successive one-electron reductions. The peak A1 appears at potentials at which the oxidation of Cu+–chloride complexes to the corresponding Cu2+-chloride complexes occurs. Given the high concentration of NaCl in the supporting electrolyte solution, it is conceivable that these signals reflect the formation of Cu+–chloride complexes when the deposit of metallic copper generated in the cathodic process C2 is oxidized in the oxidative dissolution process A2. Interestingly, the peak current per mole of copper of process C2 is the same (within the range of experimental uncertainty) for all four tested complexes. This feature suggests that in the case of the binuclear complexes, both metal centres are reduced simultaneously and independently.
This voltammetry can be interpreted in terms of the coexistence of two reductive pathways, the first one involving the one electron reduction of the parent CuIIL and CuII2L complexes to analogue CuIL and CuI2L ones (C1 peak). As judged from previous studies on Cu–receptor complexes,19,41–43 the progress from the process C1 to the process C2 involves some previous coordinative rearrangement of the CuI intermediate. Alternatively, this species can undergo disproportionation so that the peak C2 is equivalent to the two-electron reduction of the CuIIL and CuII2L complexes to Cu metal. The apparent formal electrode potential of the CuII/CuI couple was calculated as the half peak potential of the signal C1. The calculated apparent formal electrode potentials are representative of the potential SOD activity of the complexes. These values remain intermediate between −0.33 V and +0.89 V vs. NHE as desired, but presenting significant variations between the different species, being more positive for the binuclear complexes (+0.10 V for Cu2L1 and −0.05 V vs. NHE for Cu2L2 and Cu2L3) than for the corresponding mononuclear species (−0.10 V for CuL1 and −0.15 V vs. NHE for CuL2 and CuL3).
Evaluation of the superoxide dismutase activity
In view of the interesting SOD activity of the analogous complexes of the ligands L4 and L5 containing pyridine instead of 1H-pyrazole rings, and taking into account the ability of the uncoordinated pyrazole nitrogen to form hydrogen bonds (vide supra), we made preliminary assays about the potentiality of the Cu2+ complexes of L1–L3 to behave as SOD mimics. SOD activity was evaluated at physiological pH by means of the indirect enzymatic assay McCord–Fridovich,44 which allows calculating the IC50 and catalytic constant (kcat) values for both the mononuclear and binuclear Cu2+ complexes of L1, L2 and L3 as well as for the free L1, L2 and L3 macrocycles. Table 6 shows these values along with the previously reported ones for the Cu2+–L3 and Cu2+–L4 pyridine-based analogous binuclear systems,19 and for the native CuZn-SOD enzyme and the free Cu2+ ion in aqueous solution.As we can see in Table 6, both the mononuclear and binuclear Cu2+–L1, Cu2+–L2 and Cu2+–L3 systems exhibit SOD activity. However, in all three systems the binuclear complexes show clearly higher antioxidant activity than the mononuclear ones. The non-saturated coordination spheres of the metal ions in the binuclear complexes and the close proximity between the metal ions should likely contribute to the enhanced activity of these complexes. Furthermore, the activity found for the binuclear systems of L2 and L3 is very remarkable with IC50 and kcat values close to those reported for the native enzyme, constituting two of the as far as we know best results reported in the literature. In order to facilitate the discussion of the results, Fig. 12 plots the kcat values for the mononuclear and binuclear Cu2+–L1, Cu2+–L2 and Cu2+–L3 systems, as well as those of the binuclear complexes of the pyridinaphanes L4 and L5. Interestingly, the replacement of the pyridine moiety by the pyrazole one leads to a slight enhancement in the SOD activity of the systems.
 | ||
| Fig. 12 Plot of the kcat values for the mononuclear and binuclear Cu2+–L1, Cu2+–L2, Cu2+–L3, Cu2+–L4 and Cu2+–L5 systems. | ||
One plausible explanation might be the presence of the N–H in the 1H-pyrazole moiety, which might be involved in hydrogen bonding with the incoming superoxide anion. On the other hand, the imidazole of the His-61 residue in the native CuZn-SOD enzyme seems to play a key role during the catalytic pathway, allowing the formation and breaking of an imidazolate bridge.21,22
Finally, preliminary studies on hydrogen peroxide removal performed using the xylenol orange method45 show moderate capacity of the binuclear complexes. A more thorough analysis of the ROS scavenging capacity of these systems is currently under study.
Conclusions
The studies presented here show that the polyamine length and the number of nitrogen atoms of the bridges in pyrazolaazacyclophanes regulate the endo- or exo-coordinate binding modes of the bis(monodentate) pyrazolate moiety. While shorter tetraamine bridges give rise to exo-coordination with the formation of dimeric species of 2:1, 3:2 or 4:2 Cu2+:L stoichiometry, longer pentaamine or hexaamine bridges lead to the formation of discrete monomeric binuclear complexes. In these complexes a single pyrazolate bridge connects the metal ions which complete their coordination spheres with the nitrogen atoms of the bridges and, if necessary, with exogenous ligands. This has been proven both in solution by a variety of experimental techniques and by solid state by X-ray diffraction analysis. The presence of a single pyrazolate bis(monodentate) bridge places the metal ions at distances close to those shown by copper enzymes participating in ROS removal, at the time that leave enough room to allow the binding of the substrates to the metal centre. This situation has produced two of the potential SOD mimics with the highest activity so far reported in the literature with IC50 values approaching those displayed by the native enzymes. Further studies on the biological capacity of these complexes and derivatives are currently underway.
Experimental section
The synthesis of L2 and L3 was performed as described in ref. 16. The compounds gave satisfactory elemental microanalysis and spectroscopic characterization results. 1H-3,5-Bis(chloromethyl)pyrazole and 3,5-bis-(chloromethyl)-1-(tetrahydropyran-2-yl)-pyrazole were prepared as described in the literature.26,46 The tosylated amine 1,5,8,11,15-penta(p-tosylsulfonyl)-pentaazapentadecane was prepared as described in ref. 47.:L molar ratios varying from 2:1 to 1:2 were titrated with NaOH with Cu2+ concentrations ranging from 2.0 × 10−4 M to 1.1 × 10−3 M. The different titration curves for each system were treated as separated curves without significant variations in the values of the stability constants. Finally, the sets of data were merged together and treated simultaneously to give the final stability constants. When more than one model fits the experimental data, the most reliable chemical model was chosen by performing F tests at the 0.05 confidence level.52,53
56 was used as front-end for solving and refining. The initial structure was solved with direct methods using SHELXS and then refined with SHELXL2018.57 Initially, an isotropic refinement was performed on the non-hydrogen atoms. Then, anisotropic refinement was done.
:1 and 2:3 molar ratios were acquired in the positive ion mode using a Triple TOF 5600 hybrid quadrupole time-of-flight (TOF) mass spectrometer. N2 was used as a curtain and nebulizing gas. The experiments were performed at a voltage of 5300 V and GS1 and GS2 (35 psi) ion source gas at 723.15 K. The AB SCIEX Peak View software was used for the analysis of the data.
:1 and 2:1 molar ratios (1.0 × 10−3 M) were recorded with an Agilent 8453 spectrometer at 298.15 K.
:L2 at 1:1 molar ratio were recorded under non-saturating conditions on a Bruker ER 200 D spectrometer equipped with a helium cryostat.
Data availability
Crystallographic data for compounds 1 and 2 have been deposited at the CCDC repository under the accession numbers 2240668 and 2215780.†1H and 13C NMR spectra of compounds L1, L2 and L3, mass spectra of the Cu2+ complexes, distribution diagrams of the free ligands and metal complexes, and protonation and stability constants are included as a part of the ESI.†
Author contributions
I. Bonastre-Sabater: investigation, writing – original draft, and writing – review & editing; A. Lopera: investigation; A. Martínez-Camarena: formal analysis and writing – original draft; Salvador Blasco: formal analysis; A. Doménech-Carbó: formal analysis, writing – original draft, and writing – review & editing; H. R. Jimenez: investigation, writing – original draft and writing – review & editing; B. Verdejo: investigation and writing – review & editing; E. García-España: conceptualization, formal analysis, funding acquisition, methodology, project administration, supervision, validation, and writing – review & editing; M. P. Clares: conceptualization, project administration, supervision, validation, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Spanish Ministerio de Economía y Competitividad (Project PID2019-110751RD-I00) and the Conselleria de Innovación, Universidades, Ciencia y Sociedad Digital of the Generalitat Valenciana (PROMETEO Grant CIPROM/2021/030) is acknowledged. A. M.-C. wants to acknowledge the support received under the “Margarita Salas” post-doctoral program (grant MS21-095) funded by the Ministerio de Universidades from the Spanish Government and the European Union – NextGenerationEU.References
- (a) J. Elguero, in Pyrazoles in Comprehensive Heterocycle Chemistry II; A Review of the Literature 1982–1995, ed. A. R. Katrizky, C. V. Ress and E. F. V. Scriven, Pergamon New York, 1997, vol. 3, pp. 1–75 Search PubMed; (b) R. Muckheerjee, Coord. Chem. Rev., 2000, 203, 151–258 CrossRef.
- (a) J. Bedi, V. Muelas-Ramos, M. Peñas-Garzón, A. Gómez-Avilés, J. J. Rodríguez and C. Belver, Catalysts, 2019, 9, 52 CrossRef; (b) Y. Li, R. Wang, X. Liu, K. Li and Q. Xu, Nanotechnology, 2023, 34, 202002 CrossRef PubMed; (c) K. Wang, X.-L. G Lv, D. Feng, J. Li, S. Chen, J. Sun, L. Song, Y. Xie, J.-R. Li and H.-C. Zhou, J. Am. Chem. Soc., 2016, 138, 914–919 CrossRef CAS PubMed; (d) S. Saha, M. Das, K. S. Das, R. Datta, S. Bala, J.-L. Liu, P. P. Ray and R. Mondal, Cryst. Growth Des., 2023, 23, 1104–1118 CrossRef CAS; (e) S. Posada-Pérez, J. Poater, N. Bahri-Laleh and A. Poater, Catalysts, 2023, 13, 317 CrossRef; (f) M. El Boutaybi, A. Taleb, R. Touzani and Z. Bahari, Mater. Today: Proc., 2020, 31, S96–S102 CAS.
- (a) R. K. Gupta, P. Kallem, G. G. Luo, P. Cui, Z. Wang, F. Banat, C.-H. Tung and D. Sun, Chem. Rev., 2023, 497, 215436 Search PubMed; (b) A. Lancheros, S. Goswami, M. R. Mian, X. Zhang, X. Zarate, E. Schott, O. K. Farha and J. T. Hupp, Dalton Trans., 2021, 50, 2880–2890 RSC.
- (a) S. Hamood, S. Azzam and M. A. Pasha, European J. Pharm. Med. Res., 2020, 7, 867–887 Search PubMed; (b) L. M. R. Orlando, G. C. Lechuga, L. S. Lara, B. S. Ferreira, C. N. Pereira, R. C. Silva, M. S. Santo and M. C. S. Pereira, Molecules, 2021, 26, 6742 CrossRef CAS; (c) S. Kumari, S. Paliwal and R. Chauhan, Synth. Commun., 2014, 44, 1521–1578 CrossRef CAS.
- (a) R. H. Holm, P. Kennepohl and E. I. Solmon, Chem. Soc. Rev., 1996, 96, 2239–2314 CrossRef CAS PubMed; (b) E. I. Solomon, U. M. Sundaram and T. E. Machonkin, Chem. Rev., 1996, 98, 2563–2605 CrossRef PubMed; (c) S. Ferguson-Miller and G. T. Babcock, Chem. Rev., 1996, 96, 2889–2907 CrossRef CAS PubMed; (d) H. Decker, T. Schweikardt and F. Tuczek, Angew. Chem., Int. Ed., 2006, 45, 4546–4550 CrossRef CAS PubMed; (e) N. Kitajima, K. Fujisawa, C. Fujimoto, Y. Morooka, S. Hashimoto, T. Kitagawa, K. Toriumi, K. Tatsumi and A. Nakamura, J. Am. Chem. Soc., 1992, 114, 1277–1291 CrossRef CAS; (f) Y. Sheng, I. A. Abreu, D. E. Cabelli, M. J. Maroney, A.-F. Miller, M. Teixeira and J. S. Valentine, Chem. Rev., 2014, 114, 3854–3918 CrossRef CAS.
- (a) P. Chaudhuri, V. Kataev, B. Düchner, H. Kaluss, B. Kersting and F. Meyer, Coord. Chem. Rev., 2009, 253, 2261–2283 CrossRef CAS; (b) J. L. Vlugt, S. Demeshko, S. Dechert and F. Meyer, Inorg. Chem., 2008, 47, 1576–1585 CrossRef PubMed; (c) A. Prokofieva, A. Prikohod`ko, E. Enyedy, E. Farkas, W. Maringele, S. Demeshko, S. Dechert and F. Meyer, Inorg. Chem., 2007, 46, 4298–4307 CrossRef CAS PubMed; (d) A. Eisenwiener, M. Neuberger and T. Kaden, Dalton Trans., 2007, 218–223 RSC; (e) S. Brooker, T. C. Davidson, S. J. Hay, R. J. Kelly, D. K. Kennepohl, P. G. Plieger, B. Moubaraki, K. S. Murray, E. Bill and E. Bothe, Coord. Chem. Rev., 2001, 216–217, 3–30 CrossRef CAS; (f) H. Weller, L. Siegfred, M. Neugerger, M. Zhender and T. A. Kaden, Helv. Chim. Acta, 1997, 80, 2315–2328 CrossRef CAS; (g) A. Eisenwiener, M. Neuberger and T. A. Kaden, Dalton Trans., 2007, 218–223 RSC.
- (a) M. Kumar, V. J. Arán and P. Navarro, Tetrahedron Lett., 1993, 34, 3159–3162 CrossRef CAS; (b) V. J. Arán, M. Kumar, J. Molina, L. Lamarque, P. Navarro, E. García-España, J. A. Ramírez, S. V. Luis and B. Escuder, J. Org. Chem., 1999, 64, 6135–6146 CrossRef; (c) C. Miranda, F. Escartí, L. Lamarque, E. García-España, P. Navarro, J. Latorre, F. Lloret, H. R. Jiménez and M. J. R. Yunta, Eur. J. Inorg. Chem., 2005, 189–208 CrossRef CAS; (d) C. Miranda, F. Escartí, L. Lamraque, M. J. R. Yunta, P. Navarro, E. Gracía-España and M. L. Jimeno, J. Am. Chem. Soc., 2004, 126, 823–833 CrossRef CAS; (e) J. Pitarch, M. P. Clares, R. Belda, R. D. Costa, P. Navarro, E. Ortí, C. Soriano and E. García-España, Dalton Trans., 2010, 39, 7741–7746 RSC.
- (a) M. Kumar, V. J. Arán and P. Navarro, Tetrahedron Lett., 1995, 12, 2161–2164 CrossRef; (b) L. Lamarque, C. Miranda, P. Navarro, F. Escartí, E. García-España, J. Latorre and J. A. Ramírez, Chem. Commun., 2000, 1337–1338 RSC; (c) L. Lamarque, P. Navarro, C. Miranda, V. J. Arán, C. Ochoa, F. Escartí, E. García-España, J. Latorre, S. V. Luis and J. F. Miravet, J. Am. Chem. Soc., 2001, 123, 10560–10570 CrossRef CAS PubMed.
- J. Pitarch-Jarque, R. Belda, S. Blasco, P. Navarro, R. Tejero, J. M. Junquera-Hernández, V. Pérez-Mondéjar and E. García-España, New J. Chem., 2016, 40, 5670–5674 RSC.
- (a) F. Escartí, C. Miranda, L. Lamarque, J. Latorre, E. García-España, M. Kumar, V. J. Arán and P. Navarro, Chem. Commun., 2002, 936–937 RSC; (b) J. Pitarch-Jarque, K. Rissanen, S. García-Granda, A. Lopera, M. P. Clares, E. García-España and S. Blasco, New J. Chem., 2019, 43, 18915–18924 RSC.
- J. Pitarch-Jarque, R. Belda, F. Lloret, J. Ferrando-Soria, P. Navarro, A. Lopera and E. García-España, Dalton Trans., 2015, 44, 3378–3383 RSC.
- J. Pitarch-Jarque, R. Belda, L. García-España, J. M. Llinares, F. F. Pen, K. Rissanen, P. Navarro and E. García-España, Dalton Trans., 2015, 44, 7761–7764 RSC.
- M. I. Rodríguez-Franco, P. San Lorenzo, A. Martínez and P. Navarro, Tetrahedron, 1999, 55, 2763–2772 CrossRef.
- R. Belda, J. Pitarch-Jarque, C. Soriano, J. M. Llinares, S. Blasco, J. Ferrando-Soria and E. García-España, Inorg. Chem., 2013, 52, 10795–10803 CrossRef CAS PubMed.
- A. Lopera, A. Gil-Martínez, J. Pitarch-Jarque, B. Verdejo, S. Blasco, M. P. Clares, H. R. Jiménez and E. García-España, Dalton Trans., 2020, 49, 8614–8624 RSC.
- A. Lopera, J. A. Aguilar, R. Belda, B. Verdejo, J. W. Steed and E. García-España, Soft Matter, 2020, 16, 6514–6522 RSC.
- P. J. Altman and A. Pothig, J. Am. Chem. Soc., 2018, 138, 13171–13174 CrossRef PubMed.
- (a) M. P. Clares, S. Blasco, M. Inclán, L. del Castillo Agudo, B. Verdejo, C. Soriano, A. Doménech, J. Latorre and E. García-España, Chem. Commun., 2011, 47, 5988–5990 RSC; (b) M. P. Clares, C. Serena, S. Blasco, A. Nebot, L. del Castillo, C. Soriano, A. Domènech, A. V. Sánchez-Sánchez, L. Soler-Calero, J. L. Mullor, A. García-España and E. García-España, J. Inorg. Biochem., 2015, 143, 1–8 CrossRef CAS PubMed; (c) C. Serena, E. Calvo, M. P. Clares, M. L. Diaz, J. U. Chicote, R. Beltrán-Debon, R. Fontova, A. Rodriguez, E. García-España and A. García-España, PLoS One, 2015, 10, 1–12 CrossRef PubMed; (d) M. Merino, M. D. Sequedo, A. V. Sánchez-Sánchez, M. P. Clares, E. García-España, R. P. Vázquez-Manrique and J. L. Mullor, Int. J. Mol. Sci., 2022, 23, 8936 CrossRef CAS PubMed; (e) M. Merino, S. González, M. C. Tronch, A. V. Sánchez-Sánchez, M. P. Clares, A. García-España, E. García-España and J. L. Mullor, Int. J. Mol. Sci., 2023, 24, 15153 CrossRef CAS PubMed.
- R. Belda, S. Blasco, B. Verdejo, H. R. Jiménez, A. Doménech-Carbó, C. Soriano, J. Latorre, C. Terencio and E. García-España, Dalton Trans., 2013, 42, 11194–11204 RSC.
- (a) A. Martínez-Camarena, E. Delgado-Pinar, C. Soriano, J. Alarcón, J. M. Llinares, R. Tejero and E. García-España, Chem. Commun., 2018, 54, 3871–3874 RSC; (b) A. Martínez-Camarena, J. M. Llinares, A. Domènech-Carbó, J. Alarcón and E. García-España, RSC Adv., 2019, 9, 41549–41560 RSC.
- (a) R. Sandhir, A. Yadav, A. Sunkaria and N. Singhal, Neurochem. Int., 2015, 89, 209–226 CrossRef CAS PubMed; (b) H. Zhao, R. Zhang, X. Yan and K. Fan, J. Mater. Chem. B, 2021, 9, 6939–6957 RSC.
- (a) K. M. Lincoln, T. E. Richardson, L. Rutter, P. González, J. Simpkins and K. N. Green, ACS Chem. Neurosci., 2012, 3, 919–927 CrossRef CAS PubMed; (b) K. M. Lincoln, P. González, T. E. Richardson, D. A. Julovich, R. Saunders, J. W. Simpkins and K. N. Green, Chem. Commun., 2013, 49, 2712–2714 RSC; (c) K. Lincoln, M. E. Offutt, T. D. Hayden, R. E. Saunders and K. N. Green, Inorg. Chem., 2014, 53, 1406–1416 CrossRef CAS PubMed.
- A. Martínez-Camarena, M. Merino, A. V. Sánchez-Sánchez, S. Blasco, J. M. Llinares, J. L. Mullor and E. García-España, Chem. Commun., 2022, 58, 5021–5024 RSC.
- A. Martínez-Camarena, P. A. Sánchez-Murcia, S. Blasco, L. González and E. García-España, Chem. Commun., 2020, 56, 7511–7514 RSC.
- (a) J. E. Richman and T. J. Atkins, J. Am. Chem. Soc., 1974, 96, 2268–2270 CrossRef CAS; (b) A. Bencini, M. I. Burguete, E. García-España, S. V. Luis, J. F. Miravet and C. Soriano, J. Org. Chem., 1993, 58, 4749 CrossRef CAS.
- J. C. Röder, F. Meyer and H. Pritzkow, Organometallics, 2001, 20, 811–817 CrossRef.
- P. Gans, A. Sabatini and A. Vacca, Talanta, 1996, 43, 1739 CrossRef CAS PubMed.
- L. Alderighi, P. Gans, A. Ienco, D. Peters, A. Sabatini and A. Vacca, Coord. Chem. Rev., 1999, 184, 311–318 CrossRef CAS.
- G. Yagil, Tetrahedron, 1967, 23, 2855–2861 CrossRef CAS PubMed.
- J. Catalán, R. M. Claramunt, J. Elguero, J. Laynez, M. Menéndez, F. Anvia, J. H. Quian, M. Taagepera and R. W. Taft, J. Am. Chem. Soc., 1988, 110, 4105–4111 CrossRef.
- (a) A. Bencini, A. Bianchi, E. García-España, M. Micheloni and J. A. Ramírez, Coord. Chem. Rev., 1999, 188, 97–156 CrossRef CAS; (b) A. Bianchi, B. Escuder, E. García-España, S. V. Luis, V. Marcelino, J. F. Miravet and J. A. Ramírez, J. Chem. Soc., Perkin Trans. 2, 1994, 1254–1259 Search PubMed.
- J. Pitarch-Jarque, H. R. Jiménez, E. Kalenius, S. Blasco, A. Lopera, M. P. Clares, K. Rissanen and E. García-España, Dalton Trans., 2021, 50, 9010–9015 RSC.
- C. E. Castillo, J. González-Garcia, J. M. Llinares, M. A. Mañez, H. R. Jiménez, E. García-España and M. G. Basallote, Dalton Trans., 2013, 42, 6131–6141 RSC.
- B. P. Hay and R. D. Hancock, Coord. Chem. Rev., 2001, 212, 61–78 CrossRef CAS.
- A. Bencini, A. Bianchi, P. Paoletti and P. Paoli, Coord. Chem. Rev., 1992, 120, 51–85 CrossRef CAS.
- C. Yuste, A. Bentama, S. E. Stiriba, D. Armentano, G. De Munno, F. Lloret and M. Julve, Dalton Trans., 2007, 5190–5200 RSC.
- T. E. Mastropietro, N. Marino, D. Armentano, G. De Munno, C. Yuste, F. Lloret and M. Julve, Cryst. Growth Des., 2013, 13, 270–281 CrossRef CAS.
- (a) B. J. Hathaway and D. E. Billing, Coord. Chem. Rev., 1970, 5, 143–202 CrossRef CAS; (b) B. J. Hathaway and A. A. G. Tomlinson, Coord. Chem. Rev., 1970, 5, 1–43 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 7, 1349–1356 RSC.
- P. M. Wood, Biochem. J., 1988, 253, 287–289 CrossRef CAS PubMed.
- A. Doménech-Carbó, E. García-España, S. V. Luis, V. Marcelino and J. F. Miravet, Inorg. Chim. Acta, 2000, 229, 238 CrossRef.
- A. Doménech-Carbó, E. García-España, P. Navarro and C. Miranda, J. Chem. Soc., Dalton Trans., 2006, 4926–4935 RSC.
- L. Guijarro, M. Inclán, J. Pitarh, A. Doménech-Carbó, J. U. Chicote, S. Trefler, E. García-España and B. Verdejo, Inorg. Chem., 2017, 56, 13748–13758 CrossRef CAS PubMed.
- (a) J. M. McCord and I. J. Fridovich, J. Biol. Chem., 1969, 244, 6049–6055 CrossRef CAS PubMed; (b) C. Beauchamp and I. J. Fridovich, Anal. Biochem., 1971, 44, 276–287 CrossRef CAS PubMed; (c) L. W. Oberley and D. R. Spitz, Methods Enzymol., 1984, 105, 457–464 CAS.
- C. Gay, J. Collins and J. M. Gebicki, Redox Rep., 1999, 4, 327–328 CrossRef CAS PubMed.
- L. Iturrino, P. Navarro, I. Rodríguez-Franco, M. Contreras, J. A. Escario, A. Martínez and M. R. Pardo, Eur. J. Med. Chem., 1987, 22, 445–451 CrossRef CAS.
- J. Aguilar, P. Díaz, F. Escartí, E. García-España, L. Gil, C. Soriano and B. Verdejo, Inorg. Chim. Acta, 2002, 339, 307–316 CrossRef CAS.
- E. García-España, M.-J. Ballester, F. Lloret, J. M. Moratal, J. Faus and A. Bianchi, J. Chem. Soc., Dalton Trans., 1988, 101–104 RSC.
- M. Fontanelli and M. Micheloni, In Proceedings of the First Spanish−Italian Congress on Thermodynamics of Metal Complexes. Diputación de Castellón, Spain, 1990 Search PubMed.
- G. Gran, Analyst, 1952, 77, 661–672 RSC.
- F. J. C. Rossotti and H. Rossotti, J. Chem. Educ., 1965, 42, 375–378 CrossRef CAS.
- W. C. Hamilton, Statistics in Physical Science. Estimation, Hypothesis Testing, and Least Squares. The Roland Press Co., New York, 1964 Search PubMed.
- L. Bologni, A. Sabatini and A. Vacca, Inorg. Chim. Acta, 1983, 69, 71–75 CrossRef CAS.
- H. E. Gottlieb, V. Kotlyar and A. Nudelman, J. Org. Chem., 1997, 62, 7512–7515 CrossRef CAS PubMed.
- A. K. Covington, M. Paabo, R. A. Robinson and R. G. Bates, Anal. Chem., 1968, 40, 700–706 CrossRef CAS.
- O. Dolomanov, O. L. Bourhis, R. Gildea, A. L. Richard, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef PubMed.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, Gaussian 9, 2009 Search PubMed.
- G. Schaftenaar and J. H. Noordik, J. Comput. Mol. Des., 2000, 14, 123–134 CrossRef CAS PubMed.
- Schrodinger LLC, The PyMOL Molecular Graphics System Version 2.0, (Schrödinger, LLC, 2015), 2015 Search PubMed.
- (a) B. H. J. Bielski and H. W. Richter, J. Am. Chem. Soc., 1977, 99, 3019–3023 CrossRef CAS; (b) R. F. Pasternack and B. Halliwell, J. Am. Chem. Soc., 1979, 101, 1026–1031 CrossRef CAS; (c) S. Durot, C. Policar, F. Cisnetti, F. Lambert, J.-P. Renault, G. Pelosi, G. Blain, H. Korri-Youssoufi and J.-P. Mahy, Eur. J. Inorg. Chem., 2005, 2005, 3513–3523 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Text figures, tables, and physical measurements. CCDC 2240668 and 2215780. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01236d |
| This journal is © The Royal Society of Chemistry 2024 |