
A facile method to prepare reduced graphene oxide with nano-porous structure as electrode material for high performance capacitor†
Nian Yanga,
Xiaoyang Xua,
Lingzhi Lia,
Heya Naa,
Huan Wanga,
Xuefang Wanga,
Fubao Xing*ab and
Jianping Gao*ab
aSchool of Science, Tianjin University, Tianjin 300072, PR China. E-mail: fbxing@sohu.com; jianpinggaols@126.com; Fax: +86 2227403475; Tel: +86 15522461762
bCollaborative Innovation Center of Chemical Science and Engineering, Tianjin 300072, PR China
First published on 20th April 2016
Abstract
A facile method to prepare reduced graphene oxide (M-rGO) nanosheets with enhanced electrochemical performance has been established by using Mg powder as the reductant. The structural, morphological and electrochemical properties of the M-rGO have been characterized. The prepared M-rGO possesses well-developed nano-porous structure and superior surface characteristics. Electrochemical analyses show that the M-rGO has a maximum specific capacitance of 577.4 F g−1 at the scan rate of 1 mV s−1, and an excellent rate performance of 324.9 F g−1 at 1 V s−1. The CV curves of M-rGO can keep rectangular in shape even at a scan rate of 500 mV s−1 without obvious distortion. Such outstanding electrochemical performances of M-rGO can be attributed to both the presence of nano-pores in M-rGO and the pseudocapacitance arising from residual oxygen-containing functional groups.
1. Introduction
Supercapacitors have recently attracted a great deal of attention as one of the most promising energy storage devices because they feature both high energy density of batteries and fast power delivery of capacitors. However, the energy density of supercapacitors is generally an order of magnitude lower than conventional batteries, and thus restricts their applications which require both high power density and long cycle life.1 Considerable efforts have been devoted to developing high-performance supercapacitors based on various carbon materials and carbon based composites.2,3The electrochemical performance of a supercapacitor is mainly determined by the properties and structure of the electrode material.4 Carbon materials such as activated carbon, mesoporous carbon and carbon nanotubes have been extensively investigated as supercapacitor electrode due to their good conductivities, excellent chemical stability, tailored pore structure and surface chemical properties.5–7 Graphene, as a two dimensional carbon material, is particularly attractive for its excellent mechanical and electronic properties as well as its extraordinary high surface area.8
Graphene-based materials fabricated from graphene oxide (GO) can be obtained at relatively low cost in a large scale. However, aggregation and restacking inevitably occur in the reduction process of GO as a result of intersheet van der Waals attractions between the adjacent graphene layers.9 These phenomena will have a serious impact on the ion-accessible surface area, and also bring a large ion diffusion resistance between the adjacent graphene layers thus lead to significant compromise of the electrochemical properties of the electrode. Making graphene into porous structures is an effective strategy to solve the problem and also a promising strategy to obtain graphene materials with high surface area and high capacitance value.10 The research on the influence of specific surface area or the micropore volume on the capacitance behavior of graphene-based material has been extensively conducted, and typically the higher the surface area is, the higher the capacitance value is.11 The previously reported porous graphene with a very high specific surface area showed a specific capacitance of 303 F g−1.12 However, some deviations suggest that there are other factors that influence the capacitance such as pore size distribution.13 A suitable number of mesopores can guarantee the fast ions penetration inside the electrode and thus help the charging and discharging of the double layer,14 and the presence of micropores plays an essential part in charging and discharging process. However, not all the nanopores are electrochemically accessible and thus cannot contribute to the overall double layer capacitance.15 Therefore, in order to obtain supercapacitor with excellent electrochemical performance, well-developed nano-pores is of great importance.
In addition, the chemical state is also an important factor that should be taken into consideration.16 The oxygen-containing functional groups presented in graphene-based supercapacitors enhances the diffusion of the electrolyte, introducing pseudocapacitive via faradic redox reactions and finally results in an improved capacitance.17 However, different kinds of oxygen groups function in different ways, and only the CO-type oxygen-containing functional groups make positive contribution to the capacitance.18 So less-agglomerated graphene-based electrodes, with suitable pore sizes distribution and surface characteristic are highly demanded.
Inspired by these results, herein we report a graphene material with well developed nano-porous structure and suitable surface characteristic, which was simply prepared by chemical reduction of GO by Mg powder and denoted as M-rGO. As we know, Mg is a highly abundant element in the Earth's crust and also a clean metal with relatively low cost. It is highly reactive and can reduce the hydrogen ions in hot water, thus the reduction of GO by Mg powder can be completed in the absence of acid. Since the hydrogen spilled from water is limited, a moderated reduction can be anticipated. The prepared M-rGO showed a high specific capacitance of 577.4 F g−1 at a scan rate of 1 mV s−1, and the M-rGO could retain 95.3% of its initial capacitance at a current density of 8 A g−1 even after 3000 cycles. The high capacitive performance and good cycling property make M-rGO very attractive as high-performance electrode materials for supercapacitor applications.
2. Experimental
2.1. Materials
Graphite was obtained from Qingdao Graphite Factory, potassium permanganate (KMnO4), sodium nitrate (NaNO3), magnesium powder (Mg), concentrated sulfuric acid (H2SO4), hydrogen peroxide (H2O2, 30%), hydrochloric acid (HCl), potassium hydroxide (KOH), polytetrafluoroethylene emulsion (PTFE 60%), and other reagents were all purchased from Tianjin Chemical Reagent Co. All the chemicals were analytical grade and used without further purification.2.2. Preparation of GO
GO was prepared from natural graphite by the modified Hummers method.192.3. Preparation of M-rGO
First, 5 mg magnesium power was added to the GO suspension (1 mg mL−1) of 20 mL. Then the mixture was reacted at 95 °C for 5 h with continuously stirring to obtain reduced GO. After complete removal of impurities, the sample was dried at 60 °C to obtain M-rGO. The energy dispersive X-ray spectrometry (EDX) spectra of M-rGO is presented in Fig. S1,† which suggests a Mg content of 0.06 at%. A control was prepared with the same procedure by using 3 mL hydrazine (85%) as the reductant.2.4. Characterization
The UV-vis absorption spectra during the reduction process were recorded with a TU-1901 UV-vis spectrophotometer. The Raman and X-ray diffraction (XRD) spectra of the M-rGO and GO were measured using a Raman microscope (RENISHAW, UK) and an X-ray diffractometer (BDX3300, reference target: Cu Kα radiation, voltage: 30 kV, current: 30 mA, steps: 2° min−1), respectively. Thermogravimetric analysis (TGA) diagrams were recorded with a Rigaku-TD-TDA analyzer with a heating rate of 10 °C min−1, the samples were dried in a vacuum at 60 °C for 2 days before TGA test. Elemental analyses were carried out with an X-ray photoelectron spectrometer (XPS, PHI1600 ESCA System, Perkin-Elmer, US). Scanning electron microscopy (SEM) was conducted using a JEOL-6700F ESEM microscope. Transmission electron microscopy (TEM) was performed using a Tecnai G2F20 microscope. The sample for TEM was prepared by dropping the aqueous suspension onto a carbon-coated copper grid. Then it was dried under ambient conditions before being introduced into the TEM chamber. Nitrogen adsorption isotherm of M-rGO was recorded at 77 K with a autosorb iQ instrument (Quantachrome U.S.). The total surface area was calculated using both the Brunauer–Emmett–Teller (BET) and t-plot method, the pore size distribution data was calculated with the Density Functional Theory (DFT) method based on the adsorption and desorption data. The sample was degassed at 90 °C for 6 h prior to the N2 physisorption experiment.2.5. Electrochemical measurement
Electrochemical performance of the M-rGO electrode was firstly measured using a three-electrode system, in which a platinum mesh was used as counter electrode while saturated calomel electrode (SCE) electrode was used as reference electrode. The working electrode was obtained by dropping 5 μL active material onto a glassy carbon electrode (GCE) with a diameter of 3 mm. The active material was prepared by mixing 4 mg M-rGO with 1.9 mL ethanol and 0.1 mL Nafion solution under ultrasonic treatment. The typical mass loading of the electroactive material is 0.01 mg.In a two-electrode configuration, the symmetric supercapacitors were assembled into a device composed of two glass plates, where two identical M-rGO electrodes were separated by a porous separator. The working electrode was prepared by mixing the active material (75%), carbon black (15%) and PTFE (10%) in ethanol. Then the mixture was pressed onto a nickel mesh (100 mm2). The M-rGO electrodes separated by a porous separator were steeped in 2.0 M KOH for 12 h before test. The total mass loading of the electroactive materials is 4 mg.
The gravimetric specific capacitance (C1, F g−1) measured in a three-electrode configuration was calculated using the eqn (1):
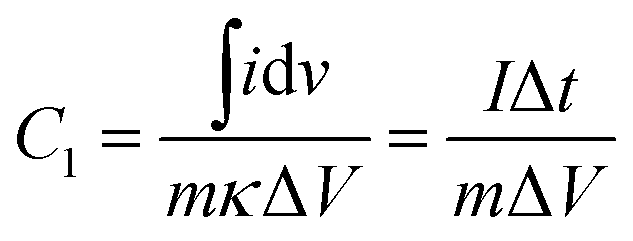 | (1) |
And the capacitance (C2, F g−1) measured in a symmetrical two-electrode configuration was calculated using the eqn (2):
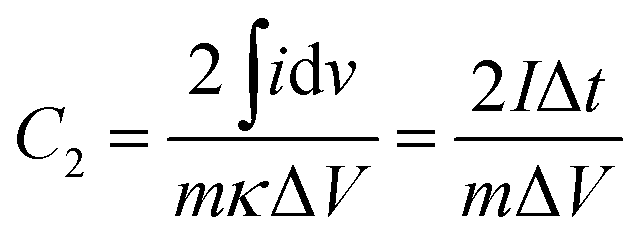 | (2) |
3. Results and discussion
3.1. The reaction between GO and Mg powder
The GO was obtained using a modified Hummer's method. It was then dispersed in deionized water with ultrasonic treatment before use. The Mg powder was added to reduce GO at 95 °C, and the product was then characterized with UV-vis, Raman, XRD, TGA, XPS, SEM, TEM and BET analyses.It has been reported that GO can be reduced by metals in two different mechanisms. One is the direct reduction of GO by metals, and the other is the reduction of GO via hydrogen spillover mechanism, in which hydrogen (produced by reaction of acid with metals) is the real reductant instead of metals.20 To produce sufficient hydrogen, excess acids need to be used. In this work, Mg powder itself can react with the hydrogen in water, thus the reduction of GO by Mg powder can be completed in the absence of acid. Since there is no need of large amounts of acids to produce sufficient hydrogen, the present method is beneficial to the environmental protection. The effect of Mg/GO ratio on the reduction was also investigated and the results are shown in Fig. S2.† The reaction rate slowed down as the Mg/GO ratio decreased, so more time was required to reach the same reduction degree when less Mg powder was applied. It is observed that the absorption red-shifted to 266 nm only, which suggest a moderate reduction of GO. Compared to other hydrogen spillover mechanism, the hydrogen spilled from water is limited thus resulted in a moderated reduction. Partly reduced rGO can introduce additional pseudo-capacitive effects without sacrificing its good electronic conductivity (compared with that of GO) which can be confirmed by the Raman results of M-rGO (Fig. 1(a)). The G-band at 1595 cm−1 is related to the E2g mode of the sp2 carbon atoms whereas the D band at 1350 cm−1 arises from the vibrations of the sp3 carbon atoms in the disordered graphene nanosheets. The intensity ratio of D and G corresponds to the electronic conjugation state of GO and also the degree of GO reduction. The D/G ratio increases after reaction, which indicates the formation of sp2 domains and the reduction of GO. In order to establish a cost effective method, Mg/GO ratio of 1/4 was chosen in the following discussion.
 | ||
| Fig. 1 (a) Raman spectra (b) XRD patterns and (c) TGA curves of GO and M-rGO. | ||
X-ray diffraction analysis was conducted to study the crystal structure of M-rGO. The XRD patterns of GO and M-rGO are shown in Fig. 1(b). It is obvious that the peak of GO at 10.7° (d-spacing is 0.78 nm) disappeared in M-rGO, which demonstrates the extensive reduction of GO. The large interlayer distance of GO should be attributed to the presentation of hydroxyl, epoxy and carboxyl groups which allows water molecules to penetrate into the layers thus increases the distances between the graphene layers. The peak of M-rGO at 24° (d-spacing is 0.35 nm) suggests that the interlayer distance decreased during the reduction process which can be ascribed to the removal of oxygen-containing functional groups and the reestablishment of the conjugated graphene network (sp2 carbon). However, the peaks shifts slightly lower than that of many other chemical reduced graphene,21–23 which can be resulting from both the existence of residual oxygen-containing functional groups on M-rGO sheets and also a larger distance between the graphene layers.24
TGA technique was used to analyze the thermal behavior of the prepared M-rGO and GO. The results shown in Fig. 1(c) indicates that there are two major mass losses in the TGA curve of GO, a mass loss at temperatures below 150 °C mainly ascribed to the evaporation of adsorbed water, and a mass loss around 200 °C, probably due to the decomposition of some oxygen-containing functional groups.25 In contrast, no sharp weight loss was observed for M-rGO at the same temperature range but a slow and lower-mass loss at the whole temperature range, demonstrating the removal of most oxygen-containing functional groups during the reduction.26
The reduction of GO was quantitatively proved by XPS. The percentages of different carbon-oxygen groups and the C/O ratio for GO and M-rGO are presented in Table 1. It is observed that the atomic ratio of C to O of M-rGO increases to 5.3 as compared to GO (2.9). This increase suggests the reduction of GO, which is consistent with the XRD and TGA results. This C/O ratio of M-rGO is comparable to those reduced by NaBH4 or urea,23,27,28 indicating a fairly good reduction of GO sheets. The C 1s spectra of M-rGO and GO are shown in Fig. 2 and S3,† respectively. The peak at 284.5 eV should be attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, other peaks at 286.2 eV and 288.0 eV belongs to C–O and CO, respectively. Compared to GO, the M-rGO shows a decrease in intensity at 286.2 eV and a simultaneous increase at 284.5 eV. These changes again indicate the removal of most oxygen-containing functional groups.
C, other peaks at 286.2 eV and 288.0 eV belongs to C–O and CO, respectively. Compared to GO, the M-rGO shows a decrease in intensity at 286.2 eV and a simultaneous increase at 284.5 eV. These changes again indicate the removal of most oxygen-containing functional groups.
| Sample | Analysis | |||
|---|---|---|---|---|
| CC |
C–O | CO |
C/O ratio | |
| GO | 52.07 | 42.69 | 5.24 | 2.3 |
| M-rGO | 66.25 | 22.83 | 7.46 | 5.3 |
 | ||
| Fig. 2 (a) XPS and (b) C 1s spectra of M-rGO. | ||
The morphology of the M-rGO was studied by TEM and SEM (Fig. 3). TEM images of the as-prepared M-rGO sample are shown in Fig. 3(a) and (b). The M-rGO presents partially overlapped morphology with a low contrast, indicating a thickness of few layers. The high resolution TEM (HRTEM) image also confirms the above conclusion. The M-rGO nanosheets restacks with each other to stabilize into thick layers as a result of their thermodynamic instability and yield micro or macro wrinkling and folding as showed in the SEM images in Fig. 3(c).29
 | ||
| Fig. 3 (a, b) Typical TEM and (c) SEM images of M-rGO. | ||
The N2 adsorption–desorption isotherms at 77 K and pore size distribution plot of the prepared M-rGO are shown in Fig. 4. The adsorption volume of M-rGO samples greatly increases when the relative pressure is below 0.04, which suggest the existence of micropores in M-rGO samples. And an apparent hysteresis loop in N2 adsorption/desorption isotherms at the relative pressure range of 0.4 to 0.9 indicates the existence of mesopores. The micropores, with pore sizes of 0.6–0.8 nm and 1.2–1.7 nm, mainly come from the defects in the graphene sheets, while the mesopores can be ascribed to the overlap, entanglement and incompact stacking of the graphene sheets.30,31 A pore volume analysis suggests that about 80% of the total pore volume is contributed by nanopores with a pore size below 5 nm (Fig. 4(b)). The corresponding pore size distribution of M-rGO shows the prominent mesopores at 3.8 nm, coupled with the presence of micropores. Previous work of Gryglewicz and his co-workers proves that mainly micropores play an essential part in charging an electrical double layer and the presence of mesopores is extremely helpful for ions transportation.32 Thus, the nanopores in M-rGO are beneficial for electrochemical performance of M-rGO electrode. The micropores presented in M-rGO will enhance the charging and discharge of an electrical double layer and the mesopores will facilitate the ions transportation.
 | ||
| Fig. 4 (a) N2 adsorption–desorption isotherms at 77.3 K and (b) pore width distribution of M-rGO. | ||
3.2. Electrochemical capacitance performance of M-rGO as electrode materials
The cyclic voltammetry (CV) curves of M-rGO electrode measured in 2 M KOH at different scan rates from 1 to 100 mV s−1 were displayed in Fig. 5(a). The CV curves of M-rGO are nearly rectangular with a pair of broadened faradic peaks (Faradic redox reactions happened in carbon materials with oxygen-containing functionalities such as the conversion between C–O/CO),33 evidently implying the presence of both EDLC and pseudocapacitance, which can be attributed to the surface ion adsorption and the remaining oxygen-containing functional groups in M-rGO, respectively.34 It is noted that the nearly rectangular shaped CV curves of M-rGO electrode can be maintained without big distortion even at a high scan rate of 500 mV s−1 (Fig. S4†), indicating a low resistance for ion transfer and also good charge transfer of ions between the electrolyte and the electrode material.35
 | ||
| Fig. 5 Capacitance performance of M-rGO measured in a three-electrode system. (a) CV of M-rGO electrode recorded between −1 and 0 V at different scan rates; (b) scan rate dependent specific capacitance of M-rGO electrode; (c) GV of M-rGO at different current densities; (d) current density dependent specific capacitance of M-rGO electrode; (e) Nyquist spectra of M-rGO. | ||
The specific capacitances based on the mass of the M-rGO are calculated and shown in Fig. 5(b). The M-rGO exhibits excellent electrochemical behavior in a wide range of scan rates from 1 to 1000 mV s−1. It should be noted that the specific capacitance of the M-rGO can still reach 324.9 F g−1 even at a scan rate of 1 V s−1. The specific capacitance of M-rGO electrode decreased with increase in the scan rates (Fig. 5(b)), which should be attributed to the fact that at high scan rates, the electrolyte ions cannot effectively reach the inner surfaces of the active material for the reduced diffusion time at higher scan rates.36 The maximum specific capacitance of 577.4 F g−1 was achieved from the M-rGO electrode at the scan rate of 1 mV s−1, which is much higher than most graphene-based material and is well comparable to the recently reported 3D FMG-based supercapacitors (three-dimensional functionalized multilayer graphene annealed at different temperatures).34,37,38 Such a high specific capacitance of M-rGO can be attributed to the synergistic effect of unique nano-porous structure, larger distance between adjacent graphene layers, suitable surface characteristic and also the improved wettability. The electrochemical performance of rGO prepared by hydrazine reduction is presented in Fig. S5.† The capacitance calculated from CV of rGO is 57.4 F g−1.
The galvanostatic charge/discharge (GV) curves of the prepared M-rGO electrode recorded at different current densities are shown in Fig. 5(c). The GV curves based on M-rGO electrode show a linear triangular shape with humps, which indicates the existence of both pseudocapacitance and electrical double layer-capacitance.39 However, longer discharge time (compared to the charging time) is observed in the GV curves. This phenomenon is also observed in previous papers.40–43 It may be attributed to the occurrence of additional electrode reactions, such as decomposition of the electrolyte in the discharging process. And this phenomenon can happen more often in electrode with pseudocapacitance.
A maximum capacitance of 367.7 F g−1 can be obtained at a current density of 0.5 A g−1, which is much higher than 321 F g−1 of 3D hollow porous graphene balls,44 and exceeds most graphene-based electrode with capacitance of 100-250 F g−1 in aqueous electrolyte. However, the capacitance of M-rGO electrode decreased at high current densities (Fig. 5(d)), which resulted from the poor accommodation of ions at the inner surface of active electrode material.
To further understand the capacitive behavior of the M-rGO electrodes, EIS test was conducted in the frequency range from 100 kHz to 0.1 Hz under open circuit voltage conditions with the amplitude of 5 mV. The result is showed in Fig. 5(e). The Nyquist plot of M-rGO showed excellent capacitive behavior, which can be concluded by the nearly vertical line at the low frequency ranges. The low semicircle diameter at the high frequency ranges suggests low electronic resistance of the M-rGO electrode which can be attributed to the presence of oxygen-containing functional groups. The oxygen-containing functional groups in M-rGO can improve the wettability of the electrode, facilitate the diffusion of ions and thus can lower the electronic resistance.
To fully reveal the pseudocapacitive effects of M-rGO and to find the most suitable aqueous electrolytes for M-rGO, more tests were carried in 1 M H2SO4. The CV curves of M-rGO electrode recorded in 1 M H2SO4 at different scan rates from 5 to 100 mV s−1 are presented in Fig. 6(a). It can be observed that each CV curves of M-rGO exhibit rectangular shape along with a pair of redox peaks (the typical conversion between quinone/hydroquinone states in carbon materials with oxygen-containing functional groups), resulting in the improved capacitance of M-rGO.45 The galvanostatic charge–discharge curves of M-rGO at different current densities are shown in Fig. 6(b). The specific capacitance values obtained in 1 M H2SO4 are a bit lower than that obtained in 2 M KOH (as shown in Fig. S6†). The decreased specific capacitance of M-rGO is probable arising from the different mechanism of electrode reaction. Thus 2 M KOH was chosen as the electrolyte in the following cyclic stability and two-electrode tests.
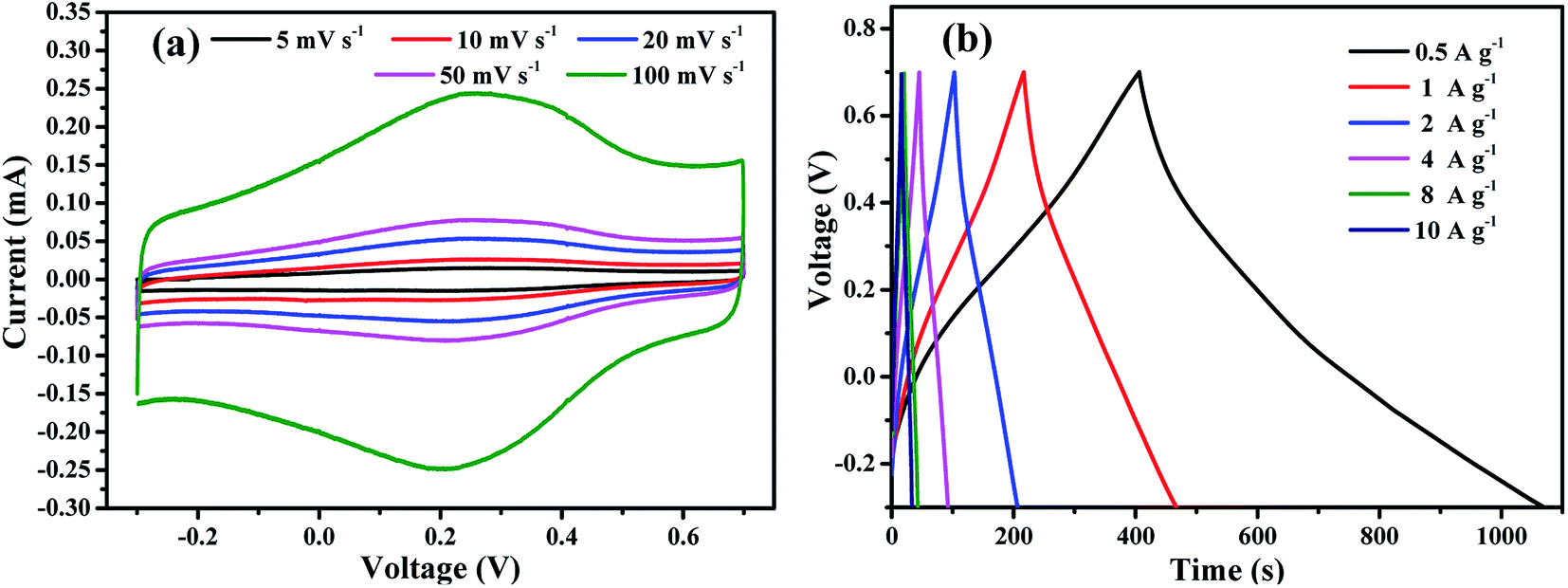 | ||
| Fig. 6 Capacitance performance of M-rGO measured in a three-electrode system in 1 M H2SO4. (a) CV trace of M-rGO electrode recorded between −0.3 and 0.7 V at different scan rates; (b) galvanostatic charge–discharge curves of M-rGO at different current densities. | ||
Fig. 7 shows the charge–discharge cyclic stability of the M-rGO at a constant current density of 8 A g−1 measured in 2 M KOH. After 150 charge–discharge cycles, the capacitance value of M-rGO remained stable. The capacitance retention after 3000 cycles was 93.3%, indicating that the M-rGO electrode is electrochemical stable. Capacitance loss with extended cycling is a common phenomenon in supercapacitors.46,47 The decreased specific capacitance can be ascribed to the structure damage in the electrode material. This high capacitance retention indicates that the M-rGO electrode can be very durable in practical application.
 | ||
| Fig. 7 Capacitance retention of the M-rGO electrode at a constant current density of 8 A g−1. | ||
To get more reliable capacitance value, further measurements were conducted in a two-electrode configuration with 2 mol L−1 KOH as the electrolyte. As shown in Fig. S7(a),† the CV curves of M-rGO electrode are nearly rectangular, and a maximum capacitance of 347.0 F g−1 was obtained at 5 mV s−1. Further measurements were carried out to study the rate capability and stability of M-rGO, the results were showed in Fig. S7(b) and (c).† The results suggested a much lower rate performance and stability than those measured in three-electrode configuration, which can be ascribed to the difference between the electrode preparation methods.
4. Conclusions
A simple and green method for the chemical reduction of GO using Mg powder as a reductant has been developed. The reduction of GO was achieved with only a small amount of Mg in the absence of acid. The M-rGO showed well-developed nano-porous structure and superior surface functionalities. As electrode materials in supercapacitors, M-rGO showed excellent electrochemical performance with a maximum specific capacitance of 577.4 F g−1 at the scan rate of 1 mV s−1. This high specific capacitance is attributed to its unique nano-porous structure and the residual oxygen-containing functional groups in M-rGO. Furthermore, M-rGO electrode demonstrates high durability. These results indicate potential application of M-rGO for high performance supercapacitors.Acknowledgements
This work was supported by the National Science Foundation of China (21276181, 51202158).References
- Y. W. Zhu, S. Murali, M. D. Stoller, K. J. Ganesh, W. W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz, M. Thommes, D. Su, E. A. Stach and R. S. Ruoff, Science, 2011, 332, 1537–1541 CrossRef CAS PubMed.
- F. Stoeckli and T. A. Centeno, Phys. Chem. Chem. Phys., 2012, 14, 11589–11591 RSC.
- M. X. Chen, C. C. Zhang, L. Z. Li, Y. Liu, X. C. Li, X. Y. Xu, F. L. Xia, W. Wang and J. P. Gao, ACS Appl. Mater. Interfaces, 2013, 5, 13333–13339 CAS.
- L. Zhao, Y. Qiu, J. Yu, X. Deng, C. Dai and X. Bai, Nanoscale, 2013, 5, 4902–4909 RSC.
- A. Lewandowski, M. Zajder, E. Frackowiak and F. Beguin, Electrochim. Acta, 2001, 46, 2777–2780 CrossRef CAS.
- Z. J. Li, B. C. Yang, K. Li, H. Y. Wang, X. W. Lv, Y. Z. Guo, Z. F. Zhang and Y. L. Su, RSC Adv., 2014, 4, 51640–51647 RSC.
- E. Frackowiak, K. Metenier, V. Bertagna and F. S. Beguin, Appl. Phys. Lett., 2000, 77, 2421–2423 CrossRef CAS.
- S. Park and R. S. Ruoff, Nat. Nanotechnol., 2010, 5, 217–224 CrossRef.
- Q. Wang, N. Plylahan, M. V. Shelke, R. R. Devarapalli, M. Li, P. Subramanian, T. Djenizian, R. Boukherroub and S. Szunerits, Carbon, 2014, 68, 175–184 CrossRef CAS.
- X. Zhang, H. Zhang, C. Li, K. Wang, X. Sun and Y. Ma, RSC Adv., 2014, 4, 45862–45884 RSC.
- S. Yoon, S. Lim, Y. Song, Y. Ota, W. Qiao, A. Tanaka and I. Mochida, Carbon, 2004, 42, 1723–1729 CrossRef CAS.
- H. J. Wang, X. X. Sun, Z. H. Liu and Z. B. Lei, Nanoscale, 2014, 6, 6577–6584 RSC.
- D. Lozano-Castelló, D. Cazorla-Amorós, A. Linares Solano, S. Shiraishi, H. Kurihara and A. Oya, Carbon, 2003, 41, 1765–1775 CrossRef.
- D. Y. Qu and H. Shi, J. Power Sources, 1998, 74, 99–107 CrossRef CAS.
- L. Zhang, X. Yang, F. Zhang, G. K. Long, T. F. Zhang, K. Leng, Y. W. Zhang, Y. Huang, Y. F. Ma, M. T. Zhang and Y. S. Chen, J. Am. Chem. Soc., 2013, 135, 5921–5929 CrossRef CAS PubMed.
- C. T. Hsieh and H. Teng, Carbon, 2002, 40, 667–674 CrossRef CAS.
- Y. Chen, X. O. Zhang, D. C. Zhang, P. Yu and Y. W. Ma, Carbon, 2011, 49, 573–580 CrossRef CAS.
- M. J. Bleda-Martínez, J. A. Maciá-Agulló, D. Lozano-Castelló, E. Morallon, D. Cazorla-Amorós and A. Linares-Solano, Carbon, 2005, 43, 2677–2684 CrossRef.
- G. R. Desiraju, Acc. Chem. Res., 2002, 35, 565–573 CrossRef CAS PubMed.
- B. K. Barman, P. Mahanandia and K. K. Nanda, RSC Adv., 2013, 3, 12621–12624 RSC.
- X. G. Mei, H. Q. Zheng and J. Y. Ouyang, J. Mater. Chem., 2012, 22, 9109–9116 RSC.
- H. Feng, R. Cheng, X. Zhao, X. Duan and J. Li, Nat. Commun., 2013, 4, 1539 CrossRef PubMed.
- Z. B. Lei, L. Lu and X. S. Zhao, Energy Environ. Sci., 2012, 5, 6391–6399 CAS.
- T. T. Li, N. Li, J. W. Liu, K. Cai, M. F. Foda, X. M. Lei and H. Y. Han, Nanoscale, 2015, 7, 659–669 RSC.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- X. Y. Xu, T. Wu, F. L. Xia, Y. Li, C. C. Zhang, L. Zhang, M. X. Chen, X. C. Li, L. Zhang, Y. Liu and J. P. Gao, J. Power Sources, 2014, 266, 282–290 CrossRef CAS.
- H. J. Shin, K. K. Kim, A. Benayad, S. M. Yoon, H. K. Park, I. S. Jung, M. H. Jin, H. K. Jeong, J. M. Kim, J. Y. Choi and Y. H. Lee, Adv. Funct. Mater., 2009, 19, 1987–1992 CrossRef CAS.
- S. Zhang, Y. Y. Shao, H. G. Liao, M. H. Engelhard, G. P. Yin and Y. H. Lin, ACS Nano, 2011, 5, 1785–1791 CrossRef CAS PubMed.
- R. J. Seresht, M. Jahanshahi, A. Rashidi and A. A. Ghoreyshi, Appl. Surf. Sci., 2013, 276, 672–681 CrossRef.
- Z. B. Lei, N. Christov and X. S. Zhao, Energy Environ. Sci., 2011, 4, 1866–1873 CAS.
- B. Xu, S. F. Yue, Z. Y. Sui, X. T. Zhang, S. S. Hou, G. P. Cao and Y. S. Yang, Energy Environ. Sci., 2011, 4, 2826–2830 CAS.
- G. Gryglewicz, J. Machnikowski, E. Lorenc-Grabowska, G. Lota and E. Frackowiak, Electrochim. Acta, 2005, 50, 1197–1206 CrossRef CAS.
- W. Q. Tian, Q. M. Gao, Y. L. Tan, Y. L. Zhang, J. D. Xu, Z. Y. Li, K. Yang, L. H. Zhu and Z. P. Liu, Carbon, 2015, 85, 351–362 CrossRef CAS.
- J. Yang and S. Gunasekaran, Carbon, 2013, 51, 36–44 CrossRef CAS.
- D. W. Wang, Y. G. Min, Y. H. Yu and B. Peng, Electrochim. Acta, 2014, 141, 271–278 CrossRef CAS.
- J. G. Wang, Y. Yang, Z. H. Huang and F. Y. Kang, J. Mater. Chem., 2012, 22, 16943–16949 RSC.
- Y. Fang, B. Luo, Y. Y. Jia, X. L. Li, B. Wang, Q. Song, F. Y. Kang and L. J. Zhi, Adv. Mater., 2012, 24, 6348–6355 CrossRef CAS PubMed.
- Z. Li, Z. W. Xu, H. L. Wang, J. Ding, B. Zahiri, C. M. B. Holt, X. H. Tan and D. Mitlin, Energy Environ. Sci., 2014, 7, 1708–1718 CAS.
- Q. Hao, X. Xia, W. Lei, W. Wang and J. Qiu, Carbon, 2015, 81, 552–563 CrossRef CAS.
- I. I. Misnon, N. K. M. Zain, R. Abd Aziz, B. Vidyadharan and R. Jose, Electrochim. Acta, 2015, 174, 78–86 CrossRef CAS.
- Rusi and S. R. Majid, Electrochim. Acta, 2015, 175, 193–201 CrossRef CAS.
- Y. Chen, X. O. Zhang, D. C. Zhang, P. Yu and Y. W. Ma, Carbon, 2011, 49, 573–580 CrossRef CAS.
- F. Barzegar, A. Bello, D. Momodu, M. J. Madito, J. Dangbegnon and N. Manyala, J. Power Sources, 2016, 309, 245–253 CrossRef CAS.
- X. J. He, H. B. Zhang, H. Zhang, X. J. Li, N. Xiao and J. S. Qiu, J. Mater. Chem. A, 2014, 2, 19633–19640 CAS.
- Y. Chen, X. Zhang, D. Zhang, P. Yu and Y. Ma, Carbon, 2011, 49, 573–580 CrossRef CAS.
- Q. Q. Zhou, Y. R. Li, L. Huang, C. Li and G. Q. Shi, J. Mater. Chem. A, 2014, 2, 17489–17494 CAS.
- Y. Xu, Z. Lin, X. Huang, Y. Liu, Y. Huang and X. Duan, ACS Nano, 2013, 7, 4042–4049 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra02997c |
| This journal is © The Royal Society of Chemistry 2016 |