DOI:
10.1039/C5RA22079C
(Paper)
RSC Adv., 2016,
6, 2361-2367
A rapid spectrofluorimetric method for the determination of malondialdehyde in human plasma after its derivatization with thiobarbituric acid and vortex assisted liquid–liquid microextraction
Received
21st October 2015
, Accepted 21st December 2015
First published on 23rd December 2015
Abstract
A rapid, simple and sensitive methodology has been developed for the determination of trace levels of malondialdehyde (MDA) in human plasma. The method is based on the derivatization of MDA using thiobarbituric acid (TBA) to form a highly fluorescent compound, “MDA-TBA” at acidic pH. The TBA-derivatized MDA formed was preconcentrated using vortex assisted liquid–liquid microextraction into 90 μL of n-heptanol as extractant solvent and measured spectro-fluorimetrically. Parameters such as nature of the extraction solvent, extraction time, sample volume, temperature, derivatization reaction time and pH of the sample solution were studied and optimized. The developed protocol was found to yield a linear calibration curve in the concentration range between 1.0–100.0 μg L−1 with a limit of detection of 0.32 μg L−1. The enrichment factor for the analyte was calculated to be 65. The repeatability of the method was satisfactory (RSD ≤ 1.6%). Total extraction time was less than 10 min. The developed method was successfully applied to the determination of MDA in a human plasma sample.
1. Introduction
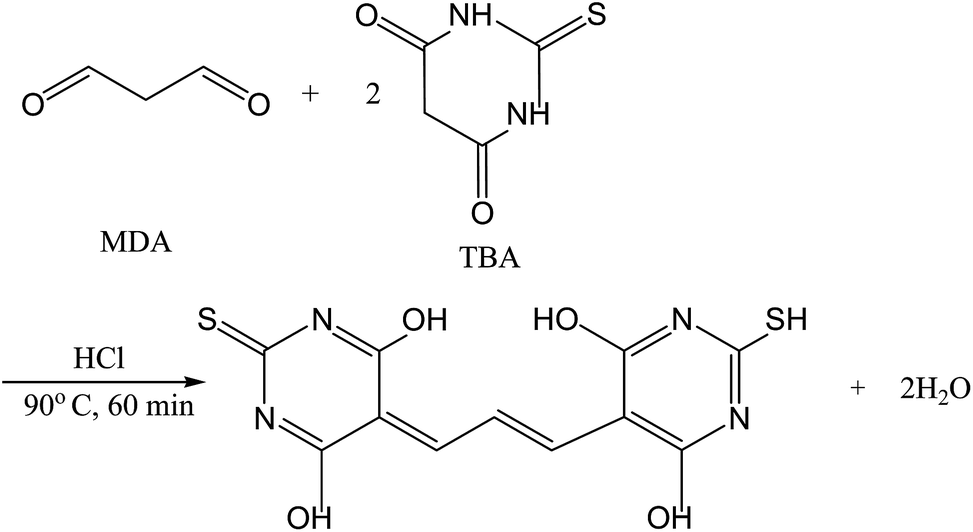
Oxidative stress instigated by reactive oxygen species damages DNA, lipids and proteins1,2 and plays a main role in the pathogenesis of a variety of myocardial illnesses.3 Following peroxidation of polyunsaturated fatty acids, lipid peroxides are improved by sequential reactions of oxidation, rearrangement and scission into more stable carbonyl compounds, containing malondialdehyde (MDA).4 Determination of this compound in human plasma is of great importance because of its role as a biomarker of lipid peroxidation in biological and medical sciences.5 MDA is widely used as an indirect indicator of various diseases including cancers,6 diabetes7,8 and cardiovascular,9 thus necessitating its accurate determination. The concentration of MDA is rather low and there are also many interfering factors in biological samples.10 Therefore a simple, sensitive, and selective method for determination of MDA is desired. The most common method of MDA analysis is based on its specific reaction with 2-thiobarbituric acid (TBA) in acidic media (Fig. 1)11,12 and measuring its absorbance by spectrophotometer13 or HPLC.14 Other derivatization reagents which has been used for this purpose are 2,4-dinitrophenylhydrazine,15 dansyl hydrazine,16,17 rhodamine B hydrazide,1 diamino-naphthalene18 and hydralazine.5
 |
| | Fig. 1 Reaction scheme of malondialdehyde (MDA) with thiobarbituric acid (TBA). | |
Due to the trace amount of MDA in body fluids, as sensitive tools, HPLC19,20 and GC21 are commonly used for its detection after derivatization. However, these instruments are relatively expensive and cannot be purchased by small laboratories, especially in less developed areas. They are also not too fast. Moreover, HPLC consumes highly priced pure organic solvents. There is also a method based on high performance capillary electrophoresis,22 which can directly determine MDA in plasma without derivatization but it is too expensive for routine use. Contrary to these methods, spectrofluorimetry represents an attractive, common technique adequate for solving many analytical problems due to its suitable precision and accuracy, associated with its lower cost compared to the above mentioned techniques. The critical point against the use of spectrofluorimeter for determination of MDA is generally associated with its low sensitivity and impossibility of direct determination without sample preparation. On the other hand, due to very complex matrix of human plasma, removal of interfering elements is characteristic for all developed methods of assay of MDA, other than chromatographic techniques. Beside elimination of interferences, preconcentration enhances the detection limits, increases the sensitivity and improves the accuracy of the results.3,5,21,23,24 Considering the advantages and disadvantages of the existing techniques the set objective is the development of a robust, simple, economical and green MDA determination method that can be used in any laboratory. In the present study, a new vortex assisted dispersive liquid–liquid microextraction (VADLLME) technique was investigated as a sample pre-treatment method for spectrofluorimetric analysis of MDA in human plasma. VADLLME which was introduced in 2010 by Yiantzi and co-workers25 is based on the formation of tiny droplets of extraction solvent and a mild emulsification procedure then restoring the floating extractant phase to its initial single-drop shape for the following instrumental analysis. Its simplicity and fastness are probably the most attractive benefits of this technique. In this technique, a 5- or 10 mL canonical centrifuge tube is used as an extraction, separation and preconcentration container. The mixture in the tube was then vigorously shaken using a vortex agitator then a cloudy solution was formed. Then, the organic solvent droplets were separated and floated on the surface of the aqueous solution by centrifugation. This method is simpler, rapid and has short equilibrium time compared with other dispersive method.26,27
To our best of knowledge, there is no literature available regarding combination of VADLLME and fluorescence sensing up to now. In our study, we established a modern, simple, fast, low cost, efficient and green method for preconcentration and separation of malondialdehyde from human plasma and its spectrofluorimetric determination with satisfactory sensitivity and detection limit. The effects of various experimental parameters on the extraction were investigated for this method and operating conditions were also optimized.
2. Experimental
2.1. Chemicals and reagents
All reagents were of analytical grade and used as received. Milli-Q® water (18.3 MΩ cm) was used throughout the experiment after filtering through 0.22 μm nylon membrane. Sodium chloride, acetic acid, trichloroacetic acid (TCA), hydrochloric acid, n-heptanol (HPLC grade) and thiobarbituric acid (TBA) were purchased from Merck KGaA (Darmstadt, Germany) and 1,1,3,3-tetramethoxypropane (99%) was obtained from Sigma-Aldrich (St. Louis, MO, USA).
2.2. Apparatus
A PerkinElmer spectrofluorimeter, Model LS 45 (Waltham, USA) was used for fluorescence measurements and recording the spectra. This instrument was equipped with two 10 μL microcells (Starna, UK; part number 16.10-F4/Q/10). A Denver (Germany) model UB-10 pH meter was used for pH measurements. Instrument excitation and emission slits both were 10 nm widths during measurements. The relative fluorescence intensity (RFI) was measured at wavelengths of 533 and 560 nm as excitation and emission of the MDA-TBA respectively.
2.3. Preparation of MDA and TBA standard solutions
1,1,3,3-Tetramethoxypropane (TMP) was used to make a malondialdehyde stock solution. A volume of 10 μL of TMP was accurately diluted in 10 mL of 0.1 M hydrochloric acid (HCl) and incubated in boiling water bath for 5 min and then rapidly cooled with tap water.26 This stock solution was stored at 4 °C and freshly prepared weekly. Working standard solutions were prepared by serial dilutions of this solution prior to analysis. A standard solution of MDA was prepared by pipetting 1 mL of the hydrolysed acetal into a 100 mL volumetric flask and diluted to volume with ultrapure water. The working solution was 5.25 × 10−5 mol L−1 acetal or 3.78 mg L−1 malondialdehyde. TBA solution (0.6% w/v, 42 mM) was prepared by dissolving 0.6 g of TBA in 4 M acetic acid.
2.4. Vortex assisted dispersive liquid–liquid microextraction (VADLLME) procedure
Before doing microextraction, derivatization of MA with TBA was performed under acidic conditions in a 10 mL test tube with screw cap. To the 4 mL of the sample solution containing MDA, 250 μL of 0.6% TBA, 700 μL of 5 M HCl and 0.04 g of NaCl were added and the mixture was incubated at 90 °C for 60 min. After cooling down to room temperature, VADLLME was performed by adding 90 μL of n-heptanol as extracting solvent to the sample. The mixture was then vigorously shaken on a Labnet vortex agitator for 1 min at max rate. As a result, a fine dispersed suspension solution was formed. After this step, the mixture was kept constant for 4 min to let dispersed fine droplets stick together. 16 μL of the pink upper organic phase was taken using a sampler and transferred into a micro-litter volume quartz cell. Then, the cell was placed in spectrofluorimeter to obtain its florescence against a blank solution which was prepared in the same way, containing no MDA.
3. Results and discussion
3.1. Fluorescence spectra of MDA-TBA
In order to find the relative fluorescence intensity for the MDA-TBA, the excitation and emission spectra of it was determined at the wavelength range of 450 to 650 nm against the reagent blank (Fig. 2). The results indicate that the maximum absorption and emission wavelengths are 533 and 560 nm, respectively. Accordingly, these wavelengths were selected as the wavelengths for further experiments. During all of the following experiments, the blank absorbance of all reagents was corrected.
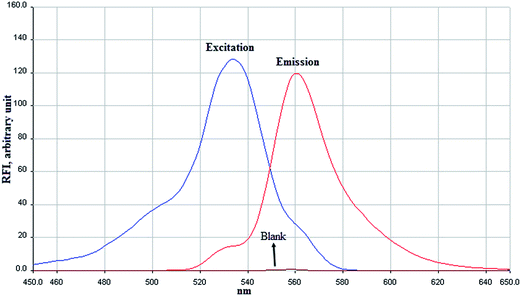 |
| | Fig. 2 Excitation and emission spectra of MDA after its derivatization with TBA and VADLLME extraction. Conditions are: MDA, 50 ng L−1; TBA, 2.63 mmol L−1; HCl concentration 0.87 mol L−1; sample volume, 4 mL; n-heptanol volume, 90 μL; vortex time, 1 min. The fluorescence background is indicated as blank. | |
3.2. MDA-TBA vortex assisted dispersive liquid–liquid microextraction optimization
To obtain the maximal extraction efficiency, important experimental parameters which can potentially influence the enrichment performance, such as kind and volume of extractant solvent; temperature, volume and pH of sample solution; effect of salt addition; have been investigated in detail for VADLLME method. The univariant method was used to simplify the optimization procedure. A series of experiments were designed for this goal as discussed below. Number of replicates of analysis was at least three for each experiment.
3.2.1. Selection of type and volume of the extracting solvent. The selection of an appropriate extraction solvent is a major challenge for the optimization of all DLLME processes.27 The criteria for selection of an adequate extraction solvent are: low solubility in water, transparency at the excitation and emission wavelengths of the analyte, formation of tiny droplets during vortex, low level of toxicity and of course high extraction capability for the target compound.28,29 To find the best solvent to extract the MDA-TBA derivative from the solution, a number of organic solvents including n-butanol, n-pentanol, n-heptanol, n-octanol, nitrobenzene, n-hexane, n-heptane, cyclohexane, methyl cyclohexane, toluene, methyl toluene, anisole and t-butyl acetate were examined. The choice of solvent was appraised for the extraction of 4 mL of a standard solution containing 50 μg L−1 of the analyte in deionized water. Experiments showed that only three of them, n-pentanol, n-heptanol and n-octanol can extract the compound of interest. That's probably because of the existence of hydrogen bonding between these solvents and S, N and OH groups of MDA-TBA adduct. Among them, n-heptanol was found to be the premier solvent.For selecting the best volume of the solvent, different volumes of n-heptanol were investigated in the range of 30–170 μL. The RFI reached maximum when the volume of the extracting solvent was 90 μL, and then decreased with the increase of n-heptanol volume. It was because before the optimum point, the amount of solvent was not enough to extract all of the analyte and after that, the target analyte was diluted with the increase of the solvent volume.29 On the basis of the results (shown in Fig. 3), solvent volume of 90 μL was chosen for the future experiments.
 |
| | Fig. 3 Effect of volume of solvent in VADLLME of MDA. Extraction condition: aqueous sample volume, 4 mL; TBA concentration 1.05 mmol L−1; HCl concentration 0.75 mol L−1; time and temperature, 80 °C for 60 min; vortex time, 15 s. | |
3.2.2. Effect of sample temperature and time of extraction. The reaction of MDA with TBA requires high temperatures.21,30 Therefore, the effects of the reaction time and temperature on RFI were also studied. A temperature range of 30–100 °C was examined to study the optimum derivatization temperature. The results showed that RFIs of MDA-TBA adduct increases with temperature increase up to 80 °C and then because of the completion of the reaction remains almost constant (Fig. 4). Since the highest RFI was observed at 90 °C, the derivatization temperature of subsequent experiments was set at this point.
 |
| | Fig. 4 Effect of temperature on fluorescence intensity. Extraction condition: aqueous sample volume, 4 mL; TBA concentration 1.05 mmol L−1; HCl concentration 0.75 mol L−1; time, 60 min; volume of extraction solvent, 90 μL; vortex time, 15 s. | |
The effect of time on the derivatization was also examined in the range of 10–80 min at 90 °C. The obtained results showed that after 60 min, reaction reaches to its equilibrium state with no further change in the RFI. Results are illustrated in Fig. 5.
 |
| | Fig. 5 Effect of residence time in 90 °C on fluorescence intensity. Extraction condition: aqueous sample volume, 4 mL; TBA concentration 1.05 mmol L−1; HCl concentration 0.75 mol L−1; temperature, 90 °C; volume of extraction solvent, 90 μL; vortex time, 15 s. | |
3.2.3. Effect of hydrochloric acid concentration. The MDA-TBA adduct is stable at acidic pHs but dissociates slowly at neutral or alkaline media.31 Therefore, the influence of hydrochloric acid concentration over the range of 0.125–1.125 mol L−1 on the MDA-TBA derivatization was studied. The results which are shown in Fig. 6 indicate that the RFIs of MDA-TBA increases with increasing the acid concentration in solution up to 0.75 mol L−1 and then becomes almost constant due to the completion of the reaction. To be sure of having the highest analytical signal, 0.88 mol L−1 concentration of HCl was considered as the suitable extraction acidity.
 |
| | Fig. 6 Influence of HCl concentration on on fluorescence intensity. Extraction condition: aqueous sample volume, 4 mL; TBA concentration 2.63 mmol L−1; time and temperature, 90 °C for 60 min; volume of extraction solvent, 90 μL; vortex time, 15 s. | |
3.2.4. Effect of the derivatizing reagent concentration. The amount of the derivatizing reagent is an important chemical variable that affect the efficiency of extraction. By adding appropriate volumes of MDA stock solution, TBA concentration in the range of 0.53–4.20 mM was generated in situ within the solution. As can be seen in Fig. 7, concentration of 2.63 mM gives the highest intensity. Hence, 2.63 mM was chosen as optimum concentration of TBA. Addition of MDA solution more than this amount, dilutes the solution and a decrease in the signal observes.
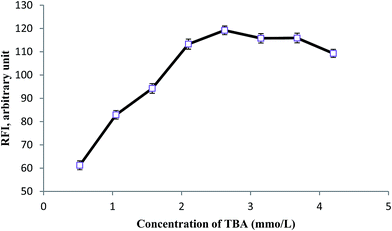 |
| | Fig. 7 Effect of TBA concentration on VADLLME of MDA. Extraction condition: aqueous sample volume, 4 mL; HCl concentration 0.75 mol L−1; time and temperature, 90 °C for 60 min; volume of extraction solvent, 90 μL; vortex time, 15 s. | |
3.2.5. Influence of vortex time. Without dispersive solvents, vortex turbulence is a simple and short way to increase the extraction efficiency. The vortex mixer leads to revolution of fluid and facilitates the dispersion of the extraction solvent into the aqueous solution because the fine droplets could extract analytes to equilibrium more rapidly.25,32,33 The time of vortex, as an important factor, was varied in the range of 10–80 s, whereas the vortex agitator was in the highest speed. According to the results obtained, 60 s were applied in subsequent experiments. At low agitation times, n-heptanol cannot be disperse well in the aqueous solution, which resulted in low extraction efficiency. Meanwhile, at times more than 60 s, droplets of the extracting solvent became so tiny that they cannot be re-collected to each other very fast.
3.2.6. Ionic strength influence. The salting-out effect is widely applied in traditional liquid–liquid extraction because it makes the solubility of target analytes in the aqueous phase decrease, thus more analytes enter into the extracting phase.34 Here, the effect of salt on the extraction efficiency is studied by adding different amounts of sodium chloride ranging from 0 to 0.07 g mL−1 of the sample solution to investigate the effect of ionic strength on the extraction efficiency. It was found that the extraction efficiency was increased by adding NaCl at the concentration range of 0–0.01 g mL−1. At higher NaCl concentrations, the extraction efficiency was decreased. The reason for the decrease of RFI signal may be due to the increase of the aqueous solution viscosity which prevents effective agitation. Thus, further extractions were carried out at a NaCl concentration of 0.01 g mL−1.
3.2.7. Effect of sample volume. The effect of sample volume on the extraction efficiency was evaluated by increasing of the sample volume from 50 to 2000 μL in a fixed 4 mL solution while the MDA content was kept constant (50 μg mL−1). Maximum peak responses were obtained when 100 μL of sample solution was under extraction and the responses decreased as its volume increased. With further increasing of the aqueous phase volume, there is a process that sample solution may dissolve n-heptanol droplets, which has adverse influence on extraction efficiency and consequently causes signal loss. Therefore, the aqueous phase sample volume of subsequent experiments was set at 100 μL.
3.3. Linear range, limit of detection and precision
Analytical figures of merit for the proposed method were obtained under optimal conditions (i.e. using 90 μL of n-heptanol as extractant solvent with a derivatization temperature of 90 °C for 60 min at concentration of 2.625 mmol L−1, 0.875 mmol L−1 and 1 × 10−2 mg L−1 for TBA, HCl and NaCl respectively. Vortex was performed for 60 s) and results are summarized in Table 1. Detection limits were obtained based on a signal-to-noise ratio of 3. The repeatability of the method, expressed as relative standard deviation (RSD), was calculated for eight replicates of the standard at 60 μg L−1 concentration of the MDA. An enrichment factor (EF) of 65 was obtained which was calculated as the ratio between the analyte concentration in the n-heptanol phase (Corg) and the initial concentration of analyte (C0) within the sample (eqn (1)).35 Triplicate measurements were performed.
Table 1 Analytical figure of merit for VADLLME extraction of MDA-TBAc
| Parameter |
Analytical feature |
| LOD, was based on 3σ criterion for 10 blank measurements. RSD, relative standard deviation, for eight replicate measurements of 60 μg L−1 MDA. F, fluorescence intensity (arbitrary units) CMDA, concentration of MDA (μg L−1). |
| Equation of calibration curve |
F = 2.49CMDA + 22.14 |
| Dynamic range (μg L−1) |
1.0–100.0 |
| R2 (determination coefficient) |
0.986 |
| Repeatabilityb (RSD%, n = 8) |
1.60 |
| Limit of detectiona (μg L−1) |
0.32 |
| Enrichment factor (fold) |
65 |
| Total extraction time (min) |
≤10 |
3.4. Analysis of real sample
To evaluate performance of the proposed method, microextraction and determination of TBA in a plasma sample was carried out under the optimal conditions. In a 1.5 mL polypropylene tube, a 250 μL aliquot of plasma sample was transferred and 100 μL of NaOH 6 mol L−1 was added. This solution was mixed thoroughly and incubated for 30 min at 60 °C in a water bath. 200 μL of 20% trichloroacetic acid was added to this solution in order to precipitate its proteins and then centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min. 100 μL of supernatant was taken and 250 μL of 0.6% TBA and 700 μL of 5 mol L−1 HCl were added to it successively. After that, 0.04 g NaCl was added and solution was diluted with 2950 μL of ultrapure water. After mixing, this solution was heated at 90 °C for 60 min in a water bath. After cooling, the solution was analyzed both by HPLC and by the suggested VADLLME method which showed that it is free from the analyte. To assess matrix effects, blood plasma samples were spiked with two levels of MDA concentration (20 and 40 μg L−1) and analyzed with the suggested method. Good relative recoveries of 95.5% and 96.8% were obtained for 20 and 40 μg L−1 of added MDA, respectively, which indicate that the matrix effect is negligible. In order to investigate precision, six replicate analysis were performed for each level of spiking. Excellent relative standard deviation of 1.45% and 1.51% were respectively attained for 20 and 40 μg L−1 of added MDA.
000 rpm for 10 min. 100 μL of supernatant was taken and 250 μL of 0.6% TBA and 700 μL of 5 mol L−1 HCl were added to it successively. After that, 0.04 g NaCl was added and solution was diluted with 2950 μL of ultrapure water. After mixing, this solution was heated at 90 °C for 60 min in a water bath. After cooling, the solution was analyzed both by HPLC and by the suggested VADLLME method which showed that it is free from the analyte. To assess matrix effects, blood plasma samples were spiked with two levels of MDA concentration (20 and 40 μg L−1) and analyzed with the suggested method. Good relative recoveries of 95.5% and 96.8% were obtained for 20 and 40 μg L−1 of added MDA, respectively, which indicate that the matrix effect is negligible. In order to investigate precision, six replicate analysis were performed for each level of spiking. Excellent relative standard deviation of 1.45% and 1.51% were respectively attained for 20 and 40 μg L−1 of added MDA.
3.5. Comparison of the suggested method with other techniques
Table 2 compares the characteristic data of the present method with those using different preconcentration techniques for MDA determination, reported in the literature recently. The limit of detection and precision reported by the current method is in the same order of magnitude of the other methods, while the presented method has an advantage of a wider dynamic range. In addition, simplicity of operation, low cost, high enrichment factor, rapidity and low sample volume are some other advantages of the proposed methods. No special devices for extraction are needed as for SPE, and the total analysis time is less than 10 minutes. Spectrofluorimetric instrumentation also own its merits of simplicity, cheapness, portability and so on.
Table 2 Comparison of the proposed method with other methods for the determination of MDA
| Extraction technique |
Detection technique |
Linear range (μg L−1) |
LOD (μg L−1) |
RSD (%) |
Ref. |
| Headspace solid phase microextraction. Liquid–liquid microextraction. Not mentioned. Solid phase extraction. |
| HS-SPMEa |
GC/MS |
5–100 |
0.4 |
<8% |
39 |
| LLMEb |
Spectrophotometry |
NMc |
4.54 |
<0.8 |
24 |
| HS-SPME |
GC |
NM |
0.742 |
— |
38 |
| SPEd |
HPLC |
0 to 720.6 |
— |
4.8 |
36 |
| SPE |
LC-MS/MS |
0.72–72.06 |
0.115 |
<10 |
40 |
| — |
Fluorimetry |
NM |
1.08 |
<3.8 |
43 |
| SPE |
HPLC |
10–5000 |
10 |
9 |
41 |
| VADLLME |
Spectrofluorimetry |
1–100 |
0.325 |
<1.6 |
This work |
4. Conclusions
Until now there are no reports of using liquid–liquid microextraction coupled with spectrofluorimetry in the literature. Moreover, only a few reports21,24,36–42 can be find in which an extraction technique was used for preconcentration of MDA before its determination. In this study, the combination of vortex assisted liquid–liquid microextraction with micro-volume spectrofluorimetry has been performed for the determination of MDA in human serum sample after a derivatization reaction. The proposed method has enough simplicity and sensitivity to be employed for routine analysis of serum samples in any lab. Additional advantages of the developed method are low instrumental costs and easy operation. No centrifugation step was required during extraction and the whole analysis time of extraction was less than 10 minutes which means many samples can be analysed in a clinical laboratory in each day. Moreover, there is no need for spending time to obtain chromatograms or wasting precious HPLC solvents.
References
- P. Li, G. Ding, Y. Deng, D. Punyapitak, D. Li and Y. Cao, Free Radical Biol. Med., 2013, 65, 224–231 CrossRef CAS PubMed.
- S. Mao, A. Zhang and S. Huang, Renal Failure, 2014, 36, 994–999 CrossRef PubMed.
- G. A. Cordis, N. Maulik and D. K. Das, J. Mol. Cell. Cardiol., 1995, 27, 1645–1653 CrossRef CAS PubMed.
- O. Korchazhkina, C. Exley and S. Andrew Spencer, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2003, 794, 353–362 CrossRef CAS.
- Z. Rezaei, A. Jamshidzadeh and E. Sanati, Anal. Methods, 2013, 5, 2995–2999 RSC.
- E. Bakan, S. Taysi, M. F. Polat, S. Dalga, Z. Umudum, N. Bakan and M. Gumus, Jpn. J. Clin. Oncol., 2002, 32, 162–166 CrossRef PubMed.
- N. Dierckx, G. Horvath, C. van Gils, J. Vertommen, J. van de Vliet, I. de Leeuw and B. Manuel-y-Keenoy, Eur. J. Clin. Nutr., 0000, 57, 999–1008 CrossRef PubMed.
- G. Loguercio and A. Federico, Free Radical Biol. Med., 2003, 34, 1–10 CrossRef PubMed.
- M. C. Polidori, K. Savino, G. Alunni, M. Freddio, U. Senin, H. Sies, W. Stahl and P. Mecocci, Free Radical Biol. Med., 2002, 32, 148–152 CrossRef CAS PubMed.
- D. Del Rio, A. J. Stewart and N. Pellegrini, Nutr., Metab. Cardiovasc. Dis., 2005, 15, 316–328 CrossRef PubMed.
- D. R. Janero, Free Radical Biol. Med., 1990, 9, 515–540 CrossRef CAS PubMed.
- M. Wada, M. Nagano, H. Kido, R. Ikeda, N. Kuroda and K. Nakashima, J. Oleo Sci., 2011, 60, 579–584 CrossRef CAS PubMed.
- J. Lovrić, M. Mesić, M. Macan, M. Koprivanac, M. Kelava and V. Bradamante, Period. Biol., 2008, 110, 63–68 Search PubMed.
- A. de las Heras, A. Schoch, M. Gibis and A. Fischer, Eur. Food Res. Technol., 2003, 217, 180–184 CrossRef CAS.
- M. Armenteros, M. Heinonen, V. Ollilainen, F. Toldrá and M. Estévez, Meat Sci., 2009, 83, 104–112 CrossRef CAS.
- H. L. Lord, J. Rosenfeld, V. Volovich, D. Kumbhare and B. Parkinson, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2009, 877, 1292–1298 CrossRef CAS PubMed.
- J. M. Anderson, Anal. Biochem., 1986, 152, 146–153 CrossRef CAS PubMed.
- J.-P. Steghens, A. L. van Kappel, I. Denis and C. Collombel, Free Radical Biol. Med., 2001, 31, 242–249 CrossRef CAS PubMed.
- Z. Wei, X. Li, D. Thushara and Y. Liu, J. Food Eng., 2011, 107, 379–384 CrossRef CAS.
- F. Karatas, M. Karatepe and A. Baysar, Anal. Biochem., 2002, 311, 76–79 CrossRef CAS PubMed.
- E. M. Gioti, Y. C. Fiamegos, D. C. Skalkos and C. D. Stalikas, J. Chromatogr. A, 2007, 1152, 150–155 CrossRef CAS PubMed.
- D. W. Wilson, H. N. Metz, L. M. Graver and P. S. Rao, Clin. Chem., 1997, 43, 1982–1984 CAS.
- C. Fucile, V. Marini, M.
L. Zuccoli, S. Leone, L. Robbiano, A. Martelli and F. Mattioli, Clin. Lab., 2013, 59, 837–841 CAS.
- A. H. S. Muñoz, M. P. Puga, K. Wrobel, M. E. G. Sevilla and K. Wrobel, Microchim. Acta, 2004, 148, 285–291 CrossRef.
- E. Yiantzi, E. Psillakis, K. Tyrovola and N. Kalogerakis, Talanta, 2010, 80, 2057–2062 CrossRef CAS PubMed.
- W.-Y. Chang, C.-Y. Wang, J.-L. Jan, Y.-S. Lo and C.-H. Wu, J. Chromatogr. A, 2012, 1248, 41–47 CrossRef CAS PubMed.
- G. Leng, G. Lui, Y. Chen, H. Yin and D. Dan, J. Sep. Sci., 2012, 35, 2796–2804 CrossRef CAS PubMed.
- J. Tsaknis, S. Lalas, V. Tychopoulos, M. Hole and G. Smith, Analyst, 1998, 123, 325–327 RSC.
- M. Kaykhaii and E. Ghasemi, Anal. Methods, 2013, 5, 5260–5266 RSC.
- T. Sakai, A. Habiro and S. Kawahara, J. Chromatogr. B: Biomed. Sci. Appl., 1999, 726, 313–316 CrossRef CAS.
- S. Wong, J. Knight, S. Hopfer, O. Zaharia, C. N. Leach and F. Sunderman, Clin. Chem., 1987, 33, 214–220 CAS.
- K. Seebunrueng, Y. Santaladchaiyakit and S. Srijaranai, Talanta, 2015, 132, 769–774 CrossRef CAS PubMed.
- A. Gure, F. J. Lara, A. M. García-Campaña, N. Megersa and M. del Olmo-Iruela, Food Chem., 2015, 170, 348–353 CrossRef CAS PubMed.
- M. Kaykhaii and M. H. Ghalehno, Anal. Methods, 2013, 5, 5622–5626 RSC.
- M. Kaykhaii and M. Sargazi, Spectrochim. Acta, Part A, 2014, 121, 173–179 CrossRef CAS PubMed.
- J. Suttnar, J. Čermák and J. E. Dyr, Anal. Biochem., 1997, 249, 20–23 CrossRef CAS PubMed.
- A.-M. Hoberg, M. Otteneder, L. J. Marnett and H. E. Poulsen, J. Mass Spectrom., 2004, 39, 38–42 CrossRef CAS PubMed.
- K. Fujioka and T. Shibamoto, J. Agric. Food Chem., 2005, 53, 4708–4713 CrossRef CAS PubMed.
- H.-S. Shin, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2009, 877, 3707–3711 CrossRef CAS PubMed.
- J.-L. Chen, Y.-J. Huang, C.-H. Pan, C.-W. Hu and M.-R. Chao, Free Radical Biol. Med., 2011, 51, 1823–1829 CrossRef CAS PubMed.
- R. Bakalova, M. Mileva, C. Kotsev, V. Bardarov and S. Ribarov, Methods Finds Exp. Clin. Pharmacol., 2000, 22, 267–269 CrossRef CAS.
- S. Marcinčák, J. Sokol, P. Turek, P. Popelka and J. Nagy, Chem. Listy, 2006, 100, 528–532 Search PubMed.
- D. Del Rio, N. Pellegrini, B. Colombi, M. Bianchi, M. Serafini, F. Torta, M. Tegoni, M. Musci and F. Brighenti, Clin. Chem., 2003, 49, 690–692 CAS.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.