DOI:
10.1039/C4RA04832F
(Paper)
RSC Adv., 2014,
4, 33826-33839
Preparation and characterization of zinc oxide and nanoclay reinforced crosslinked starch/jute green nanocomposites
Received
22nd May 2014
, Accepted 23rd July 2014
First published on 23rd July 2014
Abstract
In the present work, ZnO nanoparticles (ZNP) and ZNP in combination with nanoclays are reported as reinforcing agents for the preparation of ‘Green’ nanocomposites based on glutaraldehyde (GA) crosslinked starch/jute fabric. A solution-induced intercalation method has been used for the successful fabrication of the nanocomposites. Both ZNP and nanoclay are successfully incorporated into the composite as revealed by X-ray diffractometry (XRD), transmission electron microscopy (TEM), scanning electron microscopy (SEM), and Fourier-transform infrared spectroscopy (FT-IR). The thermal and mechanical properties of the nanocomposites are studied using thermogravimetric analysis (TGA) and mechanical tests, respectively. The study reveals significant changes in the observed properties of the synthesized composites with the amount of nanofillers. Interestingly, the flame retarding properties, UV-resistance, and dimensional stability of the nanoparticle filled composites are found to be much better than those of the unfilled one. The study reveals a strong interfacial interaction between the filler and the matrix within the synthesized green nanocomposites.
1. Introduction
The current interest in the conservation of natural resources and recycling has encouraged researchers in different countries to focus more on renewable raw materials. Due to the increasing global awareness, environmental legislation has resulted in restriction on the use of traditional composites made of glass, carbon, aramid fibres, etc. Therefore, the interest for greener and more sustainable technologies has focused attention in bio-based products and their development to reduce the dependence on fossil fuel and move to a sustainable materials basis.1–3 One of such potential material is the biopolymers, which exist abundantly worldwide. Biopolymers provide several fundamental and practical advantages such as they are organic in nature and a host for other useful materials and chemicals, particularly for the production of “greener” materials.4
Among the various biodegradable polymers, starch is one of the most exciting and promising raw material for the production of biodegradable products. Starch is produced by many plants as a source of storage energy. In global scenario, it is also the second most abundant biomass material.5 Starch mainly comprises of two key polysaccharides, amylose and amylopectin along with some minor components such as lipids and proteins. Amylose is defined as a linear polymer of (1 → 4)-linked α-D-glucopyranosyl units with minor amount of (1 → 6)-α-branches. Amylopectin is a highly branched molecule composed of chains of α-D-glucopyranosyl residues linked together mainly by (1 → 4)-linkages along with (1 → 6)-linkages at the branched points.6 Starch based films are widely used in food packaging, agricultural mulching industries, foamed materials for loose-fill packing and most importantly in health sciences.7–9 Besides having various advantages, starch based materials exhibit certain disadvantages too such as high hydrophilicity and low moduli of elasticity which reduces its industrial applications.10 Starch plastics possess good biodegradability but their application is limited due to their poor flexibility. However, plasticizer like glycerol has been used to improve the flexibility of starch but is reported to produce lower mechanical and high water sensitivity.11 Glutaraldehyde (GA) can be used as a crosslinking agent, thereby improving its thermo mechanical properties and also reducing its hydrophilicity.12 Moreover, biofibres evolved as one of the potential materials in improving the mechanical strength of such material. The noteworthy advantage of biofibres over their conventional counterparts include comparatively low cost and weight, high specific modulus, easy availability, comparative safety and less abrasive with respect to processing tools. The applications of biofibres in furniture, packing materials, construction, and automobiles sectors are now highly demanding.13 Among different types of biofibres, jute fibre can also be used as a reinforcing agent for the synthesis of polymer composites. Jute, which consists of cellulose and lignin, is an extensively used material in textile and other such industries. The most attractive features of jute fibres are that they are inexpensive, widely available, and most importantly they can be modified to a cost-effective and environmentally benign green products with improved physical properties.14
The combination of starch polymer and jute fibres crosslinked with GA lead to biopolymer based composites whose properties improved by dispersing the nanofillers into their matrix. Among the various nanoparticles predecessors, nanoclay is most widely used for the synthesis of polymer nanocomposites. The addition of nanoclay to the composite enhances the mechanical, thermal and other requisite properties.15–17 As the crosslinked starch/jute (S/J) composite are used in different indoor and outdoor applications, it is very important to study and modulate the ultraviolet (UV) resistant property of the composites. Clay nanoparticles are widely used to shield UV irradiation of polymer composite.
In recent years, most of the researchers have examined the role of ZNPs in exterior coatings to improve photostability,18 as a component of UV coatings for nanocomposites.19 Further ZNPs also enhances the mechanical, thermal, as well as other relevant properties of polymer nanocomposites.20,21 Keeping in mind the advantages of biopolymers, nanofillers and most importantly the increasing demand for greener technologies, herein we demonstrated the preparation and characterization of biopolymer based nanocomposites consisting of starch, jute, glutaraldehyde, ZNP, and nanoclay with high thermo-mechanical and UV-resistance property.
2. Experimental
2.1. Materials
Jute fabrics (0.450 g cm−2) have been purchased from local market, Tezpur, Assam, India. Starch soluble extra pure [(bulk density 300 kg m−3), pH value 6.0–7.5 (20 g L−1, H2O, 25 °C)], Glutaraldehyde (GA) 25% (w/v), benzene (purity > 99%), glycerol (AR grade) and NaOH pellets (purity > 97%) are obtained from Merck Private Limited (Mumbai, India). ZnO nanopowder (<100 nm) and Montmorillonite K-10 clay (purity 99.99%) are obtained from Sigma Aldrich (USA). All these chemicals received as such are used without any further treatment.
3. Methods
3.1. Surface modification of jute
Jute fabrics (J) are first treated with 2% liquid detergent (Rankleen, M/S Rankem, India. pH: 6.5–7.5) at 70 °C for 1 h, washed with distilled water and finally dried in a vacuum oven at 70 °C till attainment of constant weight. The washed fabrics are dewaxed by treatment with a mixture of alcohol and benzene (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) for 72 h at 50 °C. It is washed with distilled water and dried till attainment of constant weight. The fabrics are then treated with 5% (w/v) NaOH solution for 30 min at 30 °C and washed with distilled water for several times to leach out the absorbed alkali. The fabrics are finally kept immersed in distilled water for overnight and are washed repeatedly to avoid the presence of any trace amount of alkali. The alkali treated fabrics are dried in a vacuum oven at 50 °C to obtain constant weight and stored at ambient temperature in a desiccator.
:2) for 72 h at 50 °C. It is washed with distilled water and dried till attainment of constant weight. The fabrics are then treated with 5% (w/v) NaOH solution for 30 min at 30 °C and washed with distilled water for several times to leach out the absorbed alkali. The fabrics are finally kept immersed in distilled water for overnight and are washed repeatedly to avoid the presence of any trace amount of alkali. The alkali treated fabrics are dried in a vacuum oven at 50 °C to obtain constant weight and stored at ambient temperature in a desiccator.
3.2. Preparation of the slurry
Starch powders are first mixed thoroughly with deionized water at 1:10 ratio (by w/w) at room temperature for 1 h to obtain a suitable resin for fabrication of composites. In absence of plasticizer starch is found to be very brittle, weak and difficult to process into suitable films. Therefore, 5% glycerol (w/w of dry starch) is added as a plasticizer. The slurry containing starch and glycerol (5% w/w of dry starch) is transferred to a 500 mL round bottom flask and stirred with a mechanical stirrer maintaining the temperature in the range 55–60 °C for 4 h and subsequently cooled to 35 °C. The resultant mixture is further stirred for another 5 min at that temperature with subsequent addition of GA. This stage is referred to as pre-cured resin. In order to prepare ZNP filled resin, desired amount of ZNP {1–5% (w/w of dry starch)} is first taken in a beaker containing water, stirred for 8 h using mechanical stirrer followed by sonication for 30 min under 0.5 cycle and a wave amplitude of 140 μm using a sonicator (Heilscher UP200S, Germany) and finally added to the slurry containing starch and glycerol. Similarly, for the ZNP and nanoclay filled resin, desired amount of ZNP (5% w/w of dry starch) and nanoclay (1–5% w/w of dry starch) is first taken in a beaker containing water. Then the mixture is stirred for 8 h using a mechanical stirrer and sonicated for 30 min. The mixture is then poured in the pre-cured resin system. After adding to the pre-cured resin system, the whole resin mixture is stirred for another 1 h with the help of high speed mechanical stirrer followed by sonication for 30 min so that the ZNP and nanoclay particles are well dispersed inside the resin matrix.
3.3. Impregnation of jute fabrics & fabrication of composites
The prepared pre-cured resin is first poured in a bowl and then surface modified jute fabric is dipped into the pre-cured resin slurry. The resultant mixture is kept for 24 h at 25 °C. The impregnated jute fabrics are placed in a Teflon coated glass plate and dried over a heating plate at 30–40 °C for 12 h. The weight of jute fabrics before impregnation is noted as W1.
Composite laminates are prepared by the method of film stacking. The layers of impregnated jute fabric are placed on one another on 15 levels in a metal mould of thickness 3 mm. The laminates are then compression moulded in a compression moulding press (Santec, New Delhi) at 80 °C under a pressure of 100 MPa. The laminates are 3 mm in thickness. The final weight of the composite is noted as W2. The calculations are made as per the following equations:
| | |
Percentage of jute in the composite = (W1/W2) × 100
| (1) |
| | |
Percentage of resin in the composite = {(W2 − W1)/W2} × 100
| (2) |
where,
W1 = weight of jute fabrics before impregnation,
W2 = weight of the final composite.
3.4. Bacterial media
The preparation of mineral salt medium for bacterial growth is carried out according to the following composition: 4.75 g of KH2PO4, 2.0 g of (NH4)2SO4, 2.0 g of Na2HPO4, 1.2 g of MgSO4·7H2O, 100 mg of CuSO4·7H2O, 100 mg of MnSO4·5H2O, 70 mg of ZnSO4·7H2O, 10 mg of MoO3, 10 mg of H3BO3·5H2O, 1 mg of FeSO4·7H2O, and 0.5 mg of CaCl2·2H2O dissolved in 1000 mL of demineralized water. 3 mL of this liquid culture medium is then transferred into a 50 mL conical test tube and it is sterilized by autoclave at 121 °C and 15 lbf pressure for 15 min. The autoclaved media are then allowed to cool to room temperature and composite samples are added into the media under sterile condition inside a laminar air flow hood. Media which contained S/J/GA50 composites were cultured as negative control. The S/J composites are exposed to fungal colonization and degradation for 90 days.
3.5. Bacterial strains
Bacillus sp. Cd-3 culture has been grown using nutrient broth at 37 °C for 18 h. 1 mL of bacterial cultures is centrifuged at 6000 rpm for 20 min at room temperature and the pellets are washed with 0.9% NaCl and resuspended in 1 mL of mineral salt medium. After that 0.5 mL of the culture medium containing 1 × 108 mL−1 microbes is inoculated to the test tube containing 50 mL of media for each test. The test tubes are then incubated under sterile condition at 37 °C and 100 rpm for the degradation study.
4. Physical Measurements
4.1. X-ray diffractometry (XRD) study
XRD measurements are carried out in a Rigaku X-ray diffractometer (Miniflex, UK) using Cu Kα (λ = 0.154 nm) radiation at a scanning rate of 2° min−1 with an 2θ ranging from 2° to 70° to study the degrees of ZNP and clay intercalation in the synthesized composite.
4.2. Transmission electron microscope (TEM) study
The study of the dispersion of ZNP alone and in combination with nanoclay particles in jute reinforced starch composite and morphology were studied by using High Resolution Transmission Electron Microscope (HRTEM) (JEOL, JEM 2100) at an accelerating voltage of 200 kV. For TEM investigation the samples are embedded with epoxy resin following the method suggested by Devi et al.22 Ultrathin sections (approximately 100 nm thick) of the transverse film surfaces have been sectioned using an ultramicrotome fitted with a diamond knife. The sections are then stained with 1 wt% uranyl acetate for sufficient contrast.
4.3. Scanning electron microscopy (SEM) study
The surface morphology of some of the prepared samples was studied by scanning electron microscopy (SEM) before it is subjected to mechanical test. The morphology of the fracture surface of the samples obtained from flexural test is also studied by SEM. All the samples were coated on Pt for analysis. SEM analysis is performed by using a JEOL scanning electron microscope (model JSM-6390LV) at an accelerating voltage of 15 kV.
4.4. Fourier-transform infrared spectroscopy (FT-IR) study
FT-IR spectra of the post cured composite samples are recorded in FT-IR (Nicolet, Impact 410 spectrophotometer, USA) spectrophotometer. Small quantities of finely powdered composite samples were thoroughly grounded with exhaustively dried KBr and subsequently pellets were prepared by compression under vacuum.
4.5. Thermogravimetric analysis (TGA) study
Thermogravimetric analysis of the composite samples were done by using Shimazdu TGA 50, thermal analyzer under the nitrogen flow rate of 30 mL min−1 at the heating rate of 10 °C min−1 from 30 °C to 600 °C. The sample weight is maintained 5 mg and each set is recorded thrice to check the consistency of the observation.
4.6. Mechanical property study
The tensile properties of the composites are determined in accordance with ASTM D-638 at a crosshead speed of 2 mm min−1. The samples were cut into blocks of 100 mm × 3 mm × 20 mm (longitudinal × radial × tangential). For the flexural test, the specimens were cut into dimension of 3 mm × 10 mm × 100 mm (radial × tangential × longitudinal) and examined as per ASTM D-790 at a crosshead speed of 2 mm min−1. Both the tensile and flexural strength of the samples were measured by Universal Testing Machine-HOUNSEFIELD, England (model H100K-S) at room temperature. Ten samples were recorded for each set and the mean value is reported. The synthesized composite are cut into blocks of 3 mm × 10 mm × 25 mm (radial × tangential × longitudinal) for dimensional stability test. All the data are expressed as means ± standard deviation.
4.7. UV resistance study
The UV degradation study of the S/J composite samples were carried out using a UV chamber (Model S.L.W, voltage, 230 V, power, 1000 W; Advanced Research Co., India) employing a mercury arc lamp system that produces a collimated and highly uniform UV flux in the 254–365 nm range. Specimen's dimensions of (25 × 25 × 3) mm3 are exposed in the UV chamber at room temperature and characterized at definite time intervals. The exposure period is varied from 0 to 60 days. The weight loss is measured using an analytical balance and is calculated as follows:23| | |
% weight loss = {(Wt − W0)/W0} × 100
| (3) |
where, Wt is the specimen weight at time t, and W0 is the specimen weight before UV exposure. The chemical degradation of the samples were studied by FT-IR analysis. The intensity of the carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) stretching peaks at 1650 cm−1 corresponding to starch of the composite is measured. The net peak heights are determined by subtracting the height of the baseline directly from the total peak height. The same baseline is taken for each peak before and after exposure to UV radiation.24 The S/J composites degradation are studied by carbonyl index which is calculated by equation:
O) stretching peaks at 1650 cm−1 corresponding to starch of the composite is measured. The net peak heights are determined by subtracting the height of the baseline directly from the total peak height. The same baseline is taken for each peak before and after exposure to UV radiation.24 The S/J composites degradation are studied by carbonyl index which is calculated by equation:| | |
carbonyl index = IC/IR × 100
| (4) |
where IC is the intensity of the carbonyl absorption band (1650–1750 cm−1) and IR is the intensity of the reference band (2900–2950 cm−1). This peak is chosen as reference due to its least change during irradiation. Mechanical properties are also studied after UV exposure.
4.8. Dimensional stability study
Dimension of oven dried samples are measured and conditioned at room temperature (30 °C) and 30% relative humidity. The samples are then kept in humidity chamber for 72 h at room temperature maintaining relative humidity of 65%. The specimens are taken out from the humidity chamber after stipulated time period and volumes are measured with the help of slide calipers. Swelling is considered as a change in volume and expressed as percentage of volume increase compared to oven dried samples. The percentage of swelling in water vapour is calculated as per formula given below:| | |
% swelling = {(Vt − V0)/V0} × 100
| (5) |
where, ‘Vt’ is volume of the sample after “t” time. ‘V0’ is initial volume of composite sample before water vapour absorption.
4.9. Limiting oxygen index (LOI)
Limiting oxygen index (LOI) is defined as the minimum concentration of oxygen, expressed as percent volume, in a flowing mixture of oxygen and nitrogen that will support flaming combustion of a material initially at room temperature. The specimens were cut into 100 mm × 6 mm × 3 mm (longitudinal × tangential × radial) according to ASTM-D 2863 and placed vertically in the flammability tester (S.C. Dey Co., Kolkata). The total volume of the gas mixture (N2 + O2) is kept fixed at 18 cm3. The volume of nitrogen gas and that of oxygen gas are kept initially at a maximum and minimum level, respectively. Thereafter, the volume of nitrogen gas is decreased and that of oxygen gas is increased gradually. However, the total volume of gas mixture is kept fixed at 18 cm3 during the experiment. The ratio of nitrogen and oxygen at which the sample continued to burn for at least 30 s is recorded. Limiting Oxygen Index is calculated as:| | |
Limiting oxygen index (LOI) = {volume of O2/volume of (O2 + N2)} × 100
| (6) |
4.10. Biodegradation study
The microbial degradation is studied spectrophotometrically by using a UV visible spectrophotometer (Shimadzu Corporation UV2450 spectrophotometer, Kyoto, Japan) at 600 nm against blank culture media under sterile condition.
5. Results and discussion
The codification and compositions of the obtained nanocomposites (each approximately 3 mm thick) are reported in Table 1. The samples as defined in Table 1, has been prepared by keeping the weight (%) of the components viz. starch, glycerol, glutaraldehyde and jute fixed while varying the weight percentages of ZNPs and nanoclay.
Table 1 Codification and filler content of the nanocomposites based on crosslinked starch/jute with zinc oxide nanoparticles and nanoclay (wt%)
| Sample |
Starch (S) |
Glycerol |
Glutaraldehyde (GA) |
Jute (J) |
ZnO (Z) |
Nanoclay (M) |
| S/J/GA50 |
100 |
5 |
50 |
75 |
— |
— |
| S/J/GA50/Z1 |
100 |
5 |
50 |
75 |
1 |
— |
| S/J/GA50/Z3 |
100 |
5 |
50 |
75 |
3 |
— |
| S/J/GA50/Z5 |
100 |
5 |
50 |
75 |
5 |
— |
| S/J/GA50/Z5/M1 |
100 |
5 |
50 |
75 |
5 |
1 |
| S/J/GA50/Z5/M3 |
100 |
5 |
50 |
75 |
5 |
3 |
| S/J/GA50/Z5/M5 |
100 |
5 |
50 |
75 |
5 |
5 |
5.1. XRD study
As an indirect but non-invasive technique, XRD analysis is accomplished to obtain comprehensive information regarding crystal structure of the nanoparticles (i.e. ZNP and nanoclay) and the synthesized composites. Fig. 1 represents the diffractograms of jute, starch, ZNP, S/J/GA50, nanoclay and the nanocomposites. Jute (curve a) shows peaks at 2θ = 22° (002 plane of cellulose I) and 19° (101 plane of cellulose II).12 Curve (b) implies the diffractograms of starch macromolecules. A little broader hump connoting the amorphous nature of starch is noticed at 2θ = 19°. Curve (c) shows the X-ray diffraction patterns of bare ZNPs. From Fig. 1c, a series of characteristic peaks at 2θ = 32° (100), 34.5° (002), 36.4° (101), 47.6° (102), 56.7° (110), 63° (103), 66.5° (200), 68° (112) and 69° (201) are observed and they are in accordance with the zincite phase of ZnO (International Centre for Diffraction Data, JCPDS 5-0664). Curve (d), the diffractograms of composites without ZnO (i.e. S/J/GA50) exhibits a small broad diffraction peak corresponding to jute and starch in the 2θ range 1–20°. Curve (e–g) are for jute based crosslinked starch nanocomposites (S/J/GA50/Z1, S/J/GA50/Z3, and S/J/GA50/Z5) with different percentage of ZNPs (1–5% w/w of dry starch). The crystalline peak intensities of jute and starch matrix appeared in the range 2θ = 19–22° were found to diminish after the introduction of ZNPs within the composite. With the increase in the concentration of ZNPs, the peak intensity for jute and starch is further decreased.25 However, at a higher amount of ZNP loading, the peaks corresponding to ZNPs appeared with higher intensity. A strong diffraction peak for nanoclay at 2θ = 9° and a small peak at 2θ = 24° is observed in curve (h). The peak at 9° resembles to a d-spacing of about 1.2 nm in pure clay. The diffraction peak of the nanoclay tactoids is absent in the X-ray diffractograms for 1% and 3% clay incorporated composites (Fig. 1i and j). The d001 peak of the clay within the synthesized nanocomposites is completely vanished, signifying the development of a delaminated structure in the nanocomposites. It can be said that either the full expansion of the nanoclay gallery occurs, which is not possible to identify by XRD, or the nanoclay layers are delaminated due to which no crystal diffraction peak appears. In S/J/GA50/Z5/M5 composites, the position of the characteristic peak of nanoclay at 2θ = 9° is found to shift to lower 2θ values (i.e. 2θ = 7°) with less intensity representing an occurrence of agglomeration of clay within the nanocomposite.
 |
| | Fig. 1 XRD pattern of (a) Jute, (b) Starch, (c) ZNP, (d) S/J/GA50, (e) S/J/GA50/Z1, (f) S/J/GA50/Z3, (g) S/J/GA50/Z5, (h) Nanoclay, (i) S/J/GA50/Z5/M1, (j) S/J/GA50/Z5/M3 and (k) S/J/GA50/Z5/M5. | |
5.2. TEM study
The morphological features of the synthesized nanocomposites are provided by HRTEM investigation. Fig. 2(a–e) are the micrographs of the S/J/GA50, S/J/GA50/Z1, S/J/GA50/Z5, S/J/GA50/Z5/M1 and S/J/GA50/Z5/M5 samples, respectively. Fig. 2a represents the TEM micrographs of composite without ZNPs and nanoclay. Dark spots (shown by arrow marks) are appeared for ZNPs (Fig. 2b and c).26 From the Fig. 2b and c, it is clearly observed that the nanoparticles are well dispersed within the synthesized nanocomposites comprising of starch, and jute. TEM micrographs corresponding to S/J/GA50/Z5/M1 sample shows the dispersion of nanoclay in the composite. The threadlike dark lines (indicated with arrows) signify the intersections of the clay layers, and the bright areas symbolize the S/J composite matrix. The presences of threadlike line suggest that nanoclay is not homogeneously distributed within the composite indicating the existence of partial compatibility between polymer, ZNPs and nanoclay surface. TEM micrographs clearly reveal the loss of stacking structure of clay layers and eventually dictates the disordered dispersion within the composite. However, at higher concentration of the nanoclay, the thickness of dark slices of nanoclay increases and as a result agglomeration takes place (Fig. 2e). The results obtained from HRTEM analysis are in accordance with our XRD analysis. Both the results suggest the formation of the partially exfoliated nanocomposites.
 |
| | Fig. 2 TEM micrograph of (a) S/J/GA50, (b) S/J/GA50/Z1, (c) S/J/GA50/Z5, (d) S/J/GA50/Z5/M1 and (e) S/J/GA50/Z5/M5. | |
5.3. SEM study
Scanning electron microscopy is considered as a vital tool for studying surface morphology of composite materials. Fig. 3 shows SEM micrographs of S/J composites with or without ZNP and nanoclay, and different percentage of ZNP and nanoclay. The fractured surface of some selective samples is considered for this study. S/J/GA50 sheet shows a relatively smooth surface (Fig. 3a). Fig. 3b inferred that the fracture surface of S/J/GA50/Z1 displays a homogenous structure suggesting a relatively uniform distribution of the ZNPs within the composite. However, as the ZNP content increased, i.e., the fracture surface of S/J/GA50/Z5 containing higher amount of ZNP (Fig. 3c) exhibits a relatively rough surface, indicating relatively high interfacial adhesion among starch, jute and ZNPs. Moreover, upon addition of nanoclay into the ZNP loaded nanocomposites (i.e. S/J/GA50/Z5), the roughness is found to enhance (Fig. 3d, e and f).14 This might be due to the fact that the clay particles enhance the interaction with the starch matrix and jute surface. The roughness of fractured surface of composite having 3% nanoclay is more compared with those of composite prepared with 5% nanoclay. This indicates that agglomeration of nanoclay particles may take place within the composite at higher concentration of clay due to which interaction of clay particles with starch and jute are diminished and less rough surface is appeared.
 |
| | Fig. 3 SEM micrographs of (a) S/J/GA50, (b) S/J/GA50/Z1, (c) S/J/GA50/Z5, (d) S/J/GA50/Z5/M1, (e) S/J/G50/Z5/M3 and (f) S/J/GA50/Z5/M5. | |
The fracture surface of ZNPs loaded composites taken for SEM study is also investigated by energy dispersive X-ray elemental analysis (EDX) simultaneously. Fig. 4 shows the EDX analysis of jute reinforced starch composite. The presence of Zn along with C and O confirmed the successful incorporation of ZNP into the composite. Elements such as Al, Na and Si, which are mainly from the silicate nanoclay, are also detected (Fig. 4d) along with Zn, indicating the successful incorporation of nanoclay and ZNPs into the composite.
 |
| | Fig. 4 Energy dispersive X-ray analysis of S/J/GA50/Z5 and S/J/GA50/Z5/M5. | |
5.4. FT-IR study
The interactions among starch, jute, glycerol, GA, ZNPs and clay are studied by FT-IR spectroscopy. FT-IR spectra of starch, jute clay and ZNPs are presented in Fig. 5. The FT-IR spectrum of starch shows the absorption bands at 571, 981, 1161, 1653, 2921 and 3450 cm−1 confirming the carbohydrate nature.27 The typical saccharide bands appeared in the range 1180–950 cm−1 was considered as the stretching mode of C–C and C–O vibration and the bending mode of C–H bond vibration.28 The peak at 2921 cm−1 is characteristic of the C–H stretching vibration of methylene group.29 Jute shows the presence of peaks in the range 3440 cm−1 for –OH stretching, 1737 cm−1 for CO stretching vibration of ester groups of hemicelluloses, 1642 cm−1 for CO stretching, 1254 cm−1 for –C–O–C– bond in cellulose chain and 1057–1116 cm−1 for C–O stretching.30 Clay exhibits peaks at 3450 cm−1 (–OH stretching), 1619 cm−1 (–OH bending), 1030–460 cm−1 (oxide bonds of metals like Si, Al, Mg etc.). FT-IR spectrum of ZNP is presented in Fig. 5d. Absorption peaks at 3429 and 1633 cm−1 in the spectra is due to –OH stretching and –OH bending vibrations while the high intensity broad band at approximately 421 cm−1 resulting from the stretching of the zinc and oxygen bond.31
 |
| | Fig. 5 FTIR spectra of (a) Starch, (b) Jute, (c) Nanoclay and (d) ZNP. | |
FT-IR spectra for the composite S/J/GA50, S/J/GA50/Z1, S/J/GA50/Z3, S/J/GA50/Z5, S/J/GA50/Z5/M1, S/J/GA50/Z5/M3 and S/J/GA50/Z5/M5 are shown in Fig. 6. The FT-IR spectra for all the samples show peaks in the region 3400–3250 cm−1, which could be attributed to the –OH stretching vibrations. The typical saccharide bands of starch appeared in the region of 1180–950 cm−1. Similarly the characteristic peaks for jute has been appeared in all the spectrum at 1254 and 1116–1057 cm−1. The entire spectra show the band around 900 cm−1 due to the β-(1 → 4)-glycosidic linkages. The position of –OH and CO stretching is shifted in the crosslinked composite (S/J/GA50) suggesting the interaction between jute and starch. From the IR spectrum, it is found that, with the increase in ZNP concentration the peak intensities of –OH band are decreased and slightly shifted to lower wavenumber. This behaviour can be ascribed to the interaction between the free –OH groups of starch and jute with –OH groups of ZNPs. The FT-IR spectra indicated the existence of a strong binding but no obvious formation of covalent bonds between the S/J/GA50 composites and ZNPs.26 Upon the addition of nanoclay, the peaks in the ∼3400 cm−1 range are further reduced in intensity and shifted to lower wavenumber. The peak intensities in the ranges of 1030–460 cm−1 and 1620 cm−1 are found to be reduced upto a considerable extent. These results confirm the participation of hydroxyl group of clay with S/J/GA50/Zn composites.32
 |
| | Fig. 6 FTIR spectra of (a) S/J/GA50, (b) S/J/GA50/Z1, (c) S/J/GA50/Z3, (d) S/J/GA50/Z5, (e) S/J/GA50/Z5/M1, (f) S/J/GA50/Z5/M3 and (g) S/J/GA50/Z5/M5. | |
5.5. TGA study
The influence of ZNP and nanoclay on the thermal properties of the synthesized green nanocomposites is investigated by TGA as shown in Fig. 7. Fig. 7 displays the thermograms of S/J/GA50, S/J/GA50/Z1, S/J/GA50/Z3, S/J/GA50/Z5, S/J/GA50/Z5/M1, S/J/GA50/Z5/M3, and S/J/GA50/Z5/M5. Table 2 (derived from Fig. 7) shows the initial decomposition temperature (Ti), maximum pyrolysis temperature (Tm), decomposition temperature (Td) at different weight loss (%) and residual weight (RW, %) of the nanocomposites upto 600 °C. All the thermograms show an initial weight loss around 100 °C due to the loss of trapped water molecules. From the thermograms it is clearly inferred that all the values of the nanocomposites (Table 2) are found to increase with the increase in ZNPs concentration. This might be ascribed to the heat shielding effect of ZNPs.33 Nanoclay is widely used to enhance the thermal stability of composite materials. After the incorporation of nanoclay, the thermal stability of the ZNP loaded nanocomposites are further improved. This increase in thermal stability of the synthesized nanocomposites is attributed to the hindered diffusion of volatile decomposition products within it. This might also be due to the physico-chemical absorption of the volatile degradation products on the silicate surface of nanoclay. The volatilization of the degraded products originated by carbon–carbon bond scission in the composite is delayed by tortuous path provided by the silicate layers. Clay treated composites show a subsidiary enhancement in RW values over clay untreated one.34 At higher concentration of clay, the agglomeration probably decreased the interaction and can give rise to the reduction in the thermal stability. Therefore, it is concluded that the thermal stability of S/J composites is increased on addition of upto a certain concentration of ZNP and nanoclay.
 |
| | Fig. 7 TGA thermograms of (a) S/J/GA50, (b) S/J/GA50/Z1, (c) S/J/GA50/Z3, (d) S/J/GA50/Z5, (e) S/J/GA50/Z5/M1, (f) S/J/GA50/Z5/M3 and (g) S/J/GA50/Z5/M5. | |
Table 2 Thermal Properties of (a) S/J/GA50, (b) S/J/GA50/Z1, (c) S/J/GA50/Z3, (d) S/J/GA50/Z5, (e) S/J/GA50/Z5/M1, (f) S/J/GA50/Z5/M3 and (g) S/J/GA50/Z5/M5d
| Sample particulars |
Tia |
Tmb |
Tmc |
Temperature of decomposition at different weight loss (%) |
RW% at 600 °C |
| 20 |
30 |
40 |
50 |
60 |
| Ti: initial decomposition temperature. Tm: maximum pyrolysis temperature value for 1st step. Tm: maximum pyrolysis temperature value for 2nd step. Each value in the table represents average value of three samples. |
| S/J/GA50 |
199 ± 2 |
321 ± 1 |
367 ± 2 |
285 ± 1 |
304 ± 1 |
314 ± 2 |
321 ± 2 |
324 ± 1 |
6 ± 2 |
| S/J/GA50/Z1 |
205 ± 1 |
325 ± 1 |
371 ± 1 |
294 ± 2 |
310 ± 2 |
320 ± 1 |
325 ± 1 |
347 ± 2 |
9 ± 2 |
| S/J/GA50/Z3 |
208 ± 2 |
327 ± 2 |
376 ± 1 |
305 ± 1 |
313 ± 1 |
325 ± 2 |
331 ± 3 |
366 ± 1 |
12 ± 2 |
| S/J/GA50/Z5 |
214 ± 3 |
333 ± 2 |
381 ± 2 |
311 ± 1 |
318 ± 1 |
329 ± 1 |
335 ± 2 |
372 ± 3 |
14 ± 1 |
| S/J/GA50/Z5/M1 |
218 ± 2 |
338 ± 1 |
388 ± 3 |
314 ± 1 |
325 ± 3 |
332 ± 2 |
340 ± 1 |
378 ± 2 |
15 ± 3 |
| S/J/GA50/Z5/M3 |
231 ± 1 |
349 ± 1 |
403 ± 2 |
324 ± 3 |
333 ± 1 |
343 ± 1 |
354 ± 1 |
397 ± 1 |
17 ± 2 |
| S/J/GA50/Z5/M5 |
224 ± 2 |
343 ± 2 |
394 ± 1 |
319 ± 2 |
328 ± 1 |
337 ± 2 |
347 ± 2 |
385 ± 2 |
16 ± 2 |
5.6. Mechanical property study
The mechanical properties of S/J composites reinforced with ZNPs and nanoclay are studied at room temperature. From these measurements, we have determined the flexural strength, flexural modulus, tensile strength and tensile modulus. Experimental data are deposited in Table 3. A noticeable reinforcing effect is observed upon filler addition, as displayed by the increase in both the modulus and strength for composite compared to the unfilled composites. The increase in the concentration of ZNP might have the direct effect in increasing the mechanical properties of the nanocomposites.31 As the ZNPs content increased, both the tensile and flexural properties of the nanocomposites also increased. The interaction of ZnO with crosslinked starch and jute through its surface hydroxyl group may stiffen the composites and results in enhancement of mechanical properties. Considering the 5% concentration of the ZNPs as optimum, we have further modified the composites viz. S/J/GA50Zn5/M1. S/J/GA50/Zn5/M3 and S/J/GA50/Zn5/M5 having different percentage of nanoclay ranging from 1–5% (w/w of starch). With the incorporation of nanoclay, composites show a significant improvement in the mechanical properties with respect to the nanoclay untreated one. The reason for such improvement in mechanical properties of the composites may be the high aspect ratio of the nanoclay which generates a large surface area for the polymer chains absorption. Clay acts as a rigid reinforcement to the polymer matrix. The interaction of the polymer chains into the galleries of nanoclay restricts its mobility.12 However, such improvement in mechanical properties with the incorporation of nanoclay may only be realized up to a certain clay loading. In our work, the enhancement in mechanical properties is found up to clay loading of 3%, and beyond that it declines on further addition of nanoclay. At higher concentration of nanoclay, agglomeration of clay particles may takes place and therefore the interaction between polymer–clay is reduced. This led to the reduction in the extent of enhancement of reinforcement efficiency with the addition of higher amount of nanoclay.
Table 3 Comparison of tensile and flexural properties of unfilled and filled jute based crosslinked starch nanocomposites before UV treatmenta
| Composite |
Tensile properties |
Flexural properties |
| System |
Strength (MPa) |
Modulus (MPa) |
Strength (MPa) |
Modulus (MPa) |
| Each value in the above table represents average of ten samples. Values in the parenthesis represent the standard deviation. |
| S/J/GA50 |
22.23 (±2.35) |
1272.7 (±14.74) |
42.41 (±1.43) |
2425.2 (±12.47) |
| S/J/GA50/Z1 |
30.45 (±1.63) |
1685.9 (±11.53) |
50.42 (±2.62) |
3076.3 (±09.84) |
| S/J/GA50/Z3 |
36.78 (±1.13) |
1857.4 (±12.22) |
60.68 (±1.78) |
3987.7 (±10.54) |
| S/J/GA50/Z5 |
41.23 (±2.42) |
2019.6 (±16.56) |
68.45 (±0.94) |
4867.8 (±11.67) |
| S/J/GA50/Z5/M1 |
49.57 (±1.86) |
2258.7 (±11.85) |
83.75 (±1.54) |
5789.6 (±14.24) |
| S/J/GA50/Z5/M3 |
59.65 (±2.27) |
2648.3 (±13.42) |
99.57 (±2.25) |
6656.1 (±13.49) |
| S/J/GA50/Z5/M5 |
54.89 (±1.73) |
2414.8 (±12.61) |
90.42 (±2.11) |
6027.3 (±15.32) |
5.7. UV test results
The weight loss of S/J composite and S/J composite loaded with ZNPs and nanoclay is shown in Fig. 8. Weight losses of the samples are determined at room temperature as a function of exposure time and found linear variation with exposure time. Initially a small increase of weight is found due to moisture uptake by the samples, which is greater than the material loss induced by the degradation in the early stage. The rate of weight loss is lowest for S/J/GA50/Z5/M3 followed by S/J/GA50/Z5/M5, S/J/GA50/Z5/M1, S/J/GA50/Z5, S/J/GA50/Z3, S/J/GA50/Z1 and S/J/GA50. The ZNPs and nanoclay unfilled composite shows maximum weight losses. After 60 days of exposure, the weight losses in S/J/GA50, S/J/GA50/Z1, S/J/GA50/Z3, S/J/GA50/Z5, S/J/GA50/Z5/M1, S/J/GA50/Z5/M3 and S/J/GA50/Z5/M5 are −4.41% ± 0.4%, −3.76% ± 0.5%, −3.12% ± 0.3%, −2.54% ± 0.3%, −1.67% ± 0.2%, −1.02% ± 0.2%, and −1.28% ± 0.2% respectively. Results presented are the average of four specimens. The lower weight loss (%) for S/J/GA50/Z5/M3 composites after UV exposure is due to the UV shielding ability of ZNPs and nanoclay. On exposure to UV radiation, the chain scission followed by decrease in the density of the entanglements of the polymer chains occurred. This resulted in the decrease in the weight of the synthesized composites.
 |
| | Fig. 8 Weight loss vs. exposure time for (a) S/J/GA50, (b) S/J/GA50/Z1, (c) S/J/GA50/Z3, (d) S/J/GA50/Z5, (e) S/J/GA50/Z5/M1, (f) S/J/GA50/Z5/M3 and (g) S/J/GA50/Z5/M5. | |
Fig. 9 represents the FT-IR spectra of (a) S/J/GA50, (b) S/J/GA50/Z1, (c) S/J/GA50/Z3, (d) S/J/GA50/Z5, (e) S/J/GA50/Z5/M1, (f) S/J/GA50/Z5/M3 and (g) S/J/GA50/Z5/M5 after UV exposure, respectively. The intensity of the carbonyl peaks is found to increase after irradiation of the samples for 60 days. Upon exposing the samples to UV-radiation, chain scission of the polymers takes place and the carbonyl index value increases as shown in Fig. 10. The S/J/GA50/Z5/M3 has lowest carbonyl index value whereas S/J/GA50 has the highest one. ZNP plays an important role of stabilizing the jute based crosslinked starch composites by acting as screen and slows down the photo degradation process. ZNP absorbs the UV radiation and hence reduces the UV intensity required for the oxidation of the synthesized nanocomposites.35 The presence of nanoclay in the composite also has a screening effect which further delays the photo degradation process. Grigoriadou et al.36 has observed an increase in UV stability of HDPE after incorporating montmorillonite clay. S/J/GA50/Z5/M5 exhibits lower protection against UV with respect to S/J/GA50/Z5/M3. This might be due to the agglomeration of nanoclay particles within the composite material which provides lower protection against photodegradation.
 |
| | Fig. 9 Change in carbonyl peak intensity of (a) S/J/GA50, (b) S/J/GA50/Z1, (c) S/J/GA50/Z3, (d) S/J/GA50/Z5, (e) S/J/GA50/Z5/M1, (f) S/J/GA50/Z5/M3 and (g) S/J/GA50/Z5/M5. | |
 |
| | Fig. 10 Carbonyl index value of (a) S/J/GA50, (b) S/J/GA50/Z1, (c) S/J/GA50/Z3, (d) S/J/GA50/Z5, (e) S/J/GA50/Z5/M1, (f) S/J/GA50/Z5/M3 and (g) S/J/GA50/Z5/M5. | |
It is well known that UV-radiation has a deterioration effect on many plastic materials. These materials, when exposed to the outdoor environment, undergo significant changes, namely, photo-degradation, causing loss of mechanical properties. Therefore, the changes in the mechanical properties of the composites after the UV treatment are presented in Table 4. Both the flexural and tensile properties reduced after UV treatment. The loss of mechanical properties of unfilled composites is more significant compared to nanoparticles filled composites. S/J/GA50 is more prone to UV attack and hence it shows maximum loss of mechanical properties. The values presented in Table 4 suggest that the interfacial interactions among starch, jute, GA, ZNPs and nanoclays are strong enough to decay the massive sheer due to rupture. Therefore, the resistance of jute based crosslinked starch composite to UV instability, i.e., photodegradation embrittlement, can be improved significantly with the addition of nanoparticles.
Table 4 Changes in the mechanical properties of unfilled and filled jute based crosslinked starch nanocomposites after UV exposurea
| Composite |
Tensile properties |
Flexural properties |
| System |
Strength (MPa) |
Modulus (MPa) |
Strength (MPa) |
Modulus (MPa) |
| Each value in the above table represents average of ten samples. Values in the parenthesis represent the standard deviation. |
| S/J/GA50 |
14.05 (±1.11) |
1022.2 (±15.23) |
34.52 (±1.32) |
2099.2 (±12.82) |
| S/J/GA50/Z1 |
25.68 (±1.79) |
1412.2 (±11.24) |
44.34 (±1.57) |
2689.4 (±11.53) |
| S/J/GA50/Z3 |
30.84 (±1.52) |
1589.8 (±13.56) |
55.25 (±1.68) |
3556.2 (±15.67) |
| S/J/GA50/Z5 |
36.56 (±1.78) |
1798.5 (±15.31) |
64.57 (±1.25) |
4511.9 (±10.84) |
| S/J/GA50/Z5/M1 |
44.72 (±1.65) |
1948.5 (±11.78) |
76.31 (±1.54) |
5425.7 (±11.48) |
| S/J/GA50/Z5/M3 |
56.34 (±1.13) |
2494.2 (±14.54) |
95.58 (±1.11) |
6396.5 (±13.43) |
| S/J/GA50/Z5/M5 |
50.61 (±2.11) |
2124.4 (±12.56) |
85.74 (±2.25) |
5765.8 (±12.22) |
5.8. Dimensional stability study
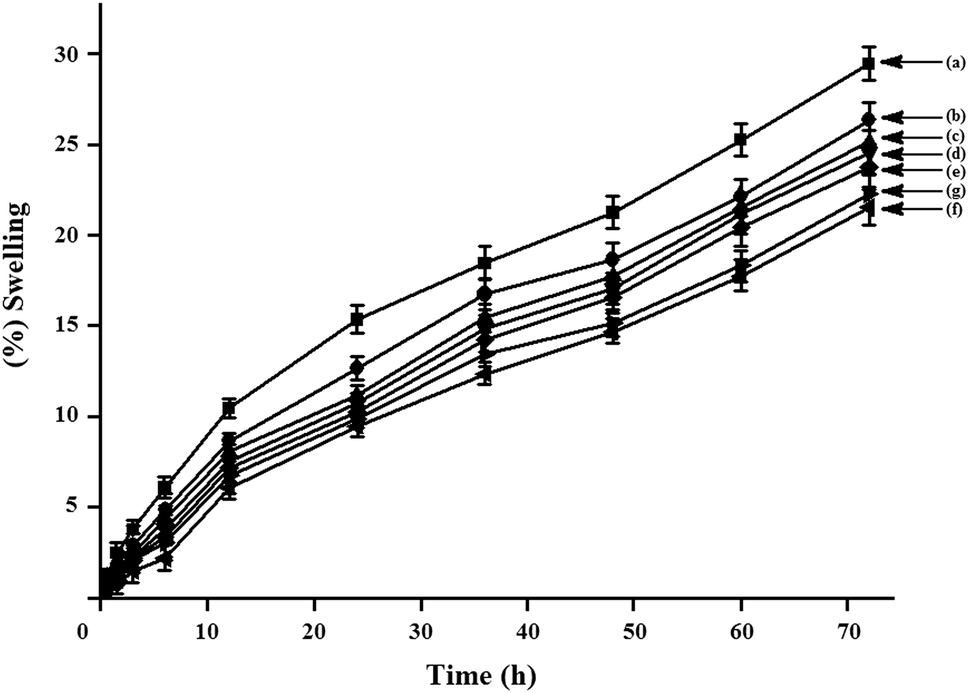
The water vapour absorption study was carried out at room temperature (∼30 °C) with 65% relative humidity for different time period is shown in Fig. 11. Form the figure, it is seen that for all the composites, the % swelling is increased with increase in time. Water vapour absorption is found to decrease due to the presence of dispersed phase of ZNPs in the composite system. Water vapour absorption of S/J/GA50/Z composites decreased with the increase in ZNP content.25 ZNPs act as a barrier to the passage of water as the strong affinity of water molecules towards ZNPs restricts its free motion and reduces the diffusion co-efficient of water. The well dispersed phase of ZNPs improves the resistance of the composite and retards the motion of water molecules through it. The better the distribution of nanoparticles, the higher is the barrier property. Further, the dimensional stability of the S/J/GA50/Z composite is improved with the incorporation of nanoclay. This may be due to the fact that the silicate layers of nanoclay provide a tortuous path which hinders the diffusivity of water particles through the nanocomposite.14 As the nanoclay concentration is increased to 5%, the superfluous filler congregates easily, decreasing the effective content of filler and therefore has little effect on decreasing of water vapour absorption.
 |
| | Fig. 11 Swelling behavior of (a) S/J/GA50, (b) S/J/GA50/Z1, (c) S/J/GA50/Z3, (d) S/J/GA50/Z5, (e) S/J/GA50/Z5/M1, (f) S/J/GA50/Z5/M3 and (g) S/J/GA50/Z5/M5. | |
5.9. Limiting oxygen index (LOI) study
LOI values of jute based starch composites with different percentage of ZNP and nanoclay are depicted in Table 5. From the table, it is observed that all the samples produce small localised flame and nanoclay filled composites produces higher char than that of the nanoclay unfilled one. The LOI test assumes that inherently less flammable materials require greater oxygen concentrations to produce the heat necessary for the continuous production of flammable volatiles and flame propagation. It is also observed that the LOI value increases with the increase in the concentration of ZNPs in the composites. The surface hydroxyl group of ZNPs may interact with starch, jute and GA and thus restricts the accessibility of oxygen for the production of degradable components from the composites and hence improvement in LOI value is found.37 However, on addition of nanoclay into ZNP incorporated composites, further enhancement in LOI value is observed. The incorporation of the nanoclay into the synthesized composite produces a silicate char on the surface of it and hence improves their flame resistance property. The silicate rich surface has better barrier property to heat and oxygen transport due to which ignition of the composite delays.38 The LOI value is increased upto the addition of 3% of nanoclay. At higher concentration of nanoclay (5%), the LOI value is decreased. At higher clay loading condition, the agglomeration of nanoclay results in decrease of interaction with concomitant reduction in barrier property as well as LOI value.
Table 5 Limiting oxygen indices (LOI) and flaming characteristics of the prepared compositesa
| Samples |
LOI (%) |
Flame description |
Smoke & fumes |
Char |
| Each value represents average five samples. |
| S/J/GA50 |
41 (±1.0) |
Small localised flame |
Small and black smoke |
Little |
| S/J/GA50/Z1 |
46 (±2.0) |
Small localised flame |
Small and black smoke |
Higher |
| S/J/GA50/Z3 |
51 (±2.0) |
Small localised flame |
Small and black smoke |
Higher |
| S/J/GA50/Z5 |
56 (±1.0) |
Small localised flame |
Small and black smoke |
Higher |
| S/J/GA50/Z5/M1 |
61 (±3.0) |
Small localised flame |
Small and black smoke |
Higher |
| S/J/GA50/Z5/M3 |
68 (±2.0) |
Small localised flame |
Small and black smoke |
Higher |
| S/J/GA50/Z5/M5 |
64 (±2.0) |
Small localised flame |
Small and black smoke |
Higher |
5.10. Biodegradation study
S/J composites are exposed to cellulolytic bacterial strain directly in broth culture medium for the biodegradation study. After one month of incubation of the samples in broth culture media, the growth of bacteria and degradation of the composite samples is clearly detectable. The bacterial growth of the samples with respect to time is represented by Fig. 12. The microbial attack is more in case of ZNP free composite compared to the composite filled with ZNP and nanoclay. Initially, the growth of the bacterial strains increased quite steadily with bacterial exposure time, but the rate of growth slightly decreased after 3 month of incubation. As the composites are mainly comprised of starch and jute which are basically carbohydrate and cellulose, these two are very good carbon source for the bacteria due to which higher rate of bacterial growth is observed. The powerful cellulolytic and pectinolytic activity of bacteria could be a reason for the enhancement of bacterial growth.39 After 3 months, the rate of microbial growth is slightly diminished due to the production of toxic metabolites by the microbes. In our study, the growth of bacteria and degradation of the samples are revealed by SEM study (Fig. 13). SEM is used to observe the extent of physical breakdown of S/J polymer composites. SEM shows different levels of degradation, colonized nanoclay S/J composite sometimes only shows superficial degradation whereas, ZNP incorporated S/J composites show less degradation as ZnO sometimes acts as a fungicide and protective agent.40 The degradation is more in the unfilled S/J composites as starch and jute are gradually breakdown in the broth culture media. The lower biodegradability of filled S/J nanocomposites is sacrificed considering the enhanced thermal and mechanical properties. However, in case of nanoclay incorporated composites, the rate of degradation increases with increasing the concentration of nanoclay in S/J composites. The catalytic role plays by the nanoclays may be the cause of this degradation.41,42 In bacterial media the degradation of S/J polymer composites is a complex process involving four main phenomena: (i) water adsorption, (ii) bond cleavage and formation of oligomer fragments, (iii) solubilisation of oligomer fragments, and finally (iv) diffusion of soluble oligomers by bacteria.43,44 Therefore, the factor that increases the hydrolysis tendency of S/J composites eventually controls the degradation of the composites. In case of nanoclay incorporated composites, the presence of unreacted terminal hydroxyl group may be the responsible factors for this behaviour.45
 |
| | Fig. 12 Bacterial growth of (a) S/J/GA50, (b) S/J/GA50/Z5/M5, (c) S/J/GA50/Z5/M3, (d) S/J/GA50/Z5/M1, (e) S/J/GA50/Z1, (f) S/J/GA50/Z3 and (g) S/J/GA50/Z5. | |
 |
| | Fig. 13 SEM micrographs of samples after microbial test on (a) S/J/GA50, (b) S/J/GA50/Z1, (c) S/J/GA50/Z3, (d) S/J/GA50/Z5, (e) S/J/G50/Z5/M1, (f) S/J/GA50/Z5/M3, and (g) S/J/GA50/Z5/M5. | |
6. Conclusion
Renewable resources based green nanocomposites comprising of starch, jute, GA, ZNPs and nanoclay are developed by using solution induced intercalation method and characterized by various physicochemical and spectroscopic techniques. Effect of the ZNPs and nanoclay particles are studied. The incorporation of ZNPs improved the physicochemical properties such as thermal, mechanical, flame retardancy, UV-resistance and dimensional stability via the interaction of the surface hydroxyl group of ZNPs with the hydroxyl group of starch and jute. The composite with 5% ZNPs is found to exhibit better physical properties than those of either unfilled composites or composites containing lower amount of ZNP. The incorporation of nanoclay results in further improvement of the physicochemical properties of the synthesized material. FT-IR study shows a strong interaction among jute fabric, starch, GA, ZNPs and nanoclay. TEM and XRD studies reveal the distribution of ZNPs and silicate layers of nanoclay in S/J composites. Composites containing 5% ZNPs and 3% nanoclay produce maximum improvement in several physicochemical properties. Thus, ZNPs in combination with nanoclay can considerably improve the physical properties of natural resources based composites. Consequently, such kind of ZNP and nanoclay filled S/J composites are eco-friendly and can receive applications in newer domains.
Acknowledgements
M.I. thanks Council of Scientific and Industrial Research (CSIR), New Delhi, INDIA for Senior Research Fellowship (SRF). We thank all the reviewers for their helpful comments.
References
- G. W. Coates and M. A. Hillmyer, Macromolecules, 2009, 42, 7987 CrossRef CAS.
- J. K. Pandey, A. P. Kumar, M. Misra, A. K. Mohanty, L. T. Drazal and R. P. Singh, J. Nanosci. Nanotechnol., 2005, 5, 497 CrossRef CAS PubMed.
- A. Espert, F. Vilaplana and S. Karlsson, Composites, Part A, 2004, 35, 1267 CrossRef PubMed.
- K. G. Satyanarayana, G. G. C. Arizaga and F. Wypych, Prog. Polym. Sci., 2009, 34, 982 CrossRef CAS PubMed.
- D. L. Corre, J. Bras and A. Dufresne, Biomacromolecules, 2010, 11, 1139 CrossRef PubMed.
- P. J. Halley, Thermoplastic starch biodegradable polymers, in Biodegradable Polymers for Industrial Applications, ed. R. Smith, CRC Press, Washington, DC, 2005, vol. 1, p. 140 Search PubMed.
- G. Q. Chen and M. K. Patel, Chem. Rev., 2012, 112, 2082 CrossRef CAS PubMed.
- I. U. Unalan, G. Cerri, E. Marcuzzo, C. A. Cozzolino and S. Farris, RSC Adv., 2014, 4, 29393 RSC.
- R. L. Quirino, T. F. Garrison and M. R. Kessler, Green Chem., 2014, 16, 1700 RSC.
- W. Ban, J. Song, D. S. Argyropoulos and L. A. Lucia, J. Appl. Polym. Sci., 2006, 100, 2542 CrossRef CAS PubMed.
- H. Li and M. A. Huneault, J. Appl. Polym. Sci., 2011, 119, 2439 CrossRef CAS PubMed.
- M. Iman and T. K. Maji, Carbohydr. Polym., 2012, 89, 290 CrossRef CAS PubMed.
- A. K. Bledzki and J. Gassan, Prog. Polym. Sci., 1999, 24, 221 CrossRef CAS.
- M. Iman, K. K. Bania and T. K. Maji, Ind. Eng. Chem. Res., 2013, 52, 6969 CrossRef CAS.
- F. Hussain, M. Hojjati, M. Okamoto and R. E. Gorga, J. Compos. Mater., 2006, 40, 1511 CrossRef CAS PubMed.
- H. Goesmann and C. Feldmann, Angew. Chem., Int. Ed., 2010, 49, 1362 CrossRef CAS PubMed.
- S. S. Ray and M. Okamoto, Prog. Polym. Sci., 2003, 28, 1539 CrossRef CAS PubMed.
- Y. Yu, Z. Jiang, G. Wang and Y. Song, Holzforschung, 2010, 64, 385 CAS.
- F. Weichelt, R. Emmler, R. Flyunt, E. Beyer, M. R. Buchmeiser and M. Beyer, Macromol. Mater. Eng., 2010, 295, 130 CAS.
- M. M. Demir, M. Memesa, P. Certignolles and G. Wegner, Macromol. Rapid Commun., 2006, 27, 763 CrossRef CAS PubMed.
- N. Lu, X. Lu, X. Jin and C. Lu, Polym. Int., 2007, 56, 138 CrossRef CAS PubMed.
- R. R. Devi and T. K. Maji, Ind. Eng. Chem. Res., 2012, 51, 3870 CrossRef CAS.
- W. Fa, C. Yang, C. Gong, T. Peng and L. Zan, J. Appl. Polym. Sci., 2010, 118, 378 CrossRef CAS PubMed.
- N. M. Stark and L. M. Matuana, Polym. Degrad. Stab., 2004, 86, 1 CrossRef CAS PubMed.
- P. J. P. Espitia, N. de F. F. Soares, R. F. Teófilo, J. S. dos, R. Coimbra, D. M. Vitor, R. A. Batista, S. O. Ferreira, N. J. de Andrade and E. A. A. Medeiros, Carbohydr. Polym., 2013, 94, 199 CrossRef CAS PubMed.
- X. Ma, P. R. Chang, J. Yang and J. Yu, Carbohydr. Polym., 2009, 75, 472 CrossRef CAS PubMed.
- Aboubakar, Y. N. Njintang, J. Scher and C. M. F. Mbofung, J. Food Eng., 2008, 86, 294 CrossRef CAS PubMed.
- H. Liu, D. Chaudhary, S. Yusa and M. O. Tade, Carbohydr. Polym., 2011, 83, 1591 CrossRef CAS PubMed.
- J. M. Fang, P. A. Fowler, J. Tomkinson and C. A. S. Hill, Carbohydr. Polym., 2002, 47, 245 CrossRef CAS.
- M. Iman and T. K. Maji, J. Appl. Polym. Sci., 2013, 127, 3987 CrossRef CAS PubMed.
- B. K. Deka and T. K. Maji, J. Appl. Polym. Sci., 2012, 124, 2919 CrossRef CAS PubMed.
- M. Darder, M. Colilla and E. R. Hitzky, Chem. Mater., 2003, 15, 3774 CrossRef CAS.
- R. R. Devi and T. K. Maji, Ind. Eng. Chem. Res., 2012, 51, 3870 CrossRef CAS.
- L. Pranger and R. Tannenbaum, Macromolecules, 2008, 41, 8682 CrossRef CAS.
- H. Zhao and R. K. Y. Li, Polymer, 2006, 47, 3207 CrossRef CAS PubMed.
- I. Grigoriadou, K. M. Paraskevopoulos, K. Chrissafis, E. Pavlidou, T.-G. Stamkopoulos and D. Bikiaris, Polym. Degrad. Stab., 2011, 96, 151 CrossRef CAS PubMed.
- N. Wu, C. Ding and R. Yang, Polym. Degrad. Stab., 2010, 95, 2589 CrossRef CAS PubMed.
- L. Urbanczyk, S. Bourbigot, C. Calberg, C. Detrembleur, C. Jérôme, F. Boschini and M. Alexandre, J. Mater. Chem., 2010, 20, 1567 RSC.
- C. A. Clausen, Int. Biodeterior. Biodegrad., 1996, 37, 101 CrossRef.
- C. Lykidis, G. Mantanis, S. Adamopoulos, K. Kalafata and I. Arabatzis, Wood Mater. Sci. Eng., 2013, 8, 242 CrossRef CAS.
- B. K. Deka, M. Mandal and T. K. Maji, Polym. Bull., 2011, 67, 1875–1892 CrossRef CAS.
- U. Konwar, N. Karak and M. Mandal, Polym. Degrad. Stab., 2009, 94, 2221 CrossRef CAS PubMed.
- J. Lunt, Polym. Degrad. Stab., 1998, 59, 145 CrossRef CAS.
- R. E. Drumright, P. R. Gruber and D. E. Henton, Adv. Mater., 2000, 12, 1841 CrossRef CAS.
- S. S. Ray, K. Yamada, M. Okamoto and K. Ueda, Nano Lett., 2002, 2, 1093 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.