 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffective photocatalysis of functional nanocomposites based on carbon and TiO2 nanoparticles†
Chan
Lin
ab,
Yang
Song
b,
Lixin
Cao
c and
Shaowei
Chen
*b
aCollege of Chemistry and Chemical Engineering, Ocean University of China, Qingdao, Shandong 266100, China
bDepartment of Chemistry and Biochemistry, University of California, 1156 High Street, Santa Cruz, California 95064, USA. E-mail: shaowei@ucsc.edu
cInstitute of Materials Science and Engineering, Ocean University of China, Qingdao, Shandong 266100, China
First published on 10th April 2013
Abstract
A unique nanocomposite C–TiO2 was prepared by the growth of TiO2 on carbon nanoparticles using a simple hydrothermal procedure. Transmission electron microscopic (TEM) measurements showed that the nanocomposites exhibited an average core diameter of approximately 5 nm with a rather well-defined lattice space (0.4 nm) that was somewhat larger than that (0.38 nm) of the (100) crystalline planes of anatase TiO2. This lattice expansion was accounted for by the formation of surface defect dipoles of the nanosized TiO2 particles. X-ray photoelectron spectroscopic (XPS) measurements suggested that partial charge transfer occurred from carbon nanoparticles to TiO2 by the interfacial Ti–O–C linkages, which led to effective diminishment of the C–TiO2 photoluminescence as compared to that of pure TiO2 or carbon nanoparticles, suggesting intimate electronic interactions between the carbon and TiO2 components in the nanocomposites. Such unique characteristics were then exploited for the effective photocatalytic degradation of organic pollutants, as exemplified by methylene blue, by C–TiO2 under UV photoirradiation. Experimental measurements showed that the photocatalytic activity of C–TiO2 nanocomposites was about twice that of TiO2 alone, whereas little activity was observed with carbon nanoparticles. This was attributed to the electron-accepting sites on the carbon nanoparticles that facilitated interfacial charge separation.
Introduction
Titanium dioxide (TiO2) has been extensively studied for potential applications in photocatalysis due to its low cost, non-toxicity, high redox ability and stability.1–3 However, with an intrinsic bandgap close to ∼3 eV, TiO2 typically exhibits low efficiency in light utilization and fast recombination of electron–hole pairs, which greatly limit the photocatalytic performance. Therefore, in the past few decades, a number of effective approaches have been developed to further engineer the TiO2 structures and properties, such as coupling with other semiconductor materials,4–7 modification with metal and nonmetal elements and structures,8–12 and surface sensitization by dye molecules.13–15 Among these, formation of functional composites with carbon nanomaterials represents one of the most popularly used methods, largely motivated by the unique optical and electronic properties of carbon nanostructures. There are various kinds of carbon modifications, such as carbon-doped TiO2,16–20 carbon-coated TiO2,21–24 and mounting of TiO2 on activated carbon.25–27 For instance, Chu et al.17 prepared carbon-doped nanostructured TiO2 films (the average size of the nanoparticles was about 40 nm) with oxalic acid and tetrabutylammonium bromide as the carbon sources and the carbon-doped into the lattice sites of TiO2, leading to apparent narrowing of the TiO2 bandgap and hence improved efficiency of photoenergy utilization. In another study,20 carbon-doped TiO2 nanotubes, nanowires and nanorods were prepared by utilizing the nanoconfinement of hollow titanate nanotubes. The procedure entailed two steps, adsorption of ethanol onto the inner walls of the nanotubes and thermal treatment of the complex in an inert nitrogen atmosphere. The resulting catalysts exhibited enhanced photocatalytic activity in the degradation of toluene in the gas phase under both visible and simulated solar light irradiation, as compared to that of commercial Degussa P25. Carbon dopants have also been incorporated into TiO2 micro/nanospheres and nanotubes by chemical vapor deposition in an inert atmosphere, and the resulting TiO2 exhibited much higher photocurrents than commercial P25.28,29 In these studies, the key motivation is to engineer the TiO2 bandgap by carbon dopants so that the efficiency of light absorption and utilization can be improved. However, the synthetic procedure typically requires rather harsh experimental conditions and/or sophisticated instrumentation, and the lack of structural stability of the dopants might compromise the durability of the photocatalysts.Another effective strategy is to prepare carbon and TiO2 nanocomposites. For instance, carbon-coated anatase TiO2 catalysts (>20 nm in diameter) have been prepared by heating a mixture of poly(vinyl alcohol) and commercial TiO2 at elevated temperatures (700–900 °C), which exhibited markedly enhanced photocatalytic activity in the degradation of methylene blue, as compared to that of the original commercial TiO2.30 This was ascribed to the improved crystalline structure of the anatase TiO2 after the heating treatments as well as the suppression of phase transformation from anatase to rutile by the carbon coating layer. In another study,27 effective photocatalysts were prepared by homogeneous dispersion of TiO2 on activated carbon cloths by a process of dip-coating and subsequent annealing in a nitrogen atmosphere, and the photocatalytic activity was assessed quantitatively by the photodegradation of methylene blue under UV irradiation. Yet, in these early studies, the TiO2 nanoparticles are typically rather large (diameter of the order of tens of nm), and whereas the photocatalytic activity of the nanocomposites might be apparently improved, the impacts of the electronic interactions between carbon and TiO2 are mostly ignored.
In the present study, we report the facile synthesis of a functional nanocomposite based on carbon and TiO2 nanoparticles. The carbon nanoparticles were prepared by refluxing the combustion soot of natural gas in nitric acid, and then mixed with titanium(IV) n-butoxide to form C–TiO2 composite nanoparticles by using a simple hydrothermal process. XPS and photoluminescence studies of C–TiO2 suggested that there were effective electronic interactions between the carbon and TiO2 components in the nanocomposites, as compared to pure TiO2 nanoparticles. This unique characteristic led to a marked improvement of the photocatalytic activity of the resulting C–TiO2 nanocomposites in the photodegradation of methylene blue, which was about twice that of the TiO2 nanoparticles alone.
Experimental section
Chemicals
Nitric acid (HNO3, 69.8%, Fisher), sodium carbonate anhydrous (Na2CO3, 99%, Fisher Scientific), 3-chloroaniline (99%, ACROS), titanium(IV) n-butoxide (99%, ACROS), oleic acid (Fisher Scientific), and methylene blue (ACROS) were used as received without further treatment. Water was supplied by a Barnstead Nanopure Water System (18.3 MΩ cm).Synthesis of carbon nanoparticles (CNP)
The preparation of carbon nanoparticles has been reported in our previous studies.31,32 In brief, carbon soot was collected from the inside wall of a glass beaker by placing the beaker upside-down above the flame of a natural gas burner. Then 100 mg of soot was refluxed in 10 mL of HNO3 (5 M) for 12 h. After cooling down to room temperature, the brownish yellow supernatant was filtered and then neutralized by Na2CO3. Finally, the sample was dialyzed against Nanopure water through a dialysis membrane for 3 days, affording purified carbon nanoparticles (CNP). Transmission electron microscopic (TEM) measurements showed that the nanoparticles exhibited a mixture of amorphous and graphitic structures and an average core diameter of 4.8 ± 0.6 nm.31,32Synthesis of TiO2 nanoparticles
TiO2 nanoparticles were prepared by a hydrothermal method.33 Briefly, 0.1 mL of 3-chloroaniline was added into 15 mL of Nanopure water, and then transferred to a Teflon-lined stainless-steel autoclave. Subsequently, 0.17 g of titanium(IV) n-butoxide and 1.0 mL of oleic acid were dissolved in 10 mL of toluene and then transferred to the autoclave. The mixed solution was then subject to hydrothermal reactions at 180 °C for 12 h. The white precipitates were collected and extensively rinsed with methanol.Synthesis of carbon hybridized TiO2 nanoparticles (C–TiO2)
The preparation and processing of carbon hybridized TiO2 was the same as that of TiO2 described above. The only difference was to add 15 mg of carbon nanoparticles to the water phase at a concentration of 1 mg mL−1 prior to the hydrothermal procedure. The resulting C–TiO2 nanocomposite contained about 27 wt% of carbon.Characterization
Transmission electron microscopy (TEM) studies were carried out with a JOEP JEM-2010 TEM microscope operated at 200 kV. The samples were prepared by suspending the powder sample in toluene and dropcasting onto a Cu grid. UV-vis absorption spectra of the samples were measured with an ATI Unicam UV4 spectrometer by using a 1 cm quartz cuvette with a resolution of 2 nm. Fluorescence spectra were acquired with a PTI photoluminescence spectrometer. X-ray photoelectron spectra (XPS) were recorded with a PHI 5400 XPS instrument equipped with an Al Kα source operated at 350 W and at 10−9 Torr. The spectra were charge-referenced to the Au4f7/2 peak (83.8 eV) of sputtered gold.Photocatalysis
Methylene blue at the concentration of 20 mg L−1 was used as the testing solution, and a UV lamp (365 nm, 6 W) was employed as the light source. In a typical experiment, 2 mL of the methylene blue solution was added into a quartz cuvette and 1 mg of photocatalysts was added into the solution and mixed under magnetic stirring for at least 2 h in the dark to establish an adsorption/desorption equilibrium, prior to exposure to the light source. In photocatalytic assessments, the distance between the light source and the cuvette was ca. 10 cm. The solution was under magnetic stirring during the entire experimental procedure. The photocatalytic activities were quantified by collecting the UV-vis absorption spectra of the solution at different reaction intervals.Results and discussion
Fig. 1 depicts two representative TEM micrographs of (A and B) TiO2 and (C and D) C–TiO2 nanoparticles. Despite rather extensive agglomeration in both samples (panels A and C), individual particles remained resolvable with a largely spherical shape and an average diameter of 5.20 nm and 5.05 nm for the TiO2 and C–TiO2 nanoparticles, respectively. In addition, high-resolution TEM measurements exhibited well-defined lattice fringes in both TiO2 and C–TiO2 nanoparticles, where the lattice spacing was estimated to be ca. 0.40 nm (red lines). This is somewhat larger than the d-spacing (0.38 nm) of the (100) diffracting planes of bulk anatase TiO2.34 In a previous study it was found that TiO2 exhibited an apparent linear lattice expansion with a diminishment of the grain size to the nanometer regime (<50 nm), as a result of the formation of surface defect dipoles, in contrast with metal nanoparticles that typically exhibited positive surface pressure and hence lattice contraction instead with shrinking physical dimensions.35 In the present study, the TiO2 nanoparticles were very small, only about 5 nm in diameter. Because of low coordination of the surface sites, bonding interactions with reactive molecules (e.g., H2O) likely occurred, leading to the generation of surface defect dipoles and lattice expansion, as observed above. Nevertheless, one can see that in both samples the TiO2 nanoparticles were of rather pure crystalline phase and rich in high-energy {100} facets, consistent with X-ray diffraction studies (Fig. S1†). This may be advantageous to their photocatalytic performance in the degradation of organic pollutants, as detailed below.34 | ||
| Fig. 1 Representative TEM micrographs of (A and B) TiO2 and (C and D) C–TiO2 nanoparticles. Red lines highlight the lattice fringes of TiO2. | ||
In the above TEM study, it was difficult to resolve the carbon components in C–TiO2 probably because of the low contrast and poor crystallinity as compared to TiO2.32 Yet the electronic interactions between the carbon and TiO2 components in the C–TiO2 hybrids were readily revealed by XPS measurements. Fig. 2(A) depicts the full survey spectra of the TiO2 and C–TiO2 nanoparticles. One can see that in both samples the elements of C, Ti, and O can be clearly identified, with the binding energies of C1s, Ti2p and O1s electrons at approximately 286.6 eV, 459.4 eV and 530.7 eV, respectively (the carbon signals in TiO2 were likely from the organic precursors used to prepare the nanoparticles, whereas in C–TiO2, there were additional contributions from the carbon nanoparticles). However, high-resolution scans showed that the exact binding energies of these electrons were slightly different, as revealed in panels (B) to (D). In panel (B), the Ti2p1/2 and 2p3/2 electrons can be identified at 465.1 eV and 459.4 eV for the TiO2 nanoparticles (bottom curves), and slightly lower for C–TiO2 at 465.0 eV and 459.2 eV (upper curves), suggesting a partial charge transfer from functional moieties on the carbon nanoparticle surface to the Ti(IV) centers in the oxide nanoparticles. Nevertheless, these binding energies (and their corresponding spin–orbital splitting) are highly comparable to those reported in the literature for TiO2. For instance, in a previous study, for anatase TiO2 grown on multiwalled carbon nanotubes (MWCNT) the binding energies for Ti2p electrons were found at 464.9 eV and 458.8 eV,36 whereas for bulk anatase TiO2 at 464.5 eV and 458.8 eV, respectively.37
 | ||
| Fig. 2 (A) XPS survey spectra of TiO2 and C–TiO2 nanoparticles, and high-resolution scans of the binding energies of (B) Ti2p, (C) C1s and (D) O1s electrons in TiO2 (bottom curves) and C–TiO2 (top curves) nanoparticles. | ||
Similar behaviors were observed with the binding energies of the C1s electrons. From panel (C), one can see that for TiO2 nanoparticles (bottom curves), there are two major peaks centered at 285.4 eV and 286.8 eV, which may be assigned to the 1s electrons of sp2 hybridized carbon (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, for instance, from the aniline precursors used in TiO2 nanoparticle synthesis)38 and to the formation of Ti–O–C carbonate species,36,39,40 respectively. For the C–TiO2 nanoparticles (top curves), these two peaks red-shifted slightly to 285.0 eV and 286.7 eV. The significantly greater intensity of the higher energy peak can be ascribed to the presence of carbon nanoparticles where the surface was decorated with various oxygenated functional species and they served as the anchoring sites for TiO2 nanoparticle growth.31,32 Furthermore, a small broad peak can be seen at around 290 eV, which may be assigned to the carboxylate C1s electrons on the carbon nanoparticle surface.31,32,36
C, for instance, from the aniline precursors used in TiO2 nanoparticle synthesis)38 and to the formation of Ti–O–C carbonate species,36,39,40 respectively. For the C–TiO2 nanoparticles (top curves), these two peaks red-shifted slightly to 285.0 eV and 286.7 eV. The significantly greater intensity of the higher energy peak can be ascribed to the presence of carbon nanoparticles where the surface was decorated with various oxygenated functional species and they served as the anchoring sites for TiO2 nanoparticle growth.31,32 Furthermore, a small broad peak can be seen at around 290 eV, which may be assigned to the carboxylate C1s electrons on the carbon nanoparticle surface.31,32,36
For the O1s electrons, one can see from panel (D) that both TiO2 and C–TiO2 nanoparticles exhibited an asymmetric peak within the range of 528 eV to 538 eV, which might be deconvoluted into two major subpeaks. The lower energy peak was located at 530.8 eV for TiO2 and 530.7 eV for C–TiO2 nanoparticles, which was assigned to oxygen bonded to Ti.41–45 The binding energy of the Ti2p electrons was also somewhat lower with C–TiO2 than with TiO2 (panel (B)), which suggests that partial charge transfer might occur from the carbon nanoparticles to TiO2 by Ti–O–C linkages. In addition, for TiO2, the peak at 532.1 eV is likely due to hydroxyl O on the TiO2 surfaces,41,46 whereas for C–TiO2, the second peak was markedly broader, spanning a large range of 528 to 538 eV with center at 533.3 eV, which most probably included contributions from both the TiO2 hydroxyl groups as well as various oxygenated species on the carbon nanoparticle surfaces (such as carbonyl, carboxylate, phenylate, etc.).31,32,47 This again indicates that carbon nanoparticles were indeed incorporated with TiO2 to form nanoscale composites.
The intimate electronic interactions between the carbon and TiO2 nanoparticles in C–TiO2 led to unique interfacial charge transfer upon photoirradiation. Fig. 3 shows the excitation (solid curves) and emission (dashed curves) fluorescence spectra of the carbon, TiO2 and C–TiO2 nanoparticles. It can be seen that the carbon nanoparticles (black curves) exhibited a broad excitation spectrum centered at ca. 311 nm and an equally broad emission peak at approximately 428 nm, a behavior consistent with the results reported previously that was ascribed to electronic transitions of oxygenated functional species on the nanoparticle surface.31,32 For TiO2 nanoparticles (red curves), the fluorescence features were different, where a major excitation peak might be identified at 362 nm, along with two emission peaks at 460 nm and 528 nm. Note that the excitation peak energy (3.4 eV) is somewhat greater than the bandgap energy of bulk anatase TiO2 (3.2 eV), most probably as a result of lattice expansion as observed above (Fig. 1).35 It should be mentioned that reports of photoluminescence emission of TiO2 nanoparticles are generally rather few, as titania is a well-known indirect bandgap semiconductor and in most previous studies the nanoparticles possess low crystallinity and broad size distributions.48–52 In an earlier study, using a sol–gel method we prepared TiO2 nanoparticles (5 nm in diameter) that were attached onto a gold Janus nanoparticle surface and observed apparent fluorescence characteristics with the excitation and emission peaks at ca. 400 and 609 nm, respectively, which were ascribed to electronic transitions involving trap states of the nanoparticles.53
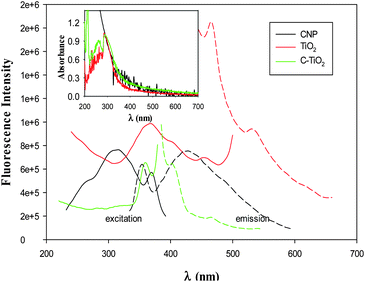 | ||
| Fig. 3 Excitation (solid curves) and emission (dashed curves) fluorescence spectra of carbon, TiO2 and C–TiO2 nanoparticles. The inset shows the corresponding UV-vis spectra of the three samples with the absorbance normalized to the respective value at 290 nm. | ||
However, for the C–TiO2 nanoparticles (green curves), the fluorescence intensity was drastically diminished in both excitation and emission measurements, with the excitation and emission peaks at 360 nm and 403 nm, respectively. Note that the former is very close to that of TiO2 nanoparticles whereas the latter appears to be part of the broad emission envelope of carbon nanoparticles. This suggests that in the composite C–TiO2 nanoparticles, efficient charge/energy transfer likely occurred from TiO2 to carbon under photoirradiation, leading to apparent quenching of the TiO2 fluorescence. This may be facilitated by the oxygenated functional moieties on the carbon nanoparticle surface that behaved as efficient electron acceptors and were suspected to be the active sites for photoluminescence.54
In addition, as manifested in the UV-vis absorption spectra (Fig. 3, inset), one can see that whereas TiO2 nanoparticles exhibited an absorption edge around 360 nm (red curve), the C–TiO2 composite nanoparticles extended the absorption to the visible range (up to 450 nm, green curve), very similar to that of the carbon nanoparticles (black curve). This again indicates that carbon nanoparticles and TiO2 nanoparticles were indeed integrated to form C–TiO2 nanocomposites.
The photocatalytic activity of the C–TiO2 nanocomposites was then examined by using the photodegradation of methylene blue as the illustrating example. Fig. 4 shows the UV-vis absorption spectra of methylene blue in water in the presence of C–TiO2 under UV photoirradiation (365 nm) for various periods of time. It can be clearly seen that within the range of 500 to 800 nm, two major absorption peaks appear at about 612 and 664 nm which were characteristic of methylene blue,55 and when exposed to UV light the absorbance diminished apparently with increasing exposure time, indicating effective photocatalytic activity of the C–TiO2 nanocomposites in the degradation of methylene blue. From the optical absorbance at 664 nm (A664), the fraction of methylene blue that was degraded could be estimated, (Ao,664 − A664)/Ao,664, where Ao,664 denotes the absorbance prior to UV light irradiation (i.e., t = 0 min). The corresponding reaction dynamics is depicted in Fig. 5 (yellow diamonds). The photocatalytic activities of the carbon, TiO2 nanoparticles and their simple mixture were also evaluated in a similar fashion and included for comparison.
 | ||
| Fig. 4 UV-vis absorption spectra of methylene blue (MB) in water as a function of reaction times catalyzed by C–TiO2 nanoparticles. The concentrations of the C–TiO2 nanoparticles and MB were 0.5 mg mL−1 and 20 mg L−1 respectively. | ||
 | ||
| Fig. 5 Photodegradation efficiency of carbon, TiO2, and C–TiO2 nanoparticles, as well as the simple mixture of carbon nanoparticles and TiO2 nanoparticles (C + TiO2) under UV light irradiation. Data were acquired from UV-vis absorption measurements as exemplified in Fig. 4. | ||
It can be seen that for carbon nanoparticles alone (black circles), there was virtually no change of the optical absorption even after four hours of UV photoirradiation of the methylene blue solution, indicating little photocatalytic activity. For TiO2 nanoparticles (red triangles), the fraction of methylene blue that was photodegraded increased almost linearly with reaction time, and after 120 min of UV light irradiation, about 70% of methylene blue was consumed. Note that when carbon and TiO2 nanoparticles were simply mixed together (with a quantity and mass ratio similar to those of C–TiO2) into the reaction solution (green squares), the reactivity in the early stage of the reactions (∼60 min) was almost identical to that of TiO2 nanoparticles, suggesting that the TiO2 nanoparticles were the major contributors in the mixture to the photocatalytic activity; at longer reaction times (>60 min), the activity of the carbon and TiO2 nanoparticle mixtures actually diminished. In sharp contrast, the activity of the C–TiO2 nanocomposites (yellow diamonds) was markedly better. After only 60 min of UV light photoirradiation, more than 75% of methylene blue was degraded already. That is, within the present experimental context, the photocatalytic activity of C–TiO2 nanocomposites was about twice that of TiO2 alone, and the performance increased in the order of carbon nanoparticles < mixture of carbon and TiO2 nanoparticles ≤ TiO2 nanoparticles < C–TiO2 nanocomposites. This indicates that intimate contacts and hence electronic interactions between the carbon and TiO2 nanoparticles in the C–TiO2 nanocomposites were likely to be the key factors that facilitated interfacial charge separation upon photoirradiation and hence led to improved photocatalytic activity, consistent with the effective diminishment of the photoluminescence emission of C–TiO2 as compared to that of TiO2 nanoparticles alone (Fig. 3).
As mentioned earlier, the carbon nanoparticle surface was decorated with various oxygenated functional moieties, including quinone derivatives, such as 9,10-phenanthraquinone, that are well-known electron acceptors.31,32,56 For instance, previously we observed that the carbon nanoparticles exhibited two pairs of voltammetric peaks with the formal potentials at −0.28 V and −0.06 V (vs. Ag/AgCl) in electrochemical measurements.54 Note that in neutral solutions, the conduction band of TiO2 is located at ca. −0.35 V (vs. Ag/AgCl) whereas the valence band is at ca. +2.65 V.57 Thus, under UV photoirradiation, the valence electrons of TiO2 were excited to the conduction band, which then crossed the carbon–TiO2 interface and were accepted by the quinone moieties of the carbon nanoparticles. Such photogenerated electrons were then used to reductively degrade methylene blue, hence leading to the diminishment of the optical absorption (the corresponding holes were likely involved in the oxidation of water). For the physical mixture of carbon and TiO2 nanoparticles, the lack of bonding interactions greatly impeded charge/energy transfer between them and thus they behaved independently, as observed above.
It should be noted that in earlier studies of carbon-supported TiO2, the enhanced photocatalytic activities were largely attributed to the enhanced dispersion and/or crystallinity (by thermal treatments) of TiO2, whereas the impacts of the chemical reactivity of the carbon support were largely ignored.21–27 In the present study, using equally nanosized TiO2 particles as the comparative example, one can see that the rich surface chemistry of carbon nanoparticles might also be exploited to facilitate charge separation upon UV photoirradiation, leading to further enhancement of the photocatalytic performance.
Conclusions
A functional C–TiO2 nanocomposite was prepared by the growth of TiO2 on carbon nanoparticles by a simple hydrothermal procedure. XPS measurements showed that partial charge transfer occurred from carbon nanoparticles to the Ti(IV) centers in the oxide nanoparticles by the Ti–O–C interfacial bonds, in comparison to pure TiO2 nanoparticles. Because of these intimate electronic interactions, the C–TiO2 nanoparticles exhibited apparent diminishment of the photoluminescence as compared to TiO2 and carbon nanoparticles, suggesting that effective electron/energy transfer occurred in the nanocomposites under photoirradiation. Such unique properties were exploited for the enhancement of the photocatalytic activity of C–TiO2 nanocomposites, as illustrated in the photodegradation of methylene blue, which was ascribed to the improved efficiency of interfacial charge separation from TiO2 to carbon because of abundant electron-accepting sites on the carbon nanoparticles.Acknowledgements
This work was supported, in part, by the National Science Foundation (CHE – 1012258, CBET – 1258839 and DMR – 0804049) and the ACS Petroleum Research Fund (49137 – ND10). C. L. acknowledged partial support of a research fellowship from the China Scholarship Council. XPS and TEM studies were carried out at the Molecular Foundry and National Center for Electron Microscopy, Lawrence Berkeley National Laboratory, as part of a user project.Notes and references
- K. Hashimoto, H. Irie and A. Fujishima, Jpn. J. Appl. Phys., 2005, 44, 8269–8285 CrossRef CAS
.
- J. Zhu, J. Yang, Z.-F. Bian, J. Ren, Y.-M. Liu, Y. Cao, H.-X. Li, H.-Y. He and K.-N. Fan, Appl. Catal., B, 2007, 76, 82–91 CrossRef CAS
- P. P. Das, S. K. Mohapatra and M. Misra, J. Phys. D: Appl. Phys., 2008, 41, 245103 CrossRef
- A. Benoit, I. Paramasivam, Y. C. Nah, P. Roy and P. Schmuki, Electrochem. Commun., 2009, 11, 728–732 CrossRef CAS
- Z.-X. Li, Y.-L. Xie, H. Xu, Y.-M. Wang, Z.-G. Xu and H.-L. Zhang, J. Photochem. Photobiol., A, 2011, 224, 25–30 CrossRef CAS
- H.-i. Kim, J. Kim, W. Kim and W. Choi, J. Phys. Chem. C, 2011, 115, 9797–9805 CAS
- H. Y. Yang, S. F. Yu, S. P. Lau, X. Zhang, D. D. Sun and G. Jun, Small, 2009, 5, 2260–2264 CrossRef CAS
- X. Chen, Y. Su, X. Zhang and L. Lei, Chin. Sci. Bull., 2008, 53, 1983–1987 CrossRef CAS
- L. Deng, S. Wang, D. Liu, B. Zhu, W. Huang, S. Wu and S. Zhang, Catal. Lett., 2009, 129, 513–518 CrossRef CAS
- P. Xu, T. Xu, J. Lu, S. Gao, N. S. Hosmane, B. Huang, Y. Dai and Y. Wang, Energy Environ. Sci., 2010, 3, 1128 CAS
- H. Zhao, Y. Chen, X. Quan and X. Ruan, Chin. Sci. Bull., 2007, 52, 1456–1461 CrossRef CAS
- K. E. deKrafft, C. Wang and W. B. Lin, Adv. Mater., 2012, 24, 2014–2018 CrossRef CAS
- J. Wang and Z. Lin, Chem. Mater., 2010, 22, 579–584 CrossRef CAS
- Y. Park, S.-H. Lee, S. O. Kang and W. Choi, Chem. Commun., 2010, 46, 2477 RSC
- T. T. Le, M. S. Akhtar, D. M. Park, J. C. Lee and O. B. Yang, Appl. Catal., B, 2012, 111–112, 397–401 CrossRef CAS
- C. Dong, A. Xian, E. H. Han and J. Shang, Solid State Phenom., 2007, 121, 939–942 CrossRef
- D. Chu, X. Yuan, G. Qin, M. Xu, P. Zheng, J. Lu and L. Zha, J. Nanopart. Res., 2007, 10, 357–363 CrossRef
- Y. Park, W. Kim, H. Park, T. Tachikawa, T. Majima and W. Choi, Appl. Catal., B, 2009, 91, 355–361 CrossRef CAS
- X. Wang, S. Meng, X. Zhang, H. Wang, W. Zhong and Q. Du, Chem. Phys. Lett., 2007, 444, 292–296 CrossRef CAS
- Z. Wu, F. Dong, W. Zhao, H. Wang, Y. Liu and B. Guan, Nanotechnology, 2009, 20, 235701 CrossRef
- S. Mozia, M. Toyoda, T. Tsumura, M. Inagaki and A. W. Morawski, Desalination, 2007, 212, 141–151 CrossRef CAS
- S. Shanmugam and A. Gedanken, Small, 2007, 3, 1189–1193 CrossRef CAS
- B. Tryba, A. W. Morawski, T. Tsumura, M. Toyoda and M. Inagaki, J. Photochem. Photobiol., A, 2004, 167, 127–135 CrossRef CAS
- T. Tsumura, N. Kojitani, I. Izumi, N. Iwashita, M. Toyoda and M. Inagaki, J. Mater. Chem., 2002, 12, 1391–1396 RSC
- B. Tryba, A. W. Morawski and M. Inagaki, Appl. Catal., B, 2003, 46, 203–208 CrossRef CAS
- B. Tryba, A. W. Morawski and M. Inagaki, Appl. Catal., B, 2003, 41, 427–433 CrossRef CAS
- D. Xu, Z.-H. Huang, F. Kang, M. Inagaki and T. H. Ko, Catal. Today, 2008, 139, 64–68 CrossRef CAS
- Y. Li, D.-S. Hwang, N. H. Lee and S.-J. Kim, Chem. Phys. Lett., 2005, 404, 25–29 CrossRef CAS
- G. Wu, T. Nishikawa, B. Ohtani and A. Chen, Chem. Mater., 2007, 19, 4530–4537 CrossRef CAS
- B. Tryba, A. W. Morawski, T. Tsumura, M. Toyoda and M. Inagaki, J. Photochem. Photobiol., A, 2004, 167, 127–135 CrossRef CAS
- Y. Song, X. Kang, N. B. Zuckerman, B. Phebus, J. P. Konopelski and S. Chen, Nanoscale, 2011, 3, 1984 RSC
- L. Tian, D. Ghosh, W. Chen, S. Pradhan, X. Chang and S. Chen, Chem. Mater., 2009, 21, 2803–2809 CrossRef CAS
- D. Pan, N. Zhao, Q. Wang, S. Jiang, X. Ji and L. An, Adv. Mater., 2005, 17, 1991–1995 CrossRef CAS
- L. Wang, L. Zang, J. C. Zhao and C. Y. Wang, Chem. Commun., 2012, 48, 11736–11738 RSC
- G. S. Li, J. Boerio-Goates, B. F. Woodfield and L. P. Li, Appl. Phys. Lett., 2004, 85, 2059–2061 CrossRef CAS
- G. An, W. Ma, Z. Sun, Z. Liu, B. Han, S. Miao, Z. Miao and K. Ding, Carbon, 2007, 45, 1795–1801 CrossRef CAS
- D. Song, J. Hrbek and R. Osgood, Nano Lett., 2005, 5, 1327–1332 CrossRef
- Y. H. Zhang, P. Xiao, X. Y. Zhou, D. W. Liu, B. B. Garcia and G. Z. Cao, J. Mater. Chem., 2009, 19, 948–953 RSC
- L. Zhao, X. Chen, X. Wang, Y. Zhang, W. Wei, Y. Sun, M. Antonietti and M.-M. Titirici, Adv. Mater., 2010, 22, 3317–3321 CrossRef CAS
- S. T. Kochuveedu, Y. J. Jang, Y. H. Jang, W. J. Lee, M. A. Cha, H. Shin, S. Yoon, S. S. Lee, S. O. Kim, K. Shin, M. Steinhart and D. H. Kim, Green Chem., 2011, 13, 3397–3405 RSC
- X.-K. Wang, C. Wang and D. Zhang, Mater. Lett., 2012, 72, 12–14 CrossRef CAS
- R. Hang, X. Huang, L. Tian, Z. He and B. Tang, Electrochim. Acta, 2012, 70, 382–393 CrossRef CAS
- J. Fang, X. Bi, D. Si, Z. Jiang and W. Huang, Appl. Surf. Sci., 2007, 253, 8952–8961 CrossRef CAS
- P. Stefanov, M. Shipochka, P. Stefchev, Z. Raicheva, V. Lazarova and L. Spassov, J. Phys.: Conf. Ser., 2008, 100, 012039 CrossRef
- C. Rath, P. Mohanty, A. C. Pandey and N. C. Mishra, J. Phys. D: Appl. Phys., 2009, 42, 205101 CrossRef
- B. Adolphi, E. Jähne, G. Busch and X. Cai, Anal. Bioanal. Chem., 2004, 379, 646–652 CrossRef CAS
-
C. D. Wagner and G. E. Muilenberg, Handbook of X-ray photoelectron spectroscopy: a reference book of standard data for use in X-ray photoelectron spectroscopy, Perkin-Elmer Corp., Physical Electronics Division, Eden Prairie, Minn, 1979 Search PubMed
- M. Niederberger, M. H. Bartl and G. D. Stucky, Chem. Mater., 2002, 14, 4364–4370 CrossRef CAS
- G. Ramakrishna and H. N. Ghosh, Langmuir, 2003, 19, 505–508 CrossRef CAS
- Y. C. Zhu and C. X. Ding, J. Solid State Chem., 1999, 145, 711–715 CrossRef CAS
- H. Tang, K. Prasad, R. Sanjines, P. E. Schmid and F. Levy, J. Appl. Phys., 1994, 75, 2042–2047 CrossRef CAS
-
J. I. Pankove, Optical processes in semiconductors, Prentice-Hall, Englewood Cliffs, NJ, 1971 Search PubMed
- S. Pradhan, D. Ghosh and S. W. Chen, ACS Appl. Mater. Interfaces, 2009, 1, 2060–2065 CAS
- L. Tian, Y. Song, X. Chang and S. Chen, Scr. Mater., 2010, 62, 883–886 CrossRef CAS
- X. W. Kang and S. W. Chen, J. Mater. Sci., 2010, 45, 2696–2702 CrossRef CAS
- J. E. Gautrot, P. Hodge, M. Helliwell, J. Raftery and D. Cupertino, J. Mater. Chem., 2009, 19, 4148–4156 RSC
- A. J. Nozik and R. Memming, J. Phys. Chem., 1996, 100, 13061–13078 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: XRD patterns of TiO2 and C–TiO2 nanoparticles. See DOI: 10.1039/c3nr01033c |
| This journal is © The Royal Society of Chemistry 2013 |