
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of cellulose nanocrystal surface chemistry and dispersion quality on latex nanocomposite stability, film formation and adhesive properties†
Julia M.
Antoniw
 a,
Vida A.
Gabriel
b,
Michael V.
Kiriakou
c,
Marc A.
Dubé
b,
Michael F.
Cunningham
d and
Emily D.
Cranston
*aef
a,
Vida A.
Gabriel
b,
Michael V.
Kiriakou
c,
Marc A.
Dubé
b,
Michael F.
Cunningham
d and
Emily D.
Cranston
*aef
aDepartment of Chemical and Biological Engineering, University of British Columbia, Vancouver, British Columbia V6T 1Z4, Canada. E-mail: emily.cranston@ubc.ca
bDepartment of Chemical and Biological Engineering, University of Ottawa, Ottawa K1N 6N5, Ontario, Canada
cAnomera Inc., Montreal, Quebec H3B 1A7, Canada
dDepartment of Chemical Engineering, Queen's University, Kingston K7L 3N9, Ontario, Canada
eDepartment of Wood Science, University of British Columbia, Vancouver, British Columbia V6T 1Z3, Canada
fUBC Bioproducts Institute, 2385 East Mall, Vancouver, British Columbia V6T 1Z4, Canada
First published on 26th January 2024
Abstract
Cellulose nanocrystals (CNCs) are sustainably sourced, non-toxic, high-strength nanoparticles most often derived from wood pulp. The incorporation of CNCs into latexes via in situ semi-batch emulsion polymerization has been shown to improve the performance of latex nanocomposites, specifically latex-based pressure sensitive adhesives (PSAs). A bench-scale study was designed to compare the effect of incorporating CNCs with different surface chemistries and dispersion quality on the final latex properties, film formation, and adhesive performance. Poly(butyl acrylate/methyl methacrylate)-CNC latex nanocomposites (at 40 wt% solids) were successfully synthesized with 1 wt% sulfated CNCs and carboxylated CNCs (DextraCel™) with different storage methods (never-dried suspension vs. dried powder). All CNCs were well-dispersed in water using probe sonication prior to being incorporated into the latex polymerization reactions. Extensive characterization revealed differences in the latex and PSA film properties, with never-dried carboxylated CNCs and dried sulfated CNCs having the highest viscosities, lowest relative colloidal stabilities by visual inspection, and most enhanced adhesive performance. Additionally, PSA films containing dried carboxylated CNCs exhibited the greatest latex particle coalescence, as measured by atomic force microscopy, which correlated to improved cohesive strength. The ability to tune latex properties with CNCs may facilitate the widespread use of “greener” water-based polymerization methods, even for applications outside of adhesives, such as paints, coatings, inks, toners and rubbers.
Introduction
Polymer nanocomposites are heterogeneous systems consisting of one or more nanoparticle types dispersed in a polymer matrix. Nanoparticles can impart desirable thermal, mechanical, optical and rheological properties without degrading polymer processability.1,2 Bio-based nanomaterials are of growing interest as they are renewable, abundant and their use supports efforts to eliminate the global dependency on fossil fuels. Cellulose nanocrystals (CNCs) are plant-derived nanoparticles that can serve as reinforcing fillers for rubber, cement, coating and adhesive applications.3 CNCs have a high aspect ratio, a high tensile strength, and are highly stable in aqueous media which makes them particularly appealing for use in latex-based nanocomposites.4,5 In addition to replacing petroleum-derived fillers, the hydrophilic nature of CNCs allows for their incorporation into hybrid acrylate-based materials using emulsion6–15 and miniemulsion16–19 polymerization (i.e., water-based synthetic routes) as opposed to solvent-based polymerization methods – eliminating the requirement for evaporation of environmentally taxing solvents.Pressure sensitive adhesives (PSAs) are a class of polymeric materials that instantaneously adhere to a surface upon the application of light pressure, most commonly they form a temporary bond which should be removable without leaving a residue.20 As industries move towards implementing “greener” chemistry practices, water-based PSAs have become more prominent in the market. Research efforts to incorporate CNCs into nanocomposites using emulsion polymerization have intensified, with the goal of achieving bond durability and bond strength proportionate to solvent-based PSAs.6–15 In 2018, we first reported that the incorporation of CNCs into nanocomposites for PSA applications caused simultaneous improvement of all three PSA performance metrics (tack, peel strength, and shear strength).7 This was unprecedented as generally reinforcing fillers which promote the improvement of one property (i.e., shear strength) cause deterioration of others (i.e., tack or peel strength) due to different mechanisms governing the properties. While we have suggested multiple mechanisms for the observed improvement in PSA properties the work is ongoing and the full range of “input parameters” has yet to be explored.
Emulsion polymers are complex, multi-phase systems consisting of aqueous dispersions of stabilized polymer particles; they are also referred to as latexes.21 The properties of latex nanocomposites can be affected by many factors including the glass transition temperature (Tg), gel content, molecular weight, particle size, particle size distribution, dispersion quality of the particles, and other additives.20 When incorporating CNCs into latexes, in situ addition has outperformed simple blending post latex synthesis. When CNCs are added in situ they are better dispersed which allows for increased interaction between the CNCs and the polymer particles.6 It is also speculated that when incorporated in situ, the CNCs may be lightly tethered to some polymer chains depending on the radical initiator used in the synthesis.7 CNCs are sensitive to pH and ionic strength which is an important consideration as emulsion polymerization contains other ionic species, including surfactants and initiators.14,19,22
Other additives which can affect the latex properties include crosslinker, buffer and chain transfer agents. A sequential design study of acrylic latex nanocomposites, which incorporated 0.5 wt% CNCs, demonstrated that adhesion decreases with high loadings of surfactant but tack and peel strength can be improved by the addition of chain transfer agents.11 In addition, CNCs can be incorporated up to 4 wt% without aggregation,13 though depending on the monomer system, the greatest improvement of all PSA properties occurs at fairly low loadings; with more hydrophilic monomers able to tolerate higher CNC loadings before aggregation than hydrophobic monomer systems.8
Latex nanocomposites targeting adhesive applications generally require a combination of hard (i.e., methyl methacrylate (MMA), polymer Tg = 105 °C) and soft (i.e., butyl acrylate (BA), polymer Tg = −54 °C) monomers to attain a low Tg.23 Based on previous work, BA and MMA combined in a 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 ratio by mass, produces latexes with Tg values around −39 °C.6,7,14,15 Another important formulation consideration is the target solids content, as it is time, energy and cost efficient to store and use latexes with high solids contents. One method of achieving industrially relevant latexes (>40 wt% solids content) is to use a seeded starved feed approach during the emulsion polymerization. This is when the majority of the monomer is fed to the reactor slowly, after the latex seed particles have already formed, which yields high solids content latexes with small-sized particles.21
:10 ratio by mass, produces latexes with Tg values around −39 °C.6,7,14,15 Another important formulation consideration is the target solids content, as it is time, energy and cost efficient to store and use latexes with high solids contents. One method of achieving industrially relevant latexes (>40 wt% solids content) is to use a seeded starved feed approach during the emulsion polymerization. This is when the majority of the monomer is fed to the reactor slowly, after the latex seed particles have already formed, which yields high solids content latexes with small-sized particles.21
The role of CNC surface group functionality on the latex synthesis and nanocomposite performance is not well-understood as sulfated CNCs (sCNCs) have been used in the majority of PSA nanocomposite research.24 Surface group functionality is primarily determined by the isolation method (i.e., the process for extracting nanoparticles from cellulosic materials such as acid hydrolysis, oxidation or enzymatic treatment)25 but can also be altered post-production.26 CNCs are commonly extracted through acid hydrolysis, whereby, the less ordered regions of cellulose are preferentially degraded, producing highly crystalline nanoparticles; when sulfuric acid is used, the CNCs possess surface sulfate half-ester groups which impart colloidal stability. In this work, sCNCs were provided by Celluforce Inc. (Montreal, Canada), a company with tonne-per-day production capacity. Anomera Inc. (Montreal, Canada) produces carboxylated CNCs (cCNCs) under the tradename DextraCel™ which have carboxylate surface groups and are produced using a more environmentally benign hydrogen peroxide oxidation method.27 Anomera Inc. produces cCNCs at pilot and demonstration plant scales up to 150 tonnes per year. Whereas sCNCs have strong acid surface groups and high surface charge densities, cCNCs have weak acid surface groups and relatively low surface charge densities.28,29 Work by our group has demonstrated that surface charge density exerts the greatest influence on CNC colloidal stability (and salt sensitivity) likely affecting networking and self-interaction capabilities of CNCs; herein cCNCs have a lower surface charge than sCNCs and are considered less colloidally stable.22 In this study, we examine how the different surface functional groups on cCNCs and sCNCs translate to latex CNC–polymer interactions, specifically as it relates to the adhesive performance of the latex.
The dispersion quality of nanoparticles has been correlated to their effect on the properties of nanocomposites, with enhanced material performance often attributed to good nanoparticle dispersion.2 cCNCs and sCNCs are commercially distributed as neutralized sodium-form suspensions in water, henceforth referred to as never-dried cCNCs and never-dried sCNCs. From a transportation and storage perspective, it is more economical to dry the suspensions into powders through spray or freeze drying, which can be later redispersed in water, as needed.30 Storage of CNCs in a dried form may also preserve their shelf-life by hindering the loss of surface charge groups following auto-catalyzed reactions.31 In this study, cCNCs and sCNCs received as powders were fully redispersed in water prior to use but are henceforth referred to as dried cCNCs and dried sCNCs referring to their storage/shipping form. Recent work has demonstrated that for cCNCs, better dispersion does not strictly equate to better performance.15 Never-dried cCNCs which were not ultrasonicated prior to their incorporation into latex nanocomposites were reported to have a higher “apparent” aspect ratio which translated to improved PSA properties.14 It has also been reported that the never-dried sCNCs have a needle-like structure (with a particle length in the nm range), whereas the structure of dried sCNCs are more flake-like (with a particle length in the μm range),32 which highlights that sometimes the drying process can permanently alter the morphology of the nanoparticles. The storage method of commercial CNCs, never-dried in suspension versus dried powder, and how this translates to their behaviour and dispersion quality within latex nanocomposites has yet to be explored.
This paper represents a comprehensive study of the semi-batch emulsion polymerization of BA/MMA latexes incorporating carboxylated/sulfated CNCs for adhesive applications at a bench scale (100 mL). Bench-scale testing is important to develop new understanding and overcome challenges (specifically in stability) without wasting large volumes of material. This work provides the framework for future scale-up studies, as prior investigations into PSA nanocomposites with CNCs have focused on larger volumes (>1 L) in a reactor.6–9,14,15 Following the successful synthesis of latexes, extensive characterization was performed to elucidate the influence that CNC surface chemistry and dispersion quality have on latex and cast film material properties which will ideally inform decisions on how to leverage CNCs to tailor PSA nanocomposites for specific applications.
Experimental
Materials
Butyl acrylate (BA), methyl methacrylate (MMA) monomers (both >99% pure and stabilized by 10–60 ppm monomethyl ether hydroquinone) and aluminum oxide (activated, basic) were purchased from Sigma Aldrich (St Louis, MO). Each monomer was passed through a packed column of aluminum oxide to remove the hydroquinone inhibitor prior to use in emulsion polymerizations. Sodium dodecyl sulfate (SDS) surfactant and potassium persulfate (KPS) initiator were purchased from Sigma Aldrich and used as received. DextraCel™ carboxylated CNCs (cCNCs) were acquired from Anomera Inc. (Montreal, Canada) in a never-dried form (4.6 wt% suspension in water) and in a dried form (spray-dried powder). The sulfated CNCs (sCNCs) were acquired from CelluForce Inc. (Montreal, Canada) in a never-dried form (6.4 wt% suspension in water) and in a dried form (spray-dried powder). The never-dried cCNCs, dried cCNCs, never-dried sCNCs, and dried sCNCs were received in sodium-salt form and were not purified. MilliQ-grade water with a resistivity of 18.2 MΩ cm was used for all experiments and characterization.Cellulose nanocrystal sample preparation
Dispersing dried cCNCs or sCNCs. Dried cCNCs (as received) in sodium form, were slowly added to water at room temperature under vigorous stirring until no visible aggregates remained. The resulting suspension was left to sit undisturbed for 1 h at room temperature. Next, the suspension was probe sonicated using a Sonifier 550 from Branson Ultrasonics (Brookfield, CT) at 60% amplitude for 15 min total (3 × 5 min) in an ice bath (8250 kJ g−1).33 To remove any contaminants the suspensions were filtered through glass microfiber filter paper with a 2.7 μm pore size. The dried sCNCs were treated following the same protocol.
Dispersing never-dried cCNCs or sCNCs. Never-dried cCNCs (4.6 wt% dispersion, as received) were diluted with water at room temperature and mixed vigorously on a magnetic plate with a stir bar for 5 min. The resulting suspension was left to sit undisturbed for 1 h at room temperature prior to being ultrasonicated. Next, the suspension was probe sonicated at 60% amplitude for 1.5 min total (3 × 0.5 min) in an ice bath (825 kJ g−1).33 To remove any contaminants the suspensions were filtered through glass microfiber filter paper with a 2.7 μm pore size. The never-dried sCNCs (6.4 wt% dispersion, as received) were treated following the same protocol.
Poly(butyl acrylate/methyl methacrylate)-CNC latex synthesis
:10 BA/MMA latexes was adapted from previous work where a reactor was used and the scale of the reactions was between 0.5–1 L.6–9,14,15 In this work, 100 mL BA/MMA 40 wt% solids content latexes, combined in a 90:10 ratio by mass of BA to MMA, were synthesized by semi-batch emulsion polymerization. The reaction was carried out in a 250 mL three neck round-bottom flask (RBF) with a 24/40 center joint, equipped with a polished glass stir shaft and a half-moon stir blade, and two 19/22 side joints. The stir shaft was rotated by a Fisherbrand™ Overhead Stirrer (Hampton, NH) which delivered constant mixing at 275 rpm. The pre-feed emulsion and initiator solution were fed separately into the RBF through needles connected to syringes which were driven by a Chemyx Fusion 4000 independent dual-channel syringe pump (Stafford, TX). In addition to synthesizing latexes which incorporated CNCs, a 40 wt% control and a 50 wt% control latex were synthesized with no CNCs.
A seeded starved feed approach was employed, where 10% of the monomer was added during the seed stage and 90% of the monomer was added during the feed stage of the reaction. The formulation and method to produce 90:10 BA/MMA latexes is described in detail in Table 1. Briefly, water, surfactant, and CNCs were added to the RBF. The RBF was made airtight by securing the stir shaft and blade through the center joint using an adapter and sealing both side joints with rubber stoppers. The sealed RBF was suspended in an oil bath at 60 °C and the suspension of water, CNCs and surfactant were stirred for 5 min. The stirring was stopped, and two needles were punctured through one of the rubber stoppers, the first to connect a nitrogen (N2) gas line and the second to provide a vent. The tip of the needle connected to the N2 line was submerged in the suspension, which was then bubbled with N2 for 30 min. Once the suspension was sufficiently purged of oxygen, the needle connected to the N2 line was moved to the headspace; where it supplied N2 for the duration of the reaction and the stirring was restarted.
:10 BA/MMA latexes which incorporate 1 wt% CNCs (or 1 part per hundred monomer) in situ
| Mixture | Description | Component | Amount (g) | |
|---|---|---|---|---|
| Abbreviations: BA, butyl acrylate; CNC, cellulose nanocrystal; KPS, potassium persulfate; MMA, methyl methacrylate; RBF, round bottom flask; rpm, revolutions per minute; SDS, sodium dodecyl sulfate. | ||||
| Seed stage | Seed emulsifier | At 60 °C the suspension of CNCs with water was mixed with SDS and was then bubbled with nitrogen for 30 min. | Water | 28 |
| SDS | 0.070 | |||
| CNC | 0.040 | |||
| Seed monomer | While stirring at 275 rpm, the seed monomer (10% of total monomer) was injected into the RBF and the headspace was purged with nitrogen for 10 min. | BA | 3.6 | |
| MMA | 0.40 | |||
| Seed initiator | Initiator dispersed in water was injected into the RBF. The seed stage is an additional hour and is under a blanket of nitrogen with stirring at 275 rpm. | Water | 2 | |
| KPS | 0.012 | |||
| Feed stage | Pre-emulsion feed | The CNCs in suspension were mixed with SDS. The combined monomers were added and stirred vigorously with a magnetic stirrer to emulsify. This mixture was fed into the reactor at a rate of 1.1 mL min−1 for 55 min. | Water | 20 |
| SDS | 0.63 | |||
| CNC | 0.36 | |||
| BA | 32.4 | |||
| MMA | 3.8 | |||
| Initiator feed | KPS was mixed with water using a magnetic stirrer. The solution was fed into the reactor at a rate of 0.17 mL min−1 for 59 min. | Water | 10 | |
| KPS | 0.11 | |||
| Cook stage | RBF was left to stir at 60 °C for one additional hour. | |||
At this point, 10% of the total monomer was injected into the RBF and water, CNCs, surfactant, and monomer was mixed. After 10 min of stirring, a KPS initiator solution (0.012 g in 2 g water) was injected into the RBF to initiate the seeding reaction. This seeding was allowed to progress for 1 h. Concurrently, a pre-emulsion was made by combining water, CNCs, and SDS using a magnetic stir bar and then slowly adding mixed BA/MMA monomer and stirring until the mixture was white in colour and opaque. A second initiator solution was also prepared during this time by combining KPS and water using a magnetic stir bar until no visible solids remained. After the 1 h seeding reaction had completed, the pre-emulsion feed and the initiator solution were fed to the reactor via needles, punctured through the second rubber stopper and connected to 60 mL syringes secured on a syringe pump. The pre-emulsion was fed to the RBF for 55 min at a rate of 1.1 mL min−1 and the initiator solution was fed for 59 min at a rate of 0.17 mL min−1. After the pre-emulsion and initiator solution were fed to the RBF, it was left to stir for an additional 1 h. To stop the reaction, the rubber stoppers on the RBF were removed and the emulsion was exposed to air quenching the KPS initiator.
Poly(butyl acrylate/methyl methacrylate)-CNC latex characterization
Poly(butyl acrylate/methyl methacrylate)-CNC film characterization
The Si wafers were first cleaned with piranha solution, a mixture of concentrated sulfuric acid and hydrogen peroxide at 3:1, then rinsed with purified water and then spin coated using a WS-650-23 Spin Coater, from Laurell Technologies (North Whales, PA) with the diluted latex samples. Each film was spin coated at 3000 rpm with 2300 rpm per s acceleration for 30 s. Height images were acquired using a Bruker Multimode 8 AFM (Santa Barbara, CA), in tapping mode at room temperature, using Al coated silicon probes with a 42 N m−1 spring constant and 350 kHz resonance frequency from NCHR probes from Asylum Research – Oxford Instruments (Santa Barbara, CA). Imaging was carried out at a scan rate of 0.25 Hz with a resolution of 512 measurements per line (512 lines). All images were processed with a standard third order polynomial flattening using the NanoScope analysis software v.8.10.
Casting the latexes into films. To prepare adhesive films for PSA testing and characterization, approximately 18 g of latex was cast using a Meyer rod (no. 50) to produce films of approximately 39 g m−1. Films were dried for 48 h at 50 ± 5% relative humidity at 23 ± 2 °C, and films were tested under these same conditions.
Peel strength. The PSTC-101 standard was employed for the peel strength test.37 Specifically, test A for 180° peel, was used. Prepared films were cut into 1 in × 12 in strips and applied along the center of a stainless-steel testing plate, adhesive side down. The strips were applied to the testing panel at approximately 10 mm s−1 and were firmly adhered to the plate using a weighted steel roller (2040 ± 45 g) by rolling the film twice in each lengthwise direction. All samples displayed adhesive, as opposed to cohesive, failure leaving no residue on the panel.
Loop tack. The PSTC-16 standard was employed for the loop tack test.37 To form a loop, the prepared films were cut into 1 in × 5 in strips and shaped with the adhesive side facing outward. The loop was secured with a 1 in piece of masking tape and this had the dual benefit of creating a thicker edge for the Instron tester grips. During the test, the loop was lowered, at a rate of 2 mm s−1, on the stainless-steel testing panel until there was 1 in of contact. The strip was lifted by the tester, at a rate of 5 mm s−1, and the maximum force required to remove the strip was recorded. All samples displayed adhesive, as opposed to cohesive, failure leaving no residue on the panel.
Shear strength. For the shear strength test, the PSTC-107A standard was modified, as using a 1 in × 1 in adhesive strip exhibited unreasonably long failure times.37 Instead strips with 0.5 in × 0.5 in testing areas were used. Adhesive strips were cut into 0.5 in × 5 in strips and placed in such a way that a 0.5 in × 0.5 in adhesive surface was in contact with the testing panel. To apply the adhesive strips to the test plate a weighted steel roller (2040 ± 45 g) was used and each strip was rolled twice in each lengthwise direction at a rate of approximately 10 mm s−1. A 500 g mass was affixed to each tape and the time to shear failure was recorded. All samples displayed cohesive, as opposed to adhesive, failure where residue remained on the panel.
Results and discussion
This work presents a bench-scale (100 mL) semi-batch emulsion polymerization study whereby 90:10 BA/MMA latexes for PSA applications were synthesized. In total, six latexes were synthesized: a 40 wt% control, a 50 wt% control, and four latexes which incorporated different CNC types – never-dried cCNCs, dried cCNCs, never-dried sCNCs, and dried sCNCs. The 40 wt% control latex is presented in all the characterizations, excluding the PSA testing, as it is a direct comparative benchmark with the 40 wt% latexes incorporating CNCs. The synthesis of the 50 wt% latex was necessitated by the observation that the 40 wt% control latex, when cast, yielded a non-uniform film, rendering it unsuitable for PSA testing. By opting for a film composition at 50 wt%, we effectively mitigated this issue and successfully cast uniform films suitable for PSA testing. We attributed this to the higher viscosity afforded by the increased number of solid particles in the 50 wt% control latex. Consequently, the results of the 50 wt% control are also incorporated into the viscosity and base characterization for a comprehensive analysis. The choice of employing a 50 wt% latex as a baseline for comparison with the other 40 wt% latexes is justifiable, because the size of the latex particles was similar (180–190 nm) and the films were cast to the same thickness (39 g m−1).
We sought to understand how the individual and coupled effects of surface chemistry and storage method/dispersion quality influence the latex quality, stability, film formation, and PSA performance. Linking the emulsion polymerization process conditions, the latex and cast film properties, and final product performance is necessary for future developments and improvement of tailored nanocomposites.
Bench-scale emulsion polymerization
A bench-scale setup was designed for the synthesis of BA/MMA latexes for PSA applications. The latexes were synthesized in a 250 mL RBF and the final volume of the formulation was 100 mL. The system design was adapted from BA/MMA latex synthesis procedures which were performed in both 1 L stainless steel and 0.5 L glass reactors.6–9,14,15 The monomer system chosen was to allow for direct comparison to previous works which have investigated CNC-latex nanocomposites for PSA applications.6,7,14,15 When using an RBF, the introduction of oxygen into the system is more likely because the openings cannot be sealed as efficiently as a larger scale reactor. During the initial latex syntheses, the monomer conversion was poor, and this was attributed to contaminating oxygen quenching the KPS initiator. Instead of only purging the system with N2, the protocol was modified such that a constant stream of N2 was flowing through the RBF for the duration of the polymerization. This improvement resulted in excellent monomer conversion (above 93%) for all reported latexes. An additional consideration when working with a bench-scale system is understanding the energy input from stirring. When the system was stirred at 250 rpm, the latex synthesis was less reproducible. Once the stir speed was increased to 275 rpm the synthesis was less likely to coagulate or yield aggregated latexes. Increasing the stir speed helped to improve the dispersion quality of the numerous components in the system which was important for maintaining colloidal stability.This hybrid emulsion polymerization proceeds by a free-radical polymerization mechanism, whereby after the initial batch or “seeding” reaction, a slow feeding of the monomer to the reactor drives high instantaneous overall monomer conversion and minimizes preferential monomer consumption.21 From our past work with CNC-nanocomposite polymer systems, we anticipate that the CNCs play a fairly passive role and remain in the water phase during emulsion polymerization. Upon drying the latexes, the CNCs appear on, around or in between polymer particles with no evidence that CNCs are encapsulated, as discussed later. Miniemulsion polymerization would also be suitable here and has been demonstrated in the past, specifically with BA/EHA for PSA applications.18 However, the additional sonication step required for miniemulsion polymerization is preferably avoided due to issues with scalability.
The formulation and method developed to produce latexes on a bench-scale was highly reproducible. The system required careful handling and a rigorous, systematic approach as it was sensitive to minor deviations in the procedure. Each latex was synthesized three times, with little variation in the characteristics and quality of the latexes, which allowed for the three latex runs to be mixed giving a final “combined” latex. A comparison of the average characterization values from the three individual latex runs, compared to the final “combined” latex highlights the low batch-to-batch variability, as shown in the ESI, Table S2.† Overall, the development of a reproducible bench-scale semi-batch emulsion polymerization protocol was the first step in studying latex nanocomposites which incorporated CNCs with different surface functionalities and storage methods.
Assessing cellulose nanocrystal dispersion quality
The CNCs were dispersed in the total available water for the polymerization reaction which allowed them to be incorporated into the latex synthesis in situ. The size by AFM, apparent (relative) particle size by DLS, and charge content of the four CNC suspension “types” (never-dried cCNCs, dried cCNCs, never-dried sCNCs, and dried sCNCs) are compared in Table 2. sCNCs are slightly larger than cCNCs and have a higher surface charge density, however the aspect ratio of all CNCs are within error which is a crucial variable for rheological performance and reinforcement potential.| Never-dried cCNCs | Dried cCNCs | Never-dried sCNCs | Dried sCNCs | |
|---|---|---|---|---|
| a Not measured but the primary particle size is assumed to be the same as the values reported for the CNCs stored as dried powders. b Average value from the seed and feed dispersion which were prepared separately, for the three latex syntheses. Standard deviation is for n = 6 repeats. | ||||
| Length (nm) | 150 ± 3029 |
180 ± 9028 |
||
| Cross section (nm) | 5 ± 229 |
6 ± 228 |
||
| Aspect ratio | 30 ± 1029 |
3128 |
||
| Charge content (mmol kg−1 CNC) | 160 ± 1022 |
141 ± 1029 |
286 ± 722 |
250 ± 1028 |
| DLS apparent sizeb (nm) | 71 ± 0.3 | 78 ± 3 | 79 ± 2 | 90 ± 4 |
A direct comparison of the apparent particle size by DLS and the particle size measured by AFM illustrates the limits of DLS with rod-like particles. Whereas DLS gives a value of ∼80 nm for dried cCNCs, by AFM their length is ∼150 nm, highlighting that DLS can give reasonable relative sizes and serve as a measure of dispersion quality.38 Past work implied that the dispersion quality of CNCs in a latex dictates the extent of the property improvements afforded by their incorporation; specifically, if CNCs are not well-dispersed they can trigger coagulation in the latex and the formation of heterogeneous films with reduced PSA performance.24 Therefore, all four CNC types were probe sonicated and filtered until they were deemed fully “nanodispersed”, with apparent particles sizes under 100 nm prior to their incorporation into the latex synthesis. Attaining size values under this 100 nm threshold required different total energy inputs for dried compared to the never-dried CNCs (3 × 5 min vs. 3 × 0.5 min). However, this difference was justified, as effective CNC dispersion is necessary to capitalize on the beneficial properties of CNCs, specifically the high aspect ratio and high surface to volume ratio, which render them effective property modifiers.24 Furthermore, this level of dispersion was needed to avoid coagulation during emulsion polymerization. All CNCs were colloidally stable with zeta potential values ca. −25 mV, in agreement with past benchmarking studies.28,29 It should be noted that the never-dried cCNCs and never-dried sCNCs are considered well-dispersed as received but were probe sonicated to ensure consistency between the treatment of the never-dried and dried samples and to mitigate any effects of sample ageing on agglomeration.
The dried sCNCs were noticeably harder to disperse in water than the dried cCNCs. When dispersing the dried sCNCs the protocol of sonicating, filtering and checking the apparent particle size was repeated more than twice to attain a suspension that was well-dispersed (particle size <100 nm). The higher charge content of sCNCs should help with redispersion but did not appear to play a big role. Previous work by Gabriel et al. noted similar difficulties where dried cCNCs dispersed completely (by eye) after 30 min of mixing with a stir bar at room temperature, whereas, dried sCNCs required two (or more) hours of vigorous mixing.15 Rationally, the sulfate half ester groups on the sCNCs are a better hydrogen bond acceptor than the carboxylate groups on the cCNCs, which creates stronger hydrogen bonding (and maybe more hornification) between dried sCNC nanoparticles – this would support more irreversible aggregation during drying for sCNCs than cCNCs. Another possible explanation for why dried sCNCs were more difficult to disperse is that they are known to bind less water on their surface (i.e., are slightly more hydrophobic) compared to their carboxylated counterparts,39 which could suggest a lower driving force for redispersion. Specifics of the drying processes to produce CNC powders will also affect aggregation/redispersibility but was outside of our control as all samples were commercially supplied. Therefore, when working with dried CNCs it is recommended to confirm proper dispersion as this can contribute to batch-to-batch variability and performance differences in the latexes.
Latex characterization
The latexes synthesized with the four CNC types were characterized to compare monomer conversion, solids content, latex particle size, polydispersity index (PDI), zeta potential and pH (Table 3). The latexes were visually indistinguishable, and all reactions proceeded similarly. The characterization data shows only minor differences between the 40 wt% control and the latexes which incorporated CNCs. Although the pKa of cCNCs is 5.1, we have shown that colloidal stability can still be maintained under the pH conditions employed during the polymerization.22 The pH of the latex was tested at two different time points during the 4 h reaction and it remained within the stable range for cCNCs at low salt concentrations (i.e., pH 3–11).22 More specifically, the pH at the end of the batch reaction was between 7–8 for all the latexes and dropped to between 3–5 by the end of the polymerization, likely due to KPS decomposition in the absence of buffer. Based on our previous study,22 we chose not to use buffer because of the extreme sensitivity of cCNCs and sCNCs to ionic strength. For cCNCs at ≤1.5 wt% the onset of colloidal instability is around 5 mM of added salt. Therefore, the addition of 0.45 mM of KPS (initiator) and 2.43 mM of SDS (anionic surfactant), which both contribute to the ionic strength, was below this 5 mM limit. In addition, past work on SDS-CNC interactions supports that this surfactant-additive combination does not contribute to instability below 100 mM for sCNCs and 20 mM for cCNCs.14,19 Overall, achieving a stable polymerization without CNC aggregation or polymer coagulation demands a fine balance of monomers, stabilizers, initiators, additives and reaction conditions. Consistent characteristics among the latexes based on Table 3 data allows us to infer that differences in the cast PSA films arise from how CNCs influence latex stability, wettability, film formation/coalescence, and drying, and not from nuanced differences in the synthesis results.| 40 wt% control (no CNCs) | 50 wt% control (no CNCs) | Never-dried cCNCs | Dried cCNCs | Never-dried sCNCs | Dried sCNCs | |
|---|---|---|---|---|---|---|
| Conversion (%) | 98 ± 0.1 | 96 ± 0.8 | 93 ± 0.1 | 93 ± 0.1 | 95 ± 0.6 | 97 ± 0.9 |
| Solids content (%) | 39 ± 0.1 | 48 ± 0.2 | 37 ± 0.1 | 37 ± 0.5 | 38 ± 0.2 | 39 ± 0.4 |
| Latex particle size (nm) | 192 ± 2 | 179 ± 1 | 185 ± 1 | 186 ± 7 | 180 ± 3 | 182 ± 2 |
| Latex PDI | 0.02 ± 0.01 | 0.03 ± 0.01 | 0.2 ± 0.04 | 0.2 ± 0.05 | 0.06 ± 0.01 | 0.2 ± 0.02 |
| Zeta potential (mV) | −24 ± 1 | −18 ± 6 | −35 ± 1 | −42 ± 2 | −49 ± 2 | −19 ± 3 |
| Latex pH | 3.4 | 3.7 | 3.8 | 3.1 | 3.6 | 3.4 |
The latex particle size by DLS was under 200 nm for all synthesized latexes, which agrees with values reported in previous BA/MMA latex work.6 Interestingly, the PDI for the 40 wt% control was monodispersed, whereas the PDI for latexes with CNCs was larger with the exception of the never-dried sCNCs. It is likely that interactions between CNCs and polymer particles are captured in this measurement as the broader PDI implies the presence of individual CNCs, polymer particles and polymer–CNC clusters. Similar PDI values have been reported for BA/MMA control latexes (PDI = 0.04–0.05),6 however, the cCNC-latex composites had higher PDI values than previously reported. Additionally, the zeta potential values were around or above the threshold (−20 mV) for latex particles in suspension to be considered colloidally stable,34 suggesting that the latexes remain stable in the presence of 1 wt% CNCs.14 However, the zeta potential of the dried sCNC latexes was statistically different from the other CNC-containing latexes, which affected the performance and stability of the latex.
All of the high-solids content latexes produced were shelf stable and remained well-dispersed for more than 6 months at room temperature by visual inspection. To compare relative stability however, the latexes were agitated using a vortex mixer for 1 min (Fig. 1). This high energy/high shear affected the colloidal stability of the samples differently. The never-dried cCNCs and the dried sCNCs (Fig. 1A and D) were visually observed to completely destabilize and became coagulated/aggregated, whereas, the dried cCNCs and never-dried sCNCs and 40 wt% control (Fig. 1B, C and E) remained fairly well-dispersed. The zeta potential of the dried sCNC latex (Table 3) was the closest to zero compared to the other CNC-containing latexes which correlates to this latex displaying the most significant coagulation. Qualitative observations were used because it was difficult to capture this information quantitatively as the coagulated latex could not be sampled for optical size measurements.
 | ||
| Fig. 1 Visual comparison of BA/MMA latex stability following 60 s of vortex mixing for (A) never-dried cCNCs, (B) dried cCNCs, (C) never-dried sCNCs, (D) dried sCNCs and (E) 40 wt% control. | ||
Using a parallel-plate rheometer, the viscosities of the latexes were measured over a range of shear rates (0.01–100 Hz); all of the latexes containing CNCs had higher viscosities than the 40 wt% control (Fig. 2). The latexes exhibited non-Newtonian shear thinning behavior, where the viscosity decreased with increasing shear rate. The latex with never-dried cCNCs had the highest viscosity which was significantly larger than all other CNC-containing latexes and even surpassed the 50 wt% control. From an application perspective, this is highly advantageous because it correlates improved surface coverage of the cast films which can translate into increased marginal value of the product. The other latex viscosities fell between the 40 and 50 wt% controls with essentially overlaid viscosity profiles at low shear values.
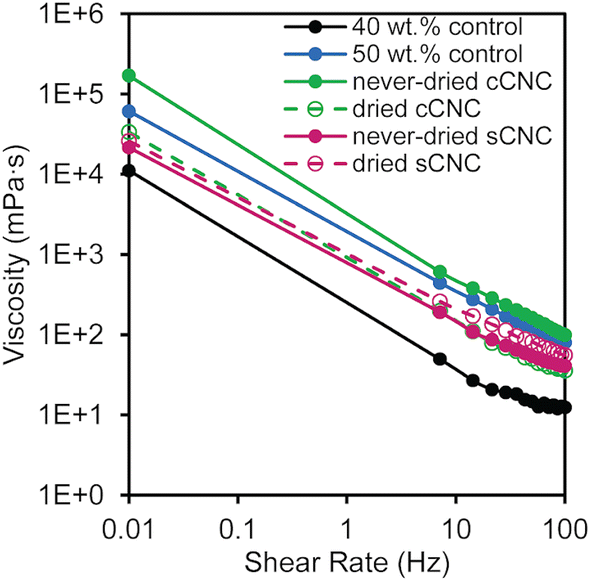 | ||
| Fig. 2 Viscosity as a function of angular frequency of BA/MMA liquid latexes containing CNCs with different surface functionalities (carboxylated vs. sulfated) and storage methods (never-dried vs. dried). | ||
The higher-than-the-control viscosities resulting from in situ incorporation of CNCs during the latex synthesis are attributed to CNC rheological behavior and CNC–polymer networking.10 The increase is more than an additive effect of latex viscosity plus CNC viscosity, since the viscosity of dispersions is generally dominated by the continuous phase. The continuous water phase here contains the CNCs, which are good rheological modifiers40 with viscosities ranging from 1–100 mPa s at 1 wt%,41 such that the order of magnitude increase in viscosity shown in Fig. 2 is reasonable. Furthermore, the viscosity behavior supports that significant CNC aggregation does not occur, as this would lead to profiles very similar to the 40 wt% control as few nanoparticles would be present throughout the suspension. Viscosity trends do not follow what would be expected based on CNC surface charge or aspect ratio alone (Table 2)29,41 which supports that CNCs may be tethered to the polymer particles. During polymerization, the presence of the KPS initiator can facilitate hydrogen abstraction on the surface of the CNCs enabling grafting-from polymerization or chain termination.42 The main contributions to viscosity are the new networked structures and reduced free space between polymer particles in the presence of CNCs.
The latexes with never-dried cCNCs and dried sCNCs have the highest viscosities at high shear (Fig. 2) which suggests that they experience the most “interactions” between CNCs and polymer particles out of the four CNC types. Initial characterization of the CNC suspensions prior to their addition to the emulsion polymerization, confirmed they were fully dispersed after sonication (Table 2). Therefore, higher latex viscosities may indirectly point to systems where CNC dispersion was best maintained through the polymerization (as aggregation would necessarily reduce the number of CNC–polymer interactions). Interestingly, the differences in the relative latex stability (Fig. 1) align with the viscosity results, suggesting that more particle–polymer interactions may also promote latex coagulation. While there are many factors that dictate the CNC–CNC and CNC–polymer interactions within the latex, the significantly different behavior for never-dried cCNCs and dried sCNCs imply dispersion and network differences. As such, we sought to investigate the latex film morphologies by AFM and the PSA performance of the nanocomposites prior to drawing conclusions about the role of the CNC properties listed in Table 2 and the solution conditions of the formulations.
CNC-latex nanocomposite films
AFM was used to study the effects of CNC surface chemistry on the film formation of CNC-latex nanocomposites. Height images of spin coated, dilute BA/MMA latexes revealed that CNC surface chemistry does affect film formation (Fig. 3). The latex particles in the film containing dried cCNCs experienced greater coalescence when compared to both the control and the film containing dried sCNCs, this suggests more CNC–polymer interactions with cCNCs. We note that the external forces exerted by spin coating can further flatten the soft latex particles and change the natural film coalescence (compared with bar coating generally used to produce PSA films), however, this dilution and film preparation technique has allowed us to elucidate interesting trends in past work.12 Clearly, the cCNCs and sCNCs in the film are interacting with the polymer particles, appearing on, within, and around the particles (Fig. 3B and C, insets), in agreement with the literature.8–10,14,36 As Table 3 suggests that the particle size and PDI of all three latexes was similar in suspension, the larger polymer “islands” in Fig. 3B are interpreted to be coalesced polymer particles where coalescence is facilitated by the presence of cCNCs. Therefore, we surmise that the onset of film coalescence/formation is earlier, ultimately leading to a more cohesive film with cCNCs compared to sCNCs, attributed to a greater degree of networking, discussed below. | ||
| Fig. 3 AFM height images of BA/MMA latex films with (A) 40 wt% control (no CNCs), (B) 1 wt% dried cCNCs and (C) 1 wt% dried sCNCs. Image has a height scale from −10 to 10 nm. | ||
The dried cCNCs were reasoned to interact more with each other in comparison to the sCNCs, because they have a lower surface charge content (Table 2), which leads to weaker electrostatic repulsion between particles. Again, this is supported by our past work showing the lower colloidal stability of these cCNCs and their salt sensitivity compared to sCNCs.22 Higher order structuring of the rigid, rod-like particles within the latex may lead to the dried cCNCs having an increased apparent aspect ratio which would enable them to bridge the gap between polymer particles and form a percolated (interconnected) network more easily (as shown previously by AFM).15 Even the addition of CNCs at 1 wt% (i.e., 1 part per hundred monomer) appears to provide sufficient cellulose–cellulose interactions to tether soft particles together, and promote particle coalescence during drying.12 End-to-end assembly of cCNCs has been reported when they are not fully redispersed from dry,15 however, here all CNCs were fully dispersed by probe sonication initially, suggesting that the assembly and networking of cCNCs may occur during latex synthesis (where solution conditions promote some agglomeration) and/or during film formation. Ultimately, this can be advantageous as it provides additional elasticity and strength to the polymer matrix which translates to the nanocomposites having improved cohesive strength (as seen in the PSA properties, discussed below).
PSA properties of BA/MMA CNC-latex nanocomposites
PSA films for performance testing were cast to the same thickness instead of the same coating weight because the strength of an adhesive is correlated with film thickness – thinner films have higher adhesive strength.43Fig. 4 shows the tack, peel strength, and shear strength results for CNC-latex films compared to two controls: a film cast from a 50 wt% BA/MMA latex without CNCs, and a post-it™ “super sticky big notes” film as a commercially available adhesive product.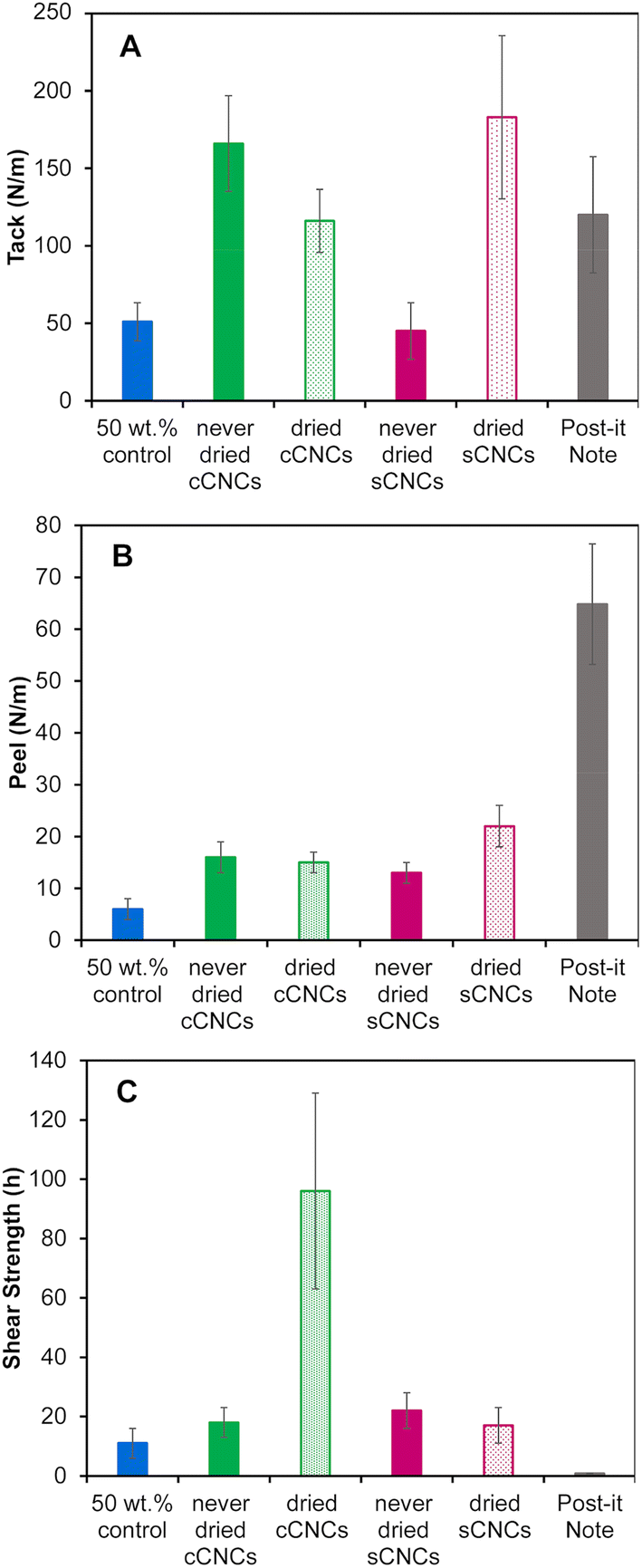 | ||
| Fig. 4 (A) Tack (B) peel strength and (C) shear strength measurements of latex films with 1 wt% incorporated CNCs having different surface chemistry (carboxylated vs. sulfated) and storage methods (never-dried vs. dried). Error bars represent the standard deviation of n ≥ 3 repeats. | ||
Unfortunately, the 40 wt% control latex was unable to form a uniform film with the required thickness due to poor surface wetting leading to the appearance of holes, hence the need for the 50 wt% control. However, this exemplifies CNCs’ ability to give a latex a higher “apparent” solids content as the 40 wt% with CNCs, behaves more like a 50 wt% control latex in terms of film casting (and viscosity, Fig. 2). Latex characterization (Table 2) of the 40 wt% and 50 wt% control shows they are equivalent in regard to conversion, particle size, PDI, zeta potential and pH, and can therefore be reasonably interchanged for PSA performance metric testing. The CNC surface chemistry and storage method clearly affected the adhesive and cohesive PSA properties with moderate to substantial improvements depending on the metric but without a clear “winner”. Statistical analysis to determine the significance of the PSA testing results was performed using an independent Student's t-test (ESI, Table S3†) and was considered in the interpretation of the results below.
Overall, the in situ addition of 1 wt% CNCs to a 40 wt% BA/MMA latex significantly improved the PSA properties (tack, peel and shear strength) of the films in comparison to the 50 wt% control (except for tack of never-dried sCNCs). This agrees with previous work which incorporated cCNCs15 and sCNCs6,7,9 into BA/MMA latexes and reported (this unique!)44 simultaneous improvement of all PSA properties. In general, cCNCs outperformed sCNCs, and never-dried CNCs outperformed dried CNCs, with some exceptions. We note that PSA improvements from CNCs in this work were less dramatic compared to published studies,7,15 attributed to the fact that we emphasized sonicating the CNC suspensions and confirming they were fully dispersed prior to their incorporation in the latex polymerization. This good starting dispersion resulted in stable/reproducible syntheses and the production of very uniform latexes (Table 3) (which are likely easier to control at the bench-scale). Disappointingly, this suggests that highly dispersed CNCs may not maximize the networking potential of rod-like nanoparticles and that some self-assembly of CNCs is an advantage. It also points to benefits emerging from larger scale polymerization reactors with CNCs incorporated without sonication, which is industrially favorable, as long as the latex synthesis can proceed without instabilities.
More specifically, the never-dried cCNCs and dried sCNCs had the highest tack (Fig. 4A) and the values were not statistically different from each other. The peel strength (Fig. 4B) follows the same trend as the tack, with the dried sCNCs having the highest tack followed closely by never-dried cCNCs and then the dried cCNCs and never-dried sCNCs. We speculate that the closer-to-zero zeta potential of the dried sCNC latexes when compared to the other CNC-containing latexes may have contributed to the improved tack and peel strength of the films, as reduced repulsion between latex particles could result in a more densely packed polymer network during the drying process. In addition, these adhesive property improvements link to the latex viscosity and shelf-life stability, where these indirect measurements supporting more CNC–polymer interactions (higher viscosity and lower relative stability) support that never-dried cCNC and dried sCNC latexes experience the greatest improvement in adhesive performance when compared to the control.
Both tack and peel strength performance are strongly dependent on latex film uniformity, the wettability of the latex on the substrate, and the ability for the dried film to have good contact with the opposing surface.20 This suggests that the never-dried cCNC and dried sCNC films are homogeneous and uniform which is an indication of good nanoparticle dispersion.24 It is difficult to discern if the behaviour of never-dried cCNCs and dried sCNCs is caused by their surface functionality, the storage method of never-dried versus dried, or is a compounded effect of both; likely, it is because these two CNC types had the least CNC aggregation and most dispersed CNCs throughout the PSA film. Unfortunately, fully assessing the location of CNCs in real PSA films (not the dilute model systems, like Fig. 3) has been found to be highly challenging because of the small quantity of CNCs and lack of contrast with the polymer in electron microscopy. Because the solution conditions under which the latexes were synthesized were generally within the colloidal stability windows for cCNCs and sCNCs we believe the surface chemistry plays a less dominant role in CNC–polymer interactions than for CNC–CNC interactions.
The shear strength (i.e., the cohesion within the film) showed the largest improvement out of all PSA properties with dried cCNCs significantly outperforming all other CNC types (Fig. 4C). Specifically, cCNC-containing films had shear strengths 5.3×, 4.4× and 5.6× higher than never-dried cCNCs, never-dried sCNCs, and dried sCNCs, respectively. These results are in line with the improved film formation/particle coalescence suggested by AFM (Fig. 3). Particle coalescence and the presence of networked structures within the film impart strength and elasticity, which translates to high shear strength.7 The shear strength of the dried cCNC nanocomposite film was 96 ± 33 h which is unprecedented, as previous work with dried cCNC-containing PSAs reported a shear strength of less than 15 h.15 The highest shear strength that has been achieved with sCNCs at 0.75 wt% is 160 h, for an acrylic latex which incorporated polymerizable surfactant.45 Surprisingly, there was no statistical difference between the shear strength of the never-dried cCNCs, never-dried sCNCs and dried sCNCs or the 50 wt% control implying the impressive performance with dried cCNCs was a synergistic combination of CNC surface chemistry and tendency to self-assemble.
Conclusions
This work detailed the set-up and protocol to study BA/MMA latexes synthesized by semi-batch emulsion polymerization at the bench-scale. The latexes were modified with either carboxylated or sulfated CNCs, which had been stored as either a never-dried suspension or as a dried powder. Prior to incorporation, the never-dried cCNCs, dried cCNCs, never-dried sCNCs, and dried sCNCs were extensively probe sonicated and DLS measurements confirmed they were similarly well-dispersed – this led to highly stable and reproducible latexes. Despite the similar starting degree of dispersion, it seems that CNCs are irreversibly changed by drying processes and these nuanced differences likely affect the tendency of nanoparticles to self-assemble during in situ latex polymerization and/or during PSA film formation.As the CNCs and latexes were colloidally stable under the conditions employed, it was difficult to pinpoint whether changes in the latex properties were triggered by the surface chemistry or the storage method; likely it was a compound effect of both. The high latex viscosity and lower visually observed relative stability suggests that the most CNC–polymer interactions occurred for never-dried cCNCs and dried sCNCs and this correlated to the most uniform PSA films with the best tack and peel strength (i.e., adhesive properties). On the other hand, dried cCNCs were inferred to have more CNC–CNC networking than dried sCNCs (attributed to their lower surface charge density), which promoted latex particle coalescence, better film formation, and a huge spike in PSA shear strength (i.e., cohesive properties). Unfortunately, without AFM images of never-dried cCNCs we cannot make the same claims for all cCNCs but PSA shear strength testing imply that never-dried cCNCs were not as effective at promoting film coalescence.
An understanding of how different CNCs alter the properties of latex nanocomposites can be leveraged to tailor PSAs for a particular application. For example, a PSA for protective films requires high cohesive strength but low adhesive strength, so as not to leave a residue, in which case, dried cCNCs would be best as they have a very high shear strength. Similarly, PSAs for permanent labels would require good adhesive strength but rely less on cohesive strength and so never-dried cCNCs and dried sCNCs, which impart good tack and peel strength, would be preferred. If you required CNC formulations with very high viscosities, then it would be advantageous to use never-dried cCNCs as their viscosity at a 40 wt% solids content mimics that of the 50 wt% solids content control latex. These examples highlight that tailoring the properties of PSA nanocomposites will allow for more widespread use of greener water-based polymerization methods, even for applications outside of adhesives, such as paints, coatings, inks, toners, and rubbers.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Anomera Inc. (Montreal, QC) for providing the DextraCel (i.e., cCNCs, carboxylated CNCs), and for technical input from their staff; specifically, Dr. Nathan Hordy, Dr. Amir Khabibullin, Dr. Timothy Morse, and Marie-Soleil Lebrun. The authors also thank the staff at BASF; specifically, Dr. Sean George, Dr. Gary Deeter, Dr. Kyle Flack, and Dr. Kevin Payne for their technical input. The authors are grateful for the support of the Natural Science and Engineering Council of Canada's (NSERC) Alliance Grant (ALLRP 560215-20) entitled “Enhancing latex-based coatings and pressure sensitive adhesives with carboxylated cellulose nanocrystals”. We express our gratitude to Dr. Megan Roberts and Dr. Fioralba Taullaj for their assistance in editing the manuscript. We thank Prof. Orlando Rojas, Prof. Johan Foster, Prof. Scott Renneckar and Prof. Feng Jiang for access to equipment and expertise. E. D. C. is grateful for support and recognition through the University of British Columbia's President's Excellence Chair initiative, the NSERC E. W. R. Steacie Memorial Fellowship alongside the Canadian Foundation for Innovation (John R. Evans Leaders Fund) used to purchase the AFM (project number 38623). We would like to acknowledge the work of Parisa Bayat, who performed PSA testing and Dr. Elina Niinivaara who characterized the CNCs by AFM.References
- K. I. Winey and R. A. Vaia, MRS Bull., 2007, 32, 314–319 CrossRef CAS.
- S. K. Kumar, B. C. Benicewicz, R. A. Vaia and K. I. Winey, Macromolecules, 2017, 50, 714–731 CrossRef CAS.
- S. Kalia, A. Dufresne, B. M. Cherian, B. S. Kaith, L. Avérous, J. Njuguna and E. Nassiopoulos, Int. J. Polym. Sci., 2011, 2011, 837875 Search PubMed.
- Y. Habibi, L. A. Lucia and O. J. Rojas, Chem. Rev., 2010, 110, 3479–3500 CrossRef CAS PubMed.
- A. Dufresne, in Monomers, Polymers and Composites from Renewable Resources, ed. M. Belgacem and A. Gandini, Elsevier, Amsterdam, 2008, pp. 401–418 Search PubMed.
- Z. Dastjerdi, E. D. Cranston and M. A. Dubé, Macromol. React. Eng., 2017, 11, 1700013 CrossRef.
- Z. Dastjerdi, E. D. Cranston and M. A. Dubé, Int. J. Adhes. Adhes., 2018, 81, 36–42 CrossRef CAS.
- A. Ouzas, E. Niinivaara, E. D. Cranston and M. A. Dubé, Macromol. React. Eng., 2018, 12, 1–10 Search PubMed.
- A. Ouzas, E. Niinivaara, E. D. Cranston and M. A. Dubé, Polym. Compos., 2019, 40, 1365–1377 CrossRef CAS.
- A. S. Pakdel, E. Niinivaara, E. D. Cranston, R. M. Berry and M. A. Dubé, Macromol. Rapid Commun., 2021, 42, 2000448 CrossRef CAS PubMed.
- A. S. Pakdel, V. Gabriel, R. M. Berry, C. Fraschini, E. D. Cranston and M. A. Dubé, Cellulose, 2020, 27, 10837–10853 CrossRef CAS.
- E. Niinivaara, A. Ouzas, C. Fraschini, R. M. Berry, M. A. Dubé and E. D. Cranston, Philos. Trans. R. Soc., A, 2021, 379, 20200330 CrossRef CAS PubMed.
- V. A. Gabriel, E. D. Cranston and M. A. Dubé, Macromol. React. Eng., 2020, 14, 2000027 CrossRef CAS.
- V. A. Gabriel, P. Champagne, M. F. Cunningham and M. A. Dubé, Can. J. Chem. Eng., 2022, 100, 767–779 CrossRef CAS.
- V. A. Gabriel, M. N. Tousignant, S. M. W. Wilson, M. D. M. Faure, E. D. Cranston, M. F. Cunningham, B. H. Lessard and M. A. Dubé, Macromol. React. Eng., 2022, 16, 2100051 CrossRef CAS.
- E. M. Dogan-Guner, F. J. Schork, S. Brownell, G. T. Schueneman, M. L. Shofner and J. C. Meredith, Polymer, 2022, 240, 124488 CrossRef CAS.
- S. Roberge and M. A. Dubé, Polymer, 2006, 47, 799–807 CrossRef CAS.
- M. Karimi Shamsabadi and M. R. Moghbeli, Int. J. Adhes. Adhes., 2017, 78, 155–166 CrossRef CAS.
- S. A. Kedzior, H. S. Marway and E. D. Cranston, Macromolecules, 2017, 50, 2645–2655 CrossRef CAS.
- R. Jovanović and M. A. Dubé, J. Macromol. Sci., Part C – Polym. Rev., 2004, 44, 1–51 CrossRef.
- P. A. Lovell and F. J. Schork, Biomacromolecules, 2020, 21, 4396–4441 CrossRef CAS PubMed.
- J. M. Antoniw, M. T. Hallman, M. V. Kiriakou, T. Morse and E. D. Cranston, Langmuir, 2023, 39, 10321–10334 CrossRef CAS PubMed.
- J. Bicerano, Prediction of Polymer Properties, CRC Press Boca Raton, 3rd edn, 2002 Search PubMed.
- S. A. Kedzior, V. A. Gabriel, M. A. Dubé and E. D. Cranston, Adv. Mater., 2021, 33, 2002404 CrossRef CAS PubMed.
- O. M. Vanderfleet and E. D. Cranston, Nat. Rev. Mater., 2021, 6, 124–144 CrossRef CAS.
- Y. Habibi, Chem. Soc. Rev., 2014, 43, 1519–1542 RSC.
- M. Andrews and T. Morse, CA Pat, CA2956661C, 2015 Search PubMed.
- M. S. Reid, M. Villalobos and E. D. Cranston, Langmuir, 2017, 33, 1583–1598 CrossRef CAS PubMed.
- G. Delepierre, O. M. Vanderfleet, E. Niinivaara, B. Zakani and E. D. Cranston, Langmuir, 2021, 37, 8393–8409 CrossRef CAS PubMed.
- S. Beck, J. Bouchard and R. Berry, Biomacromolecules, 2012, 13, 1486–1494 CrossRef CAS PubMed.
- S. Beck and J. Bouchard, Nord. Pulp Pap. Res. J., 2014, 29, 6–14 CrossRef CAS.
- K. M. Z. Hossain, V. Calabrese, M. A. Silva, J. Schmitt, S. J. Bryant, T. Islam, R. M. Felfel, J. L. Scott and K. J. Edler, Macromol. Mater. Eng., 2021, 306, 2000462 CrossRef CAS.
- M. Girard, D. Vidal, F. Bertrand, J. R. Tavares and M. C. Heuzey, Ultrason. Sonochem., 2021, 71, 105378 CrossRef CAS PubMed.
- S. Bhattacharjee, J. Controlled Release, 2016, 235, 337–351 CrossRef CAS PubMed.
- L. O. Hellström, M. Samaha, K. Wang, A. Smits and M. Hultmark, Meas. Sci. Technol., 2015, 26, 015301 CrossRef.
- S. A. Kedzior, M. Kiriakou, E. Niinivaara, M. A. Dubé, C. Fraschini, R. M. Berry and E. D. Cranston, ACS Macro Lett., 2018, 7, 990–996 CrossRef CAS PubMed.
- Test Methods for Pressure Sensitive Adhesive Tapes, Pressure Sensitive Tape Council, Naperville, Illinois, 2014 Search PubMed.
- E. J. Foster, R. J. Moon, U. P. Agarwal, M. J. Bortner, J. Bras, S. Camarero-Espinosa, K. J. Chan, M. J. D. Clift, E. D. Cranston, S. J. Eichhorn, D. M. Fox, W. Y. Hamad, L. Heux, B. Jean, M. Korey, W. Nieh, K. J. Ong, M. S. Reid, S. Renneckar, R. Roberts, J. A. Shatkin, J. Simonsen, K. Stinson-Bagby, N. Wanasekara and J. Youngblood, Chem. Soc. Rev., 2018, 47, 2609–2679 RSC.
- X. Peng, L. Manna, W. Yang, J. Wickham, E. Scher, A. Kadavanich and A. P. Alivisatos, Nature, 2000, 404, 59–61 CrossRef CAS PubMed.
- Y. C. Ching, M. E. Ali, L. C. Abdullah, K. W. Choo, Y. C. Kuan, S. J. Julaihi, C. H. Chuah and N. S. Liou, Cellulose, 2016, 23, 1011–1030 CrossRef CAS.
- S. Shafiei-Sabet, W. Y. Hamad and S. G. Hatzikiriakos, Cellulose, 2014, 21, 3347–3359 CrossRef CAS.
- S. Kalia and M. W. Sabaa, in Polysaccharide based graft copolymers, ed. S. Kalia and M. W. Sabaa, Springer Berlin, Heidelberg, 1st edn, 2013 Search PubMed.
- S. C. L. Fischer, S. Boyadzhieva, R. Hensel, K. Kruttwig and E. Arzt, J. Mech. Behav. Biomed. Mater., 2018, 80, 303–310 CrossRef CAS PubMed.
- Z. Dastjerdi, E. D. Cranston, R. Berry, C. Fraschini and M. A. Dubé, Macromol. React. Eng., 2019, 13, 1800050 CrossRef.
- J. Hamlin, M. Nuruddin, V. Tarabara and C. Szczepanski, AIChE J., 2022, 68, e17910 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The number of repeats performed per sample per test for PSA testing, latex characterization comparison between averaged results versus measured results of combined latexes, statistical analysis of the PSA testing results. See DOI: https://doi.org/10.1039/d3lp00244f |
| This journal is © The Royal Society of Chemistry 2024 |