
Alpha-metalated N,N-dimethylbenzylamine rare-earth metal complexes and their catalytic applications
Yesmin Akter
Rina
and
Joseph A. R.
Schmidt
 *
*
Department of Chemistry & Biochemistry, School of Green Chemistry and Engineering, College of Natural Sciences and Mathematics, The University of Toledo, 2801 W. Bancroft St. MS 602, Toledo, Ohio 43606-3390, USA. E-mail: Joseph.Schmidt@utoledo.edu
First published on 14th May 2024
Abstract
This perspective summarizes our group's extensive research in the realm of organometallic lanthanide complexes, while also placing the catalytic reactions supported by these species within the context of known lanthanide catalysis worldwide, with a specific focus on phosphorus-based catalytic reactions such as intermolecular hydrophosphination and hydrophosphinylation. α-Metalated N,N-dimethylbenzylamine ligands have been utilized to generate homoleptic lanthanide complexes, which have subsequently proven to be highly active lanthanum-based catalysts. The main goal of our research program has been to enhance the catalytic efficiency of lanthanum-based complexes, which began with initial successes in the stoichiometric synthesis of organometallic lanthanide complexes and utilization of these species in catalytic hydrophosphination reactions. Not only have these species supported traditional lanthanide catalysis, such as the hydrophosphination of heterocumulenes like carbodiimides, isocyanates, and isothiocyanates, but they have also been effective for a plethora of catalytic reactions tested thus far, including the hydrophosphinylation and hydrophosphorylation of nitriles, hydrophosphination and hydrophosphinylation of alkynes and alkenes, and the heterodehydrocoupling of silanes and amines. Each of these catalytic transformations is meritorious in its own right, offering new synthetic routes to generate organic scaffolds with enhanced functionality while concurrently minimizing both waste generation and energy consumption. Objectives: We aim for the research summary presented herein to inspire and encourage other researchers to investigate f-element based stoichiometric and catalytic reactions. Our efforts in this field began with the recognition that potassium salts of benzyldimethylamine preferred deprotonation at the α-position, rather than the ortho-position, and we wondered if this regiochemistry would be retained in the formation of lanthanide complexes. The pursuit of this simple idea led first to a series of structurally fascinating homoleptic organometallic lanthanide complexes with surprisingly good stability. Fundamental studies of the protonolysis chemistry of these complexes ultimately revealed highly versatile lanthanide-based precatalysts that have propelled a catalytic investigation spanning more than a decade. We anticipate that this summative perspective will animate the synthetic as well as biological communities to consider La(DMBA)3-based catalytic methods in the synthesis of functionalized organic scaffolds as an atom-economic, convenient, and efficient methodology. Ultimately, we envision our work making a positive impact on the advancement of novel chemical transformations and contributing to progress in various fields of science and technology.
 Yesmin Akter Rina | Yesmin Akter Rina was born and raised in Bangladesh, where she completed her B.S. and M.S. degrees in chemistry from Jahangirnagar University. After that, she pursued her Ph.D. at the University of Toledo under the supervision of Dr. Joseph A. R. Schmidt, focusing on rare-earth-metal catalyzed organic transformations. Her research interests involved designing and synthesizing organic scaffolds, method development, organometallic complex synthesis, and catalysis. She is passionate about solving problems in chemistry and making impactful contributions in scientific research. |
 Joseph A. R. Schmidt | Joseph A. R. Schmidt was born and raised in western Kansas, USA, and attended college at Kansas State University, completing joint B.S. degrees in Chemistry and Mathematics in 1997. He then went on to complete a Ph.D. at the University of California, Berkeley in 2002 working with Prof. John Arnold. After a post-doctoral stint with Geoff Coates at Cornell University, he began his independent research career at the University of Toledo in 2004. He has since risen to the rank of Professor and his chief research interests lie in the development of new small molecule catalysts as well as all things related to rare-earth metals. |
Introduction
Homogeneous catalysis is undoubtedly a crucial tool in modern synthetic organic chemistry and medicinal chemistry with useful applications in the development of new drugs and therapeutic agents. Within the broad field of homogeneous catalysis, there has been a reinvigorated focus on rare-earth metal catalysis along with transition metal catalysis. Buoyed by the application of rare-earth elements in various sectors such as technology, environment, economics, and everyday life, many organometallic rare-earth metal complexes find successful application as precatalysts or intermediates in a diverse array of catalytic transformations including hydrofunctionalization,1–16 various types of polymerization and copolymerization,17–28 and even the creation of thin films.29–32 Research on rare-earth elements, specifically organolanthanide chemistry, has led to significant advancements, resulting in better understanding of their distinctive properties and applications, and has been extensively reviewed. Frank T. Edelmann's annual surveys, which have been published since 1992,33–56 focusing on recent developments in organo-f-element chemistry, serve as a valuable resource in keeping researchers informed with up-to-date knowledge of the field, while also driving further developments.Since the initial report of the first homoleptic organolanthanide complexes, the tris(cyclopentadienyl) rare-earth metal complexes by Wilkinson and Birmingham in 1954,57 cyclopentadienyl ligands (both substituted and unsubstituted) have dominated the field of organolanthanide chemistry due to their ability to form readily isolable complexes.58 Much recent research has focused on the exploration of cyclopentadienyl-free organolanthanide compounds, with several reviews devoted to these non-cyclopentadienyl organolanthanide complexes.59–66 The desire to focus on non-cyclopentadienyl systems stems from the synthetic limitations inherent to the Cp ligand, most notably the overall difficulties related to incorporation of significantly electron-donating or -withdrawing groups on the cyclopentadienyl core. In this perspective, we will focus primarily on non-cyclopentadienyl lanthanide chemistry and pay particular attention to homoleptic organometallic lanthanide complexes, as there has been extensive investigation into lanthanide alkyl and aryl complexes recently. While many ligands have been explored, a select few, such as alkylsilane (–CH2SiMe3), benzyl (–CH2C6H5), silylamide (–N(SiMe3)2), and their derivatives, have emerged as viable alternatives to the commonly used cyclopentadienyl ligands. A key feature of each of these three ligand classes centers on the non-coordinating nature of these ligands upon entry into catalytic cycles via protonolysis. That is, catalyst activation (for example, treatment with a phosphine oxide, R2P(O)H) yields byproducts SiMe4, toluene, or HN(SiMe3)2 that are all non-coordinating, preventing hindering or poisoning of the resulting catalysts. The trimethylsilylmethyl complexes, Ln(CH2SiMe3)3(THF)x, proved to be readily accessible, with the yttrium complex first reported by Lappert and coworkers,67 followed by subsequent reports of samarium, terbium, erbium, thulium, ytterbium, and lutetium complexes.68–70 These Ln(CH2SiMe3)3(THF)x complexes serve as versatile starting materials for the subsequent production of cationic complexes and heteroleptic lanthanide complexes through reaction with Lewis acids, weak Brønsted acids, and other weakly acidic ligand precursors, leading to applications in various reactions, including ethylene polymerization and intramolecular hydroamination.71–74 In 1974, Lappert and coworkers made a significant breakthrough, producing the first solvent-free homoleptic lanthanide alkyl complex, Y(CH(SiMe3)2)3,75 which has proven useful subsequently.76–78 Development of the sterically larger tris(trimethylsilyl)methyl ligands [–C(SiMe3)3] has played a crucial role in stabilizing some of the most reactive lanthanides, including divalent europium and samarium, with its larger steric bulk preventing unwanted side reactions and lanthanate formation.79–81 Related explorations led to benzyl supported lanthanide complexes, such as La(CH2Ph)3(THF)3 and La(CH2-p-tolyl)3(THF)3,82 and this chemistry was later expanded to include Ce, Pr, Sm, Gd, Dy, and Er.83 In order to produce more stable compounds, benzyl ligands with pendant donor groups, such as amino groups, were also developed for rare-earth metal complexes.84–88 For example, the ortho-aminobenzyl ligands have been utilized broadly across the lanthanide series, giving the various homoleptic lanthanide complexes (o-Me2N-C6H4CH2)3Ln. These ligands proved flexible enough in both size and hapticity to accommodate the widely varying ionic radii of the full lanthanide series, as well as the related Sc and Y ions. Upon catalyst activation by protonolysis, the o-Me2N-C6H4CH3 groups proved to coordinate to lanthanides very weakly in their neutral form, minimizing catalyst poisoning. The reactivity of the array of benzyl complexes has been extensively studied with much focus on protonolysis reactions using weak Brønsted acids, polymerization, and cross-coupling of terminal alkynes with isocyanides.82,89–92 A wide range of N-donor ligands, including silylamides, N-heterocycles, pyrroles, and Schiff-base ligands, have also been utilized to stabilize and regulate the reactivity of lanthanide complexes. Among these ligands, silylamides like bis(trimethylsilyl)amide have proven useful in coordination of many rare-earth metals, including Sc3+, La3+, Ce3+, Pr3+, Nd3+, Eu2+, Eu3+, Dy3+, Er3+, Yb2+, and Yb3+.93–98 The diverse reactivity and electronic properties of these complexes make them valuable in various catalytic processes and synthetic transformations.99–101
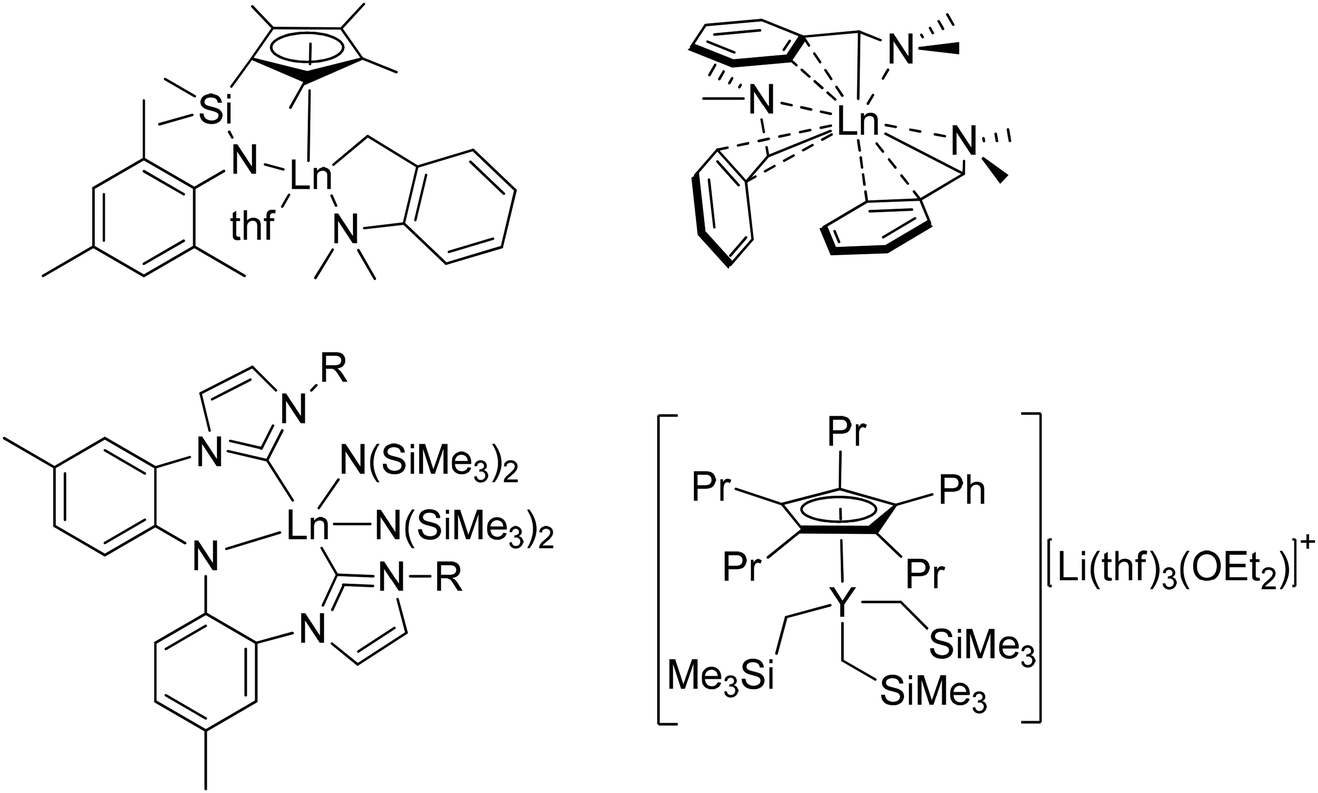
Unfortunately, despite the many successes noted above, the production of homoleptic rare-earth metal complexes remains quite challenging and suffers from significant synthetic drawbacks.65,67,83,102–107 The key step in the production of most organometallic rare-earth metal complexes involves introduction of the functionally relevant ligands either by a salt metathesis reaction between LnX3 (X = Cl, I) and lithiated organometallic species or via the protonolysis of Ln(N(SiMe3)2)3. Difficulties associated with these reactions have been well documented including the formation of lanthanate ions ([LnR4]−),68,108 the necessity to purify by careful sublimation,102 or the retention of coordinating solvent at the metal center.82 For example, reaction of LnCl3 with lithiated organometallic species frequently results in the formation of ‘ate’ salts; that is, anionic Ln species in which more than three anionic ligands remain attached to the metal center, requiring the presence of alkali metal or ammonium based counterions.69,108 The formation of ‘ate’ salts often results in difficult product isolation, as well as poor solubility in the non-polar solvents required for subsequent reactions. On the other hand, protonolysis reactions using Ln(N(SiMe3)2)3 starting materials introduce new ligands by expelling HN(SiMe3)2 as a byproduct, circumventing the issues associated with the production of ‘ate’ salts. Although these reactions work very efficiently, the difficulty in using these reagents relates to their synthesis or purchase. Ln(N(SiMe3)2)3 species, though commercially available, are very expensive. Furthermore, though quite easy to synthesize, their isolation requires a high-temperature, high-vacuum sublimation to produce materials of high purity, limiting their production to a relatively small scale in typical research laboratories.102 Therefore, the development of new lanthanide starting materials that can avoid these common synthetic pitfalls, such as the formation of lanthanate ions, purification methods, and the retention of coordinating solvents, without sacrificing the high activity associated with lanthanide catalysts, is crucial for expanding the scope of lanthanide catalysis. This area of research has become a focal point for advancing the field of lanthanide chemistry, opening up new possibilities for catalytic transformations, and driving innovation in various scientific and industrial applications. Since 2001, new homoleptic complexes featuring various ligands have been reported (Fig. 1).68,70,77,82–84,87,88,106,107,109–121 Among the various homoleptic complexes built using these ligands (LnR2 or LnR3), one critical defining feature that dictates their reactivity is their overall ability to undergo subsequent protonolysis reactions. For example, in the context of the catalysis discussed below, activation of these complexes commonly begins by protonolysis using a phosphine (R2PH) or a phosphine oxide (R2P(O)H). Those systems with ligands of significantly reduced basicity (such as SAr or CH(PPh2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NR)2) are not readily activated to enter any of the relevant catalytic cycles, while only some of those of stronger basicity are capable of activating a phosphine (R2PH), which is not particularly acidic in the first place. In contrast, the much stronger acidity of phosphine oxides has led to their deprotonation and entry into catalytic cycles with many different starting ligands using metals across the periodic table.
NR)2) are not readily activated to enter any of the relevant catalytic cycles, while only some of those of stronger basicity are capable of activating a phosphine (R2PH), which is not particularly acidic in the first place. In contrast, the much stronger acidity of phosphine oxides has led to their deprotonation and entry into catalytic cycles with many different starting ligands using metals across the periodic table.
 | ||
| Fig. 1 A selection of ligands found in recently reported homoleptic lanthanide complexes. | ||
In 2011, our group reported our first results in the field of lanthanide chemistry through the development of a new class of homoleptic trialkyl rare-earth metal complexes. Our goal was to synthesize lanthanide complexes utilizing α-metalated N,N-dimethylbenzylamine. These new benzyl ligand derivatives led to stable lanthanide complexes that were free of coordinating solvent, allowing for a straightforward isolation procedure, and exhibiting effective protonolysis reactivity. The initial report detailed a direct and robust synthetic approach to produce a series of homoleptic lanthanide complexes, Ln(DMBA)3, where DMBA represents the α-deprotonated dimethylbenzylamine ligand (Scheme 1).114 These materials were readily synthesized on a large scale using standard Schlenk techniques. Remarkably, despite starting with LnCl3 and employing THF as solvent, the formation of ‘ate’ salts was avoided, and THF did not persist at the metal center. Instead, the method allowed for the isolation of these homoleptic Ln(DMBA)3 complexes through simple crystallization, encompassing eight lanthanides (La, Ce, Pr, Nd, Sm, Gd, Ho, and Y) that were explored at that time.
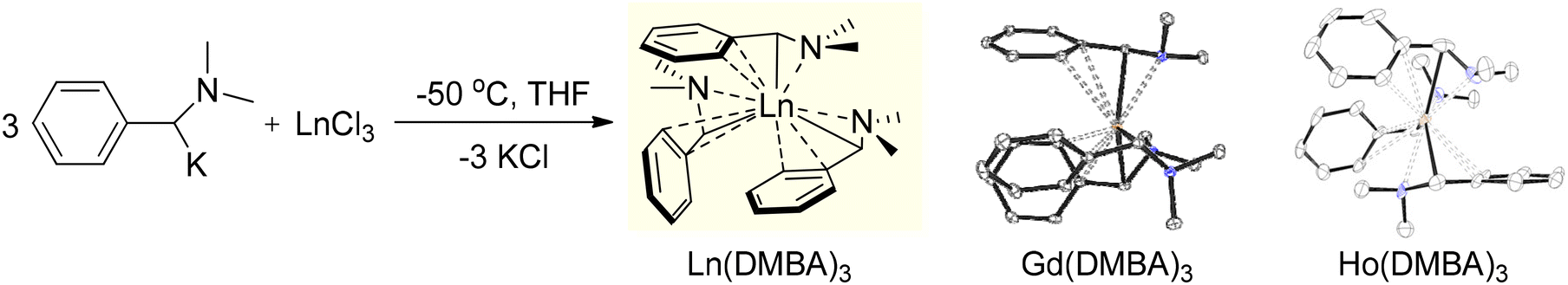 | ||
| Scheme 1 Synthesis of α-Ln(DMBA)3 complexes (Ln = La, Ce, Pr, Nd, Sm, Gd, Ho, and Y). X-ray analysis revealed two distinct isomers, as exemplified by Gd (left) and Ho (right). Adapted with permission from ref. 114. Copyright 2011 American Chemical Society. | ||
The synthesis of the homoleptic lanthanide complexes, Ln(DMBA)3 (Ln = La (1), Ce (2), Pr (3), Nd (4), Sm (5), Gd (6), Ho (7), and Y (8)), involved salt metathesis between rare-earth metal chloride and α-potassiated dimethylbenzylamine in THF at −50 °C. La(DMBA)3 (1) was stable in THF at room temperature and was isolated through recrystallization from THF/pentane at −25 °C. Y(DMBA)3 (8) was synthesized similarly to complex 1, with the THF removed under reduced pressure at 0 °C to prevent decomposition of the complex, and final isolation was accomplished via recrystallization from a concentrated pentane solution at room temperature. The other lanthanide complexes Ln(DMBA)3 (Ln = Ce (2), Pr (3), Nd (4), Sm (5), Gd (6), and Ho (7)) were synthesized similarly to complex 8, and each was recrystallized from a mixture of toluene and pentane at −25 °C. At room temperature, complex 1 displayed dynamic behavior in its NMR spectra that became sharper at −78 °C, indicative of a C3-symmetry species. At low temperature (−78 °C), the 1H-NMR spectrum of the aryl region of complex 1 exhibited ABCDX splitting with a notable upfield chemical shift of the ortho proton adjacent to the metal center, appearing at 3.10 ppm! This shift derived from η2 coordination of the phenyl ring to the lanthanum metal center, resulting in an overall η4 binding mode and a pseudoallylic arrangement for the three carbon atoms interacting with the metal center. In contrast, Y(DMBA)3, complex 8, did not display dynamic behavior at room temperature. The 1H-NMR spectrum of its aryl region displayed distinct AA′BB′C coupling patterns indicative of the presence of two independent isomers with different symmetries (C3 and CS), suggesting rapid rotation of the phenyl groups in solution. This behavior suggested an η2 coordination of the DMBA ligands to the yttrium metal center, unlike the η4 coordination observed in complex 1. The remaining lanthanide complexes 2–7, Ln(DMBA)3 (Ln = Ce (2), Pr (3), Nd (4), Sm (5), Gd (6), and Ho (7)), showed broad and uninterpretable signals in their 1H-NMR spectra due to their paramagnetic nature. X-ray crystallography was used to analyze the structure of the Ln(DMBA)3 complexes, confirming their simple monomeric and homoleptic nature. The solid-state structures of the complexes 2–6 exhibited C3 symmetry and η4 coordination to the metal centers, resembling the structure observed in complex 1. In contrast, Ho(DMBA)3 (complex 7) showed lower symmetry similar to complex 8 due to its smaller ionic radius. Overall, the two distinct bonding motifs observed, as shown in Scheme 1, readily reflected the differences in the lanthanide ionic radii.114
Catalytic applications
In the modern era, there has been a growing demand for organophosphorus compounds due to their diverse chemical reactivity, with applications in research, medicine, materials science, agriculture, and industry. Meeting this demand sustainably and with environmental responsibility is crucial, emphasizing efficient synthetic routes, waste minimization, and reduced environmental impact during production. Catalytic hydroelementation, which includes hydrophosphination, hydrophosphinylation, and hydrophosphorylation, stands out as a compelling transformation in homogeneous catalysis for the production of organophosphorus compounds, offering numerous advantages, such as high selectivity, atom efficiency, minimized waste production, and simplified purification processes. With the successful synthesis of homoleptic Ln(DMBA)3 complexes, as detailed above, we set out to address catalytic phosphination reactions by applying the lanthanum derivative, La(DMBA)3, as a catalyst in organometallic transformations. The main reason this complex was investigated first was one of practicality—its diamagnetic nature allowed for the utilization of NMR spectroscopy to more readily screen potential catalytic reactions. Goals in this project included an expansion of the understanding of fundamental lanthanide catalysis and the effective application of this catalyst as a tool for organic synthesis.Hydrophosphination of heterocumulenes
Hydrophosphination, the process of adding a P–H bond across an unsaturated organic substrate, has emerged as an efficient method for selectively forming P–C bonds, leading to the synthesis of various organophosphorus compounds while maximizing the efficient utilization of phosphorus. Although hydrophosphination can occur non-catalytically through thermal, photochemical, or radical initiation, catalytic hydrophosphination provides several advantages including reduced activation energy, wider substrate compatibility, and improved selectivity, resulting in a more efficient and controlled approach for synthesizing organophosphorus compounds. In 1990, Pringle achieved a breakthrough by reporting the first metal-catalyzed hydrophosphination, utilizing a platinum salt to synthesize tertiary phosphines from PH3 and electronically activated substrates,122 which was later extended to formaldehyde with nickel, palladium, and platinum catalysts.123 Later, in 1997, Glueck introduced the first molecular hydrophosphination catalyst, employing platinum complexes for acrylonitrile hydrophosphination.124 Subsequently, hydrophosphination catalysis has experienced remarkable progress in both catalyst and substrate development, with continuous promise for further advancements and discoveries in the future. In the field of hydrophosphination, there are several review articles available.125–130 Over the last three decades, a wide array of catalysts, including those based on alkali metal,131,132 alkaline earth metals (Mg, Ca, Sr, Ba),133–144 transition metals (Ti, Zr, Fe, Ru, Co, Rh, Ni, Pd, Pt, Zn),145–194 and f-block elements (La, Ce, Sm, Eu, Yb, Th),16,121,195–221 have been developed for hydrophosphination of alkenes, alkynes, dienes, allenes, aldehyde, heterocumulenes, and imines. Particularly, the hydrophosphination reaction of heterocumulenes has emerged as an efficient and promising strategy to produce phosphaguanidines, a valuable class of heteroatom-containing compounds with diverse applications in organic synthesis and coordination chemistry. Over the last two decades, substantial strides have been made in developing various metal catalyst systems, such as alkali–metal and alkaline-earth metal complexes,134,222 transition element complexes,154,164,223 rare-earth metal complexes,201,224–226 and actinide complexes,227–229 all facilitating the hydrophosphination reactions of heterocumulenes.In 2013, the Schmidt group demonstrated the catalytic activity of lanthanide-centered complexes, Ln(DMBA)3, in the hydrophosphination of heterocumulenes like carbodiimides, isocyanates, and isothiocyanates.201 The study reported the successful utilization of α-metalated N,N-dimethylbenzylamine rare-earth metal complexes, La(DMBA)3 and Y(DMBA)3, in various stoichiometric insertion and catalytic hydrophosphination reactions, resulting in the formation of homoleptic amidinate and phosphaguanidinate complexes. These complexes also served as effective catalyst precursors for the hydrophosphination of CN bonds, particularly the lanthanum derivative, which showed excellent activity with various heterocumulenes. Overall, catalytic performance was influenced by phosphine acidity and heterocumulene steric bulk. The research represents a notable breakthrough in the realm of alpha-metalated N,N-dimethylbenzylamine rare-earth metal catalysis, lending credence to the idea that lanthanide-based complexes, Ln(DMBA)3, could be used as precatalysts in many organometallic transformations. Furthermore, via a series of stoichiometric reactions, it was shown that neutral N,N-dimethylbenzylamine, generated by catalyst activation via protonolysis, coordinated to the resulting lanthanide centers very weakly. This was somewhat surprising, but seems well supported by the high catalytic activities observed in subsequent classes of catalysis observed with La(DMBA)3 (see below).
The simple addition of a phosphine to a diverse range of heterocumulenes demonstrated the remarkable catalytic effectiveness of La(DMBA)3, providing phosphaguanidines, phosphaureas, and phosphathioureas in yields ranging from moderate to excellent under mild reaction conditions (Scheme 2). Mechanistically, the hydrophosphination reaction is presumed to proceed with the initial formation of a metal-phosphide as the active species, followed by insertion of the heterocumulene, and subsequent generation of the hydrophosphination product through protonolysis by the phosphine.201
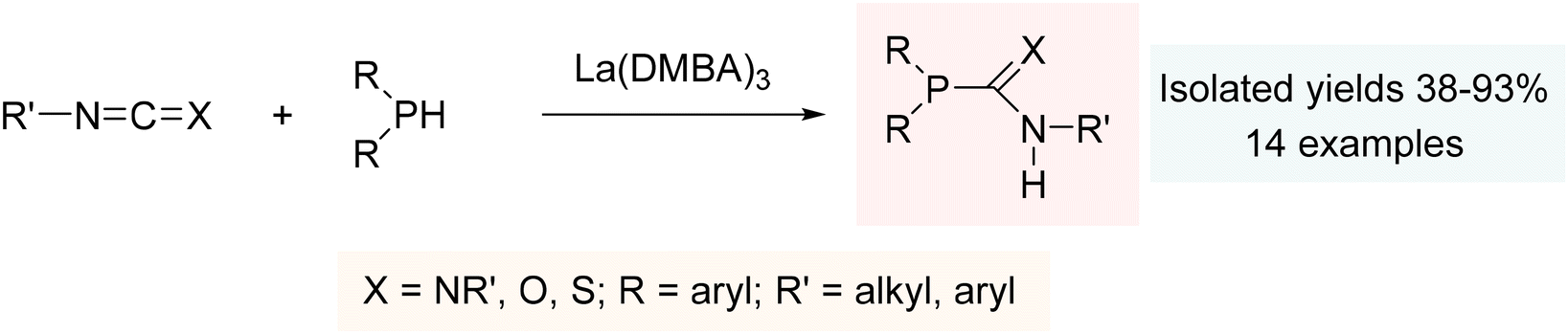 | ||
| Scheme 2 Hydrophosphination of heterocumulenes using La(DMBA)3. Adapted with permission from ref. 201. Copyright 2013 American Chemical Society. | ||
Our study using La(DMBA)3 represented the first reported hydrophosphination of heterocumulenes using rare-earth metal catalysts at room temperature.201 Prior to this, in 2008, Hou had reported the rare-earth metal catalyzed hydrophosphination of heterocumulenes utilizing half-sandwich rare-earth metal (trimethylsilyl)methyl alkyl complexes, although this required elevated temperatures (80 °C).224 Following our study, Zhang and Xi separately contributed to this area by reporting rare-earth metal catalyzed hydrophosphination of heterocumulenes using bis(NHC)-based CNC-pincer rare-earth metal amido complexes and half-sandwich rare-earth metal tris(alkyl) ate complexes, respectively (Fig. 2).225,226
 | ||
| Fig. 2 Reported rare-earth metal catalysts for hydrophosphination of heterocumulenes. | ||
Hydrophosphinylation of nitriles
The related process of hydrophosphinylation, which employs a phosphine oxide rather than a simple phosphine, has attracted substantial attention in the scientific community as an environmentally friendly and atom-economic method to produce oxidized organophosphorus compounds. Although significant and ongoing research has been dedicated to synthetic modification of organonitriles for use in organic synthesis and medicinal chemistry, the double hydrophosphinylation of nitriles was largely unexplored prior to our investigation. The Schmidt group showed that La(DMBA)3 was effective for the catalytic hydrophosphinylation of nitriles with secondary phosphine oxides.230 The reactions reported in this study were highly unusual and resulted in surprising isolated products. Unlike the expected reaction product deriving from two consecutive nucleophilic attacks on the carbon atom of the cyano group in a nitrile due to its δ+ character, the final products in this catalysis were found to have one phosphorus atom bound to each of the carbon and nitrogen atoms that initially constituted the nitrile functional group (see Scheme 3).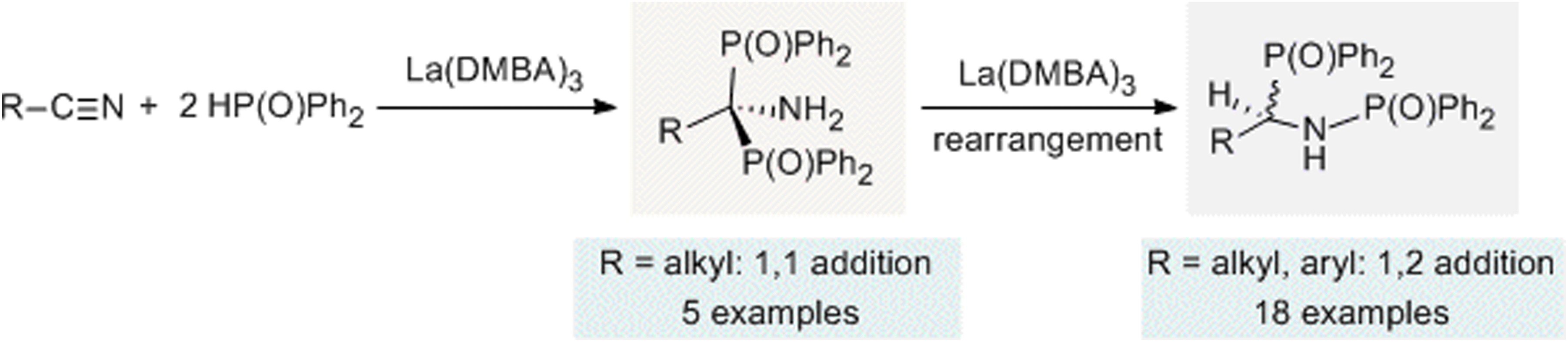 | ||
| Scheme 3 Hydrophosphinylation of nitriles using La(DMBA)3. Adapted with permission from ref. 230. Copyright 2017 American Chemical Society. | ||
In general, as a result of our investigation, it was found that for all nitriles tested, hydrophosphinylation catalyzed by La(DMBA)3 afforded the symmetric regioisomeric product with a P–C–P linkage and a primary amine moiety, where both phosphorus atoms were bound to the carbon atom of the nitrile (see 1,1-addition product; Scheme 3). For sterically unhindered primary alkyl nitriles, the reaction ends at that point, providing the initially predicted double hydrophosphinylation products. In contrast, when this catalysis was undertaken using Ph2P(O)H and various substituted aryl nitriles, as well as sterically bulkier alkyl nitriles (benzyl, cyclohexyl, and isopropyl), an isomerization reaction was observed after the catalytic addition step, leading to the generation of new regioisomeric products with a P–C–N–P linkage, where one phosphorus atom was attached to the carbon atom and the other was attached to the nitrogen atom (see 1,2-addition product; Scheme 3), resulting in products with a P–C–N–P linkage, resembling well-known bis(diphenylphosphino)ethane ligands, such as dmpe and dppe. Mechanistic investigations supported the hypothesis that the nucleophile attacked twice at the electropositive carbon atom, as expected, but this intermediate was then converted to the isolated product through a lanthanum-mediated rearrangement reaction.
The catalytic cycle for the double hydrophosphinylation of nitriles involves first protonolysis of La(DMBA)3 by 3 equiv. of diphenylphosphine oxide, producing a lanthanum-trisphosphinyl La(OPPh2)3 active species (Scheme 4). Next, the nitrile coordinates to the lanthanum center and undergoes insertion, resulting in a carbon–phosphorus and a lanthanum–amide bond, which is subsequently converted to the neutral amine by protonolysis with HP(O)Ph2. Repetition of this sequence a second time leads to the observed 1,1-addition product (the kinetic product) that ultimately isomerizes into the final unsymmetric 1,2-addition product (the thermodynamic product) in most cases. The proposed mechanism for the isomerization involves activation of a PO bond by coordination of the oxygen atom to the Lewis acidic lanthanum center. This coordination facilitates an intramolecular attack of the amine onto the phosphorus atom, leading to a three-membered cyclic intermediate. The final isomerization product then results from C–P bond cleavage, effectively causing the 1,2-shift of a phosphoryl group while simultaneously reducing the steric strain of the quaternary carbon center (Scheme 5).230 Subsequently, Panda has explored the double hydrophosphinylation of nitriles using alkali metal hexamethyldisilazides [MN(SiMe3)2] (M = Li, Na, K) as precatalysts,231 while more recently, Cai has reported a nickel-catalyzed 1,1-dihydrophosphinylation of nitriles with phosphine oxides.232
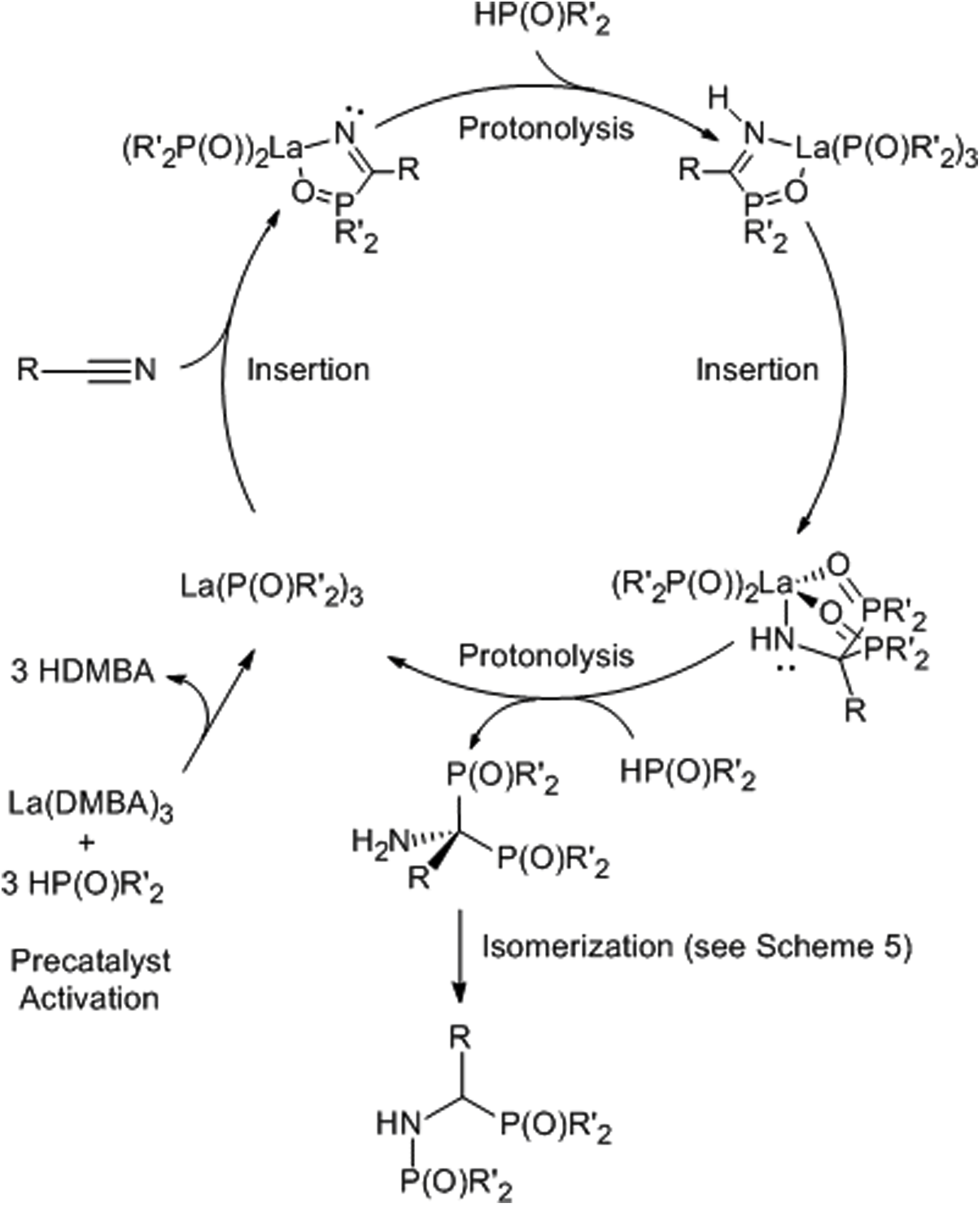 | ||
| Scheme 4 Proposed catalytic cycle for the double hydrophosphinylation (R′ = alkyl or aryl) or double hydrophosphorylation (R′ = alkoxy or aryloxy) of nitriles. Adapted with permission from ref. 230. Copyright 2017 American Chemical Society. | ||
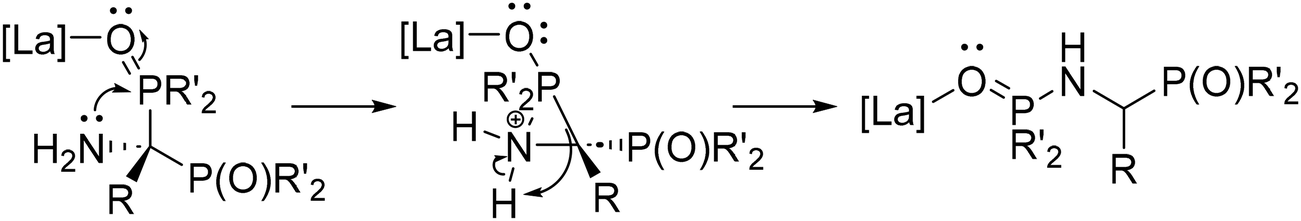 | ||
| Scheme 5 Nitrile double phosphinylation product isomerization. Adapted with permission from ref. 238. Copyright 2020 American Chemical Society. | ||
Hydrophosphorylation of nitriles
Hydrophosphorylation, addition of H–P(O)(OR)2 across an unsaturated C–C framework, typically relies on costly late transition metals such as Pd and Rh,233–237 so we felt that our rare-earth metal catalysts could offer a promising alternative for the synthesis of the biologically and synthetically important compounds known as amidophosphates. Similar to the catalytic results with phosphine oxides, La(DMBA)3 again proved to be a very effective precatalyst, facilitating the addition of diethylphosphite to various nitriles, including those bearing halides, ethers, amines, and pyridyl groups.238 The reaction, utilizing only 2.5 mol% La(DMBA)3, was carried out in either neat nitrile or environmentally friendly toluene (for solid nitriles), and led to the generation of the 1,2-addition products, N-(α-phosphoryl)amidophosphates, in all cases (Scheme 6). The catalytic cycle for this reaction likely parallels that invoked for the double hydrophosphinylation of nitriles discussed above.238 While Kaboudin has demonstrated the ZnCl2/Et3N mediated double hydrophosphorylation of nitriles to yield 1-aminobisphosphonates (1,1-addition products),239 our study was the first reported catalytic double hydrophosphorylation of nitriles to give N-(α-phosphoryl)amidophosphates (1,2-addition products).238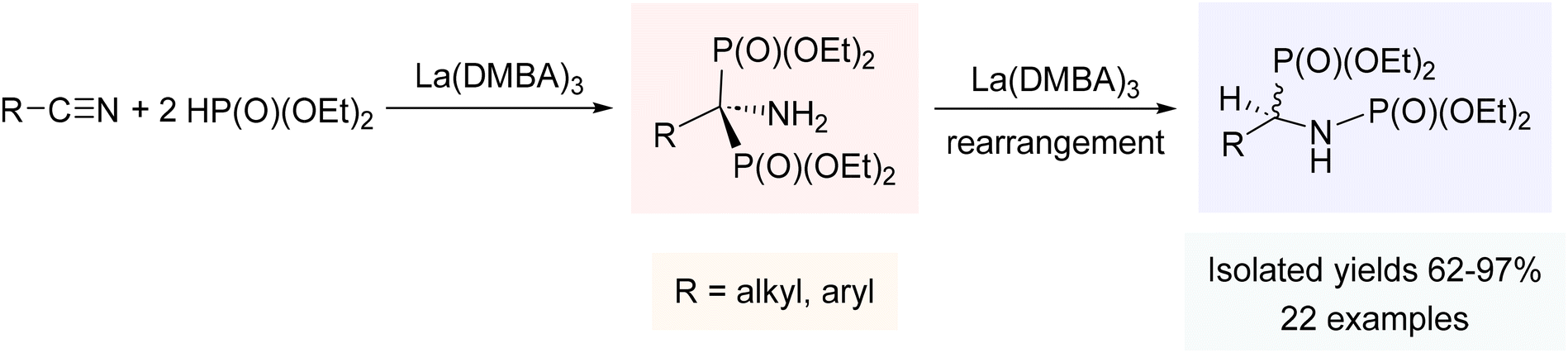 | ||
| Scheme 6 Double hydrophosphorylation of nitriles using La(DMBA)3. Adapted with permission from ref. 238. Copyright 2020 American Chemical Society. | ||
Hydrophosphination and hydrophosphinylation of hydrocarbons
With the successful catalysis observed in the addition reactions of carbodiimides, isocyanates, and nitriles, we turned our attention to the hydrophosphination and hydrophosphinylation of hydrocarbon substrates, such as alkynes and alkenes.202,240 Once again, the utilization of La(DMBA)3 as a precatalyst led to excellent reactivity, in this case revealing intriguing regio- and stereochemical outcomes in reactions with alkynes. Hydrophosphination led to mono-addition products, forming only the anti-Markovnikov product (Scheme 7). Furthermore, the stereochemical outcome could be selectively controlled to produce either the E- or the Z-isomer by simply adjusting the reaction stoichiometry. That is, when a substoichiometric amount of phosphine was employed, the reaction exclusively produced the Z-stereoisomer (Z-vinylphosphine). Conversely, an excess of phosphine led to the exclusive production of the E-vinylphosphine product, as depicted in Scheme 7.202 This catalysis was found to be second order in lanthanum, implying that the active species, lanthanum tris-phosphide, existed as a dimer in the rate-limiting alkyne activation step. Alkyne coordination, followed by a bimetallic insertion step explained the unexpected formation of the Z-isomers. Excess phosphine in the reaction mixture facilitated lanthanum-mediated isomerization into the more stable E-isomers. It is possible that this isomerization involves insertion of the Z-alkene into a La–P bond of the active species, followed by single bond rotation, and β-phosphido elimination, which ultimately leads to the E-alkene. These findings highlight the versatility of La(DMBA)3 as a catalyst, enabling control over the regio- and stereochemistry of the reaction and providing value in synthetic applications.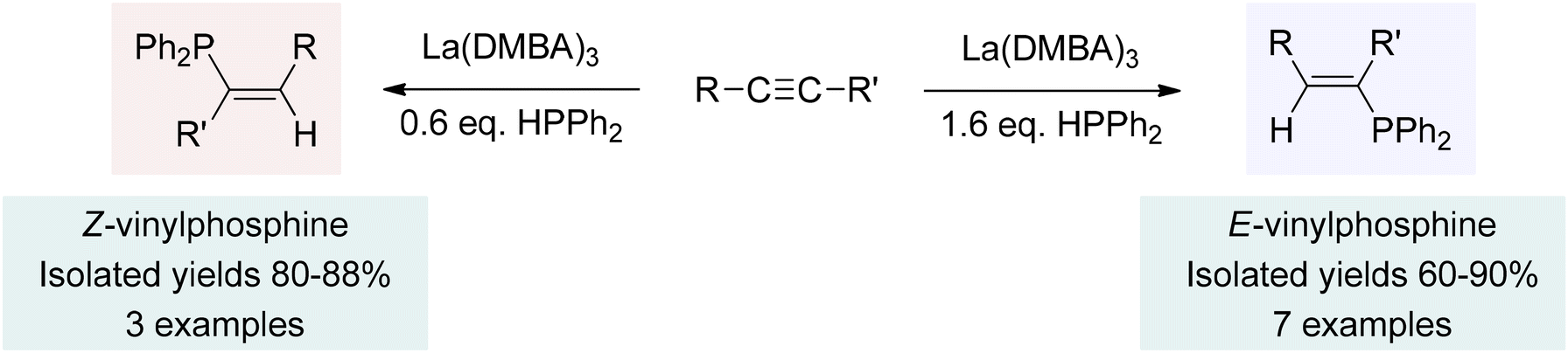 | ||
| Scheme 7 Hydrophosphination of alkynes using La(DMBA)3. Adapted with permission from ref. 202. Copyright 2019 American Chemical Society. | ||
The related hydrophosphinylation reactions using alkynes produced either the mono-addition product (a vinyl phosphine) or the bis-addition product (a 1,2-diphosphinoethane), depending on the type of alkynes used (Scheme 8). Specifically, the reaction of terminal alkynes with diphenylphosphine oxides gave only double addition products, while internal alkynes yielded both the single and double addition products (forcing conditions), which could be controlled by the stoichiometry of the phosphine oxide (Scheme 8). The steric hindrance of the alkynes seemed to play the key role in determining the product selectivity, with terminal alkynes favoring the double addition product due to lower steric hindrance, and internal alkynes yielding both single and double addition products based on the reaction conditions and stoichiometry of the phosphine oxide.202
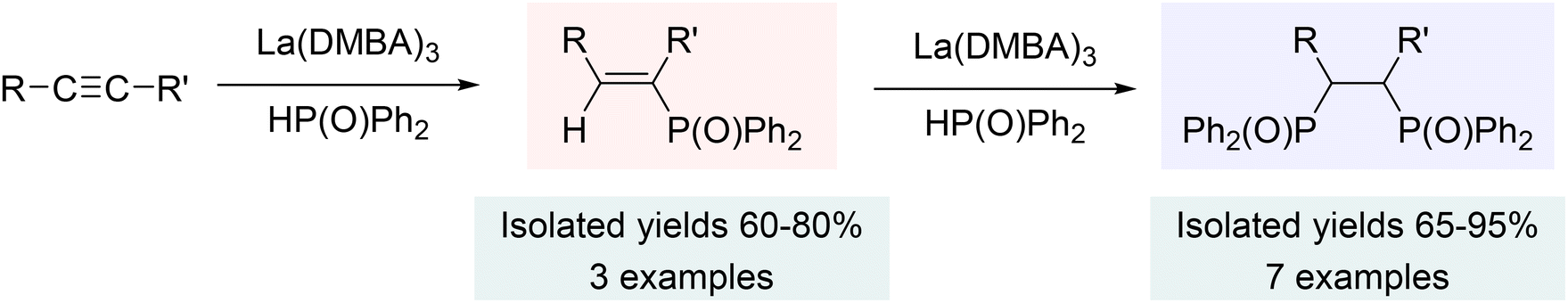 | ||
| Scheme 8 Hydrophosphinylation of alkynes using La(DMBA)3 (internal alkynes: single and double hydrophosphinylation; terminal alkynes: double hydrophosphinylation). Adapted with permission from ref. 202. Copyright 2019 American Chemical Society. | ||
Mechanistically, tautomerization of diphenylphosphine oxide plays a vital role in this reaction by reducing the steric strain at the metal center, thereby favoring the production of the double addition hydrophosphinylation products, unlike the single addition product formed with diphenylphosphine. The hydrophosphinylation catalysis likely initiates with creation of a pyridine solvated, oxygen-bound lanthanum complex: La(OPPh2)3(py)n (Scheme 9). Coordination of the alkyne, followed by a [3 + 2] cycloaddition, gives the single phosphinylation product after protonolysis by diphenylphosphine oxide. This insertion and protonolysis process repeats to form the double addition product for the less sterically hindered alkynes. Although many catalyst systems employing both early and late transition metals (Fe, Co, Ni, Cu, Pd, Pt, Rh, Mg, Re), as well as lanthanide elements (Eu, Yb), have been documented for the hydrophosphination and hydrophosphinylation of alkynes,156,163,170,189,216,241–248 this study was the first to utilize a lanthanum based catalyst. Overall, the La(DMBA)3 catalyst demonstrated remarkable performance in both hydrophosphination and hydrophosphinylation of alkynes, operating under mild conditions and exhibiting high chemo-, regio- and stereoselectivity.202
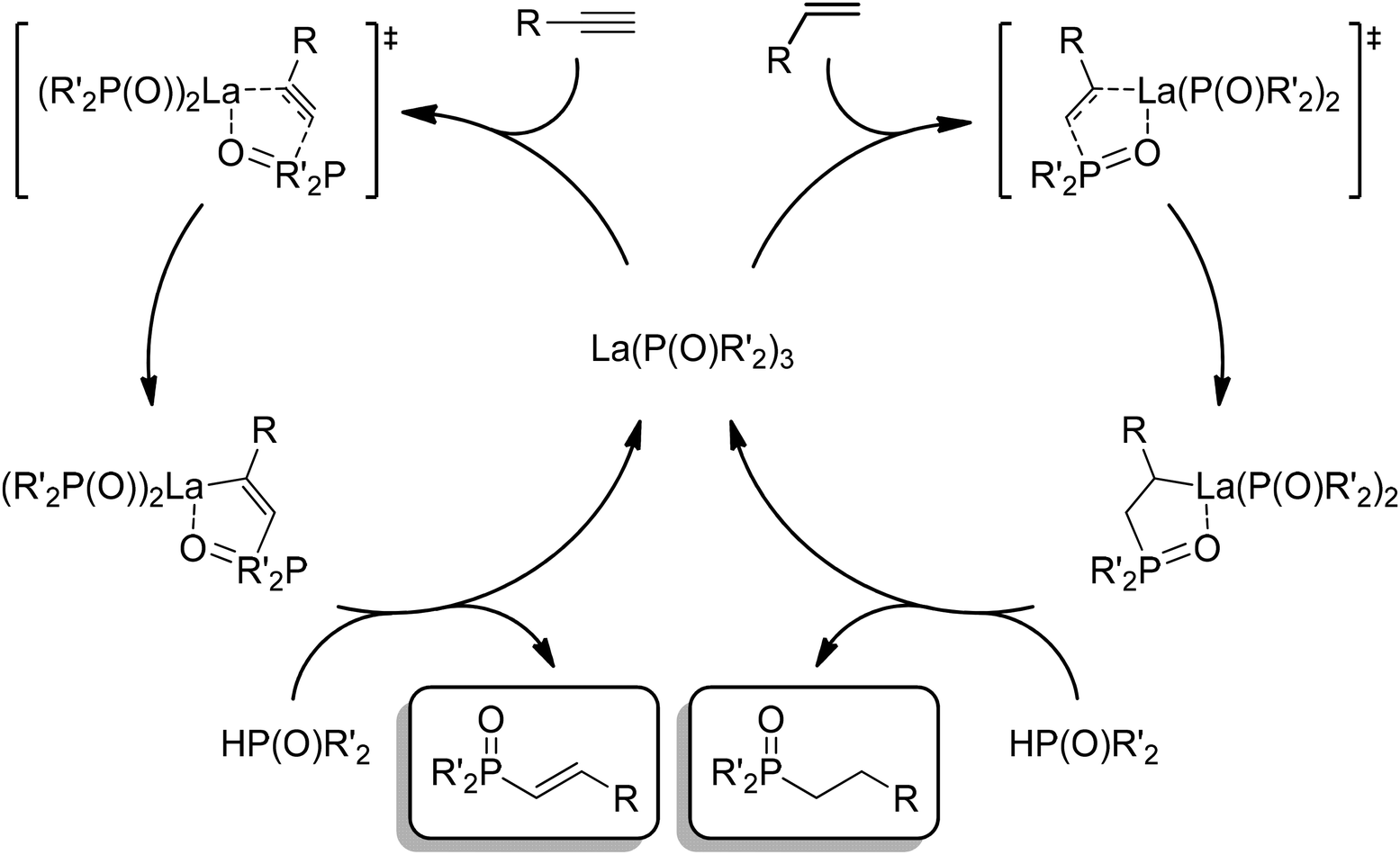 | ||
| Scheme 9 General catalytic cycles for the hydrophosphinylation of alkenes and alkynes. | ||
With similar reactivity to the alkynes, styrenes also proved to be suitable for catalytic hydrophosphinylation mediated by the La(DMBA)3 complex (Scheme 10). These reactions consistently exhibited anti-Markovnikov regiochemistry and an impressive tolerance to a wide variety of functional groups present in either the styrene or the phosphine oxide reagents. Virtually any non-acidic substituent, including halogens, NO2, and ethers, proved to be compatible with this catalytic process. Additionally, vinyl-substituted heteroarenes could be utilized in the hydrophosphinylation, leading to the formation of a more diverse set of product β-arylphosphine oxides.240 Similar to that observed for the alkynes, catalyst activation via protonolysis led to the active species, La(OPAr2)3. Then, the catalytic cycle proceeded by insertion of alkenes, and protonolysis by the phosphine oxides to produce the observed β-arylphosphine oxides.240 In subsequent work, the Hevia and Zheng groups have explored hydrophosphinylation of aryl alkenes using s-block complexes such as potassium magnesiate (PMDETA)2K2Mg(CH2SiMe3)4 [PMDETA = N,N,N′,N′,N′′-pentamethyldiethylenetriamine] and β-diketiminato n-butylmagnesium (NacNac)MgnBu.249,250
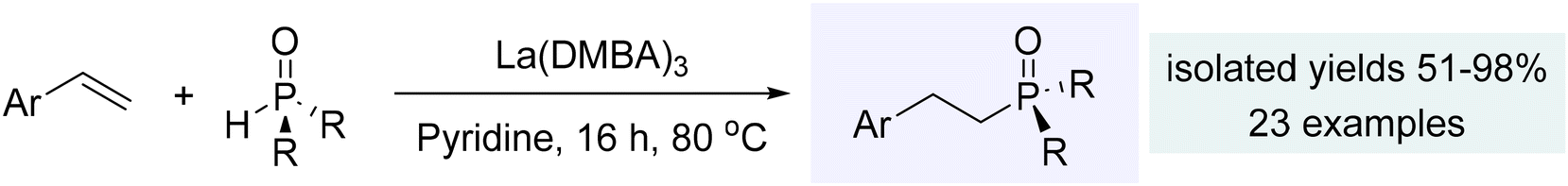 | ||
| Scheme 10 Hydrophosphinylation of styrenes using La(DMBA)3. Adapted with permission from ref. 240. Copyright 2019 American Chemical Society. | ||
Heterodehydrocoupling catalysis
The extensive breadth of catalytic chemistry discussed above demonstrates the remarkable utility of La(DMBA)3 in facilitating the addition of phosphines, phosphine oxides, and phosphites to various unsaturated systems. With an interest in expanding this system to various other catalytic transformations, we then investigated catalytic heterodehydrocoupling of silanes and amines. This efficient method for Si–N bond formation generates H2 gas as the sole byproduct. Despite initial reports of this reaction dating back to 1966, it was generally overlooked for several decades. Since the turn of the century, significant advancements have been made in catalytic dehydrocoupling of silanes and amines, utilizing s-block elements (Mg, K, Ca, Sr, and Ba),251–261 p-block elements (B and Al),262,263 and d-block elements (Ti, Zr, Cr, Fe, Ru, Rh, Pd, and others).264–274 In 1999, the first application of f-block elements (rare-earth metals) was reported for this catalytic process, with ytterbium–imine complexes utilized.275 Later, in 2012, different ytterbium complexes were also shown to catalyze this reaction,276 while more recent studies have delved into other rare-earth metals, using complexes such as [Ln{C(SiHMe2)3}2ImtBu] (Ln = Yb, Sm; ImtBu = 1,3-ditertbutylimidazol-2-ylidene) and dianionic α-iminopyridine rare-earth metal amido complexes.277,278 In 2018, Waterman reported the catalytic heterodehydrocoupling of silanes and amines using La(N(TMS)2)3(THF)2, adding further insight to this field.279 Very recently, in 2022, our research group showed that the lanthanide-based complex La(DMBA)3 was also very active for the heterodehydrocoupling of silanes and amines (Scheme 11).280 Given the ease of preparation of multigram quantities of La(DMBA)3 through salt metathesis and its isolation via straightforward recrystallization, it serves as an excellent alternative to the La(N(TMS)2)3 complex.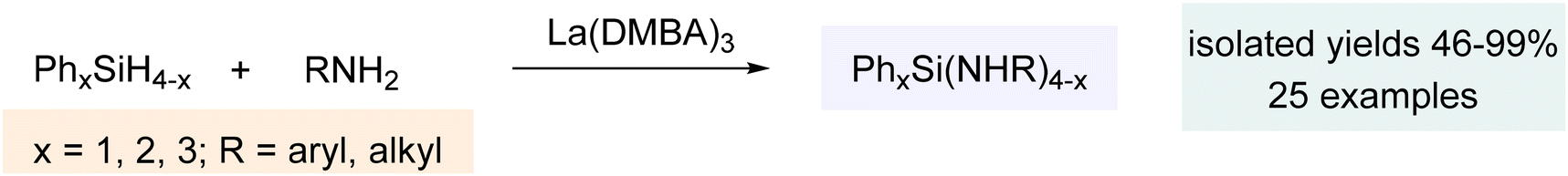 | ||
| Scheme 11 Heterodehydrocoupling of silanes and amines using La(DMBA)3. | ||
The catalytic dehydrocoupling of silanes and amines was accomplished using 2.5 mol% La(DMBA)3 in pyridine at 50 °C over 16 h. Primary cyclic and acyclic alkyl amines, as well as anilines with various ortho, para and meta substituents (including alkyl, halogen and methoxy groups), were suitable coupling partners for phenylsilane, resulting in good yields of quaternary tris(amino)silanes. Additionally, primary amines, including aniline, p-methoxyaniline, n-propylamine, cyclohexylamine, and isopropylamine, underwent heterodehydrocoupling with Ph2SiH2, providing quaternary silylamines of the form Ph2Si(NHR)2. Furthermore, nPrNH2 and iPrNH2 reacted with Ph3SiH at 50 °C and 80 °C, respectively, to form quaternary silylamines, Ph3Si(NHR). Finally, we investigated the catalytic dehydrocoupling reaction using three representative secondary amines (Et2NH, PhMeNH, and pyrrolidine) with primary, secondary, and tertiary silanes, further extending the scope of this research. Mechanistically, formation of the active catalyst, an uncharacterized lanthanum amido species (DMBA)(3−n)La(HNR)n, occurred via protonolysis of La(DMBA)3 by 1–3 equivalents of RNH2 (Scheme 12). Catalysis then proceeds by reaction with silane to yield the aminosilane and the corresponding lanthanum hydride species, (DMBA)(3−n)LaHn. Catalyst turnover is then achieved by reaction with the amine to regenerate the lanthanum amido species with the loss of dihydrogen.
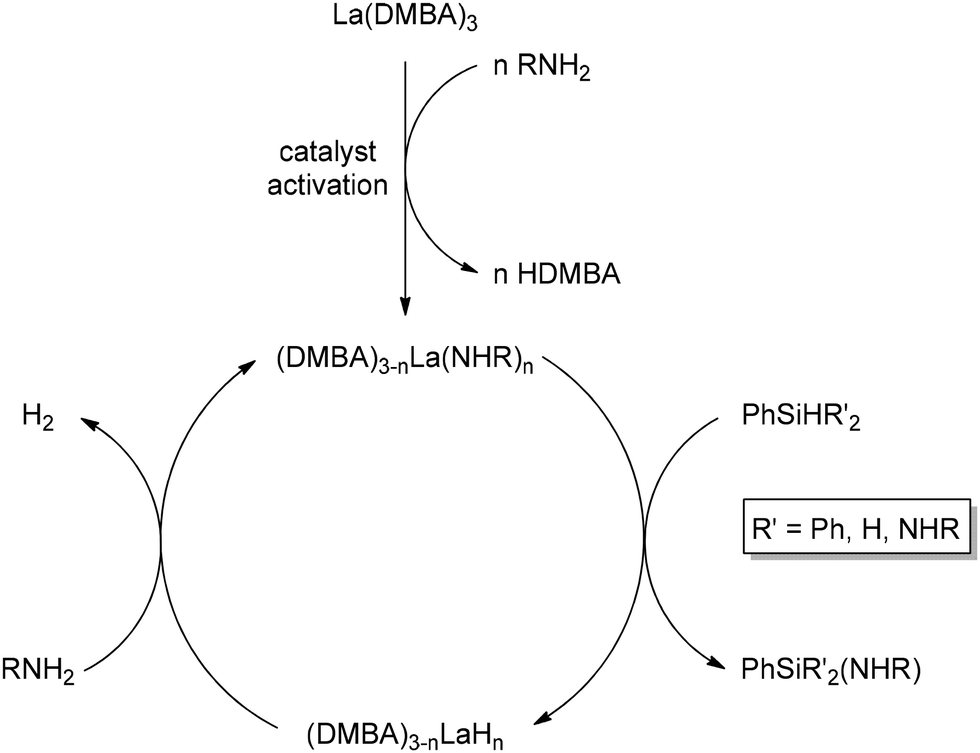 | ||
| Scheme 12 Heterodehydrocoupling catalytic cycle. Adapted with permission from ref. 280. Copyright 2022 American Chemical Society. | ||
Overall, La(DMBA)3 has demonstrated its worth as a valuable and important contribution to the field of rare-earth metal catalysis. The reactions summarized herein with La(DMBA)3 as a catalyst are just the beginning and demonstrate the potential power of this complex. Its versatility allows for a diverse range of catalytic reactions, making it an excellent platform for further advancements. Thus, ongoing efforts are focused on further expansion of the catalytic portfolio of La(DMBA)3, as well as investigation of the catalytic potential of analogous species using other lanthanide metal centers, Ln(DMBA)3.
Conclusions
The Schmidt group has built extensive expertise in lanthanide chemistry, with a focus on catalytic transformations facilitated by alpha-metalated N,N-dimethylbenzylamine rare-earth metal complexes for over a decade. In developing this chemistry, considerable success in the stoichiometric synthesis and isolation of lanthanide complexes has been achieved. Looking at the breadth of catalytic reactions studied to date, it becomes apparent that Ln(DMBA)3 complexes bring a valuable and inventive dimension to rare-earth metal catalysis. The lanthanide-based complex La(DMBA)3 has facilitated a broad spectrum of catalytic transformations, significantly impacting the synthesis of highly functionalized organic species, which find applications in many other fields, including medicine, life sciences, chemical and pharmaceutical industries, agrochemicals, materials science, organic synthesis, and catalysis. In the future, we intend to undertake a comparative study in which the other lanthanide complexes of the form Ln(DMBA)3 are utilized for catalysis and their activities compared directly with the previously published La(DMBA)3 system. Additionally, effort towards further broadening the catalytic utility of lanthanide-based complexes to include the formation of carbon–carbon bonds remains a current research topic in this laboratory.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Acknowledgement is made to the Donors of the American Chemical Society Petroleum Research Fund and various programs at the University of Toledo for support of the lanthanide catalysis chemistry in the Schmidt group over the past decade directly leading to this perspective article. We also thank Vivian R. Schmidt for contributing her artistic talents in the creation and design of the cover graphic.References
- L. J. E. Stanlake and L. L. Schafer, Organometallics, 2009, 28, 3990–3998 CrossRef CAS.
- S. Bambirra, H. Tsurugi, D. van Leusen and B. Hessen, Dalton Trans., 2006, 1157–1161 RSC.
- H. F. Yuen and T. J. Marks, Organometallics, 2009, 28, 2423–2440 CrossRef CAS.
- T. E. Müller, K. C. Hultzsch, M. Yus, F. Foubelo and M. Tada, Chem. Rev., 2008, 108, 3795–3892 CrossRef PubMed.
- J. Hannedouche and E. Schulz, Organometallics, 2018, 37, 4313–4326 CrossRef CAS.
- G. A. Molander and J. A. C. Romero, Chem. Rev., 2002, 102, 2161–2186 CrossRef CAS PubMed.
- G. A. Molander, E. D. Dowdy and B. C. Noll, Organometallics, 1998, 17, 3754–3758 CrossRef CAS.
- S. Ge, A. Meetsma and B. Hessen, Organometallics, 2008, 27, 3131–3135 CrossRef CAS.
- A. G. Trambitas, T. K. Panda, J. Jenter, P. W. Roesky, C. Daniliuc, C. G. Hrib, P. G. Jones and M. Tamm, Inorg. Chem., 2010, 49, 2435–2446 CrossRef CAS PubMed.
- D. Liu, B. Liu, Z. Pan, J. Li and C. Cui, Sci. China: Chem., 2019, 62, 571–582 CrossRef CAS.
- Y. Horino and T. Livinghouse, Organometallics, 2004, 23, 12–14 CrossRef CAS.
- A. S. Dudnik, V. L. Weidner, A. Motta, M. Delferro and T. J. Marks, Nat. Chem., 2014, 6, 1100–1107 CrossRef CAS PubMed.
- S. Seo and T. J. Marks, Chem. – Eur. J., 2010, 16, 5148–5162 CrossRef CAS PubMed.
- C. J. Weiss and T. J. Marks, Dalton Trans., 2010, 39, 6576–6588 RSC.
- Q. Yuan, S. Zhou, X. Zhu, Y. Wei, S. Wang, X. Mu, F. Yao, G. Zhang and Z. Chen, New J. Chem., 2015, 39, 7626–7632 RSC.
- I. V. Basalov, S. C. Rosca, D. M. Lyubov, A. N. Selikhov, G. K. Fukin, Y. Sarazin, J.-F. Carpentier and A. A. Trifonov, Inorg. Chem., 2014, 53, 1654–1661 CrossRef CAS PubMed.
- M. J. Stanford and A. P. Dove, Chem. Soc. Rev., 2010, 39, 486–494 RSC.
- W. Miao, S. Li, D. Cui and B. Huang, J. Organomet. Chem., 2007, 692, 3823–3834 CrossRef CAS.
- R. H. Platel, L. M. Hodgson and C. Williams, Polym. Rev., 2008, 48, 11–63 CrossRef CAS.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS PubMed.
- D. M. Lyubov, A. O. Tolpygin and A. A. Trifonov, Coord. Chem. Rev., 2019, 392, 83–145 CrossRef CAS.
- T. Song, N. Liu, X. Tong, F. Li, X. Mu and Y. Mu, Dalton Trans., 2019, 48, 17840–17851 RSC.
- D. Liu, M. Wang, Y. Chai, X. Wan and D. Cui, ACS Catal., 2019, 9, 2618–2625 CrossRef CAS.
- H. Wang, Y. Zhao, M. Nishiura, Y. Yang, G. Luo, Y. Luo and Z. Hou, J. Am. Chem. Soc., 2019, 141, 12624–12633 CrossRef CAS PubMed.
- Z. Hou and Y. Wakatsuki, Coord. Chem. Rev., 2002, 231, 1–22 CrossRef CAS.
- A. Valente, G. Stoclet, F. Bonnet, A. Mortreux, M. Visseaux and P. Zinck, Angew. Chem., Int. Ed., 2014, 53, 4638–4641 CrossRef CAS PubMed.
- M. Nishiura, F. Guo and Z. Hou, Acc. Chem. Res., 2015, 48, 2209–2220 CrossRef CAS PubMed.
- Z. Hou, Y. Luo and X. Li, J. Organomet. Chem., 2006, 691, 3114–3121 CrossRef CAS.
- F. T. Edelmann, Chem. Soc. Rev., 2009, 38, 2253–2268 RSC.
- B. S. Lim, A. Rahtu, J.-S. Park and R. G. Gordon, Inorg. Chem., 2003, 42, 7951–7958 CrossRef CAS PubMed.
- J. Päiväsaari, I. V. C. L. Dezelah, D. Back, H. M. El-Kaderi, M. J. Heeg, M. Putkonen, L. Niinistö and C. H. Winter, J. Mater. Chem., 2005, 15, 4224–4233 RSC.
- S. R. Daly, D. Y. Kim, Y. Yang, J. R. Abelson and G. S. Girolami, J. Am. Chem. Soc., 2010, 132, 2106–2107 CrossRef CAS PubMed.
- U. Kilimann and F. T. Edelmann, Coord. Chem. Rev., 1995, 141, 1–61 CrossRef CAS.
- J. Richter and F. T. Edelmann, Coord. Chem. Rev., 1996, 147, 373–442 CrossRef CAS.
- Y. K. Gonko and F. T. Edelmann, Coord. Chem. Rev., 1996, 156, 1–89 CrossRef.
- F. T. Edelmann and Y. K. Gun'ko, Coord. Chem. Rev., 1997, 165, 163–237 CrossRef CAS.
- F. T. Edelmann and V. Lorenz, Coord. Chem. Rev., 2000, 209, 99–160 CrossRef CAS.
- J.-Y. Hyeon and F. T. Edelmann, Coord. Chem. Rev., 2003, 247, 21–78 CrossRef CAS.
- J. Gottfriedsen and F. T. Edelmann, Coord. Chem. Rev., 2005, 9, 919–969 CrossRef.
- J.-Y. Hyeon, J. Gottfriedsen and F. T. Edelmann, Coord. Chem. Rev., 2005, 249, 2787–2844 CrossRef CAS.
- J. Gottfriedsen and F. T. Edelmann, Coord. Chem. Rev., 2006, 250, 2347–2410 CrossRef CAS.
- F. T. Edelmann, Coord. Chem. Rev., 2006, 250, 2511–2564 CrossRef CAS.
- J. Gottfriedsen and F. T. Edelmann, Coord. Chem. Rev., 2007, 251, 142–202 CrossRef CAS.
- F. T. Edelmann, Coord. Chem. Rev., 2009, 3, 343–409 CrossRef.
- F. T. Edelmann, Coord. Chem. Rev., 2009, 253, 2515–2587 CrossRef CAS.
- F. T. Edelmann, Coord. Chem. Rev., 2011, 15, 1834–1920 CrossRef.
- F. T. Edelmann, Coord. Chem. Rev., 2012, 23, 2641–2740 CrossRef.
- F. T. Edelmann, Coord. Chem. Rev., 2013, 7, 1122–1231 CrossRef.
- F. T. Edelmann, Coord. Chem. Rev., 2014, 261, 73–155 CrossRef CAS.
- F. T. Edelmann, Coord. Chem. Rev., 2015, 284, 124–205 CrossRef CAS.
- F. T. Edelmann, Coord. Chem. Rev., 2016, 306, 346–419 CrossRef CAS.
- F. T. Edelmann, Coord. Chem. Rev., 2016, 318, 29–130 CrossRef CAS.
- F. T. Edelmann, Coord. Chem. Rev., 2017, 338, 27–140 CrossRef CAS.
- F. T. Edelmann, Coord. Chem. Rev., 2018, 370, 129–223 CrossRef CAS.
- F. T. Edelmann, J. H. Farnaby, F. Jaroschik and B. Wilson, Coord. Chem. Rev., 2019, 398, 113005 CrossRef CAS.
- J. H. Farnaby, T. Chowdhury, S. J. Horsewill, B. Wilson and F. Jaroschik, Coord. Chem. Rev., 2021, 437, 213830 CrossRef CAS.
- G. Wilkinson and J. Birmingham, J. Am. Chem. Soc., 1954, 76, 6210–6210 CrossRef CAS.
- H. Schumann, J. A. Meese-Marktscheffel and L. Esser, Chem. Rev., 1995, 95, 865–986 CrossRef CAS.
- C. Eaborn, K. Izod and J. D. Smith, J. Organomet. Chem., 1995, 500, 89–99 CrossRef CAS.
- M. P. Hogerheide, J. Boersma and G. van Koten, Coord. Chem. Rev., 1996, 155, 87–126 CrossRef CAS.
- S. A. Cotton, Coord. Chem. Rev., 1997, 160, 93–127 CrossRef CAS.
- L. N. Zakharov and Y. T. Struchkov, J. Organomet. Chem., 1997, 536–537, 65–71 CrossRef.
- F. Nief, Coord. Chem. Rev., 1998, 178–180, 13–81 CrossRef CAS.
- F. T. Edelmann, D. M. M. Freckmann and H. Schumann, Chem. Rev., 2002, 102, 1851–1896 CrossRef CAS PubMed.
- M. Zimmermann and R. Anwander, Chem. Rev., 2010, 110, 6194–6259 CrossRef CAS PubMed.
- F. T. Edelmann, Angew. Chem., Int. Ed. Engl., 1995, 34, 2466–2488 CrossRef CAS.
- M. F. Lappert and R. Pearce, J. Chem. Soc., Chem. Commun., 1973, 126–126 RSC.
- H. Schumann, D. M. M. Freckmann and S. Dechert, Z. Anorg. Allg. Chem., 2002, 628, 2422–2426 CrossRef CAS.
- J. L. Atwood, W. E. Hunter, R. D. Rogers, J. Holton, J. McMeeking, R. Pearce and M. F. Lappert, J. Chem. Soc., Chem. Commun., 1978, 140–142 RSC.
- M. Niemeyer, Acta Crystallogr., 2001, 57, 553–555 Search PubMed.
- S. Arndt, T. P. Spaniol and J. Okuda, ChemComm, 2002, 896–897 RSC.
- B. R. Elvidge, S. Arndt, P. M. Zeimentz, T. P. Spaniol and J. Okuda, Inorg. Chem., 2005, 44, 6777–6788 CrossRef CAS PubMed.
- K. C. Hultzsch, T. P. Spaniol and J. Okuda, Angew. Chem., Int. Ed., 1999, 38, 227–230 CrossRef CAS.
- S. Bambirra, M. W. Bouwkamp, A. Meetsma and B. Hessen, J. Am. Chem. Soc., 2004, 126, 9182–9183 CrossRef CAS PubMed.
- G. Barker and M. Lappert, J. Organomet. Chem., 1974, 76, C45–C46 CrossRef CAS.
- P. B. Hitchcock, M. F. Lappert, R. G. Smith, R. A. Bartlett and P. P. Power, J. Chem. Soc., Chem. Commun., 1988, 1007–1009 RSC.
- A. G. Avent, C. F. Caro, P. B. Hitchcock, M. F. Lappert, Z. Li and X.-H. Wei, Dalton Trans., 2004, 1567–1577 RSC.
- M. Booij, N. H. Kiers, H. J. Heeres and J. H. Teuben, J. Organomet. Chem., 1989, 364, 79–86 CrossRef CAS.
- C. Eaborn, P. B. Hitchcock, K. Izod and J. D. Smith, J. Am. Chem. Soc., 1994, 116, 12071–12072 CrossRef CAS.
- C. Eaborn, P. B. Hitchcock, K. Izod, Z.-R. Lu and J. D. Smith, Organometallics, 1996, 15, 4783–4790 CrossRef CAS.
- G. Qi, Y. Nitto, A. Saiki, T. Tomohiro, Y. Nakayama and H. Yasuda, Tetrahedron, 2003, 59, 10409–10418 CrossRef CAS.
- S. Bambirra, A. Meetsma and B. Hessen, Organometallics, 2006, 25, 3454–3462 CrossRef CAS.
- A. J. Wooles, D. P. Mills, W. Lewis, A. J. Blake and S. T. Liddle, Dalton Trans., 2010, 39, 500–510 RSC.
- S. Harder, C. Ruspic, N. N. Bhriain, F. Berkermann and M. Schürmann, Z. Naturforsch., B: J. Chem. Sci., 2008, 63, 267–274 CrossRef.
- S. Harder, Angew. Chem., Int. Ed., 2004, 43, 2714–2718 CrossRef CAS PubMed.
- L. E. Manzer, J. Organomet. Chem., 1977, 135, C6–C9 CrossRef CAS.
- A. R. Petrov, K. A. Rufanov, K. Harms and J. Sundermeyer, J. Organomet. Chem., 2009, 694, 1212–1218 CrossRef CAS.
- S. Harder, Organometallics, 2005, 24, 373–379 CrossRef CAS.
- S. Bambirra, F. Perazzolo, S. J. Boot, T. J. J. Sciarone, A. Meetsma and B. Hessen, Organometallics, 2008, 27, 704–712 CrossRef CAS.
- F. Jaroschik, T. Shima, X. Li, K. Mori, L. Ricard, X.-F. Le Goff, F. Nief and Z. Hou, Organometallics, 2007, 26, 5654–5660 CrossRef CAS.
- X. Li, M. Nishiura, K. Mori, T. Mashiko and Z. Hou, Chem. Commun., 2007, 4137–4139 RSC.
- W. X. Zhang, M. Nishiura and Z. Hou, Angew. Chem., 2008, 120, 9846–9849 CrossRef.
- J. S. Ghotra, M. B. Hursthouse and A. J. Welch, J. Chem. Soc., Chem. Commun., 1973, 669–670 RSC.
- R. A. Andersen, D. H. Templeton and A. Zalkin, Inorg. Chem., 1978, 17, 2317–2319 CrossRef CAS.
- T. D. Tilley, R. A. Andersen and A. Zalkin, Inorg. Chem., 1984, 23, 2271–2276 CrossRef CAS.
- T. Fjeldberg and R. A. Andersen, J. Mol. Struct., 1985, 129, 93–105 CrossRef CAS.
- W. S. Rees Jr, O. Just and D. S. Van Derveer, J. Mater. Chem., 1999, 9, 249–252 RSC.
- W. A. Herrmann, R. Anwander, F. C. Munck, W. Scherer, V. Dufaud, N. W. Huber and G. R. J. Artus, Z. Naturforsch., B: Chem. Sci., 1994, 49, 1789–1797 CrossRef CAS.
- G. Jeske, H. Lauke, H. Mauermann, P. N. Swepston, H. Schumann and T. Marks, J. Am. Chem. Soc., 1985, 107, 8091–8103 CrossRef CAS.
- M. E. Thompson, S. M. Baxter, A. R. Bulls, B. J. Burger, M. C. Nolan, B. D. Santarsiero, W. P. Schaefer and J. E. Bercaw, J. Am. Chem. Soc., 1987, 109, 203–219 CrossRef CAS.
- Y. Li, P.-F. Fu and T. J. Marks, Organometallics, 1994, 13, 439–440 CrossRef CAS.
- D. C. Bradley, J. S. Ghotra and F. A. Hart, J. Chem. Soc., Dalton Trans., 1973, 1021–1023 RSC.
- C. T. Carver and P. L. Diaconescu, J. Am. Chem. Soc., 2008, 130, 7558–7559 CrossRef CAS PubMed.
- C. T. Carver, M. J. Monreal and P. L. Diaconescu, Organometallics, 2008, 27, 363–370 CrossRef CAS.
- D. P. Mills, O. J. Cooper, J. McMaster, W. Lewis and S. T. Liddle, Dalton Trans., 2009, 4547–4555 RSC.
- N. Meyer, P. W. Roesky, S. Bambirra, A. Meetsma, B. Hessen, K. Saliu and J. Takats, Organometallics, 2008, 27, 1501–1505 CrossRef CAS.
- S. Ge, A. Meetsma and B. Hessen, Organometallics, 2009, 28, 719–726 CrossRef CAS.
- A. D. Benischke, L. Anthore-Dalion, F. Kohl and P. Knochel, Chem. – Eur. J., 2018, 24, 11103–11109 CrossRef CAS PubMed.
- M. S. Hill and P. B. Hitchcock, Dalton Trans., 2003, 4570–4571 RSC.
- M. Roger, N. Barros, T. Arliguie, P. Thuéry, L. Maron and M. Ephritikhine, J. Am. Chem. Soc., 2006, 128, 8790–8802 CrossRef CAS PubMed.
- K. Izod, W. Clegg and R. W. Harrington, Dalton Trans., 2010, 39, 6705–6709 RSC.
- B. M. Wolf, C. Stuhl and R. Anwander, Chem. Commun., 2018, 54, 8826–8829 RSC.
- W. Huang, B. M. Upton, S. I. Khan and P. L. Diaconescu, Organometallics, 2013, 32, 1379–1386 CrossRef CAS.
- A. C. Behrle and J. A. R. Schmidt, Organometallics, 2011, 30, 3915–3918 CrossRef CAS.
- K. Yan, G. Schoendorff, B. M. Upton, A. Ellern, T. L. Windus and A. D. Sadow, Organometallics, 2013, 32, 1300–1316 CrossRef CAS.
- A. Pindwal, S. Patnaik, W. C. Everett, A. Ellern, T. L. Windus and A. D. Sadow, Angew. Chem., 2017, 129, 643–646 CrossRef.
- A. Pindwal, K. Yan, S. Patnaik, B. M. Schmidt, A. Ellern, I. I. Slowing, C. Bae and A. D. Sadow, J. Am. Chem. Soc., 2017, 139, 16862–16874 CrossRef CAS PubMed.
- R. Fischer, S. Bode, M. Köhler, J. Langer, H. Görls, M. D. Hager, U. S. Schubert and M. Westerhausen, Organometallics, 2015, 34, 23–31 CrossRef CAS.
- G. B. Deacon, P. C. Junk, R. P. Kelly and J. Wang, Dalton Trans., 2016, 45, 1422–1435 RSC.
- K. C. Boteju, A. Ellern and A. D. Sadow, Chem. Commun., 2017, 53, 716–719 RSC.
- A. N. Selikhov, G. S. Plankin, A. V. Cherkasov, A. S. Shavyrin, E. Louyriac, L. Maron and A. A. Trifonov, Inorg. Chem., 2019, 58, 5325–5334 CrossRef CAS PubMed.
- P. G. Pringle and M. B. Smith, J. Chem. Soc., Chem. Commun., 1990, 1701–1702 RSC.
- P. A. T. Hoye, P. G. Pringle, M. B. Smith and K. Worboys, J. Chem. Soc., Dalton Trans., 1993, 269–274 RSC.
- D. K. Wicht, I. V. Kourkine, B. M. Lew, J. M. Nthenge and D. S. Glueck, J. Am. Chem. Soc., 1997, 119, 5039–5040 CrossRef CAS.
- V. Koshti, S. Gaikwad and S. H. Chikkali, Coord. Chem. Rev., 2014, 265, 52–73 CrossRef CAS.
- S. Lau, T. M. Hood and R. L. Webster, ACS Catal., 2022, 12, 10939–10949 CrossRef CAS PubMed.
- A. A. Trifonov, I. V. Basalov and A. A. Kissel, Dalton Trans., 2016, 45, 19172–19193 RSC.
- B. T. Novas and R. Waterman, ChemCatChem, 2022, 14, 1–31 CrossRef.
- C. A. Bange and R. Waterman, Chem. – Eur. J., 2016, 22, 12598–12605 CrossRef CAS PubMed.
- S. A. Pullarkat, Synthesis, 2016, 48, 493–503 CrossRef CAS.
- J. L. Bookham and D. M. Smithies, J. Organomet. Chem., 1999, 577, 305–315 CrossRef CAS.
- N. T. Coles, M. F. Mahon and R. L. Webster, Chem. Commun., 2018, 54, 10443–10446 RSC.
- M. R. Crimmin, A. G. Barrett, M. S. Hill, P. B. Hitchcock and P. A. Procopiou, Organometallics, 2007, 26, 2953–2956 CrossRef CAS.
- M. R. Crimmin, A. G. Barrett, M. S. Hill, P. B. Hitchcock and P. A. Procopiou, Organometallics, 2008, 27, 497–499 CrossRef CAS.
- T. M. Al-Shboul, H. Görls and M. Westerhausen, Inorg. Chem. Commun., 2008, 11, 1419–1421 CrossRef CAS.
- B. J. Ward and P. A. Hunt, ACS Catal., 2017, 7, 459–468 CrossRef CAS.
- B. Liu, T. Roisnel, J. F. Carpentier and Y. Sarazin, Angew. Chem., Int. Ed., 2012, 51, 4943–4946 CrossRef CAS PubMed.
- S.-C. Rosca, T. Roisnel, V. Dorcet, J.-F. Carpentier and Y. Sarazin, Organometallics, 2014, 33, 5630–5642 CrossRef CAS.
- F. M. Younis, S. Krieck, T. M. Al-Shboul, H. Görls and M. Westerhausen, Inorg. Chem., 2016, 55, 4676–4682 CrossRef CAS PubMed.
- A. O. Tolpygin, A. V. Cherkasov, G. K. Fukin, T. A. Kovylina, K. A. Lyssenko and A. A. Trifonov, Eur. J. Inorg. Chem., 2019, 2019, 4289–4296 CrossRef CAS.
- B. Freitag, P. Stegner, K. Thum, C. A. Fischer and S. Harder, Eur. J. Inorg. Chem., 2018, 2018, 1938–1944 CrossRef CAS.
- E. Le Coz, H. Roueindeji, T. Roisnel, V. Dorcet, J.-F. Carpentier and Y. Sarazin, Dalton Trans., 2019, 48, 9173–9180 RSC.
- M. De Tullio, A. Hernán-Gómez, Z. Livingstone, W. Clegg, A. R. Kennedy, R. W. Harrington, A. Antiñolo, A. Martínez, F. Carrillo–Hermosilla and E. Hevia, Chem. – Eur. J., 2016, 22, 17646–17656 CrossRef CAS PubMed.
- J. Pahl, T. E. Stennett, M. Volland, D. M. Guldi and S. Harder, Chem. – Eur. J., 2019, 25, 2025–2034 CrossRef CAS PubMed.
- G. Zhao, F. Basuli, U. J. Kilgore, H. Fan, H. Aneetha, J. C. Huffman, G. Wu and D. J. Mindiola, J. Am. Chem. Soc., 2006, 128, 13575–13585 CrossRef CAS PubMed.
- A. Perrier, V. Comte, C. Moïse and P. Le Gendre, Chem. – Eur. J., 2010, 16, 64–67 CrossRef CAS PubMed.
- R. Sakae, Y. Yamamoto, K. Komeyama and K. Takaki, Chem. Lett., 2010, 39, 276–277 CrossRef CAS.
- A. J. Roering, S. E. Leshinski, S. M. Chan, T. Shalumova, S. N. MacMillan, J. M. Tanski and R. Waterman, Organometallics, 2010, 29, 2557–2565 CrossRef CAS.
- M. B. Ghebreab, C. A. Bange and R. Waterman, J. Am. Chem. Soc., 2014, 136, 9240–9243 CrossRef CAS PubMed.
- C. A. Bange, N. T. Mucha, M. E. Cousins, A. C. Gehsmann, A. Singer, T. Truax, L. J. Higham and R. J. I. Waterman, Inorganics, 2016, 4, 26 CrossRef.
- C. A. Bange and R. Waterman, ACS Catal., 2016, 6, 6413–6416 CrossRef CAS.
- C. A. Bange, M. A. Conger, B. T. Novas, E. R. Young, M. D. Liptak and R. Waterman, ACS Catal., 2018, 8, 6230–6238 CrossRef CAS.
- B. T. Novas, C. A. Bange and R. Waterman, Eur. J. Inorg. Chem., 2019, 2019, 1640–1643 CrossRef CAS.
- Y. Zhang, L. Qu, Y. Wang, D. Yuan, Y. Yao and Q. Shen, Inorg. Chem., 2018, 57, 139–149 CrossRef CAS PubMed.
- Y. Zhang, X. Wang, Y. Wang, D. Yuan and Y. Yao, Dalton Trans., 2018, 47, 9090–9095 RSC.
- M. Kamitani, M. Itazaki, C. Tamiya and H. Nakazawa, J. Am. Chem. Soc., 2012, 134, 11932–11935 CrossRef CAS PubMed.
- M. Liu, C. Sun, F. Hang, N. Sun and D. Chen, Dalton Trans., 2014, 43, 4813–4821 RSC.
- M. Itazaki, S. Katsube, M. Kamitani and H. Nakazawa, Chem. Commun., 2016, 52, 3163–3166 RSC.
- K. J. Gallagher and R. L. Webster, Chem. Commun., 2014, 50, 12109–12111 RSC.
- A. K. King, A. Buchard, M. F. Mahon and R. L. Webster, Chem. – Eur. J., 2015, 21, 15960–15963 CrossRef CAS PubMed.
- M. Espinal-Viguri, A. K. King, J. P. Lowe, M. F. Mahon and R. L. Webster, ACS Catal., 2016, 6, 7892–7897 CrossRef CAS.
- J. K. Pagano, C. A. Bange, S. E. Farmiloe and R. Waterman, Organometallics, 2017, 36, 3891–3895 CrossRef CAS.
- B. J. Ackley, J. K. Pagano and R. Waterman, Chem. Commun., 2018, 54, 2774–2776 RSC.
- H. R. Sharpe, A. M. Geer, W. Lewis, A. J. Blake and D. L. Kays, Angew. Chem., Int. Ed., 2017, 56, 4845–4848 CrossRef CAS PubMed.
- F. Jérôme, F. Monnier, H. Lawicka, S. Dérien and P. H. Dixneuf, Chem. Commun., 2003, 696–697 RSC.
- R. G. Belli, K. M. Burton, S. A. Rufh, R. McDonald and L. Rosenberg, Organometallics, 2015, 34, 5637–5646 CrossRef CAS.
- R. G. Belli, J. Yang, E. N. E. Bahena, R. McDonald and L. Rosenberg, ACS Catal., 2022, 12, 5247–5262 CrossRef CAS.
- M. P. Cibuzar, S. G. Dannenberg and R. Waterman, Isr. J. Chem., 2020, 60, 446–451 CrossRef CAS.
- H. Ohmiya, H. Yorimitsu and K. Oshima, Angew. Chem., 2005, 117, 2420–2422 CrossRef.
- J. Rajpurohit, P. Kumar, P. Shukla, M. Shanmugam and M. Shanmugam, Organometallics, 2018, 37, 2297–2304 CrossRef CAS.
- R. Nolla-Saltiel, A. M. Geer, L. J. Taylor, O. Churchill, E. S. Davies, W. Lewis, A. J. Blake and D. L. Kays, Adv. Synth. Catal., 2020, 362, 3148–3157 CrossRef CAS.
- M. Hayashi, Y. Matsuura and Y. Watanabe, J. Org. Chem., 2006, 71, 9248–9251 CrossRef CAS PubMed.
- A. M. Geer, Á. L. Serrano, B. de Bruin, M. A. Ciriano and C. Tejel, Angew. Chem., Int. Ed., 2015, 54, 472–475 CrossRef CAS PubMed.
- V. Varela-Izquierdo, A. M. Geer, J. Navarro, J. A. López, M. A. Ciriano and C. Tejel, J. Am. Chem. Soc., 2020, 143, 349–358 CrossRef PubMed.
- A. Di Giuseppe, R. De Luca, R. Castarlenas, J. J. Pérez-Torrente, M. Crucianelli and L. A. Oro, Chem. Commun., 2016, 52, 5554–5557 RSC.
- M. O. Shulyupin, M. A. Kazankova and I. P. Beletskaya, Org. Lett., 2002, 4, 761–763 CrossRef CAS PubMed.
- M. Kazankova, I. Efimova, A. Kochetkov, V. Afanas'ev and I. Beletskaya, Russ. J. Org. Chem., 2002, 38, 1465–1474 CrossRef CAS.
- R. L. Webster, Inorganics, 2018, 6, 120 CrossRef CAS.
- J. Yan, Y. B. Wang, S. Hou, L. Shi, X. Zhu, X. Q. Hao and M. P. Song, Appl. Organomet. Chem., 2020, 34, e5954 CrossRef CAS.
- S.-i. Kawaguchi, S. Nagata, A. Nomoto, M. Sonoda and A. Ogawa, J. Org. Chem., 2008, 73, 7928–7933 CrossRef CAS PubMed.
- S. Nagata, S.-i. Kawaguchi, M. Matsumoto, I. Kamiya, A. Nomoto, M. Sonoda and A. J. T. L. Ogawa, Tetrahedron Lett., 2007, 48, 6637–6640 CrossRef CAS.
- D. K. Wicht, I. V. Kourkine, I. Kovacik, D. S. Glueck, T. E. Concolino, G. P. Yap, C. D. Incarvito and A. L. Rheingold, Organometallics, 1999, 18, 5381–5394 CrossRef CAS.
- C. Scriban, I. Kovacik and D. S. Glueck, Organometallics, 2005, 24, 4871–4874 CrossRef CAS.
- C. Scriban, D. S. Glueck, L. N. Zakharov, W. S. Kassel, A. G. DiPasquale, J. A. Golen and A. L. Rheingold, Organometallics, 2006, 25, 5757–5767 CrossRef CAS.
- A. C. Chadwick, M. A. Heckenast, J. J. Race, P. G. Pringle and H. A. Sparkes, Organometallics, 2019, 38, 3871–3879 CrossRef CAS.
- A. Kondoh, H. Yorimitsu and K. Oshima, J. Am. Chem. Soc., 2007, 129, 4099–4104 CrossRef CAS PubMed.
- A. Leyva-Pérez, J. A. Vidal-Moya, J. R. Cabrero-Antonino, S. S. Al-Deyab, S. I. Al-Resayes and A. Corma, J. Organomet. Chem., 2011, 696, 362–367 CrossRef.
- N. A. Isley, R. T. Linstadt, E. D. Slack and B. H. Lipshutz, Dalton Trans., 2014, 43, 13196–13200 RSC.
- J. Yuan, L. Zhu, J. Zhang, J. Li and C. Cui, Organometallics, 2017, 36, 455–459 CrossRef CAS.
- S. G. Dannenberg and R. Waterman, Chem. Commun., 2020, 56, 14219–14222 RSC.
- T. M. H. Downie, J. W. Hall, T. P. C. Finn, D. J. Liptrot, J. P. Lowe, M. F. Mahon, C. L. McMullin and M. K. Whittlesey, Chem. Commun., 2020, 56, 13359–13362 RSC.
- D. Bissessar, J. Egly, T. Achard, P. Steffanut, M. Mauro and S. Bellemin-Laponnaz, Eur. J. Inorg. Chem., 2022, 2022, e202200101 CrossRef CAS.
- B. Zhang, X. Ma, B. Yan, C. Ni, H. Yu, Z. Yang and H. W. Roesky, Dalton Trans., 2021, 50, 15488–15492 RSC.
- B. A. Vaughan, E. M. Arsenault, S. M. Chan and R. Waterman, J. Organomet. Chem., 2012, 696, 4327–4331 CrossRef CAS.
- M. R. Douglass and T. J. Marks, J. Am. Chem. Soc., 2000, 122, 1824–1825 CrossRef CAS.
- M. R. Douglass, C. L. Stern and T. J. Marks, J. Am. Chem. Soc., 2001, 123, 10221–10238 CrossRef CAS PubMed.
- A. M. Kawaoka, M. R. Douglass and T. J. Marks, Organometallics, 2003, 22, 4630–4632 CrossRef CAS.
- A. M. Kawaoka and T. J. Marks, J. Am. Chem. Soc., 2004, 126, 12764–12765 CrossRef CAS PubMed.
- A. A. Kissel, T. V. Mahrova, D. M. Lyubov, A. V. Cherkasov, G. K. Fukin, A. A. Trifonov, I. Del Rosal and L. Maron, Dalton Trans., 2015, 44, 12137–12148 RSC.
- K. Komeyama, T. Kawabata, K. Takehira and K. Takaki, J. Org. Chem., 2005, 70, 7260–7266 CrossRef CAS PubMed.
- A. C. Behrle and J. A. R. Schmidt, Organometallics, 2013, 32, 1141–1149 CrossRef CAS.
- M. M. I. Basiouny, D. A. Dollard and J. A. R. Schmidt, ACS Catal., 2019, 9, 7143–7153 CrossRef CAS.
- P. Ji, T. Sawano, Z. Lin, A. Urban, D. Boures and W. Lin, J. Am. Chem. Soc., 2016, 138, 14860–14863 CrossRef CAS PubMed.
- J. Li, C. A. Lamsfus, C. Song, J. Liu, G. Fan, L. Maron and C. Cui, ChemCatChem, 2017, 9, 1368–1372 CrossRef CAS.
- I. V. Lapshin, A. V. Cherkasov, A. F. Asachenko and A. A. Trifonov, Chem. Commun., 2020, 56, 12913–12916 RSC.
- I. V. Lapshin, A. V. Cherkasov, K. A. Lyssenko, G. K. Fukin and A. A. Trifonov, Inorg. Chem., 2022, 61, 9147–9161 CrossRef CAS PubMed.
- Q. Liu, C. Wang, X. Zhang, M. Xue, Y. Yao, Y. Zhang and Q. Shen, New J. Chem., 2016, 40, 10447–10454 RSC.
- K. Takaki, M. Takeda, G. Koshoji, T. Shishido and K. Takehira, Tetrahedron Lett., 2001, 42, 6357–6360 CrossRef CAS.
- K. Takaki, K. Komeyama and K. Takehira, Tetrahedron, 2003, 59, 10381–10395 CrossRef CAS.
- K. Takaki, G. Koshoji, K. Komeyama, M. Takeda, T. Shishido, A. Kitani and K. Takehira, J. Org. Chem., 2003, 68, 6554–6565 CrossRef CAS PubMed.
- K. Takaki, K. Komeyama, D. Kobayashi, T. Kawabata and K. Takehira, J. Alloys Compd., 2006, 408, 432–436 CrossRef.
- K. Komeyama, D. Kobayashi, Y. Yamamoto, K. Takehira and K. Takaki, Tetrahedron, 2006, 62, 2511–2519 CrossRef CAS.
- I. V. Basalov, V. Dorcet, G. K. Fukin, J. F. Carpentier, Y. Sarazin and A. A. Trifonov, Chem. – Eur. J., 2015, 21, 6033–6036 CrossRef CAS PubMed.
- I. V. Basalov, B. Liu, T. Roisnel, A. V. Cherkasov, G. K. Fukin, J.-F. Carpentier, Y. Sarazin and A. A. Trifonov, Organometallics, 2016, 35, 3261–3271 CrossRef CAS.
- J. Yuan, H. Hu and C. Cui, Chem. – Eur. J., 2016, 22, 5778–5785 CrossRef CAS PubMed.
- H. Hu and C. Cui, Organometallics, 2012, 31, 1208–1211 CrossRef CAS.
- B. Liu, T. Roisnel, J. F. Carpentier and Y. Sarazin, Chem. – Eur. J., 2013, 19, 13445–13462 CrossRef CAS PubMed.
- M. He, M. T. Gamer and P. W. Roesky, Organometallics, 2016, 35, 2638–2644 CrossRef CAS.
- I. V. Basalov, O. S. Yurova, A. V. Cherkasov, G. K. Fukin and A. A. Trifonov, Inorg. Chem., 2016, 55, 1236–1244 CrossRef CAS PubMed.
- I. V. Lapshin, O. S. Yurova, I. V. Basalov, V. Y. Rad'kov, E. I. Musina, A. V. Cherkasov, G. K. Fukin, A. A. Karasik and A. A. Trifonov, Inorg. Chem., 2018, 57, 2942–2952 CrossRef CAS PubMed.
- M. E. Garner, B. F. Parker, S. Hohloch, R. G. Bergman and J. Arnold, J. Am. Chem. Soc., 2017, 139, 12935–12938 CrossRef CAS PubMed.
- W.-X. Zhang, M. Nishiura and Z. Hou, Chem. Commun., 2006, 3812–3814 RSC.
- M. Itazaki, T. Matsutani, T. Nochida, T. Moriuchi and H. Nakazawa, Chem. Commun., 2020, 56, 443–445 RSC.
- W. X. Zhang, M. Nishiura, T. Mashiko and Z. Hou, Chem. – Eur. J., 2008, 14, 2167–2179 CrossRef CAS PubMed.
- X. Gu, L. Zhang, X. Zhu, S. Wang, S. Zhou, Y. Wei, G. Zhang, X. Mu, Z. Huang and D. Hong, Organometallics, 2015, 34, 4553–4559 CrossRef CAS.
- W. Ma, L. Xu, W.-X. Zhang and Z. Xi, New J. Chem., 2015, 39, 7649–7655 RSC.
- I. S. Karmel, M. Tamm and M. S. Eisen, Angew. Chem., Int. Ed., 2015, 54, 12422–12425 CrossRef CAS PubMed.
- R. J. Batrice and M. S. Eisen, Chem. Sci., 2016, 7, 939–944 RSC.
- H. Liu, N. Fridman, M. Tamm and M. S. Eisen, Organometallics, 2017, 36, 3896–3903 CrossRef CAS.
- M. M. I. Basiouny and J. A. R. Schmidt, Organometallics, 2017, 36, 721–729 CrossRef CAS.
- I. Banerjee, A. Harinath and T. K. Panda, Eur. J. Inorg. Chem., 2019, 2019, 2224–2230 CrossRef CAS.
- S. Guo, W. Yan, Z. Zhang, Z. Huang, Y. Guo, Z. Liang, S. Li, Z. Fu and H. Cai, J. Org. Chem., 2022, 87, 5522–5529 CrossRef CAS PubMed.
- A. Allen Jr, D. R. Manke and W. Lin, Tetrahedron Lett., 2000, 41, 151–154 CrossRef.
- L.-B. Han, F. Mirzaei, C.-Q. Zhao and M. Tanaka, J. Am. Chem. Soc., 2000, 122, 5407–5408 CrossRef CAS.
- F. Mirzaei, L.-B. Han and M. Tanaka, Tetrahedron Lett., 2001, 42, 297–299 CrossRef CAS.
- C. Q. Zhao, L. B. Han, M. Goto and M. Tanaka, Angew. Chem., Int. Ed., 2001, 40, 1929–1932 CrossRef CAS PubMed.
- Q. Xu and L.-B. Han, Org. Lett., 2006, 8, 2099–2101 CrossRef CAS PubMed.
- Y. A. Rina and J. A. R. Schmidt, J. Org. Chem., 2020, 85, 14720–14729 CrossRef CAS PubMed.
- B. Kaboudin, H. Esfandiari, A. Moradi, F. Kazemi and H. Aoyama, J. Org. Chem., 2019, 84, 14943–14948 CrossRef CAS PubMed.
- Y. A. Rina and J. A. R. Schmidt, Organometallics, 2019, 38, 4261–4270 CrossRef CAS.
- Q. Liu, C. Wang, X. Zhang, M. Xue, Y. Yao, Y. Zhang and Q. Shen, New J. Chem., 2016, 40, 10447–10454 RSC.
- I. Wauters, W. Debrouwer and C. V. Stevens, Beilstein J. Org. Chem., 2014, 10, 1064–1096 CrossRef PubMed.
- L. Coudray and J.-L. Montchamp, Eur. J. Org. Chem., 2008, 3601–3613 CAS.
- J. Yuan, H. Hu and C. Cui, Chem. – Eur. J., 2016, 22, 5778–5785 CrossRef CAS PubMed.
- M. Espinal-Viguri, A. K. King, J. P. Lowe, M. F. Mahon and R. L. Webster, ACS Catal., 2016, 6, 7892–7897 CrossRef CAS.
- M. R. Crimmin, A. G. M. Barrett, M. S. Hill, P. B. Hitchcock and P. A. Procopiou, Organometallics, 2007, 26, 2953–2956 CrossRef CAS.
- J. J. Stone, R. A. Stockland, J. M. Reyes, J. Kovach, C. C. Goodman and E. S. Tillman, J. Mol. Catal., 2005, 226, 11–21 CrossRef CAS.
- K. I. Utegenov, V. V. Krivykh, A. M. Mazhuga, O. S. Chudin, A. F. Smol'yakov, F. M. Dolgushin, E. I. Goryunov and N. A. Ustynyuk, Organometallics, 2016, 35, 3903–3913 CrossRef CAS.
- A. W. J. Platten, A. M. Borys and E. Hevia, ChemCatChem, 2022, 14, e202101853 CrossRef CAS.
- L. Zhang, S. Bai and L. Zheng, Organometallics, 2023, 42, 2262–2268 CAS.
- S. Harder, Chem. Rev., 2010, 110, 3852–3876 CrossRef CAS PubMed.
- F. Buch and S. Harder, Organometallics, 2007, 26, 5132–5135 CrossRef CAS.
- N. V. Forosenko, I. V. Basalov, A. V. Cherkasov, G. K. Fukin, E. S. Shubina and A. A. Trifonov, Dalton Trans., 2018, 47, 12570–12581 RSC.
- J. F. Dunne, S. R. Neal, J. Engelkemier, A. Ellern and A. D. Sadow, J. Am. Chem. Soc., 2011, 133, 16782–16785 CrossRef CAS PubMed.
- N. Li and B.-T. Guan, Eur. J. Inorg. Chem., 2019, 2019, 2231–2235 CrossRef CAS.
- A. Baishya, T. Peddarao and S. Nembenna, Dalton Trans., 2017, 46, 5880–5887 RSC.
- M. Rauch, R. C. Roberts and G. Parkin, Inorg. Chim. Acta, 2019, 494, 271–279 CrossRef CAS.
- S. Anga, Y. Sarazin, J.-F. Carpentier and T. K. Panda, ChemCatChem, 2016, 8, 1373–1378 CrossRef CAS.
- M. S. Hill, D. J. Liptrot, D. J. MacDougall, M. F. Mahon and T. P. Robinson, Chem. Sci., 2013, 4, 4212–4222 RSC.
- C. Bellini, C. Orione, J.-F. Carpentier and Y. Sarazin, Angew. Chem., Int. Ed., 2016, 55, 3744–3748 CrossRef CAS PubMed.
- L. J. Morris, G. R. Whittell, J.-C. Eloi, M. F. Mahon, F. Marken, I. Manners and M. S. Hill, Organometallics, 2019, 38, 3629–3648 CrossRef CAS.
- L. Greb, S. Tamke and J. Paradies, Chem. Commun., 2014, 50, 2318–2320 RSC.
- L. K. Allen, R. García-Rodríguez and D. S. Wright, Dalton Trans., 2015, 44, 12112–12118 RSC.
- H. Q. Liu and J. F. Harrod, Organometallics, 1992, 11, 822–827 CrossRef CAS.
- E. Matarasso-Tchiroukhine, J. Chem. Soc., Chem. Commun., 1990, 681–682 RSC.
- K. A. Erickson, M. P. Cibuzar, N. T. Mucha and R. Waterman, Dalton Trans., 2018, 47, 2138–2142 RSC.
- M. B. Reuter, M. P. Cibuzar, J. Hammerton and R. Waterman, Dalton Trans., 2020, 49, 2972–2978 RSC.
- D. Gasperini, A. K. King, N. T. Coles, M. F. Mahon and R. L. Webster, ACS Catal., 2020, 10, 6102–6112 CrossRef CAS.
- D. V. Gutsulyak, S. F. Vyboishchikov and G. I. Nikonov, J. Am. Chem. Soc., 2010, 132, 5950–5951 CrossRef CAS PubMed.
- C. D. F. Königs, M. F. Müller, N. Aiguabella, H. F. T. Klare and M. Oestreich, Chem. Commun., 2013, 49, 1506–1508 RSC.
- W. Zhai, B. Li and B. Wang, Tetrahedron, 2018, 74, 1123–1128 CrossRef CAS.
- S. Itagaki, K. Kamata, K. Yamaguchi and N. Mizuno, Chem. Commun., 2012, 48, 9269–9271 CAS.
- Z. Lu, K. Murakami, S. Koge, T. Mizumo and J. Ohshita, J. Organomet. Chem., 2016, 808, 63–67 CrossRef CAS.
- P. Ríos, M. Roselló-Merino, O. Rivada-Wheelaghan, J. Borge, J. López-Serrano and S. Conejero, Chem. Commun., 2018, 54, 619–622 RSC.
- K. Takaki, T. Kamata, Y. Miura, T. Shishido and K. Takehira, J. Org. Chem., 1999, 64, 3891–3895 CrossRef CAS.
- W. Xie, H. Hu and C. Cui, Angew. Chem., 2012, 124, 11303–11306 CrossRef.
- A. Pindwal, A. Ellern and A. D. Sadow, Organometallics, 2016, 35, 1674–1683 CrossRef CAS.
- X. Zhang, S. Zhou, X. Fang, L. Zhang, G. Tao, Y. Wei, X. Zhu, P. Cui and S. Wang, Inorg. Chem., 2020, 59, 9683–9692 CrossRef CAS PubMed.
- M. P. Cibuzar and R. Waterman, Organometallics, 2018, 37, 4395–4401 CrossRef CAS.
- Y. A. Rina and J. A. R. Schmidt, Organometallics, 2022, 41, 2974–2984 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |