
Ultra-low loading Pt atomic cluster electrode with Pt–O bond as an active site with high hydrogen evolution reaction performance†
Zhandong
Ren
 *a,
Zhiqiang
Xie
a,
Li
Deng
a,
Chen
Dong
a,
Guocan
Song
a,
Xiaohui
Liu
a,
Juanjuan
Han
a,
Lin
Zhuang
b,
Yi
Liu
c and
Yuchan
Zhu
*a
*a,
Zhiqiang
Xie
a,
Li
Deng
a,
Chen
Dong
a,
Guocan
Song
a,
Xiaohui
Liu
a,
Juanjuan
Han
a,
Lin
Zhuang
b,
Yi
Liu
c and
Yuchan
Zhu
*a
aSchool of Chemical and Environmental Engineering, Wuhan Polytechnic University, Wuhan, 430023, P. R. China. E-mail: renzhandong@163.com; zhuyuchan@163.com
bCollege of Chemistry and Molecular Sciences, Hubei Key Lab of Electrochemical Power Sources, Wuhan University, Wuhan, 430072, P. R. China
cState Key Laboratory of Separation Membranes and Membrane Processes, School of Chemistry, Tiangong University, Tianjin 300387, P. R. China
First published on 29th August 2023
Abstract
Water electrolysis is the most fascinating procedure for producing pure hydrogen owing to its flexibility and convenience. Platinum (Pt) is the most effective electrocatalyst for the hydrogen evolution reaction (HER) but its high price and scarcity have greatly restricted its commercial application. Therefore, it is necessary to greatly increase the mass activity (MA) of Pt to meet practical applications. In the present study, an oxidized Pt atomic cluster-supported Au electrode (PtAC–O–Au) with an ultra-low loading was prepared by high vacuum magnetron sputtering combined with electrochemical anodic oxidation. The (PtAC–O–Au)-1 electrode has a very high mass activity (MA), reaching 49.2 A mgPt−1 at an overpotential of 50 mV, which is 41 times that of the 20 wt% Pt/C electrode and 20 times that of the 0.5 wt% Pt/C electrode. Even at such a low load, the (PtAC–O–Au)-1 electrode has excellent apparent activity and only needs an overpotential of 41 mV@10 mA cm−2, which is close to that of the 20 wt% Pt/C electrode (37 mV@10 mA cm−2). Moreover, the (PtAC–O–Au)-1 electrode has an ultra-high specific activity (SA). The SA of (PtAC–O–Au)-1 is 12–18 times higher than that of the 0.5 wt% Pt/C electrode and 36–56 times higher than commercial Pt/C electrodes. More importantly, it was confirmed by the electrochemical analysis method (cyclic voltammetry and CO adsorption–stripping) and X-ray photoelectron spectroscopy (XPS) that the active site is the oxidized platinum (Pt–O–Au) on the surface of the electrode. Density functional theory (DFT) calculations have also elucidated that the absolute value of ΔGH*(Pt) of PtAC–O–Au is close to that of Pt(111), indicating that its outstanding HER activity originates from its optimal ΔGH*(Pt) value.
Introduction
Electrochemical energy conversion technology, such as water electrolysis and CO2 electroreduction, is an ideal way to solve the energy crisis and environmental pollution.1–5 Hydrogen energy has attracted considerable attention as an ideal energy carrier and an efficient energy storage technology owing to its high energy density and carbon-free combustion emission. Water electrolysis is the most fascinating procedure for producing pure hydrogen owing to its flexibility and convenience. The development of highly active, long-term stable, and inexpensive electrocatalysts for the hydrogen evolution reaction (HER) is an attractive and challenging topic.6 Platinum (Pt) is the most effective electrocatalyst for HER; however its high price and scarcity have greatly restricted its commercial application. Therefore, it is necessary to greatly increase the mass activity (MA) of platinum to improve the utilization rate of platinum. At present, nano platinum electrodes with specific morphologies have been investigated, such as nanoparticles,7 nanoclusters,8,9 and amorphous,10 and, the loading of Pt nanoparticles on functionalized carriers is often used. Loading Pt nanoparticles onto carbon-based materials has greatly improved the MA of Pt, for example, hollow mesoporous carbon spheres,11 defective graphene,12,13 and nitrogen-doped carbon materials.14,15 Metal oxides, metal sulfides, metal carbides, and metal selenides are also used as functional carriers, such as MnO2,16 WO3−x,17,18 F-doped SnO2,19 TiO2–OV,20 TiBxOy,21 MoS3,22 VS2,23 WCx,24,25 Mo2C,26 MXene,27–29 and CdSe,30 which further enhance the activity and stability of HER. In addition, the trace Pt combined with other transition metals has also exhibited definite HER activity, such as CoPt–PtSA,31 Pt/Ni–Mo–N–O,32 PtNi,33 PtRu,34,35 PtW,36 Pt@Pd3Pb,37 PtPd,38 PtPdRuTe,39 and PtSe2.40,41In terms of Pt loading, although there have been good achievements, it is still necessary to further reduce the loading. Some electrocatalysts with low Pt loadings have been developed, such as PtW NPs/C,36 Pt3Ni2 NWs–S/C,42 Pt–MoS2,43 Pt/def-WO3@CFC,44 Pt@MoS2/NiS2,45 Mo2TiC2Tx–PtSA,46 Pt–MoO2@PC,47 and so on. However, the Pt loading in the above-mentioned electrocatalysts has exceeded 10 μg cm−2. Further research results have reduced the Pt loading to less than 10 μg cm−2, such as Pt5/HMCS,11 Pt Cs/MoO2 NSs-L,16 CNT/Pt@CdSe-OCPs,30 AL-Pt/Pd3Pb,37 Pt-GT-1,48 CDs/Pt–PANI,49 Pt@mh-3D MXene,50 Pt–GDY-2,51 Pt–HNCNT,52 Pt–Ru dimer,53etc. To date, little research has been done on the Pt loading of less than 1.0 μg cm−2, except for Pt SA/m-WO3−x,17 Pt/TiBxOy,21 Pt–ACs/CoNC,54 PtOx/TiO2,55 and Pt–SAs/WS2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 56 electrocatalysts. Most of these studies have revealed the HER activity of metallic Pt. However, some interesting phenomena regarding the oxidized Pt still require detailed attention.54–60 Therefore, the purpose of this study is to study the HER activity of oxidized Pt at very low loading (<1.0 μgPt cm−2).
56 electrocatalysts. Most of these studies have revealed the HER activity of metallic Pt. However, some interesting phenomena regarding the oxidized Pt still require detailed attention.54–60 Therefore, the purpose of this study is to study the HER activity of oxidized Pt at very low loading (<1.0 μgPt cm−2).
To obtain the ultra-low Pt electrode, the high vacuum magnetron sputtering method has been adopted in this study. The magnetron sputtering can uniformly deposit metal elements on the substrate surface in the form of atomic clusters, which can realize ultra-low load deposition through the control of the sputtering process. However, there are few applications in the preparation of electrode materials at present.61–63 The Pt loading of the (PtAC–O–Au)-1 electrode (0.48 μgPt cm−2) is much lower than the Pt loading in most reports (Table S3 (ESI†)). Through characterization analysis, it has been proved that Pt atomic clusters (PtAC) with a particle size of 0.8 nm can be deposited on the surface of the Au underlayer under the low power and high vacuum magnetron sputtering. Moreover, the Au underlayer has the characteristic of specific growth along the [111] direction. More importantly, it has been confirmed that a new and stable active site is formed on the surface of the electrode after the electrochemical oxidation. The combination of X-ray photoelectron spectroscopy (XPS) and electrochemical analysis methods (cyclic voltammetry and CO adsorption–stripping) has suggested that the active site should come from the oxidized platinum (Pt–O). In addition, density functional theory (DFT) calculations have elucidated that the ΔGH*(Pt) (absolute value) of PtAC–O–Au is close to that of Pt(111), indicating that its outstanding HER activity originates from its optimal ΔGH* value. The (PtAC–O–Au)-1 electrode not only possesses ultra-high MA as compared with the commercial Pt/C with the same loading (0.5 μgPt cm−2) but also possesses the apparent activity equivalent to the commercial 20 wt% or 40 wt% Pt/C with high loading.
Results and discussion
The preparation process of the oxidized Pt atomic cluster-supported Au (PtAC–O–Au) with ultra-low loading is shown in Scheme 1. The first step is to prepare the Au underlayer. The smooth Ti foil substrate after strict cleaning was placed in the vacuum chamber of the high-vacuum magnetron sputtering instrument. Then, using the high-purity Au as the sputtering target, the Au underlayer with the [111] preferred orientation and uniform morphology was obtained by DC sputtering. The second step is to prepare an Au underlayer modified by Pt atomic clusters. With the high-purity Pt as the sputtering target, the PtAC–Au with ultra-low Pt loading (0.48 μgPt cm−2) was prepared on the Au underlayer by the DC sputtering method with low sputtering power. The last step is to prepare the PtAC–O–Au by the electrochemical anodic oxidation. The detailed experimental process is given in the Experimental method.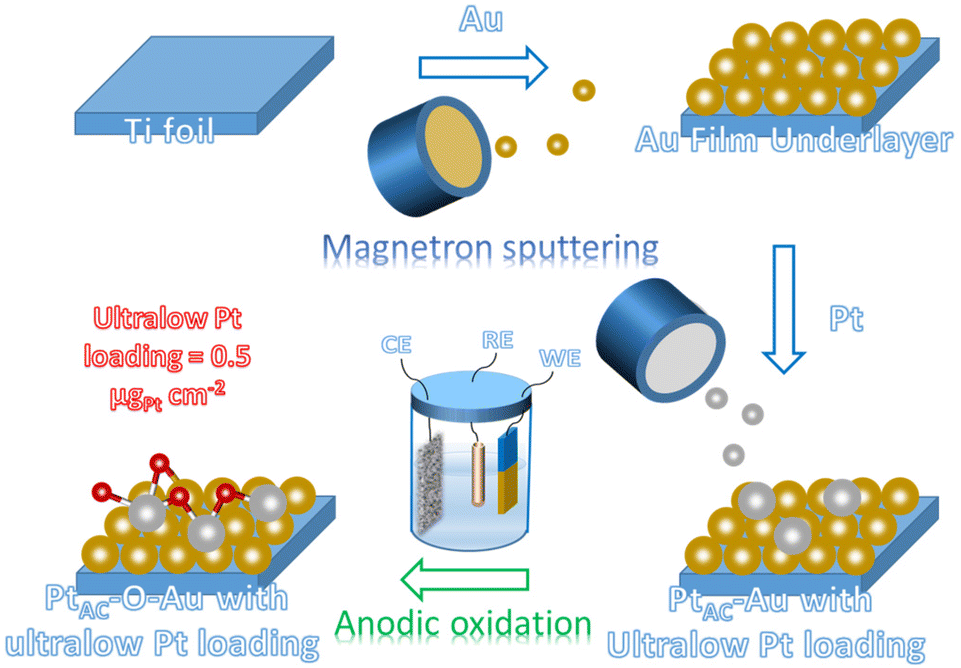 | ||
| Scheme 1 Schematic illustration of the preparation of PtAC–O–Au. | ||
PtAC–O–Au has the morphology of atomic clusters, a special crystal structure and a modified electronic structure. First, for the Au underlayer, the size of Au nanoparticles can be seen, with an average particle size of 26.8 nm (Fig. S1 and S2 (ESI†)). Fig. 1a is an AFM image of (PtAC–Au)-1, in which the particle distribution is relatively uniform. The mean particle diameter of PtAC–Au slightly increased with the enhancement of Pt loading, ranging from 28.4 ((PtAC–Au)-1, 0.482 μg cm−2) to 31.4 nm ((PtAC–Au)-4, 1.693 μg cm−2) (Fig. S1 and S2 (ESI†)). Therefore the particle size of Pt atomic clusters sputtered in the subsequent process is small, which has no significant effect on the particle size of the Au underlayer. Moreover, the analysis of the surface roughness of PtAC–Au revealed that the surface flatness was improved after loading the Pt atomic clusters. The height difference for pure Au was 7.5 nm, while the height differences for (PtAC–Au)-1, (PtAC–Au)-2, (PtAC–Au)-3 and (PtAC–Au)-4 were reduced to 6.2, 5.7, 5.2 and 4.3 nm, respectively (Fig. S3 (ESI†)). The root-mean-square roughness (Rq) of Au was 2.91 nm, while the Rq of (PtAC–Au)-1, (PtAC–Au)-2, (PtAC–Au)-3 and (PtAC–Au)-4 dropped to 2.88, 2.77, 2.65 and 2.40 nm, respectively. The decrease in the height difference and Rq indicated that the electrode surface became smooth. It may be that the fine Pt atomic clusters were filled into the gaps of the large Au particles, thereby improving the flatness of the electrode.
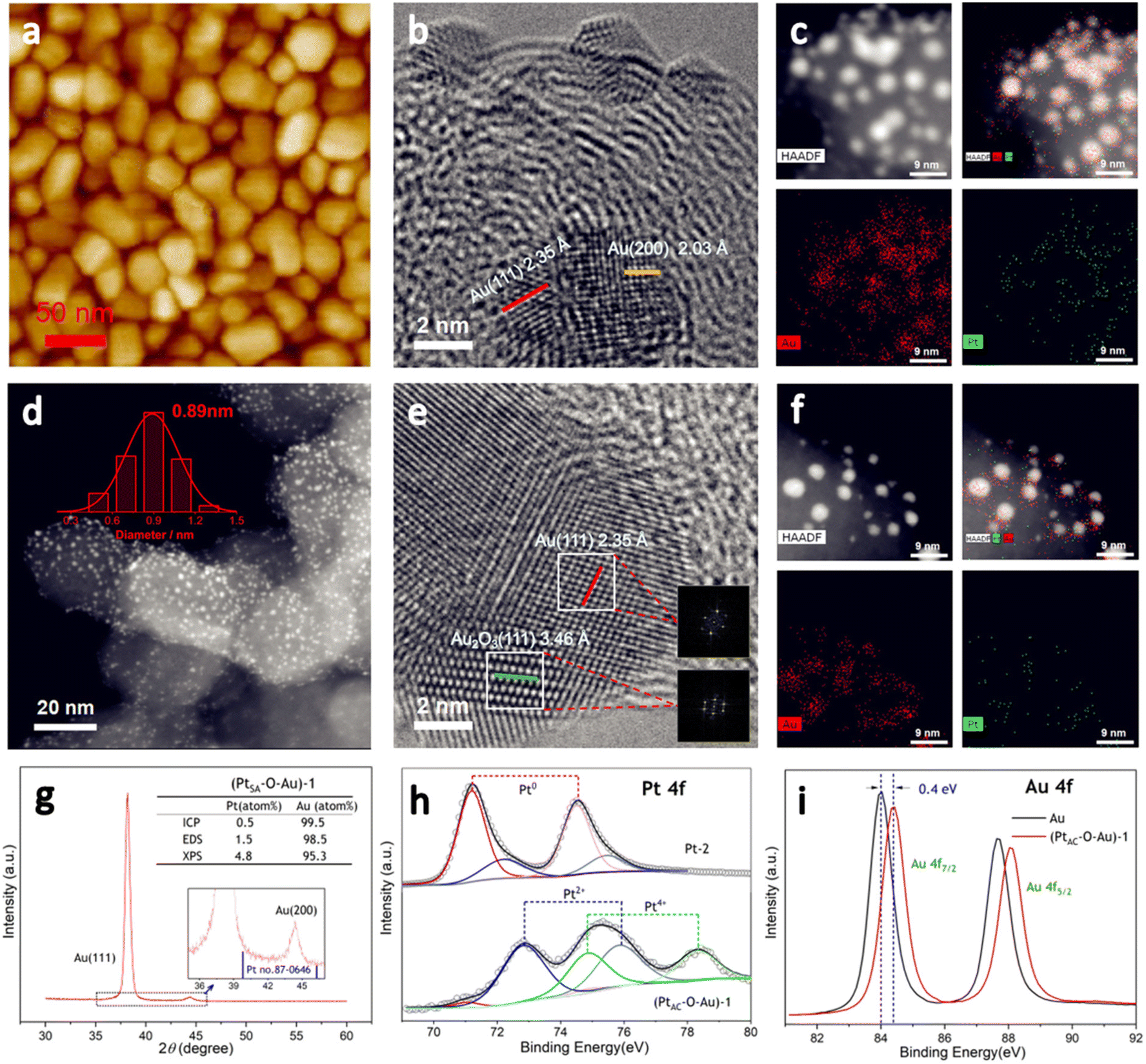 | ||
| Fig. 1 The AFM, HRTEM, HAADF-STEM images and STEM-EDS elemental maps of the (PtAC–Au)-1 electrode (a–c). The HAADF-STEM images of the Pt-1 electrode (d). The HRTEM, HAADF-STEM images and STEM-EDS elemental maps of the (PtAC–O–Au)-1 electrode (e and f). The XRD diagram of the (PtAC–O–Au)-1 electrode (g). The core-level XPS spectra of Pt 4f and Au 4f, obtained for the (PtAC–O–Au)-1, Pt-2 and Au electrodes (h and i). | ||
The HRTEM image of (PtAC–Au)-1 (Fig. 1b) facilitated the investigation of the lattice fringes of Au(111) and Au(200) but no lattice fringes of Pt were found. It was presumed that Pt exists in the form of an amorphous phase or atomic cluster. The particle size distribution of (PtAC–Au)-1 was uniform, based on the TEM images (Fig. S4 (ESI†)), which is consistent with the AFM observation results. Moreover, in the STEM-EDS elemental mapping (Fig. 1c), the distribution of Au and Pt elements was uniform. The (PtAC–Au)-1 electrode was anodized at a potential of 1.7 V for 10 min to obtain a stable (PtAC–O–Au)-1 electrode (Fig. S5 (ESI†)). In the HRTEM diagram (Fig. 1e), the lattice stripes of Au(111) and Au2O3(111) can be seen, where the generation of Au2O3 is mainly derived from the anodic oxidation process. Au and Pt are distributed evenly in the STEM-EDS elemental maps and the particle size distribution was also uniform, as observed from the HAADF-STEM image (Fig. 1f). Since the particle size of Pt is significantly less than that of Au, it is difficult to accurately observe the particle size and lattice stripe of Pt particles in Fig. 1e. Therefore, the HAADF-STEM and HRTEM analyses of Pt-1 ((PtAC–Au)-1 without Au underlayer) were performed and the results are shown in Fig. 1d. The Pt cluster is very small, only about 0.89 nm and it can be seen from the STEM diagram that the particle size distribution of Pt particles is uniform (Fig. S6 (ESI†)). After careful examination of the HRTEM image of Pt-1 (Fig. S6 (ESI†)), the lattice fringes of the Pt(111) crystal plane were observed, although most Pt particles were in the amorphous state. With the increase in sputtering time, the Pt loading increased. For example, the Pt load of the (PtAC–Au)-4 electrode was 6 times that of the (PtAC–Au)-1 electrode. For the (PtAC–Au)-4 electrode, the Pt crystal stripes were observed around Au nanoparticles even though there were also many amorphous stripes (Fig. S7 (ESI†)).
The special crystal structure of the PtAC–O–Au is analyzed in Fig. 1g and S8 (ESI†). The characteristic diffraction peaks of the (111) and (200) crystal facets of Au were observed at 38.2° and 44.4° for (PtAC–O–Au)-1 in Fig. 1g. Since the Pt loadings are very low, no diffraction peaks of Pt were observed for (PtAC–O–Au)-1, (PtAC–O–Au)-2, (PtAC–O–Au)-3 and (PtAC–O–Au)-4 (Fig. S8 (ESI†)). Only for the (PtAC–O–Au)-5 (the Pt loading = 17.58 μgPt cm−2) can the (111) crystal plane of Pt can be seen in the XRD diffraction pattern. The intensity ratio of the diffraction peaks of (111) and (200) crystal planes is about 30:1 for the (PtAC–O–Au)-1, which is far from the data from JCPDS 04-0784, indicating that the crystal structure is dominated by the [111] crystal orientation; this may be due to the preparation methods. In the magnetron sputtering technique, the sputtering power will affect the orientation of the planes of the metal. When the sputtering power is low, the close-packed plane crystal at low energy is preferentially formed. This kind of electrode with specific growth along a certain crystal direction is more convenient to coincide with the stable crystal surface selected by theoretical calculation, so we attribute this special electrode morphology to the model electrode.
The loading of Pt was investigated by ICP-OES, EDS and XPS. The elemental analysis results of (PtAC–O–Au)-1 demonstrated that the Pt content was very low (ICP-OES: 0.5 atom%; EDS: 1.5 atom%; XPS: 4.8 atom%) (Fig. 1g (inset) and Fig. S9 (ESI†)). The Pt loading increased from 0.482 to 1.693 μgPt cm−2 with sputtering time, showing a good linear correlation (Table S1 and Fig. S10 (ESI†)). The proportion of Pt in the PtAC–O–Au surface analyzed by XPS was higher than that obtained by ICP-OES. With the increase of sputtering time, the Pt content on the electrode surface increased from 4.79 to 42.4 atom% (Table S2 (ESI†)). This ratio is much higher than that of Pt in the body, indicating that Pt is highly dispersed in the electrode surface layer.
Fig. 1h and i are the electronic structure analyses of Pt and Au on the surface of the (PtAC–O–Au)-1. The survey XPS spectra of the Au, Pt-2 and (PtAC–O–Au)-1 demonstrate the presence of Pt, Au, O and C species (Fig. S11 (ESI†)). The Pt element of the Pt-2 surface mainly exists in Pt0+ and Pt2+ components in Fig. 1h. The two peaks at the binding energies of 71.3 and 74.5 eV correspond to the 4f7/2 and 4f5/2 orbitals of Pt0+ species, while the two peaks at 72.2 and 75.5 eV are attributed to the 4f7/2 and 4f5/2 orbitals of Pt2+ species. However, for (PtAC–O–Au)-1, only the Pt4+ components (74.9 and 78.3 eV)54 appeared on the surface and almost no Pt0+ components were found. In addition, the binding energies of Au 4f of the PtAC–O–Au electrode were at 84.4 and 88.1 eV (Au: 84.0 and 87.7 eV),64 indicating that the partial oxidation of Au also occurs on the surface of PtAC–O–Au.
The HER activities of (PtAC–O–Au)-1, Pt-1 and 0.5 wt% Pt/C electrodes are displayed in Fig. 2a. To ensure the fairness of the evaluation, the Pt loading of all electrodes was controlled at about 0.5 μg cm−2. The Pt-1 and 0.5 wt% Pt/C electrodes do not exhibit excellent HER activity because of their ultra-low loading of Pt. However, the (PtAC–O–Au)-1 electrode exhibited excellent HER activity, which was not affected by its low Pt loading. For instance, the overpotential at 10 mA cm−2 (η10) was 140 mV and 168 mV for the Pt-1 and 0.5 wt% Pt/C electrodes, respectively, while the η10 of the (PtAC–O–Au)-1 electrode was only 41 mV. At the same time, the pure Au electrode did not show any HER activity in the investigated potential range. The HER activity of the (PtAC–O–Au)-1 electrode was further compared with those of the commercial Pt/C catalyst (40 wt% Pt/C (40 μgPt cm−2) and 20 wt% Pt/C (20 μgPt cm−2)) electrodes (Fig. 2b). The HER activity of the (PtAC–O–Au)-1 electrode was significantly higher than that of the 20 wt% Pt/C, similar to those of 40 wt% Pt/C electrodes.
 | ||
| Fig. 2 The apparent HER activities of (PtAC–O–Au)-1, Pt-1, 0.5 wt% Pt/C and Au (a). The apparent HER activities of (PtAC–O–Au)-1, 20 wt% Pt/C and 40 wt% Pt/C (b). The MAs of (PtAC–O–Au)-1, Pt-1, 0.5 wt% Pt/C, 20 wt% Pt/C and 40 wt% Pt/C (c and d). The SAs of (PtAC–O–Au)-1, Pt-1 and 0.5 wt% Pt/C (e). The Tafel slopes of (PtAC–O–Au)-1, Pt-1 and 0.5 wt% Pt/C (f). | ||
For the noble metal Pt, the mass activity (MA) is very important, which is related to the actual usage of the noble metal. However, the comparison of MA in most reports is at present incomplete. This is because it is unreasonable to compare MA for low-load Pt catalysts and high-load commercial Pt/C alone. The Pt load of commercial Pt/C is very high (tens or hundreds of times higher than that of low-load Pt catalyst), and the deviation of the apparent activity according to mass standardization is very large. The research results of ref. 11 have already illustrated similar conclusions. Therefore, to correctly evaluate the MA of Pt catalysts with low loads, it was necessary to supplement the MA of commercial Pt/C catalysts with similar loads. The MAs of all the electrodes at different overpotentials (20, 50 and 80 mV) are shown in Fig. 2c and d. The (PtAC–O–Au)-1 electrode exhibited an ultra-high MA (3.0 A mg−1@η = 20 mV; 49.2 A mg−1@η = 50 mV; 329.0 A mg−1@η = 80 mV). The MA of the (PtAC–O–Au)-1 electrode is 20–22 times higher than that of the Pt-1 and 0.5 wt% Pt/C electrodes at η of 50 mV. The MA of the (PtAC–O–Au)-1 electrode is 60–66 times higher than that of the Pt-1 and 0.5 wt% Pt/C electrodes at the η of 80 mV. For the commercial Pt/C electrodes (20 wt% and 40 wt%), the MA is 26–41 times lower than that of (PtAC–O–Au)-1 electrode at η of 50 mV. More importantly, the MA of the (PtAC–O–Au)-1 electrode is significantly higher than that in most reports (Table S3 (ESI†)). The apparent activity depends on the electrochemical surface area (ECSA) of the electrocatalyst. The high Pt loading of the commercial Pt/C catalyst and the porous structure of the activated carbon support can make Pt highly dispersed, thus showing a great ECSA (30.8 and 15.2 cm2 in Fig. S12e and f (ESI†)). Although the Pt-2 electrode has no abundant pore structure, its high loading capacity allows its ECSA to still reach 3.09 cm2, which is 10 times larger than that of the (PtAC–O–Au)-1 electrode (Fig. S12d (ESI†)). For the (PtAC–O–Au)-1 electrode, despite the tiny particle size and high dispersion obtained by magnetron sputtering, its ECSA is very small due to its ultra-low loading (0.264 cm2 in Fig. S12b (ESI†)). The low ECSA (for the same reason) also occurs on the Pt-1 and 0.5 wt% Pt/C electrodes (Fig. S12a and c (ESI†)). The ECSA of the (PtAC–O–Au)-1 electrode is only 0.85–1.7% of the commercial Pt/C electrodes and 8.5% of the Pt-2 electrode. For the specific activity (SA), the normalized current of the commercial Pt/C (20 wt% and 40 wt%) is very low (Fig. S13†). The SAs of 0.5 wt% Pt/C and Pt-1 electrodes are relatively close (Fig. 2e) but their SAs are much lower than that of the (PtAC–O–Au)-1 electrode. The SA of the (PtAC–O–Au)-1 electrode is 12–18 times higher than that of Pt-1 and 0.5 wt% Pt/C electrodes and 36–56 times higher than that of the commercial Pt/C electrodes. Therefore, both the MA and SA of the (PtAC–O–Au)-1 electrode were greatly improved. The reason for the increase in the MA and SA activities of the HER is not only the extremely low utilization rate of Pt but also the improvement of the turnover frequency (TOF) of active sites. The TOF of the (PtAC–O–Au)-1, 0.5 wt% Pt/C and Pt-1 electrodes are shown in Fig. S14,† in which the (PtAC–O–Au)-1 electrode shows significantly increased HER activity.
The mechanism of the HER reaction was further analyzed by the Tafel slope in Fig. 2f. The Tafel slope of the (PtAC–O–Au)-1 electrode was significantly lower than that of the Pt-1 and 0.5 wt% Pt/C electrodes. The Tafel slope of the (PtAC–O–Au)-1 electrode was 28.6 mV dec−1, indicating that the reaction mechanism is the Volmer–Tafel process and the rate-determining step is the combination desorption of Had (Had + Had = H2). For the Pt-1 and 0.5 wt% Pt/C electrodes, the rate-determining step is the Heyrovsky step (H3O+ + Had + e− = H2 + H2O), according to the slope.65 The difference in the reaction mechanism determines the difference in reactivity between them.
Further attempts were made to appropriately increase the Pt loading of the PtAC–O–Au electrodes. With the continuous increase in the Pt loading, their HER activities also increased slightly. Among them, the apparent current densities of the (PtAC–O–Au)-2, (PtAC–O–Au)-3 and (PtAC–O–Au)-4 electrodes exceeded that of the Pt-2 electrode (Fig. S15a (ESI†)). It was further demonstrated that the PtAC–O–Au electrode is a very good HER electrocatalyst. The increase in the HER activity may be related to the ECSA enhancement (ECSA((PtAC–O–Au)-1) = 0.264 cm2; ECSA((PtAC–O–Au)-2) = 0.604 cm2; ECSA((PtAC–O–Au)-3) = 1.023 cm2; ECSA((PtAC–O–Au)-4) = 1.4 cm2 in Fig. S16 (ESI†)). The HER activity was further normalized by the ECSA (Fig. S15b (ESI†)). The SAs are of the order of (PtAC–O–Au)-1 > (PtAC–O–Au)-2 > (PtAC–O–Au)-3 > (PtAC–O–Au)-4. Moreover, the SAs are all much higher than that of the Pt-2 electrode. For the MA, the order is the same (Fig. S15c (ESI†)). It can be predicted that the active site of the PtAC–O–Au electrode is not necessarily on the surface of an ordinary Pt atom. It may not be reasonable to normalize the HER current with the ECSA of Pt, which will be further analyzed in detail later.
The excellent HER activity of the PtAC–O–Au electrode may be related to the surface oxidation process.66 The apparent activity of the (PtAC–Au)-1 electrode is not good enough without the anodic oxidation (Fig. S17a (ESI†)). After anodic oxidation at 1.7 V for 10 min, the apparent activity of the (PtAC–O–Au)-1 electrode increased. For instance, the η10 was 152 mV for the (PtAC–Au)-1 electrode, while the η10 of the (PtAC–O–Au)-1 electrode was only 41 mV. The enhancement in activity was not only due to the oxidation of Au because the apparent activity of the AuOx electrode was not significantly improved (Fig. S17a (ESI†)). Meanwhile, the increase in activity was not entirely from the PtOx since the apparent activity of the PtOx-1 electrode was still poor (Fig. S17a (ESI†)). Only when Au and Pt were oxidized together to form the band of Pt–O–Au could the apparent activity be remarkably improved. Therefore, the oxidation process should play an important role and needs to be studied in detail.
In the oxidation potential range of 0.7–1.1 V, the obvious underpotential deposition of H (H-UPD) and HER currents cannot be generated (Fig. 3a). When the oxidation potential increased to 1.3 V, the H-UPD and HER currents appeared at 0.2 and 0 V (0.12 mA cm−2). Once the oxidation potential was further increased to 1.5 and 1.7 V, the obvious HER current increased to 0.18 mA cm−2. Moreover, in Fig. S17b and c (ESI†), the apparent activity of (PtAC–O–Au)-1 at the anodic oxidation potential of 1.7 V was higher than that of 1.3 V. The above results have demonstrated that only when the electrode surface is completely oxidized at 1.7 V will its HER activity be significantly increased.
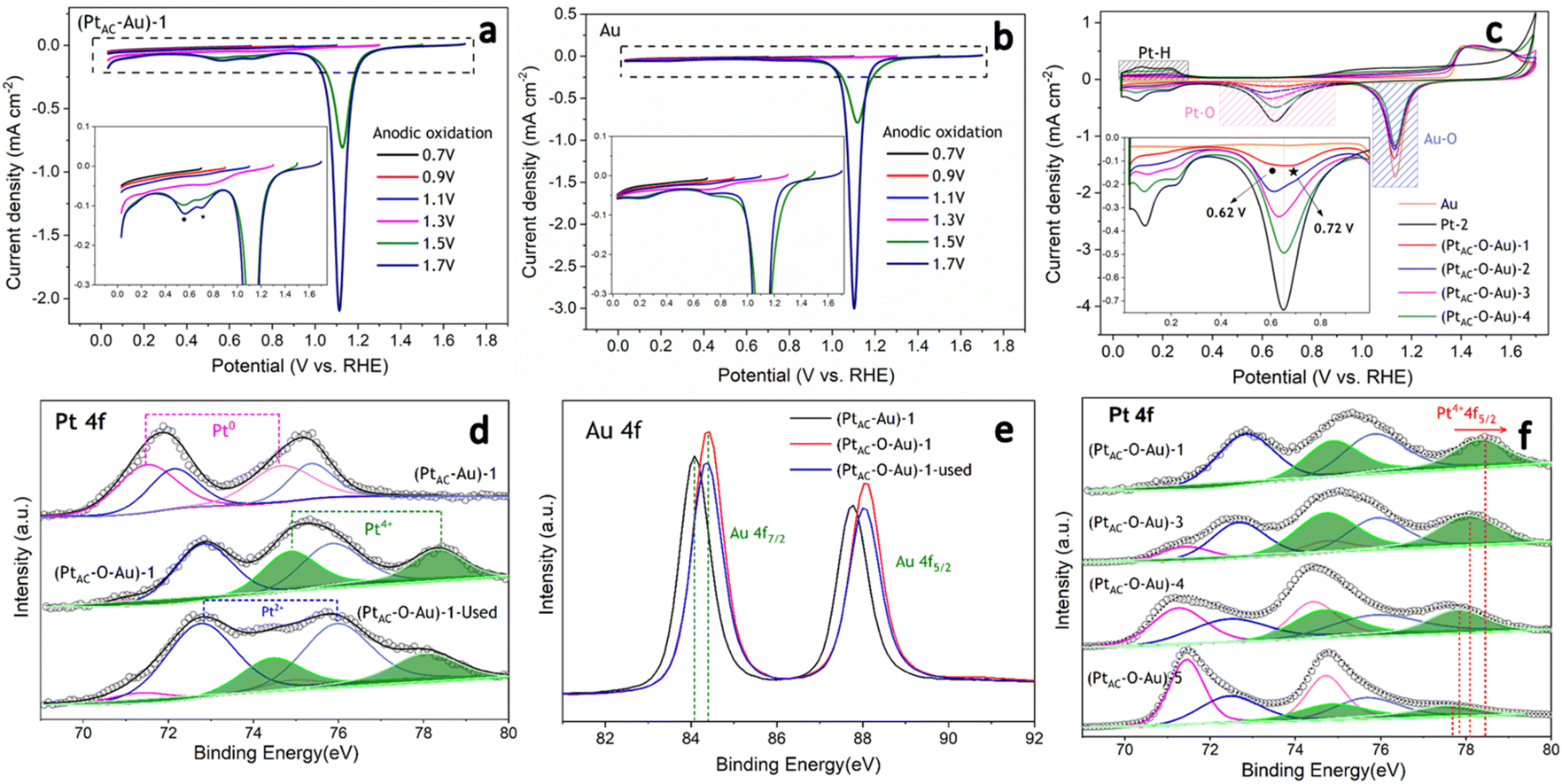 | ||
| Fig. 3 Linear sweep voltammetry curves of (PtAC–Au)-1 and Au electrodes with different anodic potentials at a sweeping rate of 5 mV s−1 (a and b). Cyclic voltammograms of (PtAC–Au)-x (x = 1, 2, 3, 4) electrodes with different Pt loadings at a sweeping rate of 20 mV s−1 (c). The XPS core-level spectra of Pt 4f and Au 4f XPS spectra obtained from used (PtAC–Au)-1, (PtAC–O–Au)-1 and (PtAC–O–Au)-1 electrodes (after the HER test for 10 h) (d and e). The XPS core-level spectra of Pt 4f obtained from (PtAC–O–Au)-x (x = 1, 3, 4, 5) electrodes with different Pt loadings (f). (Pink line – Pt0+, blue line – Pt2+, green line – Pt4+.) | ||
It was also found that when the oxidation potential was higher than 1.3 V, three reduction peaks (▲, ★, ●) appeared during the negative scanning of the potential (Fig. 3a). One belongs to the reduction peak of Au–OH (▲, 1.12 V) and the other belongs to the reduction peak of Pt–OH (●, 0.62 V). However, the reduction peak at 0.72 V (★, star peak) needs further study to determine its origin. To accurately determine the origin of the reduction peak at 0.72 V, the LSV curves of pure Au electrode oxidized at different potentials were analyzed in detail (Fig. 3b). Regardless of whether the oxidation potential is increased to 1.5 V or 1.7 V, there was no obvious “star peak”. Thus, it can be concluded that the “star peak” is not the reduction peak of Au–OH. Further analysis was carried out in the CVs of PtAC–O–Au electrodes with different Pt loadings (Fig. 3c). With the increase in the Pt loading, the reduction current at 0.62 V increased, while the reduction current of the “star peak” did not change. This indicates that the “star peak” is not due to the reduction of Pt–OH. Therefore, the “star peak” is the reduction peak for neither Au–OH nor Pt–OH. Since the “star peak” is just between the Au–OH and Pt–OH reduction peaks, it is speculated that it is the reduction peak of the newly formed “Pt–O–Au” active site in the electrochemical oxidation process, which can efficiently adsorb and desorb H atoms. Therefore, the HER apparent current was further normalized by the ECSA derived from the capacitance of the “star peak” (Fig. S18 (ESI†)), which has shown that the SA of HER is essentially the same (Fig. S19 (ESI†)), indicating that the active site is located in “Pt–O–Au” at 0.72 V (★, star peak).
The electronic structures of PtAC–Au and PtAC–O–Au electrodes were further analyzed (Fig. 3d–f and Fig. S20 (ESI†)). The Pt electronic structure of the (PtAC–Au)-1 electrode surface was mainly Pt0+ and Pt2+ components (Fig. 3d). The two peaks at binding energies of 71.5 and 74.6 eV correspond to the 4f7/2 and 4f5/2 orbitals of Pt0+, while the two peaks at 72.2 and 75.4 eV were attributed to the 4f7/2 and 4f5/2 orbitals of Pt2+, respectively. After the anodic oxidation, the Pt4+ components (74.9 and 78.3 eV) appeared on the (PtAC–O–Au)-1 electrode surface, while Pt0+ components were significantly reduced. Meanwhile, the Au in the (PtAC–O–Au)-1 electrode was partially oxidized to form AuOx, while it was Au0+ in the (PtAC–Au)-1 electrode (Fig. 3e). When the Pt load increased, the binding energy peak of Pt4+ 4f5/2 exhibited a significant negative shift (78.47 eV for the (PtAC–O–Au)-1, 78.09 eV for the (PtAC–O–Au)-3, 77.85 eV for the (PtAC–O–Au)-4 and 77.69 eV for the (PtAC–O–Au)-5 in Fig. 3f), indicating that the valence state of Pt decreased. At the same time, the number of Pt4+ components also decreased significantly with the increase in the Pt loading. Pt4+ rarely appears on the surfaces of the pure Pt-2 electrode (Fig. S21 (ESI†)); therefore, it indicates that the Pt4+ valence state of the (PtAC–O–Au)-1 electrode is at the interface between the Pt and Au atom (Pt–O–Au). The Pt loading in the (PtAC–O–Au)-5 electrode is 36.5 times that in the (PtAC–O–Au)-1 electrode and there is almost no exposed Au–Pt interface on the electrode surface, so Pt4+ rarely appears (Fig. S21 (ESI†)). Correspondingly, the anodic oxidation of Au was inhibited as the Pt loading increased. This is reflected in the negative shift of the Au 4f binding energy of the (PtAC–O–Au)-4 electrode as compared to the (PtAC–O–Au)-1 electrode or (PtAC–O–Au)-3 electrode (Fig. S22 (ESI†)). In addition, the oxidation state of the (PtAC–O–Au)-1 electrode was stable (Fig. 3d and e). After a long-term lifetime test, the valence states of Pt and Au in XPS graph remained unchanged, except for a small amount of oxidized Pt being converted into reduced Pt.
As can be seen from Fig. 4a, the CO adsorption–stripping curves of PtAC–O–Au electrodes with different Pt loadings are different. First of all, no obvious stripping peak was found on the surface of the AuOx electrode, which should correspond to the weak CO adsorption ability of AuOx. However, for the (PtAC–O–Au)-1 electrode, a clear stripping peak appeared at 1.15 V, which is attributed to the strong adsorption of CO on the surface of the electrode. The position of the stripping peak is different from that of the Pt-2 electrode (0.93 V), indicating that its adsorption active center is not just Pt. With the increase in the Pt loading, the peak potential negatively shifted to 1.0 V ((PtAC–O–Au)-2 electrode), 0.96 V ((PtAC–O–Au)-3 electrode) and 0.94 V ((PtAC–O–Au)-4 electrode), respectively; the peak area also increased, and the peak symmetry became better. In addition, during the anodic oxidation, there was no obvious peeling peak curve at the potential of 1.3 V. If the oxidation potential increased to 1.7 V, a sharp stripping peak appeared for the PtAC–O–Au electrode with different loads (Fig. S23 (ESI†)). This indicates that the anodic oxidation potential is also a very important factor. The potential of 1.7 V is generally considered the potential of the Au surface oxidation. Therefore, if the oxidation potential exceeds 1.7 V, it is possible to form Pt–O–Au active sites.
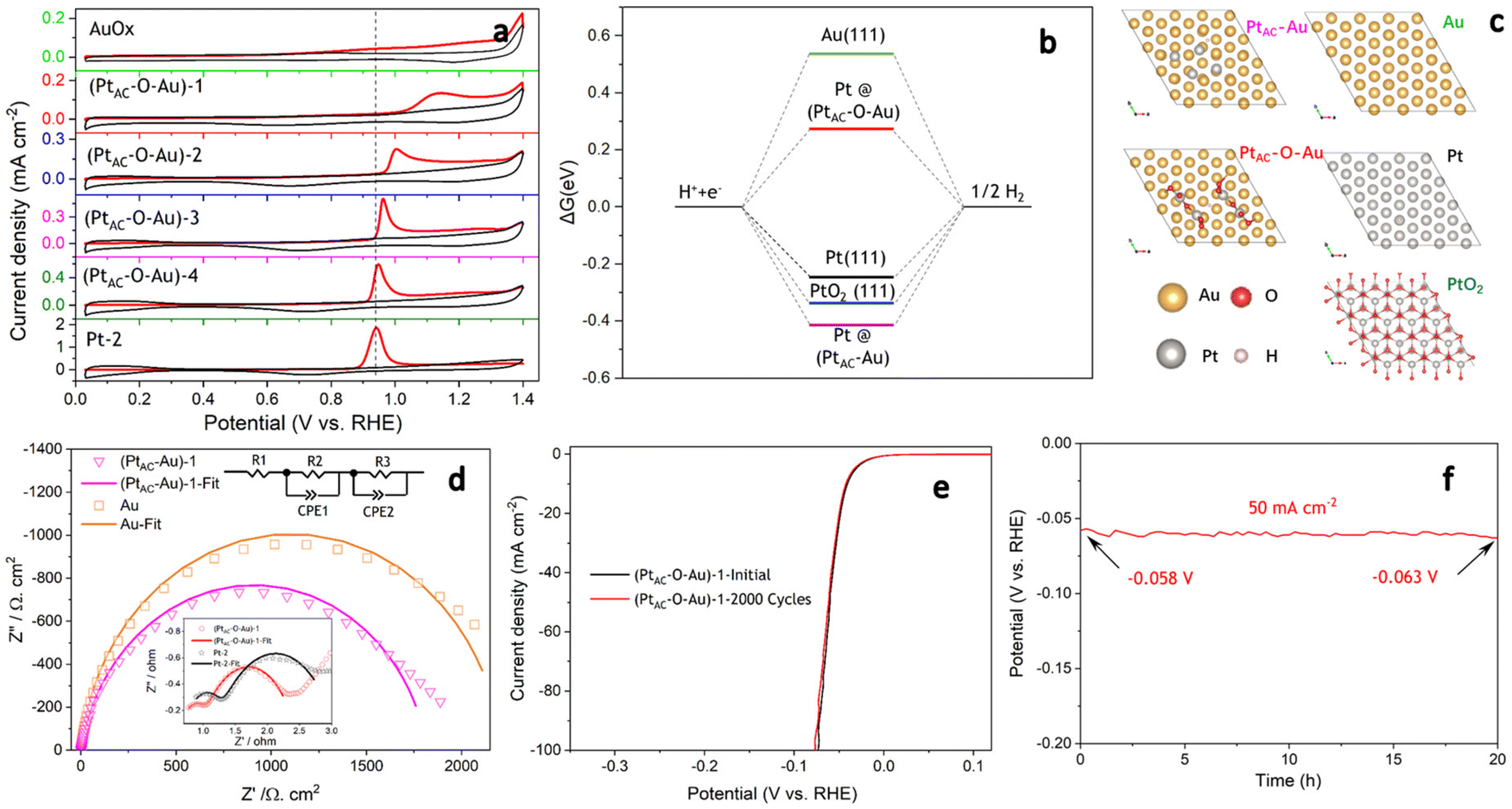 | ||
| Fig. 4 The CO adsorption–stripping curves of (PtAC–O–Au)-x (x = 1, 2, 3, 4), AuOx and Pt-2 electrodes (a). Free-energy diagram for hydrogen evolution at equilibrium (U = 0) for Au(111), Pt(111), PtO2(111), Pt@PtAC–Au and Pt@PtAC–O–Au (b). Top view atomic structure models of Au(111), Pt(111), PtO2(111), PtAC–Au and PtAC–O–Au with H* (c). Complex plane plots of impedance of (PtAC–O–Au)-1, Pt-2, (PtAC–Au)-1 and Au electrodes in 0.5 mol L−1 H2SO4 solution at the overpotential of 0.05 V (d). The long-term HER stability test for the (PtAC–O–Au)-1 electrode (e and f). | ||
To further understand the extremely high HER activity of the PtAC–O–Au electrode, DFT calculations were carried out to investigate the Gibbs free energy of hydrogen adsorption (ΔGH*). It is a widely accepted descriptor in predicting the theoretical activity of the HER. A moderate ΔGH* should be around 0 eV, which would lead to the optimal HER activity due to the balance between the adsorption and the desorption steps. Therefore, the ΔGH* of H* adsorbed on different surface models including Au(111), Pt(111), Pt@PtAC–Au and Pt@PtAC–O–Au, as well as PtO2(111), were calculated in Fig. 4b and c. The ΔGH* of Pt(111) is −0.25 eV, which is consistent with the report in the literature.67 The Au(111) and PtO2(111) surfaces were 0.55 and −0.34 eV, respectively. The ΔGH* of the Pt@PtAC–Au surface was −0.42 eV, which is too negative, implying strong hydrogen adsorption. In contrast, the ΔGH* of the Pt@PtAC–O–Au surface increased to 0.28 eV, which is beneficial for the hydrogen desorption step. The above calculation results indicate that the ΔGH* (absolute value) of the Pt@PtAC–O–Au surface is close to that of Pt(111), indicating that its outstanding HER activity originates from its optimal ΔGH* value.
The EIS is a powerful, nondestructive and informative technique used extensively to study the electrolyte–electrode interfacial properties. The Nyquist diagrams of Au, (PtAC–Au)-1, (PtAC–O–Au)-1 and Pt-2 electrodes recorded at the overpotential of 50 mV are shown in Fig. 4d. The equivalent circuit corresponding to the EIS data was fitted with a two time-constant model (Rs(RfCf)(RctCdl)), which consists of the electrolyte solution resistance Rs, the electrode film resistance Rf, the charge transfer resistance Rct in series with a parallel connection of the film capacitance and double layer capacitance Cf and Cdl. The charge transfer resistance data were obtained in the low-frequency zone, which is related to the electrocatalytic kinetics. The Rct of the (PtAC–O–Au)-1 electrode is 1.437 Ω cm2, which is similar to that of the Pt-2 electrode (1.833 Ω cm2). It is also much lower than those of the Au (2208 Ω cm2) and (PtAC–Au)-1 (1818 Ω cm2) electrodes, suggesting fast charge transport during the HER process. For the solid electrodes, the double-layer capacitance is substituted by a constant phase element (CPE). Therefore, Cdl can be calculated and the surface roughness factor (Rf) can be calculated from the formula (Rf = Cdl/20 mF cm−2). The Rf values of four electrodes are both low and similar (Rf(Au) = 3.4, Rf((PtAC–Au)-1) = 1.5, Rf((PtAC–O–Au)-1) = 2.7, Rf(Pt-2) = 2.9), indicating that they all have smooth surfaces. Accelerated CV tests were conducted to investigate the stability of the catalysts in the potential range of −0.2 to 0.1 V at a sweeping rate of 50 mV s−1. The HER activity of the (PtAC–O–Au)-1 electrode was maintained without the slightest reduction after 2000 cycles of CV measurement in Fig. 4e. Additionally, the results of chronopotentiometry showed that the overpotential generally remains stable at a current density of 50 mA cm−2 over 20 hours in Fig. 4f. Under this current density, the overpotential increased by 8.6%, suggesting that the (PtAC–O–Au)-1 electrode is durable for hydrogen evolution.
Fig. 5 shows the new active site of “Pt–O–Au” of the PtAC–O–Au electrode. For the (PtAC–O–Au)-1 electrode, the Pt loading on the Au surface is very low and highly dispersed so Pt exists in the form of the +4 valence after electrochemical oxidation (the evidence comes from the XPS diagram). At the same time, the surface of Au was partially oxidized (the evidence comes from the XPS diagram), thus forming Pt–O–Au active sites. The electrochemical evidence of the Pt–O–Au active site corresponds to the reduction peak at 0.72 V (star peak) in the CV scan, which is different from the potential of the Pt–OH reduction peak (0.62 V). The SAs normalized by the ECSA derived from the capacitance of the “star peak” are essentially the same (Fig. S19 (ESI†)), indicating that the active site is located in “Pt–O–Au” at 0.72 V (star peak). The area ratio of the reduction peak of Pt–O–Au to the reduction peak of Pt–OH is 1.75:1, which indicates that most Pt exists in the form of Pt–O–Au active sites on the (PtAC–O–Au)-1 electrode. As for the (PtAC–O–Au)-4 electrode, with the increase in the Pt loading, its nucleation grows continuously so the active sites of Pt–O–Au are relatively fewer. The area ratio of the reduction peak of Pt–O–Au to the reduction peak of Pt–OH decreased to 1:9.7. Another electrochemical evidence that can prove the formation of new Pt–O–Au active sites comes from the CO stripping curves. The adsorption of CO on the Pt surface is very sensitive, and the subtle changes in the electrode surface can be reflected in the CO stripping curve. On the surface of (PtAC–O–Au)-4, the CO oxidation stripping potential of CO is 0.94 V, which is similar to that of pure Pt. On the surface of (PtAC–O–Au)-1, the potential of CO oxidation stripping is 1.15 V, which has an obvious offset. This shows that a new active site, Pt–O–Au, was formed on the (PtAC–O–Au)-1 surface, which is different from the pure Pt surface. The reason the above electrochemical analysis can clearly show the differences between different electrodes is also due to the electrode with a flat surface obtained by high vacuum magnetron sputtering technology in this experiment. This is different from the electrode material of porous carriers used in most electrocatalyst experiments at present. However, the improvement of the intrinsic activity of electrocatalysts can be reflected more truly and reasonably by using planar electrodes.
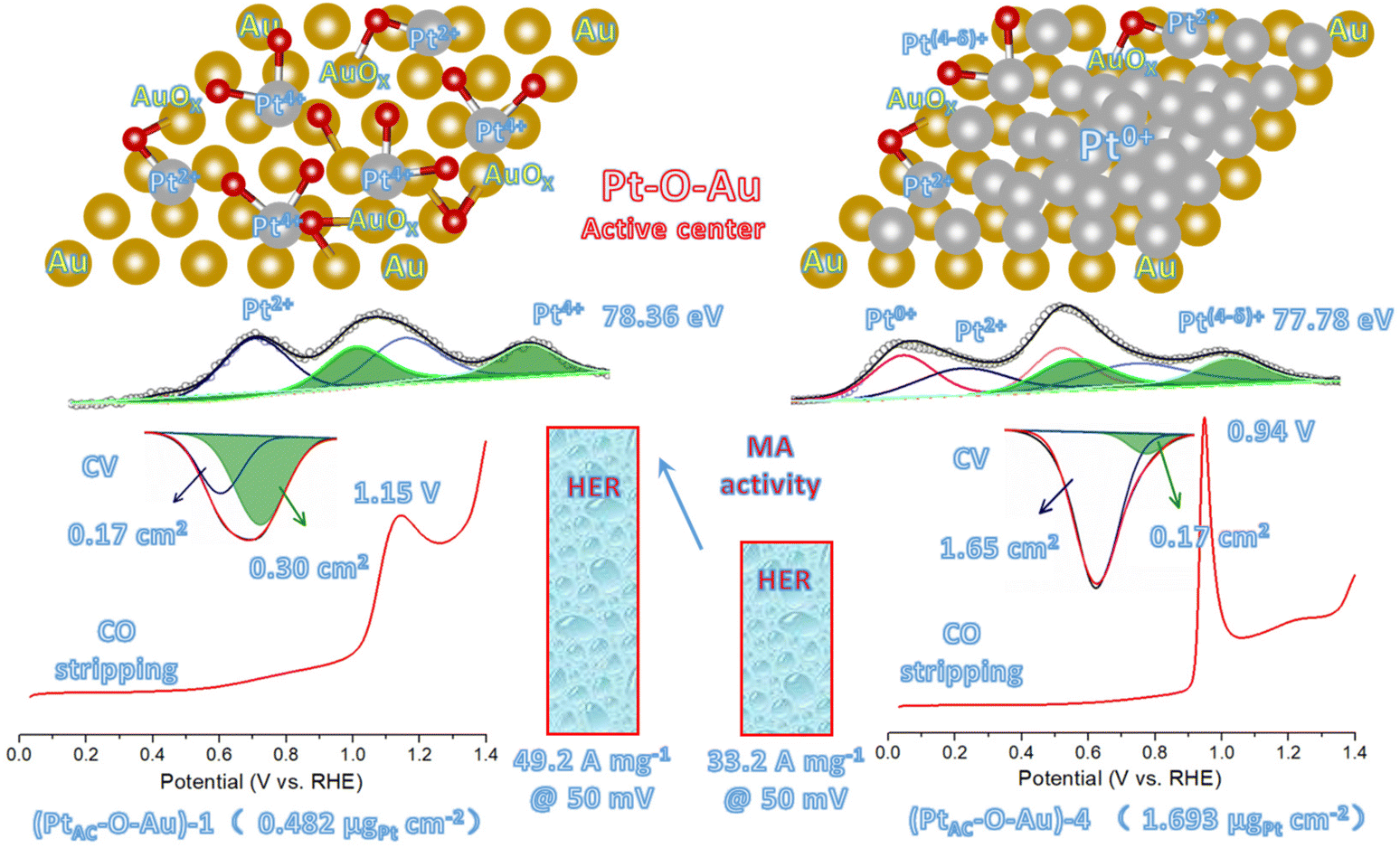 | ||
| Fig. 5 The new active site of “Pt–O–Au”. | ||
Conclusion
The PtAC–O–Au electrode with the ultra-low Pt loading was prepared by high-vacuum magnetron sputtering combined with electrochemical anodic oxidation for the first time. By controlling the sputtering time and power, the Pt loading of the (PtAC–O–Au)-1 electrode was very low with 0.48 μg cm−2. The (PtAC–O–Au)-1 electrode possesses an ultra-high MA (329 A mgPt−1@η = 80 mV), which is 66 times that of 0.5 wt% Pt/C electrode under the same loading. The MA of the (PtAC–O–Au)-1 electrode is 41 times that of the 20 wt% Pt/C electrode at the overpotential of 50 mV. Moreover, the (PtAC–O–Au)-1 electrode has good apparent activity, which is higher than that of the 20 wt% Pt/C electrode and close to that of the 40 wt% Pt/C electrode. The (PtAC–O–Au)-1 electrode has exhibited an ultra-high SA (87.7 mA cm−2@η = 50 mV), which is much higher than those of 0.5 wt% Pt/C (7.26 mA cm−2@η = 50 mV) and Pt-1 (4.77 mA cm−2@η = 50 mV). More importantly, it has been confirmed for the first time by electrochemical analysis methods (CV and CO adsorption–stripping) and XPS that a new active site of Pt–O–Au (0.72 V vs. RHE) is formed on the surface of the electrode after electrochemical oxidation. DFT calculations elucidated that the ΔGH* (absolute value) of Pt@(PtAC–O–Au) is close to that of Pt(111), indicating that its outstanding HER activity originates from its optimal ΔGH* value. In addition, the PtAC–O–Au planar electrodes are grown specifically along the [111] direction, which is beneficial to be combined with density functional theory (DFT) calculations.Experimental
Materials
The platinum target (Pt ≥ 99.99%) and gold target (Au ≥ 99.99%) were purchased from Beijing Goodwill Metal Technology Development Co. Ltd. Commercial Pt/C (5 wt%, 20 wt% and 40 wt% loading, Pt size of 2–5 nm) was obtained from Johnson Matthey Co. Ltd. Acetone (CH3COCH3, AR), sodium carbonate (Na2CO3, AR), and sulphuric acid (H2SO4, AR) were purchased from Sinopharm Chemical Reagent Co. Ltd, Shanghai, China. All reagents were analytical grade and used without further purification. Argon gas (99.999%) was purchased from Ming-Hui Company. The water (18.25 MΩ cm−1) used in all experiments was prepared by passing through an ultra-pure purification system.Preparation of the Au, PtAC–Au, and Pt electrodes
All the electrodes were prepared by magnetron sputtering (MS). A titanium plate was utilized as the electrode substrate, which was sandblasted and degreased in acetone and sodium carbonate with ultrasonication. Firstly, the vacuum chamber was vacuumed to 4.0 × 10−4 Pa, and then high-purity argon was introduced to bring the vacuum chamber pressure up to 1.0 Pa. Different electrode materials were prepared by using different metal targets and changing the sputtering power and time. The Au electrode was prepared with the Au target at a sputtering power of 20 W and a sputtering time of 10 min. The PtAC–Au electrode was obtained with the Pt target and Au base, at a sputtering power of 3 W and sputtering times of 10 s, 20 s, 30 s, 60 s and 600 s, which were denoted as (PtAC–Au)-1, (PtAC–Au)-2, (PtAC–Au)-3, (PtAC–Au)-4 and (PtAC–Au)-5. The Pt electrode was obtained from the Pt target substrate at a sputtering power of 3 W and sputtering times of 10 s and 600 s, which were denoted as Pt-1 and Pt-2.Preparation of AuOx, (PtAC–O–Au)-x and PtOx electrodes
All the electrodes were prepared by electrochemical anodic oxidation, which was carried out in a typical three-electrode electrochemical system. Carbon paper was used as the counter electrode, the reversible hydrogen electrode (RHE) as the reference electrode, and the Au, (PtAC–Au)-x and Pt electrodes as working electrodes. The anodic oxidation was carried out at a potential of 1.7 V for 10 min by chronoamperometry. The sample of the Au electrode after oxidation was recorded as AuOx. The oxidized samples of (PtAC–Au)-1, (PtAC–Au)-2, (PtAC–Au)-3, (PtAC–Au)-4 and (PtAC–Au)-5 electrodes were recorded as (PtAC–O–Au)-1, (PtAC–O–Au)-2, (PtAC–O–Au)-3, (PtAC–O–Au)-4 and (PtAC–O–Au)-5, respectively. Samples of Pt-1 and Pt-2 after oxidation were recorded as PtOx-1 and PtOx-2, respectively.Material characterization
Transmission Electron Microscopy (TEM) and High-Angle Annular Dark-Field Scanning Transmission Electron Microscopy (HAADF-STEM) images were obtained on a JEOL-2100F at an acceleration voltage of 300 kV. The samples were prepared by dropping cyclohexane dispersions of samples onto carbon-coated copper TEM grids using pipettes and drying under ambient conditions. Scanning Electron Microscope (SEM) images were taken with a Zeiss ΣIGMA field-emission SEM operated at 20 kV. Atomic Force Microscope (AFM) images were obtained by a Bruker MultiMode 8 AFM with ScanAsyst™ mode. X-ray diffraction (XRD) diagrams were collected on a Shimadzu XRD-7000 X-ray diffractometer using Cu Kα radiation (λ = 0.15405 nm), operating at 40 kV and 30 mA. The loading of Pt was determined by the inductively coupled plasma optical emission spectrometry (ICP-OES, Varian). X-ray photoelectron spectrometry (XPS: ESCALAB 250Xi, Thermo Scientific) with monochromatized Al Kα radiation was used to analyze the electronic properties and elemental composition of the samples. The fitting of the data was obtained with XPS Peak41 software.Electrochemical measurements
Electrochemical measurements were performed by using a CHI760D electrochemical workstation with a three-electrode cell. A carbon paper was used as the counter electrode and a reversible hydrogen electrode (RHE) as the reference. The working electrode was the PtAC–O–Au electrode, as well as PtAC–Au and other electrodes. Cyclic voltammetry (CV) measurements were performed from 0.03 to 1.7 V in 0.5 mol L−1 H2SO4 solution at a scan rate of 100 mV s−1. Linear sweep voltammetry (LSV) was used as a systematic and effective method to investigate the electrochemical activity of the electrocatalysts. After obtaining stable CV curves, the HER polarization curves were obtained by sweeping the potential from −0.3 to 0.2 V (vs. RHE) at a scan rate of 5 mV s−1 in Ar-saturated 0.5 mol L−1 H2SO4. The durability tests were carried out by repeating the potential scan from −0.2 to 0.1 V (vs. RHE) with 2000 cycles. Current–time responses were monitored by chronopotentiometry measurements at the current density of 50 mA cm−2 over 20 hours. All potentials were corrected with iR to the reversible hydrogen electrode (RHE). Electrochemical impedance spectroscopy was performed with the working electrode biased at a constant value of −0.05 V vs. RHE, with the frequency ranging from 100 kHz to 0.1 Hz with an amplitude of 10 mV. The EIS data were processed (electrical equivalent circuits and quantitative parameters) using a computer system with the ZView Analyst software package. For CO-stripping measurements, a cell voltage of 0.1 V was held for 30 min while the working electrode was successively flushed with 50 mL min−1 CO for the first 10 min, then with 200 mL min−1 Ar for 20 min. Finally, the CV measurements of CO-stripping were performed from 0.03 to 1.4 V at a scan rate of 50 mV s−1.DFT calculations
First-principles calculations were performed using the projector augmented wave (PAW) pseudopotential based on the density functional theory in the Vienna Ab Initio Simulation Package (VASP, version 5.3).68–72 These computations utilized the PBE (Perdew–Burke–Ernzerhof) in GGA (the Generalized Gradient Approximation) to describe the exchange–correlation interaction. A cutoff energy of 408 eV was employed for all computations. The convergence of energy and forces was set to 1 × 10−3 eV and 0.02 eV Å−1, respectively. These models were set with a vacuum gap of 15 Å to avoid the interaction of the layers. The HER calculations were performed according to previously published procedures. The free energies of the intermediates were obtained using ΔGH* = ΔEH* + ΔEZPE − TΔS, where ΔEH*, ΔEZPE and ΔS are the binding energy, zero point energy change and entropy change of adsorption H, respectively. Setting the free energy of gaseous H2 under standard conditions to 0 eV, the relative free energy of H* (absorbed atomic hydrogen) could be calculated as ΔGH* = ΔEH* + 0.24 eV.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the Natural Science Foundation of Hubei province (2020CFB777).Notes and references
- S. Chu and A. Majumdar, Opportunities and challenges for a sustainable energy future, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- Z. W. She, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Norskov and T. F. Jaramillo, Combining theory and experiment in electrocatalysis: Insights into materials design, Science, 2017, 355, eaad4998 CrossRef PubMed.
- Z.-Y. Zhang, H. Tian, L. Bian, S.-Z. Liu, Y. Liu and Z.-L. Wang, Cu-Zn-based alloy/oxide interfaces for enhanced electroreduction of CO2 to C2+ products, J. Energy Chem., 2023, 83, 90–97 CrossRef CAS.
- Z. Zhang, L. Bian, H. Tian, Y. Liu, Y. Bando, Y. Yamauchi and Z.-L. Wang, Tailoring the Surface and Interface Structures of Copper-Based Catalysts for Electrochemical Reduction of CO2 to Ethylene and Ethanol, Small, 2022, 18, 2107450 CrossRef CAS PubMed.
- P. Li, Y. Jiang, Y. Hu, Y. Men, Y. Liu, W. Cai and S. Chen, Hydrogen bond network connectivity in the electric double layer dominates the kinetic pH effect in hydrogen electrocatalysis on Pt, Nat. Catal., 2022, 5, 900–911 CrossRef CAS.
- M. Luo, J. Yang, X. Li, M. Eguchi, Y. Yamauchi and Z.-L. Wang, Insights into alloy/oxide or hydroxide interfaces in Ni–Mo-based electrocatalysts for hydrogen evolution under alkaline conditions, Chem. Sci., 2023, 14, 3400–3414 RSC.
- M. Wang, Y. Xu, C.-K. Peng, S.-Y. Chen, Y.-G. Lin, Z. Hu, L. Sun, S. Ding, C.-W. Pao, Q. Shao and X. Huang, Site-Specified Two-Dimensional Heterojunction of Pt Nanoparticles/Metal–Organic Frameworks for Enhanced Hydrogen Evolution, J. Am. Chem. Soc., 2021, 143, 16512–16518 CrossRef CAS PubMed.
- J. Han, C. Gong, C. He, P. He, J. Zhang and Z. Zhang, Sub-1 nm Pt nanoclusters on N and P co-doped carbon nanotubes for the electrocatalytic hydrogen evolution reaction, J. Mater. Chem. A, 2022, 10, 16403–16408 RSC.
- S. Kumari, T. Masubuchi, H. S. White, A. Alexandrova, S. L. Anderson and P. Sautet, Electrocatalytic Hydrogen Evolution at Full Atomic Utilization over ITO-Supported Sub-nano-Ptn Clusters: High, Size-Dependent Activity Controlled by Fluxional Pt Hydride Species, J. Am. Chem. Soc., 2023, 145(10), 5834–5845 CrossRef CAS PubMed.
- Z. Ma, C. Chen, X. Cui, L. Zeng, L. Wang, W. Jiang and J. Shi, Hydrogen Evolution/Oxidation Electrocatalysts by the Self-Activation of Amorphous Platinum, ACS Appl. Mater. Interfaces, 2021, 13, 44224–44233 CrossRef CAS PubMed.
- X.-K. Wan, H. B. Wu, B. Y. Guan, D. Luan and X. W. Lou, Confining Sub-Nanometer Pt Clusters in Hollow Mesoporous Carbon Spheres for Boosting Hydrogen Evolution Activity, Adv. Mater., 2020, 32, 1901349 CrossRef CAS PubMed.
- A. J. Shih, N. Arulmozhi and M. T. M. Koper, Electrocatalysis under Cover: Enhanced Hydrogen Evolution via Defective Graphene-Covered Pt(111), ACS Catal., 2021, 11, 10892–10901 CrossRef CAS.
- Q. Cheng, C. Hu, G. Wang, Z. Zou, H. Yang and L. Dai, Carbon-Defect-Driven Electroless Deposition of Pt Atomic Clusters for Highly Efficient Hydrogen Evolution, J. Am. Chem. Soc., 2020, 142, 5594–5601 CrossRef CAS PubMed.
- H. Jin, M. Ha, M. G. Kim, J. H. Lee and K. S. Kim, Engineering Pt Coordination Environment with Atomically Dispersed Transition Metal Sites Toward Superior Hydrogen Evolution, Adv. Energy Mater., 2023, 13, 2204213 CrossRef CAS.
- S. Zhai, D. Zhai, L. Sun, N. Feng, X. Chen, W.-Q. Deng and H. Wu, Single platinum atoms anchored on N-doped carbon materials composed of bipyridine as efficient hydrogen evolution electrocatalysts, Mater. Chem. Front., 2023, 7, 2889–2895 RSC.
- X. Li, J. Yu, J. Jia, A. Wang, L. Zhao, T. Xiong, H. Liu and W. Zhou, Confined distribution of platinum clusters on MoO2 hexagonal nanosheets with oxygen vacancies as a high-efficiency electrocatalyst for hydrogen evolution reaction, Nano Energy, 2019, 62, 127–135 CrossRef CAS.
- J. Park, S. Lee, H. Kim, A. Cho, S. Kim, Y. Ye, J. Han, H. Lee, J. H. Jiang and J. Lee, Investigation of the Support Effect in Atomically Dispersed Pt on WO3−x for Utilization of Pt in the Hydrogen Evolution Reaction, Angew. Chem., Int. Ed., 2019, 58, 16038–16042 CrossRef CAS PubMed.
- X. Fan, C. Liu, B. Gao, H. Li, Y. Zhang, H. Zhang, Q. Gao, X. Cao and Y. Tang, Electronic Structure Engineering of Pt Species over Pt/WO3 toward Highly Efficient Electrocatalytic Hydrogen Evolution, Small, 2023, 2301178 CrossRef CAS PubMed.
- T. Kim, S. B. Roy, S. Moon, S. Yoo, H. Choi, V. G. Parale, Y. Kim, J. Lee, S. C. Jun, K. Kang, S.-H. Chun, K. Kanamori and H.-H. Park, Highly Dispersed Pt Clusters on F-Doped Tin(IV) Oxide Aerogel Matrix: An Ultra-Robust Hybrid Catalyst for Enhanced Hydrogen Evolution, ACS Nano, 2022, 16, 1625–1638 CrossRef CAS PubMed.
- Z. Wu, P. Yang, Q. Li, W. Xiao, Z. Li, G. Xu, F. Liu, B. Jia, T. Ma, S. Feng and L. Wang, Microwave Synthesis of Pt Clusters on Black TiO2 with Abundant Oxygen Vacancies for Efficient Acidic Electrocatalytic Hydrogen Evolution, Angew. Chem., Int. Ed., 2023, 62, e202300406 CrossRef CAS PubMed.
- X. Cheng, B. Xiao, Y. Chen, Y. Wang, L. Zheng, Y. Lu, H. Li and G. Chen, Ligand Charge Donation–Acquisition Balance: A Unique Strategy to Boost Single Pt Atom Catalyst Mass Activity toward the Hydrogen Evolution Reaction, ACS Catal., 2022, 12, 5970–5978 CrossRef CAS.
- K. Guo, J. Zheng, J. Bao, Y. Li and D. Xu, Combining Highly Dispersed Amorphous MoS3 with Pt Nanodendrites as Robust Electrocatalysts for Hydrogen Evolution Reaction, Small, 2023, 19, 2208077 CrossRef CAS PubMed.
- J. Zhu, L. Cai, X. Yin, Z. Wang, L. Zhang, H. Ma, Y. Ke, Y. Du, S. Xi, A. Wee, Y. Chai and W. Zhang, Enhanced Electrocatalytic Hydrogen Evolution Activity in Single-Atom Pt-Decorated VS2 Nanosheets, ACS Nano, 2020, 14, 5600–5608 CrossRef CAS PubMed.
- T. Ma, H. Cao, S. Li, S. Cao, Z. Zhao, Z. Wu, R. Yan, C. Yang, Y. Wang, P. A. Aken, L. Qiu, Y.-G. Wang and C. Cheng, Crystalline Lattice-Confined Atomic Pt in Metal Carbides to Match Electronic Structures and Hydrogen Evolution Behaviors of Platinum, Adv. Mater., 2022, 34, 2206368 CrossRef CAS PubMed.
- Y. Chu, R. Peng, Z. Chen, L. Li, F. Zhao, Y. Zhu, S. Tong and H. Zheng, Modulating Dominant Facets of Pt through Multistep Selective Anchored on WC for Enhanced Hydrogen Evolution Catalysis, ACS Appl. Mater. Interfaces, 2023, 15, 9263–9272 CrossRef CAS PubMed.
- Y. Qiu, Z. Wen, C. Jiang, X. Wu, R. Si, J. Bao, Q. Zhang, L. Gu, J. Tang and X. Guo, Rational Design of Atomic Layers of Pt Anchored on Mo2C Nanorods for Efficient Hydrogen Evolution over a Wide pH Range, Small, 2019, 15, 1900014 CrossRef PubMed.
- X. Zhao, M. Chen, Y. Zhou, H. Zhang and G. Hu, Fluorinated MXenes accelerate the hydrogen evolution activity of in situ induced snowflake-like nano-Pt, J. Mater. Chem. A, 2023, 11, 5830–5840 RSC.
- J. Zhang, E. Wang, S. Cui, S. Yang, X. Zou and Y. Gong, Single-Atom Pt Anchored on Oxygen Vacancy of Monolayer Ti3C2Tx for Superior Hydrogen Evolution, Nano Lett., 2022, 22, 1398–1405 CrossRef CAS PubMed.
- Q. Dong, S. Ma, J. Zhu, F. Yue, Y. Geng, J. Zheng, Y. Ge, C. Fan, H. Zhang, M. Xiang and Q. Zhu, Ultrahigh Mass Activity for the Hydrogen Evolution Reaction by Anchoring Platinum Single Atoms on Active {100} Facets of TiC via Cation Defect Engineering, Adv. Funct. Mater., 2022, 33, 2210665 CrossRef.
- L. Najafi, S. Bellani, S. Castelli, M. P. Arciniegas, R. Brescia, R. Nunez, B. García, M. Serri, F. Drago, L. Manna and F. Bonaccorso, Octapod-Shaped CdSe Nanocrystals Hosting Pt with High Mass Activity for the Hydrogen Evolution Reaction, Chem. Mater., 2020, 32, 2420–2429 CrossRef CAS.
- W. Yang, P. Cheng, Z. Li, Y. Lin, M. Li, J. Zi, H. Shi, G. Li, Z. Lian and H. Li, Tuning the Cobalt–Platinum Alloy Regulating Single-Atom Platinum for Highly Efficient Hydrogen Evolution Reaction, Adv. Funct. Mater., 2022, 32, 2205920 CrossRef CAS.
- W. Yu, Z. Chen, Y. Fu, W. Xiao, B. Dong, Y. Chai, Z. Wu and L. Wang, Superb All-pH Hydrogen Evolution Performances Powered by Ultralow Pt-Decorated Hierarchical Ni-Mo Porous Microcolumns, Adv. Funct. Mater., 2023, 33, 2210855 CrossRef CAS.
- Y. Peng, K. Ma, T. Xie, J. Du, L. Zheng, F. Zhang, X. Fan, W. Peng, J. Ji and Y. Li, Tunable Pt–Ni Interaction Induced Construction of Disparate Atomically Dispersed Pt Sites for Acidic Hydrogen Evolution, ACS Appl. Mater. Interfaces, 2023, 15, 27089–27098 CrossRef CAS PubMed.
- K. Li, Y. Li, Y. Wang, J. Ge, C. Liu and W. Xing, Enhanced electrocatalytic performance for the hydrogen evolution reaction through surface enrichment of platinum nanoclusters alloying with ruthenium in situ embedded in carbon, Energy Environ. Sci., 2018, 11, 1232–1239 RSC.
- W. Zhao, C. Luo, Y. Lin, G.-B. Wang, H. M. Chen, P. Kuang and J. Yu, Pt–Ru Dimer Electrocatalyst with Electron Redistribution for Hydrogen Evolution Reaction, ACS Catal., 2022, 12, 5540–5548 CrossRef CAS.
- D. Kobayashi, H. Kobayashi, D. Wu, S. Okazoe, K. Kusada, T. Yamamoto, T. Toriyama, S. Matsumura, S. Kawaguchi, Y. Kubota, S. M. Aspera, H. Arai and H. Kitagawa, Significant Enhancement of Hydrogen Evolution Reaction Activity by Negatively Charged Pt through Light Doping of W, J. Am. Chem. Soc., 2020, 142, 17250–17254 CrossRef CAS PubMed.
- Y. Yao, X. Gu, D. He, Z. Li, W. Liu, Q. Xu, T. Yao, Y. Liu, H. Wang, C. Zhao, X. Wang, P. Yin, H. Li, X. Hong, S. Wei, W. Li, Y. Li and Y. Wu, Engineering the Electronic Structure of Submonolayer Pt on Intermetallic Pd3Pb via Charge Transfer Boosts the Hydrogen Evolution Reaction, J. Am. Chem. Soc., 2019, 141, 19964–19968 CrossRef CAS PubMed.
- H. Lv, X. Chen, D. Xu, Y. Hu, H. Zheng, S. L. Suib and B. Liu, Ultrathin PdPt bimetallic nanowires with enhanced electrocatalytic performance for hydrogen evolution reaction, Appl. Catal., B, 2018, 238, 525–532 CrossRef CAS.
- S. Liu, X. Mu, W. Li, M. Lv, B. Chen, C. Chen and S. Mu, Cation vacancy-modulated PtPdRuTe five-fold twinned nanomaterial for catalyzing hydrogen evolution reaction, Nano Energy, 2019, 61, 346–351 CrossRef CAS.
- Z. Li, M. Huang, J. Li and H. Zhu, Large-Scale, Controllable Synthesis of Ultrathin Platinum Diselenide Ribbons for Efficient Electrocatalytic Hydrogen Evolution, Adv. Funct. Mater., 2023, 33, 2300376 CrossRef CAS.
- X. Ping, D. Liang, Y. Wu, X. Yan, S. Zhou, D. Hu, X. Pan, P. Lu and L. Jiao, Activating a Two-Dimensional PtSe2 Basal Plane for the Hydrogen Evolution Reaction through the Simultaneous Generation of Atomic Vacancies and Pt Clusters, Nano Lett., 2021, 21, 3857–3863 CrossRef CAS PubMed.
- P. Wang, X. Zhang, J. Zhang, A. Wan, S. Guo, G. Lu, J. Yao and X. Huang, Precise tuning in platinum-nickel/nickel sulfide interface nanowires for synergistic hydrogen evolution catalysis, Nat. Commun., 2017, 8, 14580 CrossRef CAS PubMed.
- S. Ye, F. Luo, Q. Zhang, P. Zhang, T. Xu, Q. Wang, D. He, L. Guo, Y. Zhang, C. He, X. Ouyang, M. Gu, J. Liu and X. Sun, Highly stable single Pt atomic sites anchored on aniline-stacked graphene for hydrogen evolution reaction, Energy Environ. Sci., 2019, 12, 1000–1007 RSC.
- H. Tian, X. Cui, L. Zeng, L. Su, Y. Song and J. Shi, Oxygen vacancy-assisted hydrogen evolution reaction of the Pt/WO3 electrocatalyst, J. Mater. Chem. A, 2019, 7, 6285–6293 RSC.
- Y. Guan, Y. Feng, J. Wan, X. Yang, L. Fang, X. Gu, R. Liu, Z. Huang, J. Li, J. Luo, C. Li and Y. Wang, Ganoderma-Like MoS2/NiS2 with Single Platinum Atoms Doping as an Efficient and Stable Hydrogen Evolution Reaction Catalyst, Small, 2018, 14, 1800697 CrossRef PubMed.
- J. Zhang, Y. Zhao, X. Guo, C. Chen, C.-L. Dong, R.-S. Liu, C.-P. Han, Y. Li, Y. Gogotsi and G. Wang, Single platinum atoms immobilized on an MXene as an efficient catalyst for the hydrogen evolution reaction, Nat. Catal., 2018, 1, 985–992 CrossRef CAS.
- Y. Jiang, M. Yang, M. Qu, Y. Wang, Z. Yang, Q. Feng, X. Deng, W. Shen, M. Li and R. He, In situ confinement of Pt within three-dimensional MoO2@porous carbon for efficient hydrogen evolution, J. Mater. Chem. A, 2020, 8, 10409–10418 RSC.
- J. N. Tiwari, S. Sultan, C. W. Myung, T. Yoon, N. Li, M. Ha, A. M. Harzandi, H. J. Park, D. Y. Kim, S. S. Chandrasekaran, W. G. Lee, V. Vij, H. Kang, T. J. Shin, H. S. Shin, G. Lee, Z. Lee and K. S. Kim, Multicomponent electrocatalyst with ultralow Pt loading and high hydrogen evolution activity, Nat. Energy, 2018, 3, 773–782 CrossRef CAS.
- Q. Dang, Y. Y. Sun, X. Wang, W. X. Zhu, Y. Chenm, F. Liao, H. Huang and M. Shao, Carbon dots-Pt modified polyaniline nanosheet grown on carbon cloth as stable and high-efficient electrocatalyst for hydrogen evolution in pH-universal electrolyte, Appl. Catal., B, 2019, 257, 117905 CrossRef CAS.
- L. Xiu, W. Pei, S. Zhou, Z. Wang, P. Yang, J. Zhao and J. Qiu, Multilevel Hollow MXene Tailored Low-Pt Catalyst for Efficient Hydrogen Evolution in Full-pH Range and Seawater, Adv. Funct. Mater., 2020, 30, 1910028 CrossRef CAS.
- X.-P. Yin, H.-J. Wang, S.-F. Tang, X.-L. Lu, M. Shu, R. Si and T.-B. Lu, Engineering the Coordination Environment of Single-Atom Platinum Anchored on Graphdiyne for Optimizing Electrocatalytic Hydrogen Evolution, Angew. Chem., Int. Ed., 2018, 57, 9382–9386 CrossRef CAS PubMed.
- L. Zhang, Q. Wang, R. Si, Z. Song, X. Lin, M. N. Banis, K. Adair, J. Li, K. Doyle-Davis, R. Li, L.-M. Liu, M. Gu and X. Sun, New Insight of Pyrrole-Like Nitrogen for Boosting Hydrogen Evolution Activity and Stability of Pt Single Atoms, Small, 2021, 17, 2004453 CrossRef CAS PubMed.
- L. Zhang, R. Si, H. Liu, N. Chen, Q. Wang, K. Adair, Z. Wang, J. Chen, Z. Song, J. Li, M. N. Banis, R. Li, T.-K. Sham, M. Gu, L.-M. Liu, G. A. Botton and X. Sun, Atomic layer deposited Pt-Ru dual-metal dimers and identifying their active sites for hydrogen evolution reaction, Nat. Commun., 2019, 10, 4936 CrossRef PubMed.
- Y. Zhao, P. V. Kumar, X. Tan, X. Lu, X. Zhu, J. Jiang, J. Pan, S. Xi, H. Y. Yang, Z. Ma, T. Wan, D. Chu, W. Jiang, S. C. Smith, R. Amal, Z. Han and X. Lu, Modulating Pt-O-Pt atomic clusters with isolated cobalt atoms for enhanced hydrogen evolution catalysis, Nat. Commun., 2022, 13, 2430 CrossRef CAS PubMed.
- X. Cheng, Y. Li, L. Zheng, Y. Yan, Y. Zhang, G. Chen, S. Sun and J. Zhang, Highly active, stable oxidized platinum clusters as electrocatalysts for the hydrogen evolution reaction, Energy Environ. Sci., 2017, 10, 2450–2458 RSC.
- Y. Shi, Z.-R. Ma, Y.-Y. Xiao, Y.-C. Yin, W.-M. Huang, Z.-C. Huang, Y.-Z. Zheng, F.-Y. Mu, R. Huang, G.-Y. Shi, Y.-Y. Sun, X.-H. Xia and W. Chen, Electronic metal–support interaction modulates single-atom platinum catalysis for hydrogen evolution reaction, Nat. Commun., 2021, 12, 3021 CrossRef PubMed.
- F.-Y. Yu, Z.-L. Lang, L.-Y. Yin, K. Feng, Y.-J. Xia, H.-Q. Tan, H.-T. Zhu, J. Zhong, Z.-H. Kang and Y.-G. Li, Pt-O bond as an active site superior to Pt0 in hydrogen evolution reaction, Nat. Commun., 2020, 11, 490 CrossRef PubMed.
- Y. Zhan, Y. Li, Z. Yang, X. Wu, M. Ge, X. Zhou, J. Hou, X. Zheng, Y. Lai, R. Pang, H. Duan, X. Chen, H. Nie and S. Huang, Synthesis of a MoSx–O–PtOx Electrocatalyst with High Hydrogen Evolution Activity Using a Sacrificial Counter-Electrode, Adv. Sci., 2019, 6, 1801663 CrossRef PubMed.
- J. Dai, Y. Zhu, Y. Chen, X. Wen, M. Long, X. Wu, Z. Hu, D. Guan, X. Wang, C. Zhou, Q. Lin, Y. Sun, S.-C. Weng, H. Wang, W. Zhou and Z. Shao, Hydrogen spillover in complex oxide multifunctional sites improves acidic hydrogen evolution electrocatalysis, Nat. Commun., 2022, 13, 1189 CrossRef CAS PubMed.
- Q.-Q. Yan, D.-X. Wu, S.-Q. Chu, Z.-Q. Chen, Y. Lin, M.-X. Chen, J. Zhang, X.-J. Wu and H.-W. Liang, Reversing the charge transfer between platinum and sulfur-doped carbon support for electrocatalytic hydrogen evolution, Nat. Commun., 2019, 10, 4977 CrossRef CAS PubMed.
- Z. Ren, H. Jiang, M. Yuan, Z. Xie, L. Deng, J. Han, K. Lyu, Y. Zhu, L. X. Li and L. Zhuang, Si regulation of hydrogen adsorption on nanoporous PdSi hybrids towards enhancing electrochemical hydrogen evolution activity, Inorg. Chem. Front., 2023, 10, 1101–1111 RSC.
- Z. Ren, M. Tian, N. Cong, H. Jiang, H. Jiang, Z. Xie, J. Han and Y. Zhu, Promoting effect of amorphous support on ruthenium-based catalyst for electrochemical hydrogen evolution reaction, Chem. Commun., 2022, 58, 13588–13591 RSC.
- Y. Wang, Z. Ren, N. Cong, Y. Heng, M. Wang, Z. Wang, Z. Xie, Y. Liu, J. Han and Y. Zhu, The volcanic relationship between the HER activity and lattice constant of RuCo alloy nanofilms, Chem. Commun., 2022, 58, 4631–4634 RSC.
- L. Liao, S. Zhuang, C. Yao, N. Yan, J. Chen, C. Wang, N. Xia, X. Liu, M.-B. Li, L. Li, X. Bao and Z. Wu, Structure of Chiral Au44(2,4-DMBT)26 Nanocluster with an 18-Electron Shell Closure, J. Am. Chem. Soc., 2016, 138, 10425–10428 CrossRef CAS PubMed.
- N. Mahmood, Y. Yao, J.-W. Zhang, L. Pan, X. Zhang and J.-J. Zou, Electrocatalysts for Hydrogen Evolution in Alkaline Electrolytes: Mechanisms, Challenges, and Prospective Solutions, Adv. Sci., 2018, 5, 1700464 CrossRef PubMed.
- Y. Deng, Z. Liu, A. Wang, D. Sun, Y. Chen, L. Yang, J. Pang, H. Li, H. Li, H. Liu and W. Zhou, Oxygen-incorporated MoX (X: S, Se or P) nanosheets via universal and controlled electrochemical anodic activation for enhanced hydrogen evolution activity, Nano Energy, 2019, 62, 338–347 CrossRef CAS.
- L. Liu, D. Li, H. Zhao, A. Dimitrova, L. Li, Y. Fang, S. Krischok, W. Shi; and Y. Lei, Optimizing hydrogen evolution activity of nanoporous electrodes by dual-step surface engineering, Appl. Catal., B, 2019, 244, 87–95 CrossRef CAS.
- G. Kresse and J. Hafner, Ab-initio Molecular-Dynamics Simulation of the Liquid-Metal–Amorphous-Semiconductor Transition in Germanium, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Efficient Iterative Schemes for Ab-initio Total-Energy Calculations using a Plane-Wave Basis Set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- B. Hammer, L. B. Hansen and J. K. Nørskov, Improved Adsorption Energetics within Density-Functional Theory Using Revised Perdew-Burke-Ernzerhof Functionals, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 7413–7421 CrossRef.
- P. E. Blöchl, Projector Augmented-Wave Method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3qi01437a |
| This journal is © the Partner Organisations 2023 |