
Recent advances in label-free imaging of cell–matrix adhesions
Ping
Zhou
,
Lurong
Ding
,
Yajuan
Yan
,
Yafeng
Wang
and
Bin
Su
 *
*
Key Laboratory of Excited-State Materials of Zhejiang Province, Institute of Analytical Chemistry, Department of Chemistry, Zhejiang University, Hangzhou 310058, China. E-mail: subin@zju.edu.cn
First published on 27th January 2023
Abstract
Cell–matrix adhesions play an essential role in mediating and regulating many biological processes. The adhesion receptors, typically transmembrane integrins, provide dynamic correlations between intracellular environments and extracellular matrixes (ECMs) by bi-directional signaling. In-depth investigations of cell–matrix adhesion and integrin-mediated cell adhesive force are of great significance in biology and medicine. The emergence of advanced imaging techniques and principles has facilitated the understanding of the molecular composition and structure dynamics of cell–matrix adhesions, especially the label-free imaging methods that can be used to study living cell dynamics without immunofluorescence staining. This highlight article aims to give an overview of recent developments in imaging cell–matrix adhesions in a label-free manner. Electrochemiluminescence microscopy (ECLM) and surface plasmon resonance microscopy (SPRM) are briefly introduced and their applications in imaging analysis of cell–matrix adhesions are summarized. Then we highlight the advances in mapping cell–matrix adhesion force based on molecular tension probes and fluorescence microscopy (collectively termed as MTFM). The biomaterials including polyethylene glycol (PEG), peptides and DNA for constructing tension probes in MTFM are summarized. Finally, the outlook and perspectives on the further developments of cell–matrix adhesion imaging are presented.
 Ping Zhou | Ping Zhou received her BSc degree in Chemistry from Qingdao University in 2017 and PhD degree in Analytical Chemistry from Zhejiang University in 2022. She is now a postdoctoral fellow at Zhejiang University. Her current research interests are focused on ECL analysis of cell adhesive force. |
 Lurong Ding | Lurong Ding received her BSc degree in Applied Chemistry from Zhejiang University of Technology in 2020. She is currently a PhD student in the Department of Chemistry at Zhejiang University. Her research is focused on ECL analysis of cell-microenvironment interactions. |
 Yajuan Yan | Yajuan Yan received her BSc degree from the School of Chemistry and Chemical Engineering, Lanzhou University in 2021. She is currently a PhD student at Zhejiang University. Her research interests include ECL microscopy and cell analysis. |
 Yafeng Wang | Yafeng Wang received his BSc degree in Applied Chemistry from Zhejiang University of Technology in 2014 and PhD degree in Analytical Chemistry from Zhejiang University in 2020. He is now a postdoctoral fellow at Zhejiang University and his research interests are focused on unraveling ECL mechanisms and developing ultrasensitive ECL biosensors. |
 Bin Su | Bin Su received his BS, MS and PhD degrees from Jilin University, Changchun Institute of Applied Chemistry (Chinese Academy of Sciences) and Ecole Polytechnique Federale de Lausanne (EPFL), respectively. After three years of postdoctoral research at EPFL, he joined Zhejiang University in 2009 and was promoted to a full professor with tenure in 2012. His current research interests cover interfacial electrochemistry, electrochemiluminescence, biosensors and in vivo bioanalysis. |
1. Introduction
The extracellular matrix (ECM) is a highly dynamic structure that exists in all tissues, and is composed of a three-dimensional scaffold of matrix molecules, including primarily collagen, fibronectin and glycosaminoglycans.1 In addition to providing structural support for tissue integrity, the ECM continuously undergoes remodeling to maintain tissue homeostasis.2 Dysregulation of the ECM structure, composition, stiffness and function results in some disease conditions, such as fibrosis and invasive cancer.3,4 As shown in Fig. 1a, cells interact with the ECM via transmembrane receptors, typically integrin, thus forming physical connections to the cytoskeletal actin through a host of linker proteins and achieving information transmission via the signalling molecules and pathways.2 In this model, the binding of cells to the ECM introduces discrete structures at the cell surface, namely cell–matrix adhesions, which mediate direct interactions between cells and the extracellular microenvironment.5,6 These adhesive interactions are essential for the generation of adhesion-mediated signals that regulate a variety of biological processes, such as cell migration,7,8 tissue organization,9 wound healing,10,11 and tumorigenesis.12 Thus, elucidating the structure and function of cell–matrix adhesions is of great significance in the fields of biology and medicine. | ||
| Fig. 1 (a) Schematic illustration of the cell-ECM interactions and associated intracellular structures. Adapted with permission from ref. 2 (Copyright 2014 Nature publishing group). (b) Confocal fluorescence images of different adhesion structures and their component cartoons. FC, FA and FB are shown by fibroblasts stained with β3 integrin, β1 integrin or tensin, respectively. THP-1 cells and A375M cells are stained with actin/vinculin and phalloidin to define podosomes and invadopodia, respectively. Reprinted with permission from ref. 13 (Copyright 2010 Company of Biologists Ltd). | ||
Cell–matrix adhesions are formed at the specialized and elongated regions along the ventral cell membrane, which are tethered tightly to the substrate surface with a gap of only 10–15 nm.6 It has been recognized that these sites can be classified into different types in terms of their protein composition, size, life span and proteolytic properties (as shown in Fig. 1b).13 The shortest-lived adhesion structures termed as focal contacts (FCs) are typically located behind the leading edge of spreading or migrating cells, whose assembly and disassembly occur within the timescale of seconds or minutes. The cellular traction forces are propagated through the focal adhesion (FA) structures, which are composed of multiple proteins to guarantee the stability. Fibrillar adhesions (FBs), a kind of larger and more stable adhesion structure that run in parallel to fibronectin bundles in vivo, are rich in tensin and α5β1 integrin14 and also the sites of localized matrix deposition and fibronectin fibrillogenes. Podosomes commonly found in macrophages consist of a dense F-actin core and actin-binding proteins within a ring of integrins. The composition of invadopodia is similar to that of podosomes, while the former occurs only in malignant cells. The formation of adhesion structures is known to be associated with the activation of the integrin family of ECM receptors. Distinct integrin subtypes are generated by selective pairing between α and β subunits, the extracellular domains of which can bind to various ECM ligands with different affinities.15 Therefore, integrins provide a bi-directional conduit that transmits mechanochemical information across the cell membrane. During the mechanotransduction process, cells sense and respond to mechanical cues of the surrounding environment through integrin-based adhesion sites and finally adapt to the extracellular milieu. This process is crucial for a broad range of physiological events including cell migration,16 proliferation and differentiation,17 biofilm formation,18 embryonic development,19 immune response20 and so on. In particular, the cell–matrix adhesion force as a vital physical signal indeed functions in regulating cell behaviors and maintaining tissue integrity. Therefore, in-depth investigation of cell–matrix adhesions requires not only the visualization of adhesion structures and compositions but also the mapping and further quantification of cell–matrix adhesion forces, promoting synergistically the study of cell mechanobiology.
Over the past few decades, many research efforts have been made for imaging cell–matrix adhesions based on the advanced microscopy techniques and principles.13,21 A considerable part of these research studies requires immunofluorescent labelling of specific proteins in adhesion structures, which remains challenging for the dynamic analysis of living cells in the long-term. To address this limitation, the mapping of cell–matrix adhesions in a label-free manner has been conducted.22,23 This review will focus on the recent advances in label-free imaging of cell–matrix adhesions, including electrochemiluminescence microscopy (ECLM) and surface plasmon resonance microscopy (SPRM). Considering the significance of revealing when and where cell adhesive force is generated at cell–matrix interfaces, in this article we will also highlight molecular tension fluorescence microscopy (MTFM) used for mapping the cell–matrix adhesion force. Finally, we shall present an outlook and perspective on the further development of cell–matrix adhesion imaging.
2. ECL microscopy
ECL is the light emission triggered by electrochemical reactions, which possesses unique advantages of excellent spatiotemporal controllability, low background and high surface sensitivity.24–26 ECLM has recently manifested itself as a powerful tool in single cell analysis, including the identification of cellular contents and structures.22,27–29 In previous works, small molecules in cells or released from cells,30–32 (sub-)cellular structures including cell membranes,33,34 proteins,35–37 mitochondria38 and intracellular hierarchical structures such as the nucleolus, nucleus and endoplasmic reticulum39 have been visualized by ECLM.When cells adhere to the underlying electrode surface, the formed adhesions can function as the inhibitors for ECL reactions and thus appear as dark shadows in ECL images. This mode is often designated as negative imaging.40 Benefiting from the surface-confined nature of ECL-emitting regions dominated by the direct oxidative-reduction routes,41 ECLM allows imaging of cell–matrix adhesion in the vicinity of the substrate surface (Fig. 2a).42 The enhancement effects of silica nanochannel membranes (SNMs) on ECL intensity further favour a distinct visual contrast between cell adhesions and bare surfaces. With this methodology, the dynamic variation of cell–matrix adhesions during collective migration was explored. As reported recently, ECL emitting layers can be rationally modulated by changing the concentration ratio of ECL luminophore to coreactant to match with the spatial location of different cell junctions, so that cell–matrix adhesions and cell–cell junctions can be selectively imaged.43 Ino et al. have studied the difference of cell adhesions between monolayer and tube network culture for vascular endothelial cells by ECLM, providing an identification method of adhesion states during tube formation.44
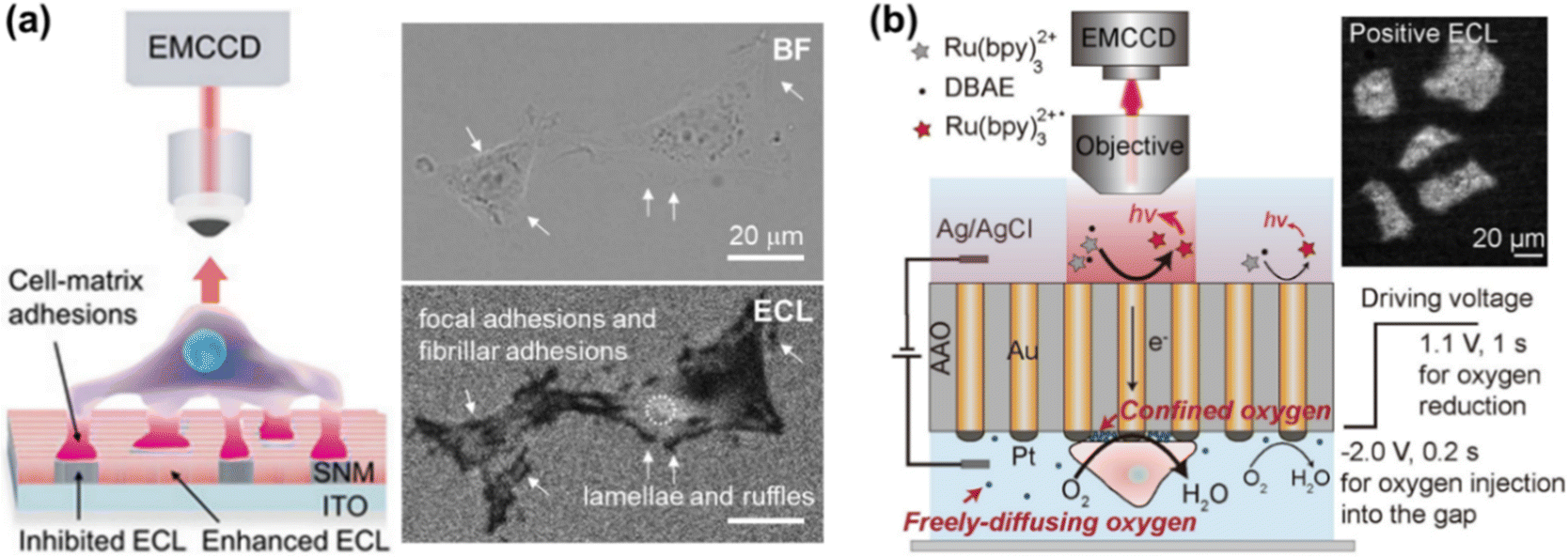 | ||
| Fig. 2 (a) Schematic illustration of imaging cell–matrix adhesions by ECLM in the negative mode (left). The bright field (BF) and corresponding ECL image of cells (right). Reproduced from ref. 42 with permission from Wiley-VCH. (b) Scheme of ECL imaging of cell–matrix adhesions in the positive mode by using a closed bipolar nanoelectrode array. Reprinted from ref. 45 with permission from Wiley-VCH. | ||
Further modifying the SNM rich in silanol groups with Arg-Gly-Asp (RGD), (3-aminopropyl)triethoxysilane (APTES), and oligo(ethylene glycol) (OEG), the influence of chemical microenvironments on cell behaviors was evaluated by imaging the cell–matrix adhesions.46 The statistical analysis of spatial distribution, area and strength of adhesions shows that the surfaces tethered with RGD and OEG groups can mediate the robust and weakest cell-microenvironment interactions, respectively, and that an intermediate adhesion is distributed on the APTES-coated surface. Moreover, in conjunction with selective immune-blocking of different integrin subunits, α6, α5, and α1 subunits are found to function in recognizing SNM, RGD/OEG and APTES surfaces, respectively.
Xia et al. have recently proposed an approach for positive imaging of cell adhesions by combined use of a closed bipolar nanoelectrode array and ECLM.45 The bipolar electrode was fabricated by electrodeposition of gold into the channels of an anodic aluminum oxide (AAO) membrane and subsequently platinizing one of the two terminals. As depicted in Fig. 2b, the reduction of oxygen beneath cells occurs at the cathodic side, which is coupled with ECL reactions at the anodic one. A high concentration of oxygen in the confined space underneath cells contributes to the enhanced ECL intensity, thus realizing the imaging of cell adhesions in a positive contrast mode. Cell morphology and adhesion strength can be also successfully imaged with this method.
3. SPR microscopy
SPRM has drawn considerable research interest since it was firstly described by Rothenhäusler et al.47 Under SPR, the surface plasmon wave travels along the metal-dielectric layer interface and decays exponentially.48 The evanescent wave is extremely sensitive to the tiny variation of physical parameters of the metal-dielectric multilayers, thus making SPR a non-invasive technique for biosensing near the metal surface.23 It has been widely used so far to study subcellular structures49–51 and dynamic cellular processes.52–56Giebel et al. analysed quantitatively the cell-substrate distance by SPRM, which was found to be 160 ± 10 nm for most parts of the cell, except for the peripheral lamellipodia (only 25 ± 10 nm).58 Subsequently renovated SPRMs, lens-imaging-type SPRM (Fig. 3a, left) and scanning localized SPRM (Fig. 3a, right), were used to measure the cell-electrode cleft gap distance.57 The former allows simultaneous large-area observation of the cellular bottom membrane, by which the strength of cell adhesions has been assessed qualitatively in terms of the reflected light intensity contrast (Fig. 3b). The latter is capable of mapping the cleft gap distance quantitatively with a high spatial resolution by selecting a region of interest (Fig. 3c), which can be used to study the difference of cell adhesions on gold surfaces coated with various peptides or proteins. It has been reported that the combination of them can contribute to observing an entire cell adhesion site and identifying regions of interest.59 The measurement accuracy of SPRM was later increased by decoupling the cell–substrate distance and the refractive index of the cytoplasm, by which the three-dimensional profiles of the basal membrane and its dynamics were reconstructed with an actual measurement accuracy of 2.3 nm.60 SPRM also allows simultaneous mapping of the cell-substrate adhesion gap, projected area, gap surface area and gap volume, thus providing a comprehensive analysis of the cell adhesion behaviors.61 In addition, the local movement of the cell membrane caused by extracellular osmotic pressure can be monitored with SPRM, so that the displacement image is considered to be the distribution of the cell adhesion strength.62 Recently, the impact of chemical fixation required for electron microscopy on the cell–substrate interface was investigated with SPRM, showing a retentive structure after chemical fixation.63
 | ||
| Fig. 3 (a) Schematic illustration of optical paths nearby the sample for lens-imaging-type (LISPM, left) and scanning localized (SLSPM, right) SPRM. (b and c) Representative LISPM (b) and SLSPM (c) images of HEK293 cells cultured on a poly-L-lysine-coated gold surface. Adapted with permission from ref. 57 (Copyright 2014 American Chemical Society). | ||
Using SPRM, the changes of cell–matrix adhesions mediated by cell–cell communication and the adhesion dynamics induced by shear stress were studied in real-time.64 A positive correlation between the FA mass and protein density was unveiled, showing a distinct difference for different cell types.65 Wavelength-scanning SPRM has also been developed for measuring the cell-substrate interaction, with a dynamic range larger than intensity-based SPRM.66 Plasmonic scattering microscopy (PSM), with a sub-micrometer spatial resolution and a microsecond temporal resolution, makes it possible to study cellular deformations induced by osmotic pressure changes or immune activation, as well as focal adhesion binding strength and rate, at the individual focal adhesion level.67,68 In addition, mapping the cell adhesion spring constants with PSM can provide an approach for the quantitative analysis of cell adhesion mechanical properties.68
4. MTFM for mapping the cell–matrix adhesion force
To date, substantial research interests have been drawn to study cell adhesive force and related physiological functions. To visualize the miniscule level of force ranging from pN to nN, different methods have been developed, such as traction force microscopy (TFM)69–71 and single molecule force spectroscopy (SMFS) including atomic force microscopy,72–74 optical or magnetic tweezers,75,76etc. Among them, TFM is only sensitive to forces at the nN level, while the SMFS methods fail to capture the mechanics of a whole cell because of their low throughput.MTFM works relying on molecular tension probes labeled with a fluorophore, fluorophore–fluorophore or fluorophore–quencher pair.77,78 Cell adhesive force transmits through the specific interactions between ligands in MTFM probes and receptors on cell membranes. For instance, the RGD peptide motif can recognize multiple integrin subtypes.79 Using MTFM, the cell–matrix adhesive force can be visualized with a sub-micrometer spatial resolution, pN force sensitivity and high throughput.80,81 Polyethylene glycol (PEG), peptide and DNA structures have been extensively used for constructing the molecular tension probes in MTFM. Corresponding information about force response and signal acquisition of molecular tension probes is summarized in Table 1.
| Tension probe | Force range or threshold/pN | Fluorophore or FRET donor | Quencher or FRET acceptor | Ref. |
|---|---|---|---|---|
| PEG | 0–20 | Alexa 647 | QSY 21 | 82 |
| ∼1–30 | Alexa 647 | QSY 21 | 85 | |
| ∼25 | Alexa 488 | AuNP | 86 | |
| ∼27 | Cy3B | AuNP | 87 | |
| Peptide | 1–7 | Alexa 546 | Alexa 647 | 83 |
| 1–5 | Alexa 546 | CoA 647 | 89 | |
| 2–7/7–11 | Alexa 546 | Alexa 647 | 90 | |
| ∼80–200 | Alexa 488/647; Cy3 | AuNP | 84 | |
| dsDNA | 12–54 | Cy3/Cy5 | — | 97 and 98 |
| 43, 54, >100 | Cy3/Cy5 | — | 99 | |
| 54 | Cy5 | BHQ2 | ||
| 12, 54 | Cy3 | BHQ2 | 100 and 106 | |
| 54, 100 | Cy3 | BHQ2 | 101 | |
| 12, 54 | Cy3, Atto647N | BHQ2 | 102 | |
| 12, 54, 160 | Cy3B | BHQ2 | 103 | |
| PNA/DNA | 12 | Alexa 488 | BHQ2 | 105 |
| 12 | Cy5 | BHQ2 | 104 | |
| DNA hairpin | 4.7 ± 1.7, 13.1 ± 2.4 | Cy3B/Cy5 | BHQ1/QSY21 | 107 |
| 4.7, 13.1, 19.3 | Cy3B | BHQ2 | 109 | |
| 12, 19, 56 | Cy3B | BHQ2, AuNP | 110 | |
| 4.7 | Cy3B, Atto647N | BHQ2 | 111 | |
| RSDTP | 4–60 | Cy3B, Atto647N | BHQ2 | 115 |
4.1 PEG-based MTFM
Thanks to the unique properties, such as well-characterized and reversible mechanical performance, biocompatibility and stability, PEG has been extensively used in MTFM. For example, PEG-based MTFM was engineered to map the cell adhesive force during the initial stages of regulatory endocytosis of the epidermal growth factor receptor (EGFR) for the first time.82 As shown in Fig. 4a, the PEG polymer is flanked with a fluorescently labeled EGF ligand and a biotin moiety for surface immobilization. When a cell exerted the force on the ligand, the PEG linker in the relaxed conformational state can be extended, thus resulting in the removal of the fluorophore from proximity to the quencher and the increase of fluorescence (FL) intensity (Fig. 4a). Punctate force signals were observed upon EGFR engagement (Fig. 4b), which was confirmed to be associated with the clathrin-mediated EGF ligand internalization. Based on the high affinity of the RGD peptide ligand to αVβ3 and α5β1 integrins, MTFM was used to study the integrin-generated force in cell adhesions, which was found to be sufficient to dissociate streptavidin–biotin tethered ligands.85 Using gold nanoparticles as a quencher, the dynamics of tensions exerted by integrin receptors during cell adhesion can be imaged, showing a translocation from the center of the cell to its periphery.86 In addition, the immobilization of tension probes on gold nanoparticles (AuNPs) through gold–thiol binding can minimize the possibility of probe rupture. Then, combining the above MTFM with block copolymer micelle nanolithography, a substrate with arrays of precisely spaced probes was fabricated to study the impact of integrin clustering on force transmission.87 Critical ligand spacing (<60–70 nm) helps in sustaining high integrin forces, thus facilitating the maturation of focal adhesion, while integrin receptors placed >100 nm apart significantly reduce the tension and destabilize the focal adhesion formation. This work realizes the simultaneous measurement of pN adhesive forces while regulating receptor nanoclustering. | ||
| Fig. 4 (a) The mechanism of PEG-based MTFM with the PEG polymer flanked by a fluorescently labeled (Alexa Fluor 647) EGF ligand and a biotin moiety for surface immobilization via streptavidin capture. (b) Representative brightfield, reflection interference contrast microscopy (RICM), FL response and force map converted from the FL image for a cell engaged to a sensor surface modified with the PEG-based tension probes. (a and b) Adapted with permission from ref. 82 (Copyright 2012 Nature publishing group). (c) Schematic composition of tension probes in peptide-based MTFM for studying the force generated by individual integrins. (d) Traction force map represented by FRET index obtained for cell spread on the surface modified with peptide-based tension probes. (c and d) Adapted with permission from ref. 83 (Copyright 2016 American Chemical Society). (e) The mechanism of the disulfide clamped I27 tension sensor in the presence of dithiothreitol (DTT) or Y-27632. (f) Representative I27-based tension signal for REF cells incubated on the sensor surface for 2 h before and after treating with 0.25 mM DTT for 10 min and subsequently with Y-27632 (40 μM) for 30 min. FL intensity profiles along the white lines are displayed in the inset of (e). (e and f) Reproduced from ref. 84 with permission from American Chemical Society. | ||
4.2 Peptide-based MTFM
Peptide molecules can also function as entropic springs for measuring the adhesive force. Grashoff et al. designed a tension-sensitive probe composed of repetitive amino-acid motifs, (GPGGA)8 peptide, which was derived from the spider silk protein flagelliform for force sensing.88 The elastic domain linked with two fluorescent proteins as a fluorescence resonance energy transfer (FRET) pair was genetically inserted into vinculin. This work revealed that stable focal adhesions required a force of ∼2.5 pN transmitted across vinculin. Using the same peptide sequence flanked with two organic dyes as the FRET pair, Dunn's group developed a molecular tension sensor to investigate the force generated by individual integrins (as seen in Fig. 4c).83,89,90 Defining the ratio of acceptor intensity to the sum of donor and acceptor intensity as FRET index, a traction force map can be drawn (Fig. 4d), which reflects the distribution of tension. In terms of the FRET values, the authors proposed that relatively modest tensions at the molecular level were sufficient to drive robust cell adhesions.83,89 Further analysis indicated that the addition of synergy sites to α5β1 integrin resulted in increased integrin recruitment to adhesions, instead of enhancing overall cellular traction generation.83 The experienced loads of integrins below their peak capacities guarantee the mechanical integrity of cells and tissues, which was uncovered by a revised model.90 Limited by the minimal dynamic range of the above-mentioned peptide-based tension probe (∼1–5 pN), the magnitude of integrin forces within cell adhesions remains unclear. A mechanically stable molecular tension probe consisting of the immunoglobulin 27th (I27) domain of cardiac titin flanked with a fluorophore and gold nanoparticle was reported (Fig. 4e).84 A threshold force of up to >100 pN is required to unfold I27.91 The integrin-mediated forces were confirmed to be more than 30–40 pN. Subsequently, a covalent disulfide bridge that can resist pN mechanical unfolding was added to I27 to “clamp” the probe (as shown in Fig. 4e). Incubation with a reducing agent (such as dithiothreitol) triggers the thiol exchange and the opening of the clamp, thus leading to full de-quenching of the fluorophore. Following Rho-associated protein kinase (ROCK) inhibition with Y-27632, the FL signal can diminish again, confirming that the opening of the clamped probes is driven by myosin and the signal is reversible (Fig. 4f). The rate of S–S reduction allows estimation of the applied integrin force in focal adhesions, which is ca. 110 ± 9 pN. This work provides a general approach for studying the high-level integrin tensions existing in stable cell adhesions.4.3 DNA-based MTFM
DNA-based MTFM has emerged as a powerful tool for sensing the cell adhesive force because of its simple and versatile design, and tunable and programmable mechanical features.78,92 The tension tolerance (Ttol) of DNA can be regulated by adjusting the DNA sequence or the force application site on the DNA backbone.93–95 The probes will rupture once the cell tension exceeds Ttol. Double-stranded DNA (dsDNA) was used for the measurement of adhesive force for the first time by Ha et al. and they termed it as a tension gauge tether (TGT).96 Subsequently, a range of TGTs with tunable tolerances have been engineered, by which the force transmitted by a single integrin–ligand during initial adhesion (a universal peak of about 40 pN) and the required force for Notch activations (less than 12 pN) can be determined.96,97 A mixture of TGTs with Ttol values of 12 pN and 54 pN labeled with Cy5 and Cy3 fluorophores, respectively, and coated on the substrate surface can be used to simultaneously map the integrin tensions, proving that integrins both inside and outside focal adhesions contribute to tensions in stable cell adhesion.98 RGD peptide is a ligand that has affinity to αVβ3 and α5β1 integrins, so the specific identification of the integrin subtype remains elusive. Recently, Kim et al. developed a novel TGT probe based on an ATN-16 ligand for specific binding to α5β1 integrin.99 As seen in Fig. 5a, a force tolerance of 43 pN or 54 pN can be controlled by the location of biotins on the bottom DNA strand, while Ttol of >100 pN was estimated by disassociation of the biotin–streptavidin bond. DNA rupture caused by integrin tension greater than Ttol resulted in the loss of the FL signal of the fluorophore (Cy3 or Cy5) conjugated to one terminal of the upper DNA strand, by which the distribution of integrin tension was imaged. As shown in Fig. 5b, αVβ3 integrin tension contributed to a ring-shaped “edge rupture” (RGD-TGT surface), whereas a “ventral rupture” was observed on the ATN-TGT surface. To evaluate the correlation between ventral rupture and invadopodia, an ATN-quenched TGT (ATN-qTGT) was developed on the basis of a fluorescent dye-quencher pair (Fig. 5c). In this configuration, the FL signal was observed when ATN-qTGT was dissociated by cellular force (Fig. 5d). Further analysis indicates that α5β1 integrin tension above 40 pN is required for the formation and maturation of invadopodia. The qTGTs with various Ttol values were also applied to map cell adhesive force in the single platelet100 and migrating keratocytes.101 The spatial resolution was calibrated to be ca. 0.4 μm. The results provide direct evidence that high-level integrin tension (>54 pN) is concentrated at the cell rear margin to detach cell adhesion, thus facilitating the cell rear retraction during the migration process.101 Ha et al. have revealed the force propagating mechanism during smooth muscle cell shortening induced by histamine through real-time imaging of FA dynamics.102 They proposed that FA reinforcement caused the time delay in cell shortening. Cell traction forces are of great significance for the development of functional cardiac muscle cells (CMCs) and the specific magnitude of integrin tension at pN level remains ambiguous. qTGT-based MTFM was used to address this knowledge gap.103 It has been shown that adhesion tethers with greater force tolerance (Ttol > 56 pN) can lead to functionally mature CMCs.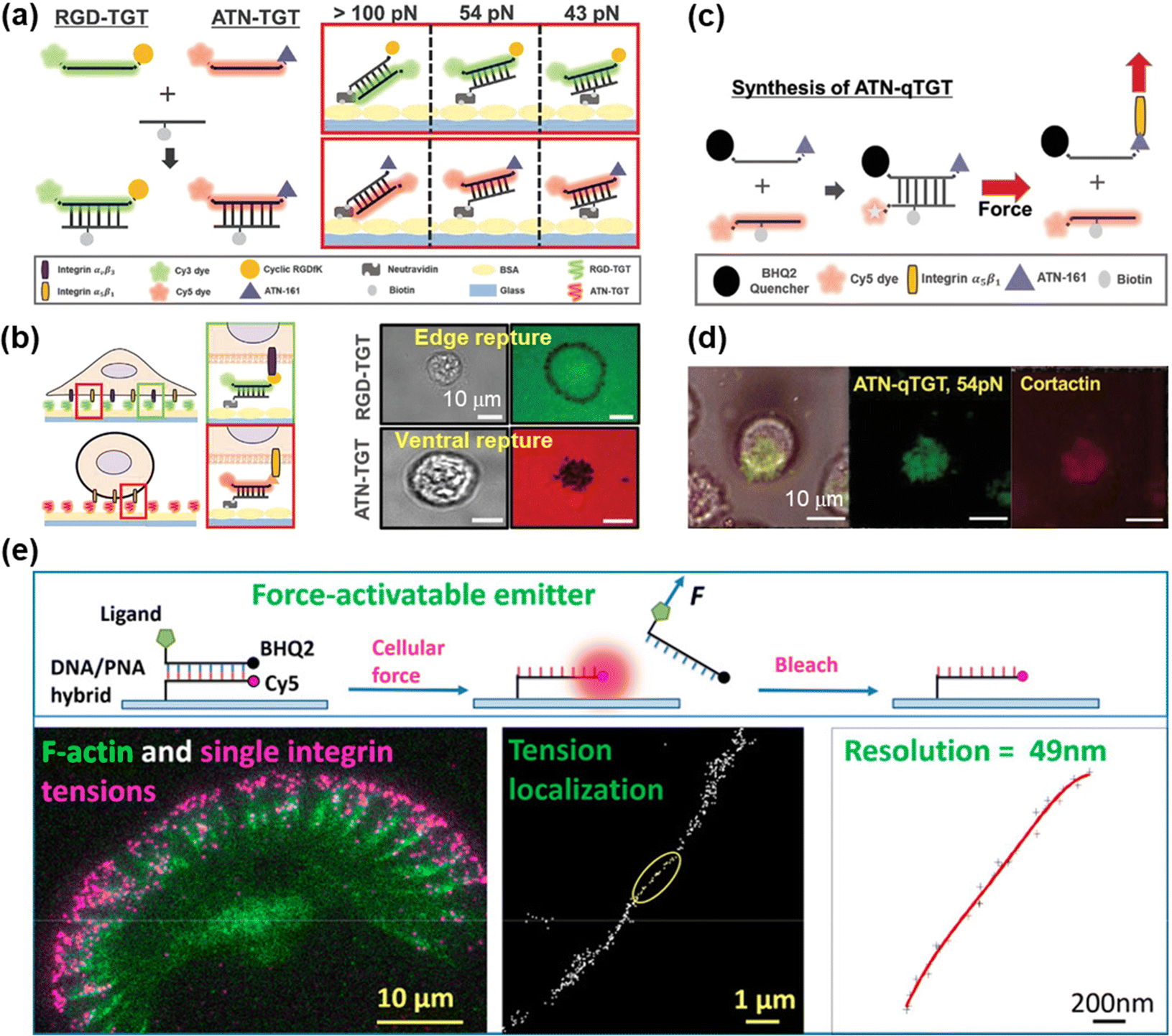 | ||
| Fig. 5 (a) Schematic diagrams of the dsDNA-based TGT systems. (b) The working principle of dsDNA-based TGT for the measurement of integrin tensions (left) and corresponding tension signals (right) during cell adhesion. (c) Illustration of the synthesis and tension-induced rupture of ANT-qTGT. (d) Invadopodia marker (cortactin) and tension response (ATN-qTGT) were co-localized. (a-d) Adapted with permission from ref. 99 (Copyright 2022 Wiley-VCH). (e) The force-activated rupture of PNA/DNA hybrid-based tension probe (top). Single integrin tension mapping in a migrating keratocyte with a calibrated resolution of 49 nm (bottom). Reprinted with permission from ref. 104 (Copyright 2020 American Chemical Society). | ||
To resist the degradation of DNA from DNase on the cell membrane, a peptide nucleic acid (PNA)/DNA hybrid-based tension probe was developed for constructing MTFM.104,105 Wang et al. have reported that the formation of a podosome is independent of extracellular integrin–ligand tensions, although they are critical for the formation of FA.105 Furthermore, PNA/DNA-based MTFM with a high spatial resolution of ca. 50 nm was developed by multiple cycles of molecular tension imaging and localization (as seen in Fig. 5e).104 Ultranarrow distribution of integrin tensions at the cell leading edge was revealed for both migratory cells and stationary cells. The irreversibility of dsDNA rupture makes it unavailable to image cell adhesive force continuously, so that the real-time force mapping can be realized by recording the time-lapse images of tension signals and then acquiring the new force map with the frame subtraction method.102,106
DNA hairpin is a single-stranded DNA consisting of intramolecular base pairs, which has been employed as a “switch” element for imaging the cell adhesive force. As shown in Fig. 6a, the fluorophore–quencher pair is conjugated to the DNA hairpin structure, thus reporting the force distribution by the FL signal when the DNA hairpin is unfolded. The tension threshold can be tuned by altering the GC content in the DNA sequence (Fig. 6b).107 Using DNA hairpin-based MTFM, highly dynamic and heterogeneous integrin tension has been visualized.107,108 As shown in Fig. 6c, the force signal is located at the cell edges and its intensity increases rapidly within minutes. Further determination of the percentage of unfolded probes by calculating the FL intensity of de-quenched dyes corresponding to a fully opened hairpin probe indicates that a large density of hairpins of up to 9% have been unzipped at these regions. A DNA hairpin-based tension probe was also used to map the force distribution during the process of platelet activation.109 A tension greater than 19 pN was produced at the central zone of the platelet, while a weak force (∼4.7–13.1 pN) was located at the cell periphery. In addition, Salaita et al. have uncovered the important role of cell adhesive force in T cell activation.110,111 In recent years, super-resolution visualization of single integrin tension has received considerable research attention.112–114 In conjunction with DNA-PAINT super-resolution microscopy,113 mechanical engagement of single integrin receptors has been studied.114 It is found that in cell adhesions the integrin receptor clustering is governed by a non-random organization with complexes spaced as close as 20–30 nm.
 | ||
| Fig. 6 (a) Schematic illustration of the DNA hairpin-based tension sensor consisting of an anchor strand immobilized onto a surface (blue), a hairpin strand (black) and a ligand strand representing an adhesive peptide (green). The fluorophore and quencher are bonded to the opposing termini of the ligand and anchoring strands. (b) The table lists the tension thresholds and quenching efficiency with the variation of the GC content. (c) Representative RICM and MTFM (4.7 pN probe) time-lapse images at the indicated time points. The percentage of unfolded tension probes (“% unfolded” channel) indicates the fraction of 22% GC hairpins that have been unfolded within each pixel. (a–c) Reproduced with permission from ref. 107 (Copyright 2014 Nature publishing group). (d) The force-application geometries of RSDTPs with different tension tolerances. (e) Time-lapse images of RICM and tension map during the spreading of NIH-3T3 cells on substrates modified by RSDTPs with different unfolding forces. (d and e) Adapted with permission from ref. 115 (Copyright 2021 Nature publishing group). | ||
In contrast to dsDNA, the opening of a DNA hairpin triggered by tensions is reversible, thus allowing the continuous imaging of the cell adhesive force. However, a broader tension range of dsDNA (10–60 pN) than the DNA hairpin (4–18 pN) makes it well suited for studying the dynamic behaviors of the cellular mechanotransduction process.77,80,81 To address the shortcoming in real-time measurements of cell force with a value of higher than 20 pN, Liu et al. have developed a reversible shearing DNA tension probe (RSDTP).115Fig. 6d displays the force-response mechanism of RSDTP, the thresholds of which can be regulated by varying both the GC content and force application site of the probe. After seeding NIH-3T3 cells on a RSDTP-functionalized surface for 1 h, the tension signal was observed at the cell edge (Fig. 6e), in agreement with that reported previously.86,107,108 Moreover, the tension pattern obtained with 56 pN RSDTPs indicates that mechanically strong integrin clusters (>56 pN) exist in FAs (Fig. 6e), which can serve as the “mechanical hotspots” to maintain the architecture of FAs and facilitate their maturation.
5. Conclusions and outlook
A range of techniques being developed currently and used successfully have opened an avenue for the studying of cell–matrix adhesions, in particular the visualization analysis of their structures and dynamics. In comparison with the mainstream fluorescence microscopy that requires immunofluorescent labeling of specific adhesive proteins, the label-free approaches are suitable for observing living cells. ECLM has manifested itself as a surface-sensitive tool for single cell analysis, by which the imaging of cell–matrix adhesions with the negative or positive mode has been realized. Dynamic variations of cell–matrix adhesions during some biological processes such as collective migration and tube formation can be mapped using ECLM. SPRM can provide a quantitative method for the measurement of cell-substrate distances with a high spatial and temporal resolution, with which the strength and dynamic evolution of cell adhesions are revealed. Combined and developed microscopy imaging methods based on SPRM can further improve the spatiotemporal resolution.Integrin-mediated cell–matrix adhesions can serve as a bi-directional conduit between the intracellular environment and extracellular matrix during cellular mechanotransduction. MTFM has emerged as a powerful sensor for visualizing and quantifying the cell adhesive force with a sub-micrometer spatial resolution, pN force sensitivity and high throughput, thus uncovering the elusive molecular mechanisms. Among these biomaterials including PEG, peptide and DNA, DNA and DNA-like structures are particularly versatile to be employed as molecular tension probes thanks to their simple and versatile designs, and tunable and programmable force response thresholds, which have drawn considerable research interest in real-time measurement of cell–matrix adhesion force. However, the current investigations show that the visualization of net orientations of averaged adhesive forces and even individual molecular tensions by MTFM is yet to be resolved and still remains challenging.
Certainly, integrins are the major proteins mediating cell–matrix adhesion, which are clustered and form patterns in various cell adhesive units such as FCs, FAs, FBs, podosomes and invadopodia. Imaging cell–matrix adhesions and mapping cell adhesive force at a single-integrin level rather than the integrin clusters will contribute to the investigation and understanding of the molecular mechanisms in adhesion dynamics. Nevertheless, the imaging methods mentioned in this article, ECLM, SPRM and MTFM, are all restricted by the optical diffraction limits, and are not suitable for imaging cell–matrix adhesions with super-high spatial resolution down to 200 nm, thus making super-resolution imaging challenging. Representative super-resolved imaging techniques such as stochastic optical reconstruction microscopy (STORM), stimulated emission depletion (STED) microscopy and photoactivated localization microscopy (PALM) have already been applied in imaging biological structures.116 It seems naturally feasible to image cell–matrix adhesions at the single-molecule level with resolution beyond the diffraction limit by combining ECLM, SPRM and MTFM with super-resolution methodologies. In addition, cryo-electron microscopy has recently made great breakthroughs in studying the adhesion structures and the transmission of mechanical force within cell adhesion,117–119 which will facilitate a profound insight into the adhesive mechanisms.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 22125405, 22074131 and 21874117).References
- C. Frantz, K. M. Stewart and V. M. Weaver, J. Cell Sci., 2010, 123, 4195–4200 CrossRef CAS PubMed.
- J. D. Humphrey, E. R. Dufresne and M. A. Schwartz, Nat. Rev. Mol. Cell Biol., 2014, 15, 802–812 CrossRef CAS PubMed.
- C. Bonnans, J. Chou and Z. Werb, Nat. Rev. Mol. Cell Biol., 2014, 15, 786–801 CrossRef CAS PubMed.
- J. Huang, L. Zhang, D. Wan, L. Zhou, S. Zheng, S. Lin and Y. Qiao, Signal Transduction Targeted Ther., 2021, 6, 153 CrossRef PubMed.
- A. L. Berrier and K. M. Yamada, J. Cell. Physiol., 2007, 213, 565–573 CrossRef CAS PubMed.
- E. Zamir and B. Geiger, J. Cell Sci., 2001, 114, 3583–3590 CrossRef CAS PubMed.
- E. K. Paluch, I. M. Aspalter and M. Sixt, Annu. Rev. Cell Dev. Biol., 2016, 32, 469–490 CrossRef CAS PubMed.
- J. R. W. Conway and G. Jacquemet, Essays Biochem., 2019, 63, 535–551 CrossRef CAS PubMed.
- E. Papusheva and C.-P. Heisenberg, EMBO J., 2010, 29, 2753–2768 CrossRef CAS PubMed.
- T. Omelchenko, J. M. Vasiliev, I. M. Gelfand, H. H. Feder and E. M. Bonder, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10788–10793 CrossRef CAS PubMed.
- M. Poujade, E. Grasland-Mongrain, A. Hertzog, J. Jouanneau, P. Chavrier, B. Ladoux, A. Buguin and P. Silberzan, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 15988–15993 CrossRef CAS PubMed.
- P. Friedl, J. Locker, E. Sahai and J. E. Segall, Nat. Cell Biol., 2012, 14, 777–783 CrossRef PubMed.
- D. C. Worth and M. Parsons, J. Cell Sci., 2010, 123, 3629–3638 CrossRef CAS PubMed.
- J. A. Green and K. M. Yamada, Adv. Drug Delivery Rev., 2007, 59, 1293–1298 CrossRef CAS PubMed.
- B.-H. Luo, C. V. Carman and T. A. Springer, Annu. Rev. Immunol., 2007, 25, 619–647 CrossRef CAS PubMed.
- C. D. Pascalis and S. Etienne-Manneville, Mol. Biol. Cell, 2017, 28, 1833–1846 CrossRef PubMed.
- R. McBeath, D. M. Pirone, C. M. Nelson, K. Bhadriraju and C. S. Chen, Dev. Cell, 2004, 6, 483–495 CrossRef CAS PubMed.
- M. Asally, M. Kittisopikul, P. Rué, Y. Du, Z. Hu, T. Çağatay, A. B. Robinson, H. Lu, J. Garcia-Ojalvo and G. M. Süel, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 18891–18896 CrossRef CAS PubMed.
- T. Mammoto and D. E. Ingber, Development, 2010, 137, 1407–1420 CrossRef CAS PubMed.
- E. Judokusumo, E. Tabdanov, S. Kumari, M. L. Dustin and L. C. Kam, Biophys. J., 2012, 102, L5–L7 CrossRef CAS PubMed.
- O. Medalia and B. Geiger, Curr. Opin. Cell Biol., 2010, 22, 659–668 CrossRef CAS PubMed.
- H. Ding, B. Su and D. Jiang, ChemistryOpen, 2022, e202200113 Search PubMed.
- M. Bocková, J. Slabý, T. Špringer and J. Homola, Annu. Rev. Anal. Chem., 2019, 12, 151–176 CrossRef PubMed.
- M. M. Richter, Chem. Rev., 2004, 104, 3003–3036 CrossRef CAS PubMed.
- M. Hesari and Z. Ding, J. Electrochem. Soc., 2015, 163, H3116–H3131 CrossRef.
- W. Miao, Chem. Rev., 2008, 108, 2506–2553 CrossRef CAS PubMed.
- J. Zhang, S. Arbault, N. Sojic and D. Jiang, Annu. Rev. Anal. Chem., 2019, 12, 275–295 CrossRef CAS PubMed.
- H. Ding, W. Guo and B. Su, ChemPlusChem, 2020, 85, 725–733 CrossRef CAS PubMed.
- C. Ma, Y. Cao, X. Gou and J.-J. Zhu, Anal. Chem., 2020, 92, 431–454 CrossRef CAS PubMed.
- R. He, H. Tang, D. Jiang and H.-Y. Chen, Anal. Chem., 2016, 88, 2006–2009 CrossRef CAS PubMed.
- J. Xu, P. Huang, Y. Qin, D. Jiang and H.-Y. Chen, Anal. Chem., 2016, 88, 4609–4612 CrossRef CAS PubMed.
- G. Liu, C. Ma, B.-K. Jin, Z. Chen and J.-J. Zhu, Anal. Chem., 2018, 90, 4801–4806 CrossRef CAS PubMed.
- S. Voci, B. Goudeau, G. Valenti, A. Lesch, M. Jović, S. Rapino, F. Paolucci, S. Arbault and N. Sojic, J. Am. Chem. Soc., 2018, 140, 14753–14760 CrossRef CAS PubMed.
- C. Ma, M.-X. Wang, H.-F. Wei, S. Wu, J.-R. Zhang, J.-J. Zhu and Z. Chen, Chem. Commun., 2021, 57, 2168–2171 RSC.
- G. Valenti, S. Scarabino, B. Goudeau, A. Lesch, M. Jović, E. Villani, M. Sentic, S. Rapino, S. Arbault, F. Paolucci and N. Sojic, J. Am. Chem. Soc., 2017, 139, 16830–16837 CrossRef CAS PubMed.
- J. Zhang, R. Jin, D. Jiang and H.-Y. Chen, J. Am. Chem. Soc., 2019, 141, 10294–10299 CrossRef CAS PubMed.
- Y. Liu, H. Zhang, B. Li, J. Liu, D. Jiang, B. Liu and N. Sojic, J. Am. Chem. Soc., 2021, 143, 17910–17914 CrossRef CAS PubMed.
- Y. Ma, C. Colin, J. Descamps, S. Arbault and N. Sojic, Angew. Chem., Int. Ed., 2021, 60, 18742–18749 CrossRef CAS PubMed.
- C. Ma, S. Wu, Y. Zhou, H.-F. Wei, J. Zhang, Z. Chen, J.-J. Zhu, Y. Lin and W. Zhu, Angew. Chem., Int. Ed., 2021, 60, 4907–4914 CrossRef CAS PubMed.
- L. Xu, Y. Li, S. Wu, X. Liu and B. Su, Angew. Chem., Int. Ed., 2012, 51, 8068–8072 CrossRef CAS PubMed.
- W. Guo, P. Zhou, L. Sun, H. Ding and B. Su, Angew. Chem., Int. Ed., 2021, 60, 2089–2093 CrossRef CAS PubMed.
- H. Ding, W. Guo and B. Su, Angew. Chem., Int. Ed., 2020, 59, 449–456 CrossRef CAS PubMed.
- H. Ding, P. Zhou, W. Fu, L. Ding, W. Guo and B. Su, Angew. Chem., Int. Ed., 2021, 60, 11769–11773 CrossRef CAS PubMed.
- K. Ino, K. Komatsu, K. Hiramoto, Y. Utagawa, Y. Nashimoto and H. Shiku, Electrochim. Acta, 2022, 415, 140240 CrossRef CAS.
- X. Qin, H.-J. Jin, X. Li, J. Li, J.-B. Pan, K. Wang, S. Liu, J.-J. Xu and X.-H. Xia, Chem. – Eur. J., 2022, 28, e202103964 CAS.
- L. Ding, H. Ding, P. Zhou, L. Xi and B. Su, Anal. Chem., 2022, 94, 10885–10892 CrossRef CAS PubMed.
- B. Rothenhäusler and W. Knoll, Nature, 1988, 332, 615–617 CrossRef.
- Y. Fang, H. Wang, H. Yu, X. Liu, W. Wang, H.-Y. Chen and N. J. Tao, Acc. Chem. Res., 2016, 49, 2614–2624 CrossRef CAS PubMed.
- W. Wang, Y. Yang, S. Wang, V. J. Nagaraj, Q. Liu, J. Wu and N. Tao, Nat. Chem., 2012, 4, 846–853 CrossRef CAS PubMed.
- A. W. Peterson, M. Halter, A. Tona and A. L. Plant, BMC Cell Biol., 2014, 15, 35 CrossRef PubMed.
- Y. Yang, H. Yu, X. Shan, W. Wang, X. Liu, S. Wang and N. Tao, Small, 2015, 11, 2878–2884 CrossRef CAS PubMed.
- R. Robelek and J. Wegener, Biosens. Bioelectron., 2010, 25, 1221–1224 CrossRef CAS PubMed.
- W. Wang, K. Foley, X. Shan, S. Wang, S. Eaton, V. J. Nagaraj, P. Wiktor, U. Patel and N. Tao, Nat. Chem., 2011, 3, 249–255 CrossRef CAS PubMed.
- X.-W. Liu, Y. Yang, W. Wang, S. Wang, M. Gao, J. Wu and N. Tao, Angew. Chem., Int. Ed., 2017, 56, 8855–8859 CrossRef CAS PubMed.
- L. Berguiga, L. Streppa, E. Boyer-Provera, C. Martinez-Torres, L. Schaeffer, J. Elezgaray, A. Arneodo and F. Argoul, Appl. Opt., 2016, 55, 1216–1227 CrossRef PubMed.
- J. Lu, Y. Yang, W. Wang, J. Li, N. Tao and S. Wang, Anal. Chem., 2016, 88, 11498–11503 CrossRef CAS PubMed.
- K. Toma, H. Kano and A. Offenhäusser, ACS Nano, 2014, 8, 12612–12619 CrossRef CAS PubMed.
- K. F. Giebel, C. Bechinger, S. Herminghaus, M. Riedel, P. Leiderer, U. Weiland and M. Bastmeyer, Biophys. J., 1999, 76, 509–516 CrossRef CAS PubMed.
- K. Watanabe, K. Matsuura, F. Kawata, K. Nagata, J. Ning and H. Kano, Biomed. Opt. Express, 2012, 3, 354–359 CrossRef PubMed.
- E. Kreysing, H. Hassani, N. Hampe and A. Offenhäusser, ACS Nano, 2018, 12, 8934–8942 CrossRef CAS PubMed.
- S. Dai, T. Yu, J. Zhang, H. Lu, J. Dou, M. Zhang, C. Dong, J. Di and J. Zhao, Biosens. Bioelectron., 2021, 174, 112826 CrossRef CAS PubMed.
- W. Wang, S. Wang, Q. Liu, J. Wu and N. Tao, Langmuir, 2012, 28, 13373–13379 CrossRef CAS PubMed.
- E. Kreysing, S. Seyock, H. Hassani, E. Brauweiler-Reuters, E. Neumann and A. Offenhäusser, Adv. Mater. Interfaces, 2020, 7, 1901991 CrossRef CAS.
- S.-H. Kim, W. Chegal, J. Doh, H. M. Cho and D. W. Moon, Biophys. J., 2011, 100, 1819–1828 CrossRef CAS PubMed.
- A. W. Peterson, M. Halter, A. Tona, A. L. Plant and J. T. Elliott, presented in part at the Conference on Plasmonics in Biology and Medicine XV, San Francisco, CA, Jan 29, 2017.
- Y. Zeng, J. Zhou, X. Wang, Z. Cai and Y. Shao, Biosens. Bioelectron., 2019, 145, 111717 CrossRef CAS PubMed.
- P. Zhang, X. Zhou, R. Wang, J. Jiang, Z. Wan and S. Wang, ACS Sens., 2021, 6, 4244–4254 CrossRef CAS PubMed.
- P. Zhang, X. Zhou, J. Jiang, J. Kolay, R. Wang, G. Ma, Z. Wan and S. Wang, Angew. Chem., Int. Ed., 2022, 61, e202209469 CAS.
- Y. Cho, E. Y. Park, E. Ko, J.-S. Park and J. H. Shin, Int. J. Precis. Eng. Man., 2016, 17, 1401–1412 CrossRef.
- S. S. Hur, J. H. Jeong, M. J. Ban, J. H. Park, J. K. Yoon and Y. Hwang, BMB Rep., 2020, 53, 74–81 CrossRef CAS PubMed.
- A. K. Nguyen and K. A. Kilian, ACS Chem. Biol., 2020, 15, 1731–1746 CrossRef CAS PubMed.
- A. Simon and M.-C. Durrieu, Micron, 2006, 37, 1–13 CrossRef CAS PubMed.
- J. Helenius, C.-P. Heisenberg, H. E. Gaub and D. J. Muller, J. Cell Sci., 2008, 121, 1785–1791 CrossRef CAS PubMed.
- K. Haase and A. E. Pelling, J. R. Soc., Interface, 2015, 12, 20140970 CrossRef PubMed.
- C. Arbore, L. Perego, M. Sergides and M. Capitanio, Biophys. Rev., 2019, 11, 765–782 CrossRef CAS PubMed.
- C. Guilluy, L. D. Osborne, L. Van Landeghem, L. Sharek, R. Superfine, R. Garcia-Mata and K. Burridge, Nat. Cell Biol., 2014, 16, 376–381 CrossRef CAS PubMed.
- Y. Tu and X. Wang, Sensors, 2020, 20, 7128 CrossRef PubMed.
- Q. Tian, P. Keshri and M. You, Chem. Commun., 2022, 58, 4700–4710 RSC.
- T. G. Kapp, F. Rechenmacher, S. Neubauer, O. V. Maltsev, E. A. Cavalcanti-Adam, R. Zarka, U. Reuning, J. Notni, H.-J. Wester, C. Mas-Moruno, J. Spatz, B. Geiger and H. Kessler, Sci. Rep., 2017, 7, 39805 CrossRef CAS PubMed.
- L. S. Fischer, S. Rangarajan, T. Sadhanasatish and C. Grashoff, Annu. Rev. Biophys., 2021, 50, 595–616 CrossRef CAS PubMed.
- Y. Liu, K. Galior, V. P.-Y. Ma and K. Salaita, Acc. Chem. Res., 2017, 50, 2915–2924 CrossRef CAS PubMed.
- D. R. Stabley, C. Jurchenko, S. S. Marshall and K. S. Salaita, Nat. Methods, 2012, 9, 64–67 CrossRef CAS PubMed.
- A. C. Chang, A. H. Mekhdjian, M. Morimatsu, A. K. Denisin, B. L. Pruitt and A. R. Dunn, ACS Nano, 2016, 10, 10745–10752 CrossRef CAS PubMed.
- K. Galior, Y. Liu, K. Yehl, S. Vivek and K. Salaita, Nano Lett., 2016, 16, 341–348 CrossRef CAS PubMed.
- C. Jurchenko, Y. Chang, Y. Narui, Y. Zhang and K. S. Salaita, Biophys. J., 2014, 106, 1436–1446 CrossRef CAS PubMed.
- Y. Liu, K. Yehl, Y. Narui and K. Salaita, J. Am. Chem. Soc., 2013, 135, 5320–5323 CrossRef CAS PubMed.
- Y. Liu, R. Medda, Z. Liu, K. Galior, K. Yehl, J. P. Spatz, E. A. Cavalcanti-Adam and K. Salaita, Nano Lett., 2014, 14, 5539–5546 CrossRef CAS PubMed.
- C. Grashoff, B. D. Hoffman, M. D. Brenner, R. Zhou, M. Parsons, M. T. Yang, M. A. McLean, S. G. Sligar, C. S. Chen, T. Ha and M. A. Schwartz, Nature, 2010, 466, 263–266 CrossRef CAS PubMed.
- M. Morimatsu, A. H. Mekhdjian, A. S. Adhikari and A. R. Dunn, Nano Lett., 2013, 13, 3985–3989 CrossRef CAS PubMed.
- S. J. Tan, A. C. Chang, S. M. Anderson, C. M. Miller, L. S. Prahl, D. J. Odde and A. R. Dunn, Sci. Adv., 2020, 6, eaax0317 CrossRef CAS PubMed.
- M. Rief, M. Gautel, F. Oesterhelt, J. M. Fernandez and H. E. Gaub, Science, 1997, 276, 1109–1112 CrossRef CAS PubMed.
- M. M. F. A. Baig, W.-F. Lai, M. F. Akhtar, A. Saleem, S. A. Ahmed and X.-H. Xia, Nano-Struct. Nano-Objects, 2020, 23, 100523 CrossRef CAS.
- C. Albrecht, K. Blank, M. Lalic-Mülthaler, S. Hirler, T. Mai, I. Gilbert, S. Schiffmann, T. Bayer, H. Clausen-Schaumann and H. E. Gaub, Science, 2003, 301, 367–370 CrossRef CAS PubMed.
- K. Hatch, C. Danilowicz, V. Coljee and M. Prentiss, Phys. Rev. E, 2008, 78, 011920 CrossRef CAS PubMed.
- M. Roein-Peikar, Q. Xu, X. Wang and T. Ha, Phys. Rev. X, 2016, 6, 011001 Search PubMed.
- X. Wang and T. Ha, Science, 2013, 340, 991–994 CrossRef CAS PubMed.
- X. Wang, Z. Rahil, I. T. S. Li, F. Chowdhury, D. E. Leckband, Y. R. Chemla and T. Ha, Sci. Rep., 2016, 6, 21584 CrossRef CAS PubMed.
- Y. Wang and X. Wang, Sci. Rep., 2016, 6, 36959 CrossRef CAS PubMed.
- K. A. Kim, S. Vellampatti, S. H. Hwang and B. C. Kim, Adv. Funct. Mater., 2022, 32, 2110637 CrossRef CAS.
- Y. Wang, D. N. LeVine, M. Gannon, Y. Zhao, A. Sarkar, B. Hoch and X. Wang, Biosens. Bioelectron., 2018, 100, 192–200 CrossRef CAS PubMed.
- Y. Zhao, Y. Wang, A. Sarkar and X. Wang, iScience, 2018, 9, 502–512 CrossRef CAS PubMed.
- M. H. Jo, B. C. Kim, K. Sung, R. A. Panettieri, S. S. An, J. Liu and T. Ha, ACS Nano, 2021, 15, 11585–11596 CrossRef CAS PubMed.
- S. A. Rashid, A. T. Blanchard, J. D. Combs, N. Fernandez, Y. Dong, H. C. Cho and K. Salaita, ACS Nano, 2022, 16, 5335–5348 CrossRef CAS PubMed.
- Y. Zhao, K. Pal, Y. Tu and X. Wang, J. Am. Chem. Soc., 2020, 142, 6930–6934 CrossRef CAS PubMed.
- K. Pal, Y. Tu and X. Wang, ACS Nano, 2022, 16, 2481–2493 CrossRef CAS PubMed.
- M. H. Jo, W. T. Cottle and T. Ha, ACS Biomater. Sci. Eng., 2019, 5, 3856–3863 CrossRef CAS PubMed.
- Y. Zhang, C. Ge, C. Zhu and K. Salaita, Nat. Commun., 2014, 5, 5167 CrossRef CAS PubMed.
- B. L. Blakely, C. E. Dumelin, B. Trappmann, L. M. McGregor, C. K. Choi, P. C. Anthony, V. K. Duesterberg, B. M. Baker, S. M. Block, D. R. Liu and C. S. Chen, Nat. Methods, 2014, 11, 1229–1232 CrossRef CAS PubMed.
- Y. Zhang, Y. Qiu, A. T. Blanchard, Y. Chang, J. M. Brockman, V. P.-Y. Ma, W. A. Lam and K. Salaita, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 325–330 CrossRef CAS PubMed.
- Y. Liu, L. Blanchfield, V. P.-Y. Ma, R. Andargachew, K. Galior, Z. Liu, B. Evavold and K. Salaita, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5610–5615 CrossRef CAS PubMed.
- R. Ma, A. V. Kellner, V. P.-Y. Ma, H. Su, B. R. Deal, J. M. Brockman and K. Salaita, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 16949–16954 CrossRef CAS PubMed.
- J. M. Brockman, A. T. Blanchard, V. Pui-Yan, W. D. Derricotte, Y. Zhang, M. E. Fay, W. A. Lam, F. A. Evangelista, A. L. Mattheyses and K. Salaita, Nat. Methods, 2018, 15, 115–118 CrossRef CAS PubMed.
- J. M. Brockman, H. Su, A. T. Blanchard, Y. Duan, T. Meyer, M. E. Quach, R. Glazier, A. Bazrafshan, R. L. Bender, A. V. Kellner, H. Ogasawara, R. Ma, F. Schueder, B. G. Petrich, R. Jungmann, R. Li, A. L. Mattheyses, Y. Ke and K. Salaita, Nat. Methods, 2020, 17, 1018–1024 CrossRef CAS PubMed.
- T. Schlichthaerle, C. Lindner and R. Jungmann, Nat. Commun., 2021, 12, 2510 CrossRef CAS PubMed.
- H. Li, C. Zhang, Y. Hu, P. Liu, F. Sun, W. Chen, X. Zhang, J. Ma, W. Wang, L. Wang, P. Wu and Z. Liu, Nat. Cell Biol., 2021, 23, 642–651 CrossRef CAS PubMed.
- S. J. Sahl, S. W. Hell and S. Jakobs, Nat. Rev. Mol. Cell Biol., 2017, 18, 685–701 CrossRef CAS PubMed.
- D. F. Kelly, D. W. Taylor, C. Bakolitsa, A. A. Bobkov, L. Bankston, R. C. Liddington and K. A. Taylor, J. Mol. Biol., 2006, 357, 562–573 CrossRef CAS PubMed.
- X. Barros-Álvarez, R. M. Nwokonko, A. Vizurraga, D. Matzov, F. He, M. M. Papasergi-Scott, M. J. Robertson, O. Panova, E. H. Yardeni, A. B. Seven, F. E. Kwarcinski, H. Su, M. C. Peroto, J. G. Meyerowitz, M. Shalev-Benami, G. G. Tall and G. Skiniotis, Nature, 2022, 604, 757–762 CrossRef PubMed.
- X. P. Xu, S. Pokutta, M. Torres, M. F. Swift, D. Hanein, N. Volkmann and W. I. Weis, eLife, 2020, 9, e60878 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |