
Stimuli-responsive polymers for interface engineering toward enhanced electrochemical analysis of neurochemicals
Shushu
Ding
 b,
Guoyue
Shi
a and
Anwei
Zhu
*a
b,
Guoyue
Shi
a and
Anwei
Zhu
*a
aSchool of Chemistry and Molecular Engineering, Shanghai Key Laboratory for Urban Ecological Processes and Eco-Restoration, East China Normal University, 500 Dongchuan Road, Shanghai, 200241, People's Republic of China. E-mail: awzhu@chem.ecnu.edu.cn; Fax: +86-21-54340042; Tel: +86-21-54340042
bSchool of Pharmacy, Nantong University, 19 Qixiu Road, Nantong, 226001, People's Republic of China
First published on 14th October 2022
Abstract
Neurochemical monitoring can provide important insights into the chemical communications in the brain and neurological diseases. Although electrochemical sensors have promoted the development of neurochemical analysis, the limited analytical performance of the existing sensors restrict our understanding of the roles that chemical signals play in the brain. The central nervous system is composed of a large number of neurochemical species. Meanwhile, it is difficult to monitor neurochemicals with high sensitivity because of the kinetic barrier of mass transport and electron/ion transfer. More importantly, to fabricate a “smart” electrochemical sensor for neurochemicals, the engineering of an electrode surface with switchable properties and a response is urgently needed. This review focuses on the construction and application of electrochemical sensors based on stimuli-responsive polymers. The response of polymers to external stimuli can not only enhance the target recognition, but also modulate the electrochemical signals, thus providing smart electrochemical sensing platform with improved analytical performance, including high selectivity, sensitivity, and controllability. In this review, we first introduce the design strategy of bio-responsive stimuli-responsive polymers and highlight the relationship between their structure and molecular recognition efficiency. Then, we summarize the electrochemical techniques with different sensing principles and emphasize the contribution that stimuli-responsive polymers made to the conversion of chemical/electrochemical reactions into electric signals. Finally, the opportunities and limitations of stimuli-responsive polymer-modified electrochemical sensors for neurochemical analysis will be discussed. Taking advantage of the development of novel materials, electrochemical techniques and microelectronic engineering, the advanced devices (e.g., antifouling, flexible, miniaturized, and multi-functional) with remarkable analytical performance will benefit the evaluation of neurochemicals, which can promote a deep understanding of brain events and the diagnosis and treatment of neurological diseases.
 Shushu Ding | Shushu Ding received her PhD degree in analytical chemistry from East China Normal University in 2018. She joined Nantong University in 2018 and was promoted to Associate Professor in 2021. Her research interests focus on designing stimuli-responsive polymers and functional materials and developing new electrochemical sensors for probing neurochemistry. |
 Guoyue Shi | Guoyue Shi received his PhD degree in analytical chemistry from East China Normal University in 1999. He worked in the Laboratory of Research and Development, BAS Inc., in Tokyo as a visiting scholar (2002–2003). Then he completed his postdoctoral studies at the University of Pittsburgh in 2005 and was promoted to Full Professor at East China Normal University in 2006. His research interests are focused on developing new analytical methods and instruments for monitoring biological species in the live brain. He also focuses on designing functional nanomaterials for biosensing. |
 Anwei Zhu | Anwei Zhu received his PhD degree in analytical chemistry from Tongji University in 2013 with Prof. Yang Tian. He is currently an associate professor of chemistry at East China Normal University. His research expertise is in vivo electrochemical measurements of biological systems, including single cell measurements and living organisms. He also focuses on designing polymers for biointerfacing and biorecognition applications. |
1. Introduction
Each neuron in the brain is endowed chemically with the presence of various molecules [e.g., neurotransmitters (dopamine, serotonin, epinephrine, etc.), neuromodulators (e.g., ascorbate), ions (H+, K+, Ca2+, etc.), reactive oxygen species (e.g., H2O2), energy suppliers (glucose, ATP, lactate, etc.), gases (NO, H2S, etc.), peptides, proteins, and nucleic acids].1–6 The complex and dynamic network of these neurochemicals defines the unique neuron/brain functions. To take an in-depth look at the brain, considerable efforts must be paid to construct analytical platforms, and then to establish the connection between neurochemistry and brain activities.Electrochemical sensors have contributed fundamental insights into neurochemical analysis due to their simple manipulation, ease of miniaturization and excellent spatiotemporal resolution.7–14 However, three obstacles limit the real-world application of electrochemical sensors to efficient analysis of neurochemicals in the brain. One is the improvement of the selectivity of sensors because biomolecules coexisting in the brain often have similar electrochemical properties and structures.1,15 Moreover, most neurochemicals are electro-inactive and thus cannot be detected by direct electrolysis. Although multiple enzyme-based electrochemical biosensors have been constructed with distinctive selectivity,16–19 the number of natural biocatalysts is limited. Another challenge is the improvement of sensitivity. The nonspecific adsorption of proteins and molecules at the electrode surface can hinder the mass transfer processes, leading to decreased sensitivity. Antifouling pretreatment was widely adopted to weaken electrode fouling, but the sensitivity of the sensors was greatly limited.20 More importantly, constructing sensors (especially implantable bioelectronic systems) with excellent switching performance is necessary for controllable, long-term stable, and on-demand monitoring of neurochemicals, which can improve the reliability of clinical diagnosis and treatment for neurological disorders. According to the above discussion, the synthesis of new molecules, the construction of functionalized surface that promote the enrichment of targets and facilitate electron transfer, and the development of new electrochemical techniques, will enable electrochemical sensors with high selectivity, sensitivity and controllability.
Stimuli-responsive polymers represent a class of “smart” materials that may undergo dynamic changes in their structures and properties (e.g., conformation, wettability, and surface charge) in response to external stimuli.21,22 Due to their unique structure and interfacial behavior, stimuli-responsive polymers have recently injected new vitality into the construction of efficient sensors.23–28 The advantages that stimuli-responsive polymers bring to electrochemical sensors include the following: (1) In comparison with small molecules, stimuli-responsive polymers are composed of repeating monomers,21 which can offer multiple interactions to enhance the efficiency of recognition. Moreover, the programmable stimuli-responsive polymers enable the incorporation of different functional monomers,29 which can provide expandable platforms for specific applications. (2) Stimuli-responsive polymers have the ability to transform microscopic molecular recognition into macroscopic properties of polymers.30,31 The change in interfacial properties can regulate reactions and then produce measurable electrical signals. (3) A reversible binding can be achieved between the stimuli-responsive polymers and targets through the synergy of noncovalent interactions,32,33 promoting the construction of a switchable system with excellent controllability. In this sense, the stimuli-responsive polymer can be regarded as a smart “sense-to-action” device (Fig. 1). Due to the above remarkable features, the electrochemical sensors based on stimuli-responsive polymers will inject fresh energy to speed up the development of neurochemical analysis.
 | ||
| Fig. 1 Schematic illustration of the signal transduction for the electrochemical sensors based on stimuli-responsive polymers. | ||
In this review, we first introduce some common stimuli-responsive polymers, followed by the discussion of a molecular design strategy, aiming to improve the performance of stimuli-responsive polymers. Then, we summarize electrochemical sensors with different analytical principles and highlight state-of-the-art examples of stimuli-responsive polymer-based electrochemical approaches for neurochemical analysis. In this section, following the elaboration of issues involved in different electrochemical techniques, possible solutions supported by stimuli-responsive polymers will be emphasized. Finally, the perspectives and challenges in this field are given for future investigation, aiming to promote the further development of advanced material and intelligent electrochemical sensors for neurochemical analysis.
2. Types of stimuli-responsive polymers
Mother nature greatly inspires us to design stimuli-responsive polymers. For example, in life systems, numerous noncovalent interactions (e.g., hydrogen bonds, hydrophobic and electrostatic interactions) mediate the biological functions with a well-tuned balance. Although noncovalent interactions are usually weaker than covalent and ionic counterparts, a stronger but tunable and reversible binding capability can be achieved through cooperativity in noncovalent interactions. Considering the important contribution that noncovalent interactions make in living systems, polyamides with hydrogen bonds have been widely used in polymer systems. For example, poly(N-isopropylacrylamide) (PNIPAAm) has been extensively explored due to its conformational transition with high reversibility derived from the competition between inter- and intra-molecular hydrogen-bonding interactions and has been utilized for building temperature-responsive sensors.34 Below the lowest critical solution temperature (LCST), PNIPAAm are hydrophilic and can form intermolecular hydrogen bonds with water molecules. When the temperature increases above the LCST, intramolecular hydrogen bonds will form between the carboxyl group and the amino group within the PNIPAAm chains, resulting in a compact and hydrophobically collapsed PNIPAAm chains (Fig. 2A). In addition to PNIPAAm, other polymers displaying conformational changes in response to temperatures have also been reported (Fig. 2B).35 Another type of stimuli-responsive polymer with outstanding flexibility is a polyelectrolyte (Fig. 2B).36,37 These polymers containing weak acids and/or bases can be ionized into different degrees towards the changes of solution pH. The electrostatic interactions between the ionizable chemical groups are able to disrupt hydrogen bonds and then drive the conformational change of polymer chains, thus altering the resulting surface wettability.38 Taking the cationic polyelectrolyte of poly(dimethylamino ethyl methacrylate) (PDMAEMA) as an example, when the pH is greater than 7, the polymer chains adopted a collapsed morphology. At pH less than 7, the polymer chains were protonated, resulting in a stretched conformation with hydrophilicity (Fig. 2A). In addition, some polymers [e.g., poly(2-(methacryloyloxy)-ethyl-trimethylammonium chloride) (PMETA)] showed unique responses to specific ions.39 To expand the applications, these polymers discussed above can work as a scaffold to be incorporated with amounts of specific monomers (recognition moieties and redox mediators). Some typical stimuli-responsive polymers and functional monomers are shown in Fig. 2B.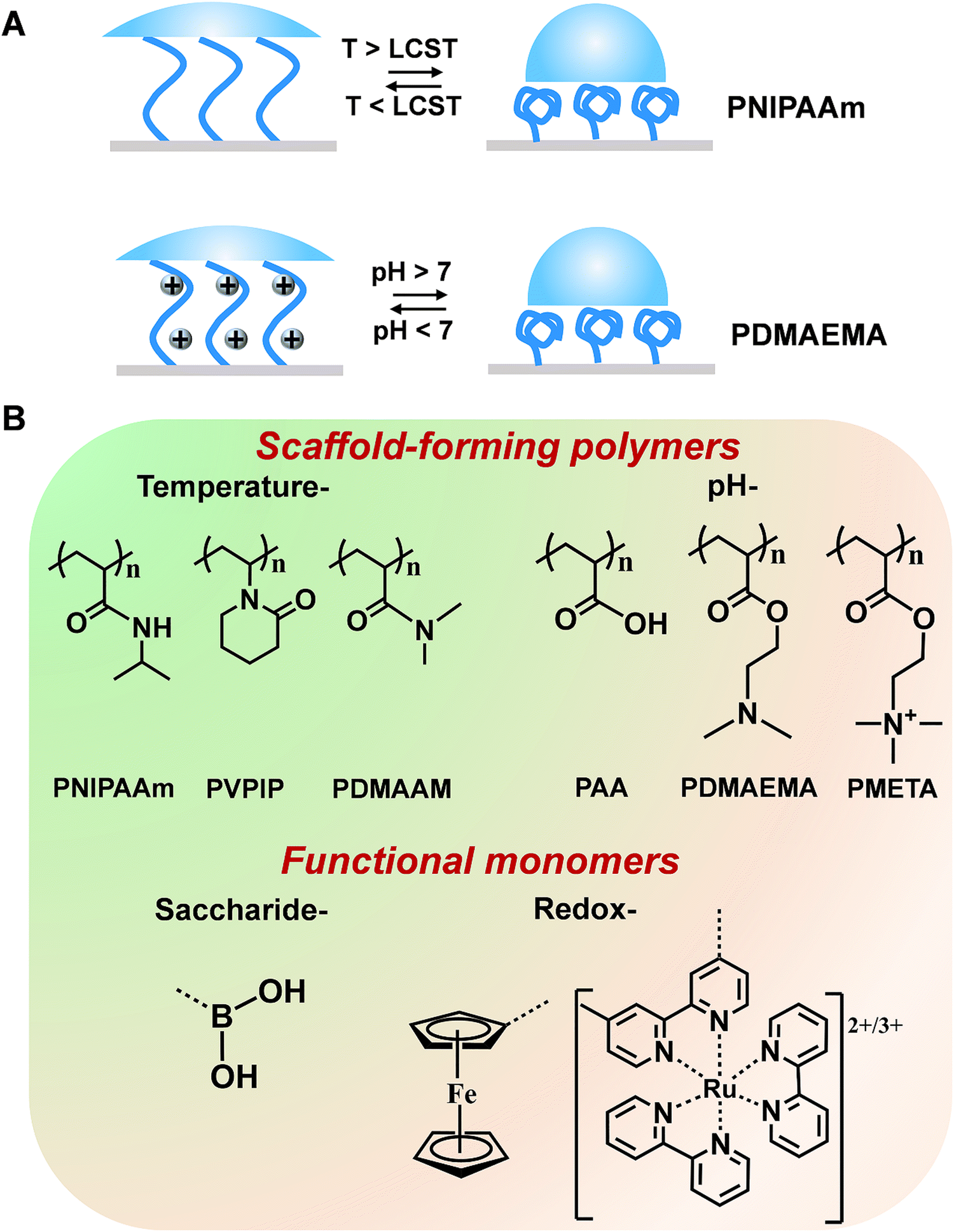 | ||
| Fig. 2 (A) Schematic illustration of reversible conformation/wettability changes of (a) PNIPAAm induced by temperature and (b) PDMAEMA induced by pH. (B) Chemical structures of some common stimuli-responsive polymers and functional monomers. | ||
3. Strategies for improving the performance of stimuli-responsive polymers
For an analytical system, stimuli-responsive polymers can work not only as the recognition elements but also as transducers. The thermodynamic and kinetic control of the behavior of stimuli-responsive polymers is dictated by not only the component but also the topology of the polymer system.21 Below, the three most studied polymer forms (1) single polymer chains, (2) two-dimensional (2D) polymer brush and (3) three-dimensional (3D) polymeric network will be introduced. In this section, we mainly introduce the molecular design strategies to improve the recognition and signal-transduction, generating a deep understanding of the requirement for interfacial chemistry in analytical platforms with high performance.3.1. Single polymer chain
In view of chemical science, the chain compositions of a polymer can be easily managed. Although the one-component polymers may modulate the receptor–ligand binding in biorecognition, more feasible strategies are required to adjust the material functions. A promising mainstay is to create the multicomponent stimuli-responsive polymers. The synergetic communication in the building components can promote the amplification of tiny biological signals to a significant extent. For example, Sun's group proposed a series of three-component polymers with an “RMF” design concept.32,33 These copolymers were comprised of recognition units, mediating units and functional switching units, which have interrelated influences on each other. The recognition unit can identify biomolecules either by hydrogen bonds or other combination ways. The functional switching unit can change the macroscopic properties of materials by conformational transitions activated by recognition. The mediating component acts as a bridge between the other two components, promoting the transformation of recognition signals into the changes in the macroscopic properties of materials (Fig. 3). | ||
| Fig. 3 The modularized design concept for “RMF” smart polymers. Reproduced with permission from ref. 33. Copyright 2017, John Wiley and Sons. | ||
Taking the smart copolymer (P(NIPAAm-co-TF-co-AAPBA)) as an example,33 the CA switched from 81° to 43° after glucose treatment (Fig. 4A(a)). However, taking a pure PNIPAAm film toward glucose recognition as the control experiments, no obvious CA changes (ΔCA ≤ 2°) can be observed (Fig. 4A(b)), indicating the weak interaction between glucose and the PNIPAAm chain. Moreover, the control copolymers of P(NIPAAm-co-AAPBA) and P(NIPAAm-co-TF) have also been explored. For P(NIPAAm-co-AAPBA), the CA only changed from 77° to 63° (Fig. 4A(c)). Although the CA of P(NIPAAm-co-TF) changed from 84° to 60°, the wettability can rapidly be restored to the original state by water triggering (Fig. 4A(d)). These results further confirmed the unique contribution of each building component in “RMF” smart polymers. The AAPBA worked as the recognition unit. Furthermore, the TF component facilitated the formation of hydrogen bonds with the other two units and promoted the surface wettability switching. Such “RMF” copolymer design breaks the limitations of conventional polymers. Furthermore, the three-component modular design endows “RMF” systems with full expandability by changing the species of three units.
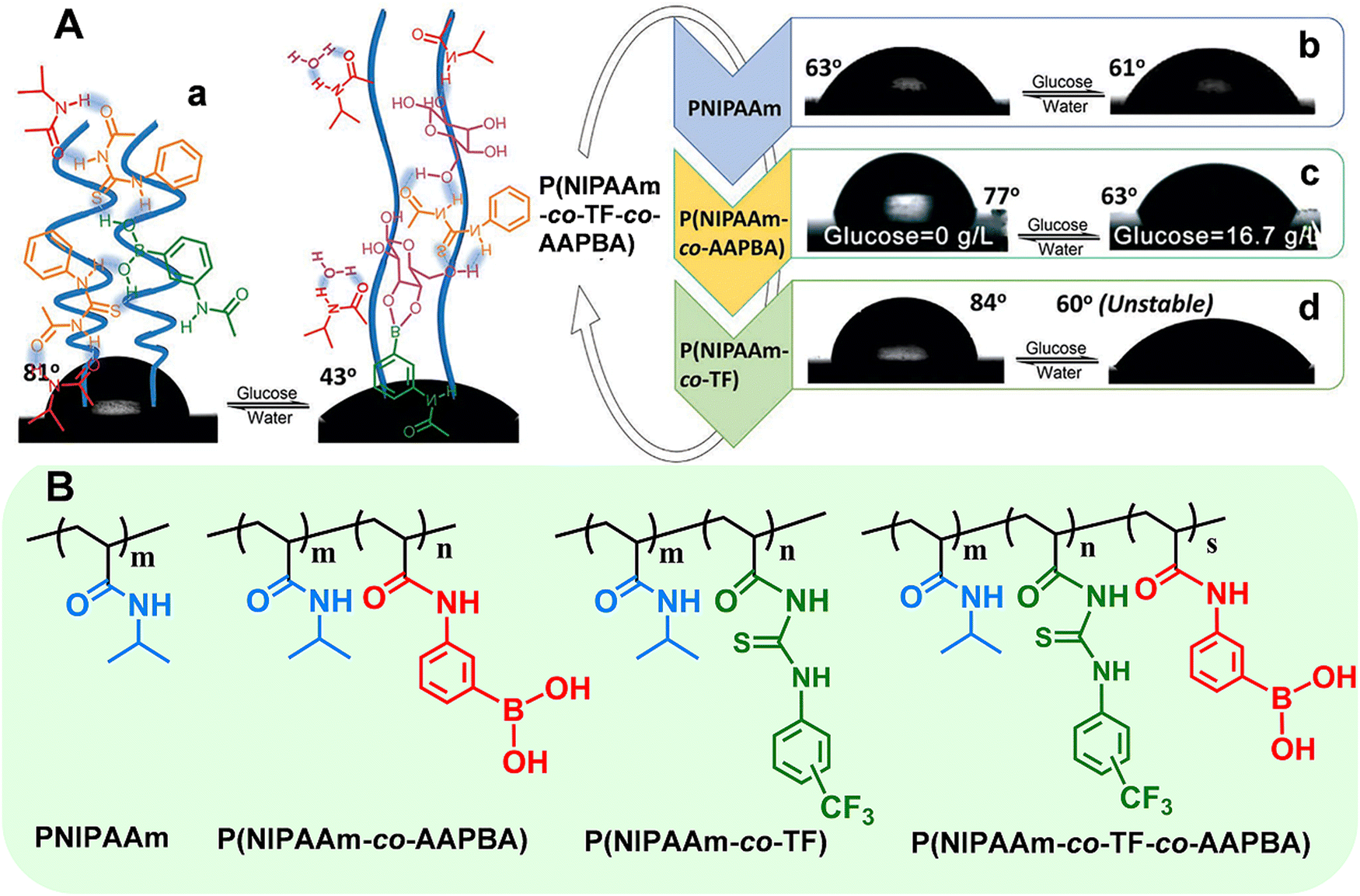 | ||
| Fig. 4 (A) Surface wettability of the polymer films deposited on flat silicon wafers: (a) P(NIPAAm-co-TF-co-AAPBA), (b) PNIPAAm, (c) P(NIPAAm-co-AAPBA), and (d) P(NIPAAm-co-TF). (B) Chemical structures of the above stimuli-responsive polymers. Reproduced with permission from ref. 33. Copyright 2017, John Wiley and Sons. | ||
Compared with small biomolecules, the diversity of proteins with complex structures restricted the specific detection. Sun's group designed a stimuli-responsive polymer which contained NIPAAm as the functional switching unit and 4-(3-acryloyl-thioureido)-benzoic acid (ATBA) as the recognition unit.40 Driven by multiple hydrogen-bonding interactions, the polymer exhibited differential complexation with non-modified peptides, singly phosphorylated and multiply phosphorylated peptides. Through the precise design of the components in polymers, the excellent expandability can help to obtain novel stimuli-responsive polymers for other proteins. In addition, several stimuli-responsive molecularly imprinted polymers (SRP-MIPs) for protein analysis have been reported.41,42 SRP-MIPs can be prepared by co-polymerization of functional monomers. After removal of the templates, recognition cavities complementary in shape, size and functionality remain. The properties of SRP-MIPs can be switched by external stimuli, which can be utilized for recognizing metabolic disorders of cells.
Although stimuli-responsive polymers have been applied for protein analysis, several challenges should be overcome. The first challenge is the design of receptor molecules with high specificity toward proteins. Multiple interactions (e.g., coordination bond and hydrogen bond) between the polymers and proteins should be explored to distinguish proteins with similar charge/structure. The second challenge is manipulating the polymer properties through external stimuli. The transition rate of stimuli-responsive polymers should be adequate for rapid analysis. Therefore, we should pay more attention to the transition mechanisms for polymer chains.
Driven by the complex functions of living systems, multi-stimuli-responsive polymers (e.g., light/pH/temperature, ionic strength/pH/temperature, and glucose/pH/temperature) have also been designed in recent years.43–46 These polymers can be synthesized with two/more types of components. However, it is difficult for these polymers to respond to multiple chemical signals simultaneously. Moreover, the multi-stimuli-responsive polymers that can be utilized for biomolecules detection were limited. These problems restrict the comprehensive elucidation of physiological and pathological phenomena in the central nervous system.47,48
3.2. Polymer brushes
Polymer brushes can be defined as polymer chains which have one end tethered to solid substrates.49–51 Due to the irreversible attachment to a surface, the chain mobility and consequently stimuli-responsiveness will be reduced.21,52 Therefore, polymer brushes exhibit very distinct behavior relative to polymers in solution. The chemical energy required for the transitions of the polymer properties will be significantly greater than that required for the polymers in solution.53 On the other hand, the behavior of polymer brushes is dictated by not only the polymer composition but also the polymer architecture and the grafting density on the substrate. The synthetic paths should therefore be carefully chosen in engineering stimuli-responsive interfaces.Currently, ‘‘living’’ free-radical polymerizations including atom transfer radical polymerization (ATRP) and reversible addition–fragmentation chain transfer (RAFT) polymerization have been widely employed.54–57 Different from the traditional radical polymerizations, these polymerization processes produce stimuli-responsive polymers with controlled molecular weight (chain length) and low polydispersity index (PDI), which allows one to probe phase transition behavior more precisely.
To create polymer brushes, two primary covalent attachment techniques (i.e., ‘‘grafting-to’’ and ‘‘grafting-from’’) were used.58 For the “grafting-to” method, end-functionalized polymer chains can covalently react with the functional groups on the surface (Fig. 5A).59 Typically, RAFT polymerization may serve as a suitable synthetic route because the obtained polymer chains possessing di-thioester or tri-thiocarbonate end groups can be utilized in further chemical reactions. Since polymer chains with well-defined structures can be pre-synthesized, the “grafting-to” method contributes to improved controllability and flexibility. For the “grafting-from” method, initiators are immobilized at the surface, followed by the polymerization of monomers from the active sites. In this approach, a higher grafting density can be achieved due to the easy penetration of monomers through grafted segments (Fig. 5B).60 Owing to the versatility of monomers and mild reaction conditions, ATRP is widely utilized in the “grafting-from” method. To better control the initiation process for ATRP and develop electrochemical sensors with excellent analytical performance, Zhang's group proposed enzyme catalyzed electron transfer atom transfer radical polymerization.61 This methodology is easy to operate, environmentally friendly, and can be prepared on a large-scale at room temperature. In addition, the in situ growth of polymers significantly contributed to the coupling efficiency of polymer chains on the electrode surface. The electrochemical sensor developed by this method has been used for a rapid analysis of DNA with the detection limit of 11.79 aM. In addition, Niu's group developed a series of electrochemical sensors based on electrochemically controlled RAFT polymerization.62–64 These methods showed great prospect as an amplification strategy for the sensitive detection of biological molecules.
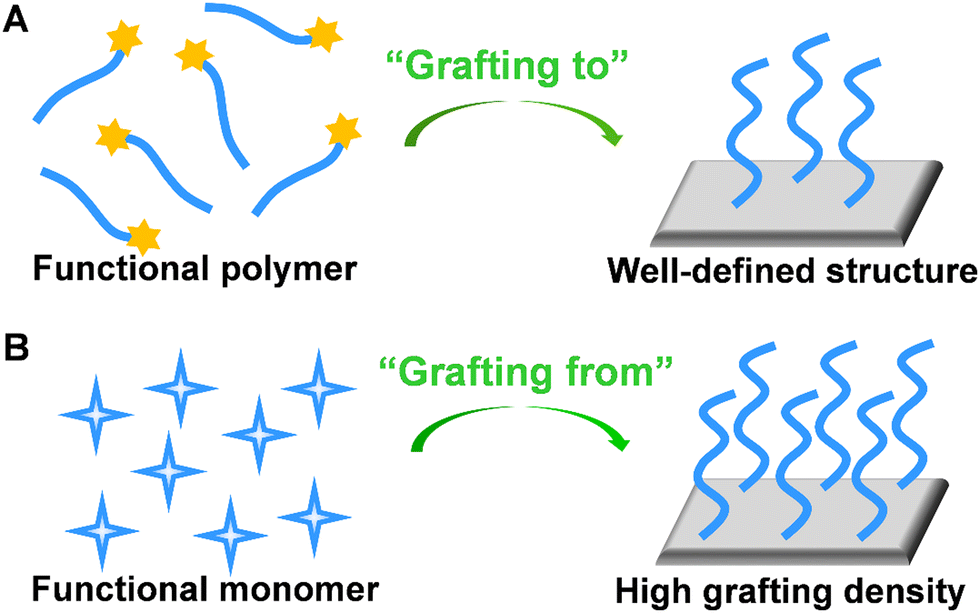 | ||
| Fig. 5 Schematic illustration of preparation of stimuli-responsive polymer brush by (A) “grafting-to” and (B) “grafting-from” approaches. | ||
3.3. Polymer networks
Three-dimensional (3D) responsive polymer-based networks are commonly generated using physical and/or chemical crosslinkers.65 Of particular interest are polymer hydrogels. The interstitial sites of hydrogel network are filled with a large amount of water, resembling the structures of biological systems. Due to the high content of water and hydrophilic nature, the polymeric hydrogels exhibit unique characteristics like biocompatibility, anti-fouling and ease of surface modification.66,67 Benefiting from the porous structure, bioreceptors (e.g. enzyme, nucleic acid, and protein) can be facially encapsulated or cross-linked into polymer matrix. Moreover, the high biocompatibility of hydrogels helps to preserve their bioactivity for practical applications.68–70Bioreceptors embedded hydrogel biosensors possess well-established molecular recognition characteristics. In the presence of external stimuli, hydrogels can undergo volume change (swelling/shrinking process) that is called volume phase transition (VPT), forming the basis of recognition in hydrogel-based sensors (Fig. 6A).71 However, for the traditional responsive hydrogels, the dense skin layer formed during the chain growth polymerization. Therefore, the diffusion of water molecules among the interior network structure of the hydrogels may be restricted, resulting in the slow swelling-shrinking rates (i.e., response rate, from several hours to days) to changes in the environmental conditions.
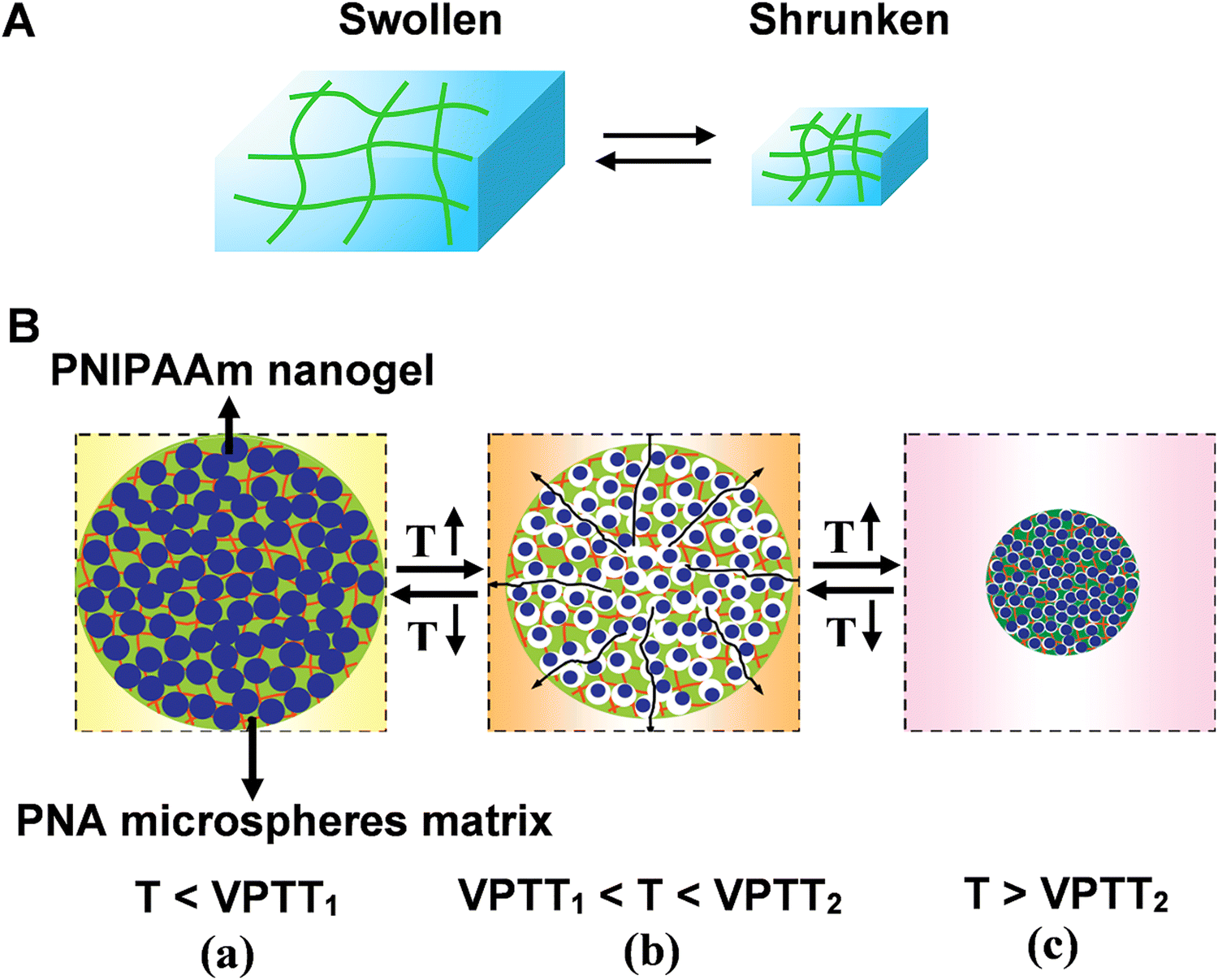 | ||
| Fig. 6 (A) Schematic illustration of reversible volume phase transition of hydrogel. (B) The rapid response process of nanocomposite smart hydrogel upon heating-up or cooling-down. Reproduced with permission from ref. 76. Copyright 2012, Elsevier. | ||
One possibility of countering the limitation is diminishing the dimensions of hydrogels. It has been verified that the response rate of hydrogels was inversely proportional to the square of a linear dimension. Microgels with the size ranging from 50 nm up to 5 μm are soft and deformable, while still retaining structural integrity. Their swelling lead to an open structure, allowing high mobility of solvent, solute molecules, chain segments as well as of the entire microgel. Therefore, there is no sharp boundary for the mass exchange between microgel and surroundings. In other words, microgels are sensitive to their environment.72–74 Because of the unique properties to undergo fast and reversible phase transitions, recently, the interest in the construction of microgel-based biosensors have significantly increased. For example, the horseradish peroxidase (HRP)-polyacrylamide (PAA)-based biosensor has been successfully used for amperometric determination of acetaminophen (APAP). PAA microgels exhibited a protective effect on the bio-catalyzer. The biosensor showed good catalytic activity with the detection limit of 3.1 × 10−6 M and the response time was 135 s.75
Moreover, the structural diversity of microgel (e.g., hollow microgels core–shell microgels) contributed to the improvement of the response rate.76 For example, a nanocomposite smart hydrogel was developed by chemically crosslinking nanogels [poly(N-isopropylacrylamide) PNIPAAm] into the microsphere matrixes [poly(N-isopropylacrylamide-co-acrylic acid) (PNA)]. The volume phase-transition temperatures (VPTT) of PNIPAAm nanogels (VPTT1) is lower compared with the VPTT2 of PNA microsphere matrixes (Fig. 6B). The different VPTTs led to hierarchical phase-transition of the microspheres, and rapid response rate could be achieved by either the cooling-down or the heating-up process: when the temperature was in the range of VPTT1 and VPTT2, the PNIPAAm nanogels were in shrunken phase whereas the PNA microsphere matrixes were in swollen phase. Hence, lots of voids and channels formed inside the nanocomposite hydrogel (Fig. 6B(b)). At this stage, the porous structure of microsphere matrixes interconnects the water pathway, facilitating the response rate.
4. Electrochemical sensing platforms based on stimuli-responsive polymers
In this section, we summarize the development and applications of electrochemical sensors based on stimuli-responsive polymers, and discuss the strategy to facilitate signal-transduction and amplification, aiming at providing promising interface materials and electrode engineering strategies for neurochemical analysis. According to the principles of signal transduction, we divided the electrochemical sensors for neurochemical detection into three groups (Fig. 7): (1) electrochemical techniques based on electron transport, (2) field-effect transistor based on carrier mobility, (3) nanochannel based on ion transport.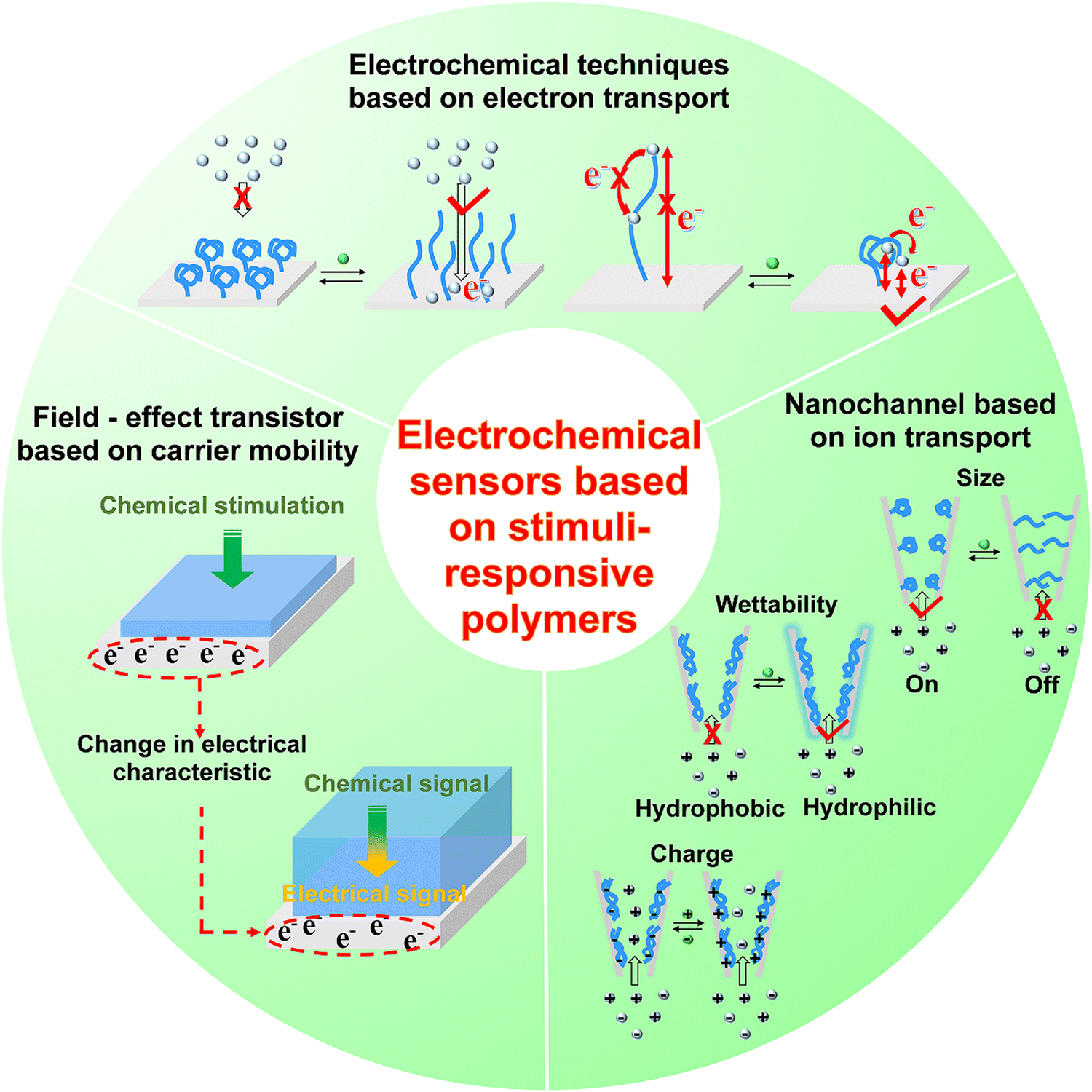 | ||
| Fig. 7 The sensing principles of stimuli-responsive polymer modified electrochemical sensors. | ||
4.1. Electrochemical techniques based on electron transport
In electrochemical sensors based on electron transport, the electrochemical reaction occurring at the inner Helmholtz layer is generally accompanied by mass transport (reactant and product) and interfacial electron conduction.77–79 For a reversible reaction, the enrichment of reactants can promote the forward reaction to accelerate the thermodynamics of the reaction process. Switching the wettability of electrodes by tailoring the surface structure can realize reactant enrichment, which will improve the electrochemical sensitivity.80 The unique properties of stimuli-responsive polymers motivated us to associate them with the electrode surface and constructed a series of electrochemical biosensors manipulating electrochemical processes based on the shrinking–swelling of polymer brushes. Firstly, we developed a stimuli-responsive copolymer/graphene hybrid-modified screen-printed carbon electrode for enantioselective determination of sugars with high sensitivity and selectivity (Fig. 8A).81 In this electrochemical sensor, the copolymer consisted of the chiral recognition center N-β-L-aspartyl-L-phenylalanine dimethyl ester, the mediating unit bis(trifluoromethyl)-modified phenylthiourea (TP) and the functional switching unit poly(N-isopropylacrylamide) (PNIPAAm). The three units were copolymerized on graphene sheets by the ATRP process. Initially, intramolecular hydrogen bond network was formed among these three components, resulting in the contracted copolymer chains and relative hydrophobicity. When exposed to monosaccharide solutions, the copolymer combined with monosaccharide through intermolecular hydrogen bonds, leading to the breakdown of intramolecular hydrogen bonds to different extents. The copolymer film exhibited swollen states with hydrophilicity. The conformation and wettability switch promoted the diffusion and enrichment of the electroactive probe [Fe(CN)6]3/4− to the electroactive surface. Therefore, the interface redox reaction was facilitated and the target could be sensitively recognized. The described method can quantify D-glucose even when the concentration was as low as 1 nM. In addition, this sensor could distinguish monosaccharides from potential interferences, and achieve chiral discrimination of monosaccharides with different spatial structures. The excellent sensing capacity enabled this platform to explore the transport mechanism of glucose enantiomers in live cells. | ||
| Fig. 8 Schematic illustration of the electrochemical biosensor based on stimuli-responsive polymers for measurement of (A) monosaccharide enantiomer and (B) sialic acid in live mouse brain. Reproduced with permission from ref. 81 and 82. Copyright 2016 and 2017, American Chemical Society. | ||
In addition, we further developed a novel three-component copolymer and constructed an electrochemical sensor for evaluating sialic acid (SA) in live mouse brain (Fig. 8B).82 The copolymer containing phenylboronic acid (PBA) as the specific recognition unit, TP as the mediating unit, and PNIPAAm as the functional switching unit was pre-synthesized with RAFT polymerization and then covalently attached onto the electrode surface via Au–S bonds. This method exhibited anomalous selectivity toward SA: (1) At the physiological pH of 7.4, PBA can form specific covalent bonding with SA with the binding constant of 37.6, which is 2–7 times higher than those for other sugars. (2) The TP unit in the copolymer can interact with the carboxylate ion (–COO−) as well as the hydroxyl groups (–OH) in SA through directional hydrogen bonds. The detection limit for SA could be achieved down to 0.4 pM. In combination with in vivo microdialysis, this electrochemical sensor with excellent analytical performance was utilized for monitoring SA levels in different brain regions of live mice with AD. This work validated the stimuli-responsive copolymer-modified electrochemical sensor as an effective platform for exploring the neurochemicals in neurodegenerative diseases. For these works, the stimuli-responsive copolymer was adopted not only as a recognition element but also as a signal amplification strategy: (1) the synergy of interactions (e.g., hydrogen bonds, covalent bonds and chirality) that the stimuli-responsive polymer presented can enhance the target recognition. Besides, the macroscopic surface changes (conformation and wettability switching) promoted the access and enrichment of targets and redox labels near the electrode surface. (2) The inherent wettability of copolymer was dramatically amplified by enhancing the surface roughness (graphene film and gold nanoflower) of the substrate, thus improving the sensitivity toward weak interactions.
For neurochemical analysis, the research must be conducted not only in vitro but also at the cellular or in vivo scale to dynamically monitor nervous process.1,6,12,15,83–93 However, the electrochemical signals for the above works are generated from the added redox labels, not suitable for the in situ and in vivo analysis. Taking advantages of the programmability of polymers, stimuli-responsive polymers with redox properties can be developed, expanding the applications of electrochemical sensors based on stimuli-responsive polymers. Recently, Sojic's group prepared one kind of stimuli-responsive hydrogel which contained PNIPAAm as the functional unit, PBA moieties as the recognition units, and [Ru(bpy)3]2+ as redox centers (Fig. 9A).94 In the presence of fructose, the hydrophilicity and charge density of the polymer chain increased due to the formation of boronate ester, which resulted in the hydrogel swelling. Consequently, the average distance of adjacent [Ru(bpy)3]2+ centers in the hydrogel matrix increased and the number of accessible electroactive sites decreased, leading to a decrease in the electrical signals. Marcisz et al. synthesized a thermoresponsive microgel based on N-isopropylacrylamide, sodium acrylate and N,N′-bisacryloylcystine. The electroactive amino-ferrocene and glucose oxidase enzyme were covalently bonded with the microgel through the classical 1-ethyl-3-(3-dimethyllaminopropyl) carbodiimide (EDC)/N-hydroxysulfosuccinimide (NHS) coupling reaction (Fig. 9B(a)).95 If the temperature is below 30 °C, the electrochemical biosensor displayed low current density. When the temperature increased to a value beyond the volume phase transition (34 °C), the microgel became shrunken state and decreased the separation distance between the enzyme and the ferrocenium group, which promoted the electron hopping in the polymer network and the efficiency of the electrocatalytic oxidation of glucose (Fig. 9B(d)). The electrochemical response was proportional to the glucose concentration in the physiological concentration range. Correspondingly, stimuli-responsive polymers functionalized with electrochemical mediators and enzymes have provided a favorable micro-environment to facilitate the catalytic reaction at the human body temperature. Zhou et al. constructed a temperature-controlled electrochemical sensor for detection of H2O2 based on a mixture of poly(N-isopropylacrylamide)-b-poly(2-acrylamidoethyl benzoate) (PNIPAAm-b-PAAE), graphene oxide (GO), and hemoglobin (Hb).96 Hb at PNIPAAm-b-PAAE/GO/Hb (PGH) film exhibited well-defined redox peaks and intrinsic electro-catalytic activity toward H2O2. Furthermore, the sensing film showed temperature-tunable catalytic activity toward H2O2. There was an obvious increase in electrocatalytic current as well as sensitivity above 32 °C in comparison with below 30 °C. These studies demonstrated the flexible regulation of electron transport among the polymer matrix and the prospect of electrochemical biosensors based on stimuli-responsive polymers for in vivo analysis. With the integration of light-sensitive monomers, the light-guided electrochemical sensors can be fabricated. Moreover, a variation of the thermo-responsive properties by illumination can even be achieved. The controlled recognition of neurochemicals can progress our understanding of the physiological and pathological events in the brain, especially for chronic brain disease.
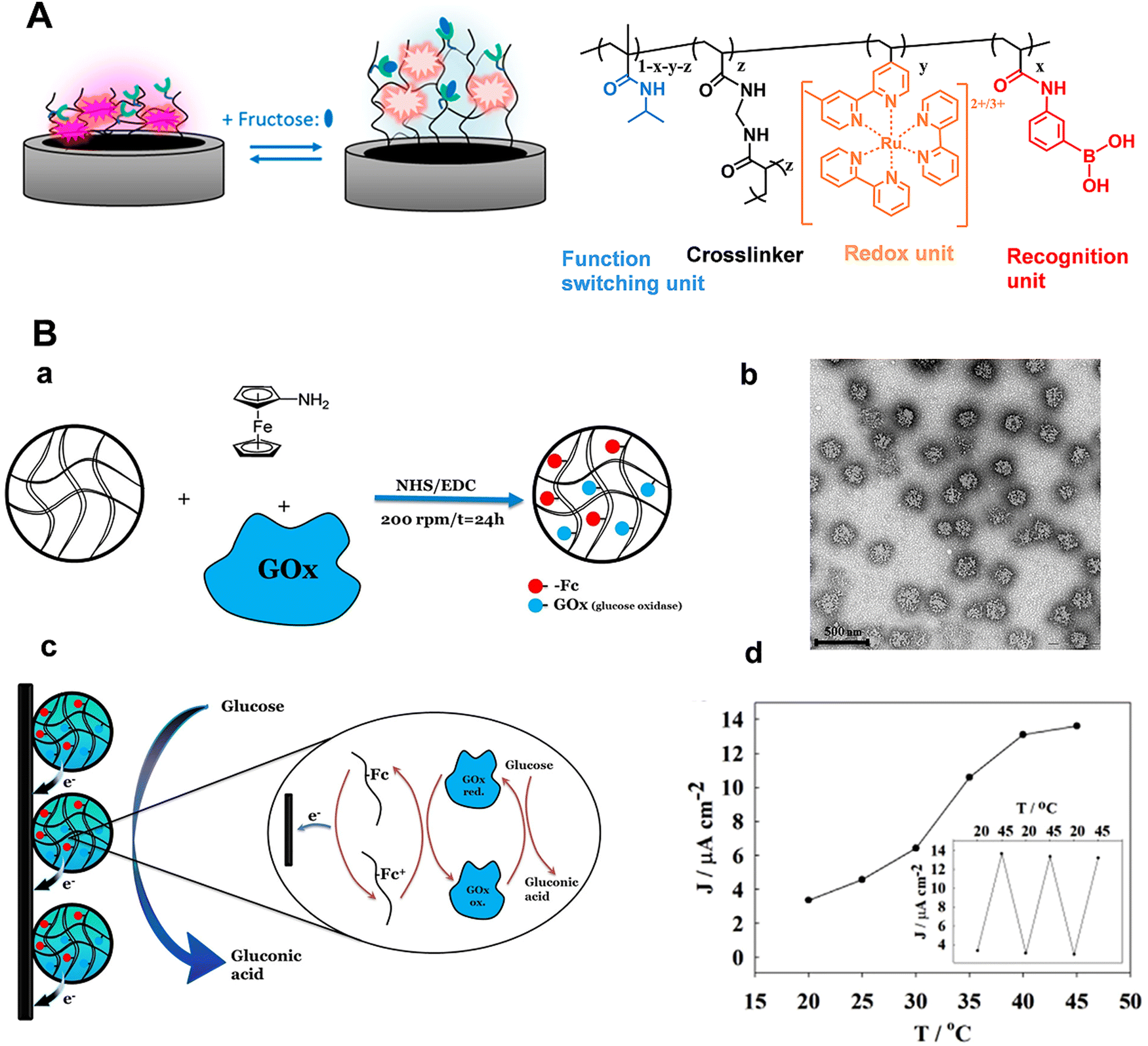 | ||
| Fig. 9 (A) Schematic illustration of the modulation of hydrogel films by fructose and chemical structure of the corresponding stimuli-responsive hydrogel. Reproduced with permission from ref. 94. Copyright 2018, American Chemical Society. (B) (a) Microgel network with amino-ferrocene and glucose oxidase. (b) TEM image of the microgel. (c) Schematic illustration of electroactive microgel modified electrode for glucose detection. (d) Influence of temperature on sensor response. Reproduced with permission from ref. 95. Copyright 2018, John Wiley and Sons. | ||
4.2. Field-effect transistor (FET) based on carrier mobility
Field-effect transistors (FET) modified with specific receptors enable direct target detection with high sensitivity and temporal resolution.97–100 The conductivity at the source–drain “channel” is proportional to its carrier density, which is sensitive to the change in the electric field in a direction perpendicular to the gate surface (Fig. 10). For bio-sensing applications, receptors are immobilized on the gate surfaces of FETs. Upon specific interactions with targets, the channel conductivity will be changed correlated to the polarity and density of charges at the gate, thus providing a basis for identification and quantification of targets (Fig. 10).101 However, the generality of FETs for real-time sensing in physiological conditions must overcome two fundamental limitations: (1) in physiological fluids, the Debye screening length (i.e., the effective sensing distance) is <1 nm, which reduces the field produced by charged macromolecules on the FET surface.102 (2) Small target molecules with few or no charges will have minimal impact on semiconductor transconductance.103 | ||
| Fig. 10 Structure and principles of Bio-FETs. | ||
To address these obstacles, stimuli-responsive polymer gel was explored as the chemical–electrical signal transducer. Upon stimulation, smart gels can evoke an abrupt volume change, accompanied by the changes of other physical parameters such as thickness, charge density and permittivity. These physicochemical changes commencing at the gel/outer aqueous media interface can geometrically propagate across the gel layer, resulting in the transport of signals beyond the ‘barrier’ of the Debye length (Fig. 11A).101,104 For example, a PBA-based polymer gel was covalently introduced into the FET gate surface (Fig. 11B).105,106 Upon treatment with glucose, the negatively charged phenylborate moiety increased the counterion osmotic pressure within the gel, resulting in the swelling of the gel. Considering the extremely high permittivity of water (ca. 80) compared to the value for condensed polymeric materials (ca. 2), the entry of water into gel matrix can alter the capacitive properties of gel/gate interface, leading to the enhancement of electrical conductance. By integrating with an inkjet-printed interdigitated capacitor (IDC), battery-free sensor was developed for the intermittent or continuous monitoring of glucose (Fig. 11B). Furthermore, the flexible polymer allowed the sensor to conform to biological tissues and living bodies.106 In addition, such a gel transition-synchronized system has been exploited for detection of Ca2+ due to the changes in charge density and permittivity evoked on the gate surface.107
 | ||
| Fig. 11 (A) Schematic illustration of a stimuli-responsive gel-mediated signal transduction enabling “Debye length-free” FET-based molecular detection. Reproduced with permission from ref. 101. Copyright 2013, Royal Society of Chemistry. (B) Battery-free glucose sensor: (a) Layered structure consisting of glucose-responsive polymer hydrogel and an inkjet-printed interdigitated capacitor. (b) Photograph of the glucose sensor conforming to chicken muscle. (c) Structure of glucose-responsive hydrogel. Reproduced with permission from ref. 106. Copyright 2021, Royal Society of Chemistry. (C) Hydrogel-gated organic field-effect transistor for dynamic measurement of pH. Reproduced with permission from ref. 108. Copyright 2018, American Chemical Society. | ||
However, in most cases, the hydrogel-modified electrode surface was constructed by drop-casting or spin-coating, leading to thick hydrogel layers and reduced response time. Taking advantage of the ability to control polymer architectures, recently, Noel's group utilized ATRP to graft a nanometric poly(acrylic acid) hydrogel (PAA, ca. 6 nm) on the gate of FET.108 Such ultrathin PAA layer can overcome the Debye length problematics. The reversible PAA swelling/deswelling processes toward pH changes were evidenced by the measurement of the drain current (Fig. 11C). Here both the swelling (τswell = 121 s) and deswelling (τswell = 75 s) time are shorter than those reported in the literature. Accordingly, the stimuli-responsive hydrogel modified FET with milliseconds resolution was demonstrated as an effective tool for neurochemical analysis.
Collectively, the stimuli-responsive gel-based FET enabled the detection of biomarkers in the body fluids (e.g., blood, urine, and cerebrospinal fluid). Moreover, this system is capable of detecting not only charged species but also neutral species. With the development of modern manufacturing techniques, advanced platform (e.g., nanometric FET and tissue-interfaced battery-free sensors) can be fabricated, which is expected to open up exciting opportunities for in vivo analysis.
4.3. Nanochannel based on ion transport
Benefiting from the advanced micro-nanofabrication technologies, solid-state nanopore/nanochannel system has emerged as a powerful electrochemical technique for biosensing.109–112 One of the main signal output strategies is based on ion current rectification (i.e., the current at a specific potential is greater than that in the opposite potential, ICR), which is inherently related to the selective transport of ions and susceptible to the surface states. When the target is passing through the nanopore/nanochannel integrated with functional elements, it will influence the surface properties (e.g., surface charge distribution, effective pore size and wettability), which can in turn effectively regulate the ion transportation within nanopores/nanochannels and finally change the ion current.113 The narrow tip and conical shape render the nanopipette a confined space that enables the enhancement of the electrochemical field within the nanopipette, and the electrochemically confined effect contributes to the signal amplification and sensitivity enhancement of the analytical system.114 Although the ion transport-based nanochannel biosensors displayed bright perspective for recognition of electro-inactive molecules, several limitations must be overcome. The ligand – (i.e., signaling-biomolecules) gated ion channels in vivo exhibited high sensitivity and precise controllability. A huge gap still existed between natural biological systems and classically manufactured ion channels. Moreover, more intelligent ion channels manipulated by specific biomolecules are urgently needed.Recently, Qing's group developed a stimuli-responsive polymer functionalized polyethylene terephthalate (PET) conical nanochannel for monitoring tyrosine phosphorylation (pTyr) (Fig. 12A).115 In this work, the polyethyleneimine-g-phenylguanidine (PEI-PG) polymer containing PG as recognition receptor and PEI as a flexible chain was designed. Initially, the PEI-PG polymer presented a swollen conformation due to the repulsive electrostatic interaction between cationic PEI and cationic guanidinium groups in PG. In the presence of pTyr, multiple interactions including hydrogen bonds and cation–π interaction can be formed between the guanidinium groups and the pTyr residue, which triggered the conformational shrinkage of PEI-PG, and the corresponding “OFF–ON” change of ion-flux. Taking advantages of strong interaction between the guanidinium group and the pTyr side-chain, apart from good selectivity toward the phosphorylated peptide (PP) in comparison to the nonmodified peptide, this nanochannel could precisely distinguish PP with different types/numbers of phosphorylated residues. Together, the nanochannel with outstanding performance was applied for real-time monitoring of pTyr process by c-Abl kinase on a peptide substrate.
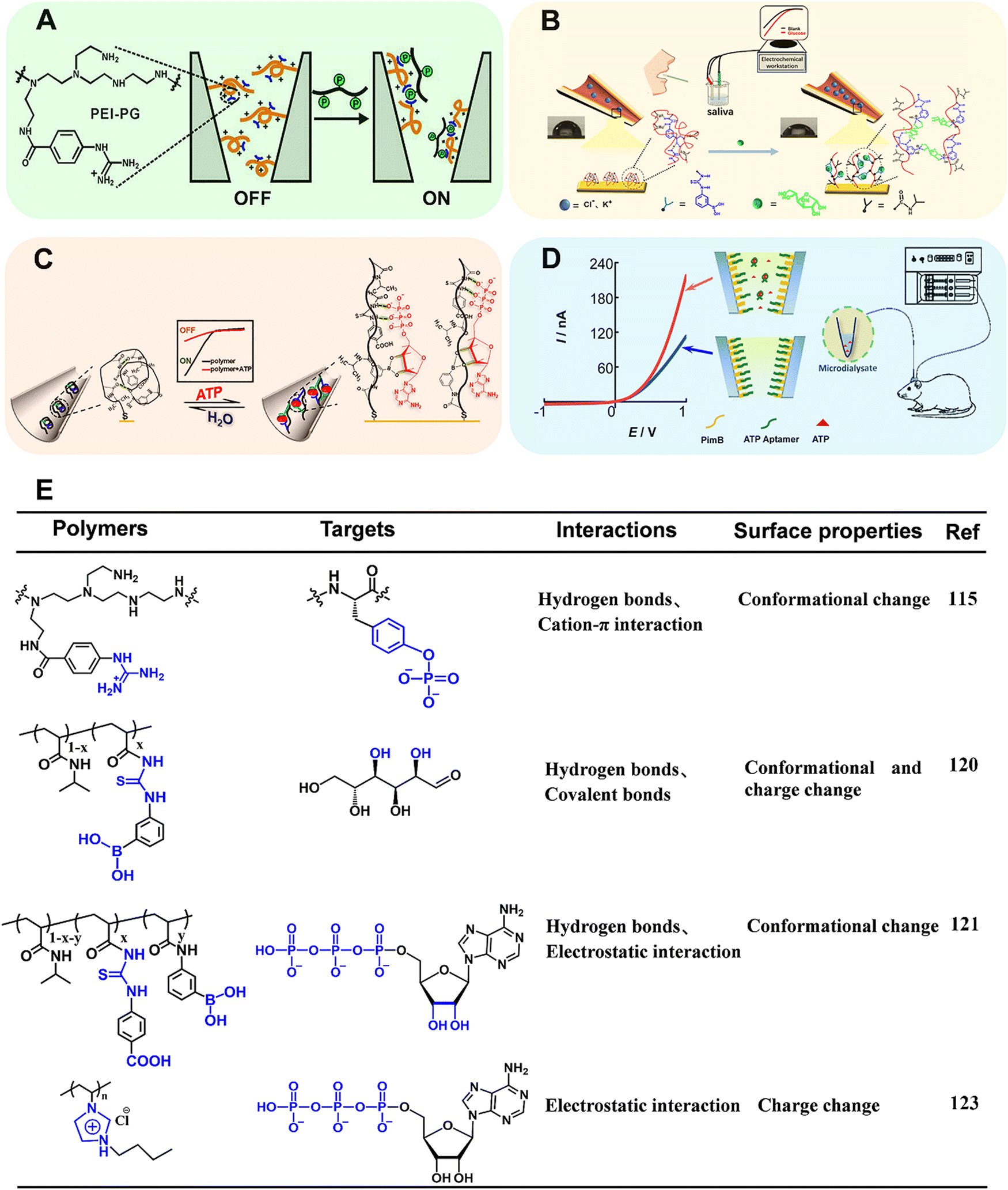 | ||
| Fig. 12 (A) Schematic illustration of copolymer-modified nanochannel for phosphorylated peptide. Reproduced with permission from ref. 115. Copyright 2020, American Chemical Society. (B) Schematic illustration of copolymer-modified glass nanopore for measurement of glucose in human saliva. Reproduced with permission from ref. 120. Copyright 2019, American Chemical Society. (C) Schematic illustration of a biomimetic ATP-sensitive potassium channel for measurement of ATP. Reproduced with permission from ref. 121. Copyright 2021, Elsevier. (D) Schematic illustration of an MICR-based glass micropipette for online measurement of cerebral ATP. Reproduced with permission from ref. 123. Copyright 2017, American Chemical Society. (E) Structure of the corresponding stimuli-responsive polymers utilized in ref. 115, 120, 121, 123. | ||
As a member of artificial solid nanochannel/nanopores, glass capillary-based nanochannel/nanopores have been widely investigated resulting from attractive properties, such as flexible preparation, stiffness and durability, surface modifiability.114,116–119 We designed a stimuli-responsive polymer modified glass nanochannel for sensitive recognition of glucose in the saliva (Fig. 12B).120 In this work, poly(3-(acryloylthioureido) phenylboronic acid-co-N-isopropylacrylamide) (PATPBA-co-PNIPAAm) was modified on the inner wall of glass nanopore via Au–S bond. The copolymer displayed good selectivity toward glucose results from multivalent boronic acid–glucose interaction and the cooperation of thiourea units. Notably, in the presence of glucose, the copolymer could undergo not only wettability switch but also charge change. The cooperation of electrostatic effect and wettability change significantly regulated the ion transport, thus improving the sensitivity with the detection limit of 1 nM. Inspired by the adenosine triphosphate (ATP)-gated ion channel in cell membranes, we have reported an ATP-regulated artificial nanochannel based on tricomponent copolymer system (denoted as PNI-PBA-CP) (Fig. 12C).121 In this copolymer, phenylthiourea (CP) was introduced to bind the phosphate units of nucleotides and phenylboronic acid (PBA) was used to combine the pentose ring of ATP. Besides, the –COOH group with electron-withdrawing properties in CP units can promote the hydrogen bonding. The ionic gating of nanochannel can be achieved by the structure changes of the copolymer after responding to ATP. Moreover, the synergistic hydrogen bonding between the ATP and the copolymer promoted the reversibility and switching performance of the ion channel. In summary, the stimuli-responsive polymers provide an efficient tool to modulate the dynamic adsorption/desorption behavior of guest biomolecules, which suggests an ideal platform for evaluating biological processes at the molecular level.
However, the relatively tiny and soft tip of glass nanopipettes renders difficulties in applying this method for real sample analysis. Recently, Mao's group has found that the ICR could be obtained at the polyelectrolyte brush-modified micropipette (e.g., 5 μm radius), essentially extending the knowledge of ICR from the nanoscale to the microscale (MICR).122 Compared with ICR at the nanoscale, MICR can be obtained in high-salt solutions and can be modulated by adjusting the polyelectrolyte length. The type of polyelectrolyte endows MICR with more designability. These unique properties made polyelectrolyte brush-modified micropipette a new platform for practical applications, such as in vivo sensing. Based on this study, Mao's group constructed a new platform for cerebral ATP assay (Fig. 12D).123 In this work, positively charged polyimidazolium (Pim) brush was first grafted onto the inner wall of micropipette by ATRP. Then, negatively charged ATP aptamer was modified by electrostatic interaction. The Pim brush not only enable the generation of MICR, but also promote enrichment of ATP in the sensing interface due to the strong affinity between Pim and the triphosphate moiety of ATP. In the presence of ATP, the aptamer was dissociated from the inner surface. The exposure of imidazole moieties led to an increase of the net surface charge and thus the rectification ratio. The detection linear range for ATP varied from 5 nM to 100 nM. In combination with in vivo microdialysis, the MICR-based micropipettes with excellent sensitivity and selectivity were used for determining cerebral ATP. Compared with the traditional nano-systems, MICR-based sensors are favored due to the ease-in-operation and relatively robust tip, opening a new way to neurochemical analysis.
In terms of scalability, the single functional micro/nanochannel demonstrate the potential for in situ detection, including single-cell and in vivo analysis. These properties will promote the full understanding of the molecular mechanism in physiological and pathological processes.
5. Conclusions and future perspectives
In summary, this review provides an outline of recent advancements about stimuli-responsive polymers functionalized electrochemical sensors. To fulfill the demanding requirements of neurochemicals measurements, considerable interest should be paid to developing novel stimuli-responsive polymers and electrochemical methods. Typically, electroactive neurochemicals can be detected by direct electrolysis. However, most neurochemicals are electro-inactive. To gain more precise neurochemical information, several strategies were proposed: first, the development of novel stimuli-responsive polymers based on multiple interaction is expected to enable the highly selective recognition of neurochemicals. The design strategies of stimuli-responsive polymers discussed in this review can be extended to more neurochemicals. Second, apart from electrochemical techniques based on electron transport, introduction of new electrochemical principles and techniques may shed new light on analysis of neutral neurochemicals. For example, stimuli-responsive gel-based field-effect transistor can transform the biomolecular recognition into electrochemical signals through the volume change of polymers accompanied by permittivity change at the gel/gate interface. In addition, as an emerging technique, nanochannel worked based on ion transportation. The stimuli-responsive polymers can flexibly regulate the ionic transportation with the transition of surface properties (e.g., charge, wettability and conformation). These techniques enabled the detection of not only electroactive neurochemicals but also electro-inactive species. For in vivo electrochemical analysis, stimuli-responsive polymers functionalized with electrochemical mediators and enzymes can be explored. In addition, rationally tailoring the structure of polymers can greatly improve the manipulation of polymer properties, and then promote the chemical–electrical signal conversion. Although notable progress has been achieved, there are still many challenges and unremitting efforts still needed in the future:1. Considerable effort must be put into the relationship between the architectures and properties of the stimuli-responsive polymers to expand the applications. Novel stimuli-responsive polymers for the simultaneous determination of multiple neurochemicals are expected to obtain more information about the central nervous system (CNS).
2. In compared with in vitro conditions, the recognition of neurochemicals in CNS may be hindered by biofouling (e.g., non-specific protein absorption, microorganism adhesion), resulting in low reliability and high signal noise. Integrating the stimuli-responsive polymers with antifouling properties may help to solve these problems and make them ideal for in vivo analysis.
3. To minimize tissue damage, advanced soft polymers possessing good electronic conductivity and biocompatibility enable the construction of flexible sensors, promoting implanted and chronical monitoring in free animals.
4. It is worth mentioning that while the electrochemical sensors based on stimuli-responsive polymers have been widely used in the recognition of various biomarkers, the sensors applied for clinical trials are rare. The limitations for clinical translation are summarized as follows: first, to guarantee the accuracy of clinical diagnosis, the electrochemical biosensor should be fabricated with high reproductivity and stability. Second, multi-functional device should be fully considered. For example, arrays of micro/nanoelectrodes can promote multimodal analysis and facilitate brain mapping study of biomolecules. In addition, wireless technique will benefit the portable diagnostics. Finally, miniaturization has been a long-term trend in the instrumentation for clinical diagnosis. Considerable effort must be made in the integration of all parts (e.g., electrodes and materials) without reducing the performance.
Overall, we believe that interdisciplinary collaboration between, e.g., advanced material, electrochemistry and microelectronic engineering will push neurochemistry analysis to new heights.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21974048, and 22104065), the Natural Science Foundation of the Jiangsu Higher Education Institutions of China (No. 21KJB150024), the Shanghai Pujiang Program (No. 21PJD020), the Science and Technology Commission of Shanghai Municipality (No. 19ZR1414600), and the Fundamental Research Funds for the Central Universities.References
- C. Xu, F. Wu, P. Yu and L. Q. Mao, ACS Sens., 2019, 4, 3102–3118 CrossRef CAS.
- T. F. Xiao, F. Wu, J. Hao, M. N. Zhang, P. Yu and L. Q. Mao, Anal. Chem., 2017, 89, 300–313 CrossRef CAS PubMed.
- D. L. Robinson, A. Hermans, A. T. Seipel and R. M. Wightman, Chem. Rev., 2008, 108, 2554–2584 CrossRef CAS PubMed.
- P. Cowen and A. C. Sherwood, J. Psychopharmacol., 2013, 27, 575–583 CrossRef.
- M. E. Rice, Trends Neurosci., 2000, 23, 209–216 CrossRef CAS.
- M. N. Zhang, P. Yu and L. Q. Mao, Acc. Chem. Res., 2012, 45, 533–543 CrossRef CAS.
- P. Sun, F. O. Laforge, T. P. Abeyweera, S. A. Rotenberg, J. Carpino and M. V. Mirkin, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 443–448 CrossRef CAS.
- Y. Z. Liu, Q. Q. Yao, X. M. Zhang, M. N. Li, A. W. Zhu and G. Y. Shi, Biosens. Bioelectron., 2015, 63, 262–268 CrossRef CAS PubMed.
- S. S. Ding, M. N. Li, H. Y. Gong, Q. Zhu, G. Y. Shi and A. W. Zhu, Anal. Chem., 2020, 92, 2543–2549 CrossRef CAS.
- S. S. Ding, Y. Z. Liu, C. R. Ma, J. Q. Zhang, A. W. Zhu and G. Y. Shi, Electroanalysis, 2018, 30, 1041–1046 CrossRef CAS.
- E. S. Bucher and R. M. Wightman, Annu. Rev. Anal. Chem., 2015, 8, 239–261 CrossRef CAS.
- P. Yu, X. L. He and L. Q. Mao, Chem. Soc. Rev., 2015, 44, 5959–5968 RSC.
- M. Ganesana, S. T. Lee, Y. Wang and B. J. Venton, Anal. Chem., 2017, 89, 314–341 CrossRef CAS.
- J. G. Roberts and L. A. Sombers, Anal. Chem., 2018, 90, 490–504 CrossRef CAS.
- L. M. Zhang and Y. Tian, Acc. Chem. Res., 2018, 51, 688–696 CrossRef CAS PubMed.
- G. S. Wilson and Y. B. Hu, Chem. Rev., 2000, 100, 2693–2704 CrossRef CAS PubMed.
- C. P. McMahon, G. Rocchitta, P. A. Serra, S. M. Kirwan, J. P. Lowry and R. D. O’Neill, Anal. Chem., 2006, 78, 2352–2359 CrossRef CAS.
- J. Zhou, C. N. Liao, L. M. Zhang, Q. G. Wang and Y. Tian, Anal. Chem., 2014, 86, 4395–4401 CrossRef CAS PubMed.
- Z. Wang, D. Liu, H. Gu, A. W. Zhu, Y. Tian and G. Y. Shi, Biosens. Bioelectron., 2013, 43, 101–107 CrossRef CAS.
- T. T. Feng, W. L. Ji, Y. Zhang, F. Wu, Q. Tang, H. Wei, L. Q. Mao and M. N. Zhang, Angew. Chem., Int. Ed., 2020, 59, 23445–23449 CrossRef CAS.
- Y. F. Gao, M. L. Wei, X. Li, W. W. Xu, A. Ahiabu, J. Perdiz, Z. N. Liu and M. J. Serpe, Macromol. Res., 2017, 25, 513–527 CrossRef CAS.
- K. Bauri, M. Nandi and P. De, Polym. Chem., 2018, 9, 1257–1287 RSC.
- T. Liu, J. M. Hu, J. Yin, Y. F. Zhang, C. H. Li and S. Y. Liu, Chem. Mater., 2009, 21, 3439–3446 CrossRef CAS.
- J. M. Hu, C. H. Li and S. Y. Liu, Langmuir, 2010, 26, 724–729 CrossRef CAS.
- M. Pita, T. K. Tam, S. Minko and E. Katz, ACS Appl. Mater. Interfaces, 2009, 1, 1166–1168 CrossRef CAS.
- I. Tokarev, M. Orlov, E. Katz and S. Minko, J. Phys. Chem. B, 2007, 111, 12141–12145 CrossRef CAS PubMed.
- X. M. Qian, J. Li and S. M. Nie, J. Am. Chem. Soc., 2009, 131, 7540–7541 CrossRef CAS.
- M. X. Zhang, G. Y. Qing, C. L. Xiong, R. Cui, D. W. Pang and T. L. Sun, Adv. Mater., 2013, 25, 749–754 CrossRef CAS.
- Z. Q. Cao and G. J. Wang, Chem. Rec., 2016, 16, 1398–1435 CrossRef CAS PubMed.
- L. Zhai, Chem. Soc. Rev., 2013, 42, 7148–7160 RSC.
- A. Lendlein and V. P. Shastri, Adv. Mater., 2010, 22, 3344–3347 CrossRef CAS PubMed.
- B. S. Chang, M. X. Zhang, G. Y. Qing and T. L. Sun, Small, 2015, 11, 1097–1112 CrossRef CAS PubMed.
- B. S. Chang, B. Zhang and T. L. Sun, Small, 2017, 13, 1503472 CrossRef.
- X. H. Wang, X. P. Qiu and C. Wu, Macromolecules, 1998, 31, 2972–2976 CrossRef CAS.
- Q. Zhang, Y. N. Zhang, Y. Wan, W. Carvalho, L. Hu and M. J. Serpe, Prog. Polym. Sci., 2021, 116, 101386 CrossRef CAS.
- F. Liu and M. W. Urban, Prog. Polym. Sci., 2010, 35, 3–23 CrossRef CAS.
- K. Kim, W. C. W. Chen, Y. Heo and Y. D. Wang, Prog. Polym. Sci., 2016, 60, 18–50 CrossRef CAS.
- N. Ayres, S. G. Boyes and W. J. Brittain, Langmuir, 2007, 23, 182–189 CrossRef CAS.
- O. Azzaroni, A. A. Brown and W. T. S. Huck, Adv. Mater., 2007, 19, 151–154 CrossRef CAS.
- G. Y. Qing, Q. Lu, X. L. Li, J. Liu, M. L. Ye, X. M. Liang and T. L. Sun, Nat. Commun., 2017, 8, 1–12 CrossRef CAS.
- W. Chen, Y. Ma, J. M. Pan, Z. H. Meng, G. Q. Pan and B. Sellergren, Polymers, 2015, 7, 1689–1715 CrossRef CAS.
- Y. Kitayama and M. Isomura, Chem. Commun., 2018, 54, 2538–2541 RSC.
- Z. F. Sun, F. C. Lv, L. J. Cao, L. Liu, Y. Zhang and Z. G. Lu, Angew. Chem., Int. Ed., 2015, 54, 7944–7948 CrossRef CAS PubMed.
- J. M. Zhuang, M. R. Gordon, J. Ventura, L. Y. Li and S. Thayumanavan, Chem. Soc. Rev., 2013, 42, 7421–7435 RSC.
- D. Roy, J. N. Cambre and B. S. Sumerlin, Chem. Commun., 2009, 2106–2108 RSC.
- Y. B. Zheng, S. Zhao, S. H. Cao, S. L. Cai, X. H. Cai and Y. Q. Li, Nanoscale, 2017, 9, 433–439 RSC.
- L. Liu, F. Zhao, W. Liu, T. Zhu, J. Z. H. Zhang, C. Chen, Z. H. Dai, H. S. Peng, J. L. Huang, Q. Hu, W. B. Bu and Y. Tian, Angew. Chem., Int. Ed., 2017, 56, 10471–10475 CrossRef CAS.
- S. Li, A. W. Zhu, T. Zhu, J. Z. H. Zhang and Y. Tian, Anal. Chem., 2017, 89, 6656–6662 CrossRef CAS PubMed.
- S. T. Milner, Science, 1991, 251, 905–914 CrossRef CAS.
- S. Edmondson, V. L. Osborne and W. T. S. Huck, Chem. Soc. Rev., 2004, 33, 14–22 RSC.
- F. Zhou and W. T. S. Huck, Phys. Chem. Chem. Phys., 2006, 8, 3815–3823 RSC.
- P. M. Mendes, Chem. Soc. Rev., 2008, 37, 2512–2529 RSC.
- T. Chen, R. Ferris, J. M. Zhang, R. Ducker and S. Zauscher, Prog. Polym. Sci., 2010, 35, 94–112 CrossRef CAS.
- P. B. Zetterlund, Y. Kagawa and M. Okubo, Chem. Rev., 2008, 108, 3747–3794 CrossRef CAS PubMed.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- X. H. Lou, M. S. Lewis, C. B. Gorman and L. He, Anal. Chem., 2005, 77, 4698–4705 CrossRef CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS PubMed.
- Z. Liu, W. Wang, R. Xie, X. J. Ju and L. Y. Chu, Chem. Soc. Rev., 2016, 45, 460–475 RSC.
- D. Menne, F. Pitsch, J. E. Wong, A. Pich and M. Wessling, Angew. Chem., Int. Ed., 2014, 53, 5706–5710 CrossRef CAS.
- M. Yang, L. Y. Chu, H. D. Wang, R. Xie, H. Song and C. H. Niu, Adv. Funct. Mater., 2008, 18, 652–663 CrossRef CAS.
- H. B. Sun, Y. L. Qiu, Y. J. Lu, J. M. Kong and X. J. Zhang, Chem. Commun., 2020, 56, 6636–6639 RSC.
- Q. Hu, J. M. Kong, D. X. Han, Y. W. Zhang, Y. Bao, X. J. Zhang and L. Niu, Anal. Chem., 2019, 91, 1936–1943 CrossRef CAS.
- Q. Hu, Y. Bao, S. Y. Gan, Y. W. Zhang, D. X. Han and L. Niu, Anal. Chem., 2020, 92, 3470–3476 CrossRef CAS PubMed.
- Q. Hu, L. F. Su, Z. H. Chen, Y. Y. Huang, D. D. Qin and L. Niu, Anal. Chem., 2021, 93, 9602–9608 CrossRef.
- M. Motornov, Y. Roiter, I. Tokarev and S. Minko, Prog. Polym. Sci., 2010, 35, 174–211 CrossRef CAS.
- A. Döring, W. Birnbaum and D. Kuckling, Chem. Soc. Rev., 2013, 42, 7391–7420 RSC.
- A. S. Hoffman, Adv. Drug Delivery Rev., 2002, 54, 3–12 CrossRef CAS PubMed.
- L. V. Sigolaeva, S. Y. Gladyr, A. P. H. Gelissen, O. Mergel, D. V. Pergushov, I. N. Kurochkin, F. A. Plamper and W. Richtering, Biomacromolecules, 2014, 15, 3735–3745 CrossRef CAS PubMed.
- L. V. Sigolaeva, O. Mergel, E. G. Evtushenko, S. Y. Gladyr, A. P. H. Gelissen, D. V. Pergushov, I. N. Kurochkin, F. A. Plamper and W. Richtering, Langmuir, 2015, 31, 13029–13039 CrossRef CAS PubMed.
- N. C. Dubey, B. P. Tripathi, M. Müller, M. Stamm and L. Ionov, ACS Appl. Mater. Interfaces, 2015, 7, 1500–1507 CrossRef CAS.
- Z. Liu, Y. Faraj, X. J. Ju, W. Wang, R. Xie and L. Y. Chu, J. Polym. Sci., 2018, 56, 1306–1313 CrossRef CAS.
- F. A. Plamper and W. Richtering, Acc. Chem. Res., 2017, 50, 131–140 CrossRef CAS PubMed.
- A. Burmistrovaa and R. V. Klitzing, J. Mater. Chem., 2010, 20, 3502–3507 RSC.
- K. Marcisz, K. Kaniewska and M. Karbarz, Curr. Opin. Electrochem., 2020, 23, 57–64 CrossRef CAS.
- M. I. González-Sánchez, J. Rubio-Retama, E. López-Cabarcos and E. Valero, Biosens. Bioelectron., 2011, 26, 1883–1889 CrossRef.
- L. L. Yue, R. Xie, J. Wei, X. J. Ju, W. Wang and L. Y. Chu, J. Colloid Interface Sci., 2012, 377, 137–144 CrossRef CAS PubMed.
- G. Trefalt, S. H. Behrens and M. Borkovec, Langmuir, 2016, 32, 380–400 CrossRef CAS.
- J. J. Bikerman, Philos. Mag., 1942, 33, 384–397 CAS.
- Q. L. Zhu, Y. M. Yang, H. X. Gao, L. P. Xu and S. T. Wang, Chem. Sci., 2022, 13, 5069–5084 RSC.
- W. W. Xu, Z. Y. Lu, X. M. Sun, L. Jiang and X. Duan, Acc. Chem. Res., 2018, 51, 1590–1598 CrossRef CAS PubMed.
- S. S. Ding, S. M. Cao, A. W. Zhu and G. Y. Shi, Anal. Chem., 2016, 88, 12219–12226 CrossRef CAS.
- S. S. Ding, S. M. Cao, Y. Z. Liu, Y. Lian, A. W. Zhu and G. Y. Shi, ACS Sens., 2017, 2, 394–400 CrossRef CAS PubMed.
- A. G. Ewing, J. C. Bigelow and R. M. Wightman, Science, 1983, 221, 169–171 CrossRef CAS.
- P. S. Cahill, Q. D. Walker, J. M. Finnegan, G. E. Mickelson, E. R. Travis and R. M. Wightman, Anal. Chem., 1996, 68, 3180–3186 CrossRef CAS.
- N. T. N. Phan, X. C. Li and A. G. Ewing, Nat. Rev. Chem., 2017, 1, 0048 CrossRef CAS.
- Q. F. Zhang, B. Liu, Q. H. Wu, B. Liu, Y. L. Li, S. H. Sun, Y. Wang, X. Wu, Z. Y. Chai, X. H. Jiang, X. Y. Liu, M. Q. Hu, Y. S. Wang, Y. T. Yang, L. Wang, X. J. Kang, Y. F. Xiong, Y. Zhou, X. K. Chen, L. H. Zheng, B. Zhang, C. H. Wang, F. P. Zhu and Z. Zhou, Neuron, 2019, 102, 173–183 CrossRef CAS PubMed.
- Y. Li, K. K. Hu, Y. Yu, S. A. Rotenberg, C. Amatore and M. V. Mirkin, J. Am. Chem. Soc., 2017, 139, 13055–13062 CrossRef CAS PubMed.
- X. K. Yang, F. L. Zhang, W. T. Wu, Y. Tang, J. Yan, Y. L. Liu, C. Amatore and W. H. Huang, Angew. Chem., Int. Ed., 2021, 133, 15937–15942 CrossRef.
- D. L. Robinson, A. Hermans, A. T. Seipel and R. M. Wightman, Chem. Rev., 2008, 108, 2554–2584 CrossRef CAS.
- F. Wu, P. Yu and L. Q. Mao, Curr. Opin. Electrochem., 2017, 5, 152–157 CrossRef CAS.
- F. Zhao, L. M. Zhang, A. W. Zhu, G. Y. Shi and Y. Tian, Chem. Commun., 2016, 52, 3717–3720 RSC.
- L. M. Zhang, Y. Y. Han, F. Zhao, G. Y. Shi and Y. Tian, Anal. Chem., 2015, 87, 2931–2936 CrossRef CAS.
- H. Gu, E. L. Varner, S. R. Groskreutz, A. C. Michael and S. G. Weber, Anal. Chem., 2015, 87, 6088–6094 CrossRef CAS PubMed.
- H. D. Li, S. Voci, V. Ravaine and N. Sojic, J. Phys. Chem. Lett., 2018, 9, 340–345 CrossRef CAS PubMed.
- K. Marcisz, K. Kaniewska, M. Mackiewicz, A. Nowinska, J. Romanski, Z. Stojek and M. Karbarz, Electroanalysis, 2018, 30, 2853–2860 CrossRef CAS.
- Y. Q. Zhou, J. Cao, J. Zhao, Y. X. Xie, J. J. Fei and Y. L. Cai, Microchim. Acta, 2016, 183, 2501–2508 CrossRef CAS.
- N. Nakatsuka, K.-A. Yang, J. M. Abendroth, K. M. Cheung, X. B. Xu, H. Y. Yang, C. Z. Zhao, B. W. Zhu, Y. S. Rim, Y. Yang, P. S. Weiss, M. N. Stojanović and A. M. Andrews, Science, 2018, 362, 319–324 CrossRef CAS.
- Q. Qing, Z. Jiang, L. Xu, R. X. Gao, L. Q. Mai and C. M. Lieber, Nat. Nanotechnol., 2014, 9, 142–147 CrossRef CAS.
- B. Tian, T. Cohen-Karni, Q. Qing, X. J. Duan, P. Xie and C. M. Lieber, Science, 2010, 329, 830–834 CrossRef CAS.
- S. J. Park, O. S. Kwon, S. H. Lee, H. S. Song, T. H. Park and J. Jang, Nano Lett., 2012, 12, 5082–5090 CrossRef CAS.
- A. Matsumoto and Y. Miyahara, Nanoscale, 2013, 5, 10702–10718 RSC.
- A. Vacic, J. M. Criscione, N. K. Rajan, E. Stern, T. M. Fahmy and M. A. Reed, J. Am. Chem. Soc., 2011, 133, 13886–13889 CrossRef CAS PubMed.
- P. S. Weiss, P. L. Trevor and M. J. Cardillo, J. Chem. Phys., 1989, 90, 5146–5153 CrossRef CAS.
- A. Matsumoto, T. Kurata, D. Shiino and K. Kataoka, Macromolecules, 2004, 37, 1502–1510 CrossRef CAS.
- A. Matsumoto, N. Sato, T. Sakata, R. Yoshida, K. Kataoka and Y. Miyahara, Adv. Mater., 2009, 21, 4372–4378 CrossRef CAS.
- H. Fujita, K. Yamagishi, W. S. Zhou, Y. Tahara, S. Y. Huang, M. Hashimoto and T. Fujie, J. Mater. Chem. C, 2021, 9, 7336–7344 RSC.
- A. Matsumoto, T. Endo, R. Yoshidaab and Y. Miyahara, Chem. Commun., 2009, 5609–5611 RSC.
- L. Fillaud, T. Petenzi, J. Pallu, B. Piro, G. Mattana and V. Noel, Langmuir, 2018, 34, 3686–3693 CrossRef CAS.
- X. Hou, W. Guo and L. Jiang, Chem. Soc. Rev., 2011, 40, 2385–2401 RSC.
- Z. Long, S. S. Zhan, P. C. Gao, Y. Q. Wang, X. D. Lou and F. Xia, Anal. Chem., 2018, 90, 577–588 CrossRef CAS PubMed.
- L. L. Yang, K. Z. Qu, J. L. Guo, H. J. Xu, Z. Q. Dai, Z. D. Gao and Y. Y. Song, Chem. Commun., 2019, 55, 14625–14628 RSC.
- G. Laucirica, Y. T. Terrones, V. Cayón, M. L. Cortez, M. E. Toimil-Molares, C. Trautmann, W. Marmisollé and O. Azzaroni, Trends Anal. Chem., 2021, 144, 116425 CrossRef CAS.
- Z. S. Siwy and S. Howorka, Chem. Soc. Rev., 2010, 39, 1115–1132 RSC.
- S. M. Lu and Y. T. Long, Trends Anal. Chem., 2019, 117, 39–46 CrossRef CAS.
- M. M. Li, Y. T. Xiong, W. Q. Lu, X. Wang, Y. H. Liu, B. Na, H. J. Qin, M. L. Tang, H. Q. Qin, M. L. Ye, X. M. Liang and G. Y. Qing, J. Am. Chem. Soc., 2020, 142, 16324–16333 CrossRef CAS PubMed.
- J. Song, C. H. Xu, S. Z. Huang, W. Lei, Y. F. Ruan, H. J. Lu, W. Zhao, J. J. Xu and H. Y. Chen, Angew. Chem., Int. Ed., 2018, 57, 13226–13230 CrossRef CAS.
- X. H. Yin, S. D. Zhang, Y. T. Dong, S. J. Liu, J. Gu, Y. Chen, X. Zhang, X. H. Zhang and Y. H. Shao, Anal. Chem., 2015, 87, 9070–9077 CrossRef CAS PubMed.
- M. Shen, Z. Z. Qu, J. DesLaurier, T. M. Welle, J. V. Sweedler and R. Chen, J. Am. Chem. Soc., 2018, 140, 7764–7768 CrossRef CAS.
- P. Hu, Y. Zhang, D. D. Wang, G. H. Qi and Y. D. Jin, Anal. Chem., 2021, 93, 4240–4245 CrossRef CAS PubMed.
- M. Yang, C. R. Ma, S. S. Ding, Y. J. Zhu, G. Y. Shi and A. W. Zhu, Anal. Chem., 2019, 91, 14029–14035 CrossRef CAS.
- Q. Liu, S. S. Ding, R. Gao, G. Y. Shi and A. W. Zhu, Anal. Chim. Acta, 2021, 1188, 339167 CrossRef CAS PubMed.
- X. L. He, K. L. Zhang, T. Li, Y. N. Jiang, P. Yu and L. Q. Mao, J. Am. Chem. Soc., 2017, 139, 1396–1399 CrossRef CAS.
- K. L. Zhang, X. L. He, Y. Liu, P. Yu, J. J. Fei and L. Q. Mao, Anal. Chem., 2017, 89, 6794–6799 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |