
Porous metal–organic frameworks for hydrogen storage
Dian
Zhao
 a,
Xinxin
Wang
a,
Lianglan
Yue
a,
Yabing
He
*a and
Banglin
Chen
*b
a,
Xinxin
Wang
a,
Lianglan
Yue
a,
Yabing
He
*a and
Banglin
Chen
*b
aKey Laboratory of the Ministry of Education for Advanced Catalysis Materials, College of Chemistry and Life Sciences, Zhejiang Normal University, Jinhua 321004, China. E-mail: heyabing@zjnu.cn
bDepartment of Chemistry, University of Texas at San Antonio, One UTSA Circle, San Antonio, Texas 78249-0698, USA. E-mail: banglin.chen@utsa.edu
First published on 26th August 2022
Abstract
The high gravimetric energy density and environmental benefits place hydrogen as a promising alternative to the widely used fossil fuels, which is however impeded by the lack of safe, energy-saving and cost-effective H2 storage systems. The use of solid adsorbents as candidate materials offers a less energy-intensive way of storing hydrogen. The exceptional diversity and tunability of the chemical composition, topological structure, and surface chemistry together with large surface area position porous metal–organic frameworks as promising hydrogen storage material candidates. In this review, we first introduce several classes of important metal–organic frameworks for hydrogen storage, and then highlight the progress associated with the key challenges to be addressed, including the improvement of hydrogen–framework interaction required for enhancing room-temperature hydrogen storage capacities, and the optimization/balance of both gravimetric and volumetric storage/working capacities. In particular, the strategies used to tune and enhance hydrogen binding energies have been comprehensively reviewed. Future development prospects and related challenges of using porous metal–organic frameworks as hydrogen storage materials are also outlined. This feature review provides a wide perspective and insightful thoughts and suggestions for hydrogen storage using metal–organic frameworks, and promotes the further development of hydrogen storage materials to realize a hydrogen economy.
 Dian Zhao | Dian Zhao was born in Hubei, China. He received his PhD in Materials Science from Zhejiang University in 2017 under the supervision of Prof. Guodong Qian. After that, he joined the Zhejiang Normal University, where he is currently an associate professor of Chemistry. His research is focused on the design and synthesis of porous metal–organic frameworks for luminescence sensing, solid-state lighting, and gas storage applications. |
 Yabing He | Yabing He earned his PhD in organic chemistry from the Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, under the direction of Prof. Lianxun Gao in 2010. After that, he worked in the group of Prof. Banglin Chen at the University of Texas at San Antonio as a postdoctoral Research Fellow during 2010–2012. In 2012, he joined the Zhejiang Normal University, where he is a professor of chemistry. His current research focuses on the design and synthesis of porous materials and study of their gas adsorption and separation properties. |
 Banglin Chen | Banglin Chen received his BS (1985) and MS (1988) degrees in chemistry from Zhejiang University in China, and his PhD from the National University of Singapore (2000). He worked with Professors Omar M. Yaghi, Stephen Lee, and Andrew W. Maverick, respectively, during 2000–2003 before joining the University of Texas-Pan American in 2003. He is now a professor at The University of Texas at San Antonio, working on multifunctional MOFs and HOFs. He is a Fellow of AAAS, FRSC and EURASC. In 2011, Dr Chen was ranked as the 15th Top Chemist over the past decade (2000 to 2010) based on the citation impact score by the Thomson Reuters. Since 2014, he was chosen annually as a highly cited researcher in Chemistry by the Clarivate Analytics. In November 2018 and October 2021, he received the Humboldt Prize and 2021 Southwest Regional ACS Award, respectively. |
1. Introduction
Currently, energy crisis and environmental pollution are two important issues facing mankind, caused by rapid industrialization and overpopulation that consume massive amounts of fossil fuels and generate huge amounts of carbon dioxide (CO2) and other greenhouse gases. The global concentration of atmospheric CO2 has crossed the unprecedented threshold of 400 ppm. To create an environmentally friendly and carbon-neutral society, finding new energy resources becomes urgent. Due to its renewability, universal abundance, pollution-free combustion (H2O as the only product without CO2 emission), and high specific energy (120 MJ kg−1vs. 44.5 MJ kg−1 for gasoline), hydrogen (H2) stands at the forefront among alternative energy carriers and is envisioned as a clean sustainable fuel of the future to replace fossil fuels. A major obstacle to the widespread implementation of H2 gas as a primary fuel source, especially for on-board mobile transportation, is its extremely low volumetric energy density (0.0813 g L−1 at 298 K and 1 bar) because of its volatility under ambient conditions. Although the energy density can be increased by liquefaction (70.8 g L−1 at 20.4 K and 1 bar) or to a lesser extent by compression (39.2 g L−1 at 298 K and 700 bar) or by the combined cryo-compression (64.1 g L−1 at 77 K and 350 bar); the above-mentioned physical densification techniques suffer from the issues of using either complex well-insulated cryogenic tanks to maintain low temperature or heavy- and thick-walled high-pressure vessels with expensive compressors to support high pressure, which involves high cost and is energy-intensive, thus preventing it from being competitive with the widely used gasoline and precluding its use in mass-market applications. Therefore, safe, efficient, and economically and technically viable storage of H2 remains a big concern for practical application.For on-board vehicle applications, the current scheme is to fabricate a fuel cell vehicle enabling a 300 mile driving range for which 5.6 kg of H2 must be stored and released in a safe and efficient manner. To encourage research in this important field and advance the move toward a H2 economy, the US DOE (United States Department of Energy) has set the 2020-year (4.5 wt%, 30 g L−1), 2025-year (5.5 wt%, 40 g L−1), and ultimate (6.5 wt%, 50 g L−1) targets for on-board storage systems. The data in brackets correspond to the storage capacities on both gravimetric and volumetric bases. It is worth noting that these targets are based on the mass and volume of the entire system including the storage tank, porous material, and all other essential accessories. Although the storage pressure is not specified, an operating pressure below 100 bar has the potential to reduce the costs associated with the storage vessel and compression while maintaining reasonable capacities. The working temperature should be at near room temperature in the range of 233–333 K.
The adsorption-based technique has received significant attention and has been proposed as an alternative approach to increasing the storage density at non-cryogenic temperatures and lower pressures. However, the success of the technique will call for the development of effective materials that are capable of storing a large amount of H2 under relatively mild conditions with small volume, light weight, and fast kinetics and high reversibility for charging and delivering H2. Although metal hydride systems displaying high volumetric storage capacity have been intensely examined in this respect, there are still unsolved issues associated with their use including high dehydrogenation temperature, low specific uptake by mass, slow kinetics, susceptibility to impurity contamination, and high cost. Besides metal hydrides, various adsorbents such as carbon materials,1 zeolites, and polymers have been studied for H2 storage, with the molecular identity of H2 being maintained, but systematic engineering of their structures is difficult. Furthermore, they do not meet the gravimetric and volumetric storage targets because of their relatively weak interaction with H2.
Metal–organic frameworks (MOFs, also known as porous coordination polymers) have emerged as an exciting class of porous solid adsorbents formed by the solution assembly of inorganic metal ions or metal-containing clusters and organic linkers that generates multi-dimensionally extended networks. In principle, variation of metal ions/clusters and organic ligands enables a multitude of design possibilities for MOF materials allowing for the fine-tuning of their porosity and pore chemistry for a wide range of potential applications. With respect to the porosity, the pore size spans from the ultra-micro-porosity to meso-porosity and even macro-porosity. Unusually high internal surface areas of more than 7000 m2 g−1 are also observed for MOFs that are not readily achieved in traditional porous materials. Regarding the pore chemistry, the desired functional groups and/or metal binding sites can be programmatically incorporated for specific guest-binding interactions. Such chemical tunability can be used to improve their properties for a desired application. Besides inherent design and synthetic flexibility, their highly crystalline nature facilitates the structural characterization via X-ray diffraction techniques and the establishment of the structure–performance correlation. These interesting properties have spurred tremendous interest in the use of MOFs as solid-state adsorbent materials for numerous applications such as fuel storage,2 mixture separation,3 molecular sensing,4 nonlinear optics,5 heterogeneous catalysis,6 and drug delivery,2e,h,7 including H2 storage discussed herein.
Since MOF-5 was reported for the first time for reversible H2 uptake in 2003 by Yaghi et al., investigation of porous MOFs for H2 storage has become one of the most active research fields in materials chemistry. At the early stage of research, the MOFs reported usually exhibited limited H2 uptake capacities due to the partial structure collapse/distortion/amorphization upon guest removal, and most studies mainly focused on the low-pressure adsorption and selective adsorption properties such as H2/N2 and H2/O2 separations.8 With the further development of MOF chemistry and the in-depth understanding of the H2 adsorption mechanism including position, occupancy and binding energy via advanced characterization techniques, together with the advent of mild activation methods such as supercritical CO2 drying9 and freeze drying10 and the availability of high-pressure adsorption measurement instruments, their full potential is being realized, overcoming the early limitations. Although important progress on H2 storage has been made and several excellent and comprehensive reviews on H2 storage have been published,2a,c,d,f,g,11 some key issues listed below are not still well addressed, necessitating further research. (1) Although the quantities of H2 adsorbed in some MOFs (for example, 10 wt% and 66 g L−1 for MOF-5 at 77 K and 100 bar12) have exceeded the DOE on-board H2 storage targets, the storage temperature of usually 77 K makes their use economically unfavourable. Nonetheless, once the storage temperature increases to room temperature, the storage capacity drops significantly and becomes quite limited, typically 1–2 wt% at 100 bar, as a result of weak nonspecific disperse-type interaction as mirrored by the adsorption enthalpies (Qst) ranging from −4 to −8 kJ mol−1, which equally limits their practical applications. Therefore, improving the room-temperature storage capacity of MOFs via enhancing the H2 binding energy is highly desirable, which however remains a long-standing challenge to be addressed. Thermodynamic considerations have indicated that the enthalpy for H2 adsorption should fall in the optimal range of −15 to −20 kJ mol−1 to have reversible storage around ambient temperature. (2) There is still a trade-off effect between the gravimetric and volumetric H2 uptake capacities, which makes it very difficult to concurrently meet the gravimetric and volumetric target capacity requirements set by US DOE. Therefore, how to balance the gravimetrical and volumetric capacities is also a crucial issue to be considered.† (3) The working/deliverable capacity is the most important consideration when evaluating adsorbents for gas fuel storage because it determines the driving range of mobile vehicles. A minimum pressure of 5 bar is assumed to be necessary for the fuel injector in the vehicle, while 100 bar is regarded as the maximum pressure that could ease tank-design constraints and safety concerns. How to improve the H2 working/deliverable capacity is also a vital issue. (4) Last but not least, it is important to understand the H2 binding characteristic and derive the structure–performance relationship, which is favourable to fabricating next-generation adsorbent materials for H2 storage. The exploration of the H2 location within MOFs and its structural behaviour during adsorption allows the delineation of the structural features that maximize H2 loading. However, such an investigation is quite scarce due to the limited availability of some advanced spectroscopic instruments in common laboratories. In this feature article, starting from the introduction of several important classes of MOFs for H2 storage, from which some valuable information and even lessons guiding the design and synthesis on H2-storage MOF materials can be gleaned, we would like to highlight the recent progress associated with the above-mentioned aspects, with the main focus placed on the chemical strategies on the optimization and improvement of the framework–H2 interactions.
2. Several types of classic MOFs for H2 storage
2.1 Zn4O-Based series
IRMOFs belong to the tetrahedral Zn4O unit-based framework family in which the organic component can be systematically varied in terms of size and functionality.14 A typical representative of this series is MOF-5 constructed from the 1,4-benzenedicarboxylate linker (Fig. 1a); its H2 adsorption properties were investigated for the first time by Yaghi et al. in 2003.13 The maximum cryogenic uptake capacity is recorded as 4.5 wt%, while the ambient-temperature one is linearly related to the storage pressure, reaching 1 wt% at 20 bar. Inelastic neutron scattering (INS) studies revealed that both the metal oxide cluster and the organic unit are the primary adsorption sites for H2 bound in MOF-5 (Fig. 1c), indicating that the H2 storage capacities of MOFs can be optimized by metal replacement and linker tuning. Although it was found later by the authors that the uptake data were incorrect, which might be caused by the presence of impurities displaying a stronger affinity compared to H2,15 this pioneering work absolutely highlighted the huge potential of porous MOFs for application in H2 storage, which has attracted the attention of many researchers in academia and industry and triggered a lot of research on MOFs for H2 storage. Besides, it is suggested that a D2 isotherm should be measured to validate that the observed uptake is due to H2 rather than other impurities. In the following year, considering that the organic linker is also one of the crucial adsorption sites in MOF-5, the same group measured H2 sorption properties at 77 K and 1 atm of a series of chemically functionalized IRMOFs including IRMOF-1, 8, 11 and 18, together with MOF-177.15 MOF-177 is another typical example of Zn4O-based MOFs constructed from the tritopic BTB (4,4′,4′′′-benzene-1,3,5-triyl-tribenzoate) linker with different topological structures.16 It is found that the H2 uptake scales with the number of organic rings per formula unit while is uncorrelated with the apparent surface area, which is different from that documented for porous nanostructured carbons.17 Obviously, this work again authenticated the importance of an organic linker on H2 adsorption. Due to the mixed results on the adsorption properties of the prototypical MOF-5 reported in the open literature, Long et al. systematically investigated the impact of preparation and handling on the H2 storage properties of MOF-5.12 Optimizing the reaction parameters including solvent, temperature, and time during the solvothermal assembly, coupled with the avoidance of water and air exposure during sample handling, furnished a material with a BET (Brunauer–Emmett–Teller) surface area of up to 3800 m2 g−1. At 77 K and 100 bar, the total gravimetric and volumetric H2 uptakes, respectively, reached 10 wt% and 66 g L−1, which are quite remarkable (Fig. 1b). In principle, ligand extension allows the formation of the expanded isoreticular analogues, but sometimes framework interpenetration occurs, which not only precludes high porosity but also hampers attaining the phase-pure sample for further investigation, as demonstrated in the case of IRMOF-8. Matzger et al. adopted the room-temperature route, instead of the solvothermal route, to synthesize phase-pure non-interpenetrated IRMOF-8, as indicated by the experimental and calculated consistency in terms of the surface area and pore size.18 H2 adsorption studies revealed the gravimetric uptake of 1.23 wt% at 77 K and 1 bar and Qst values falling in the 5.5–4.6 kJ mol−1 range.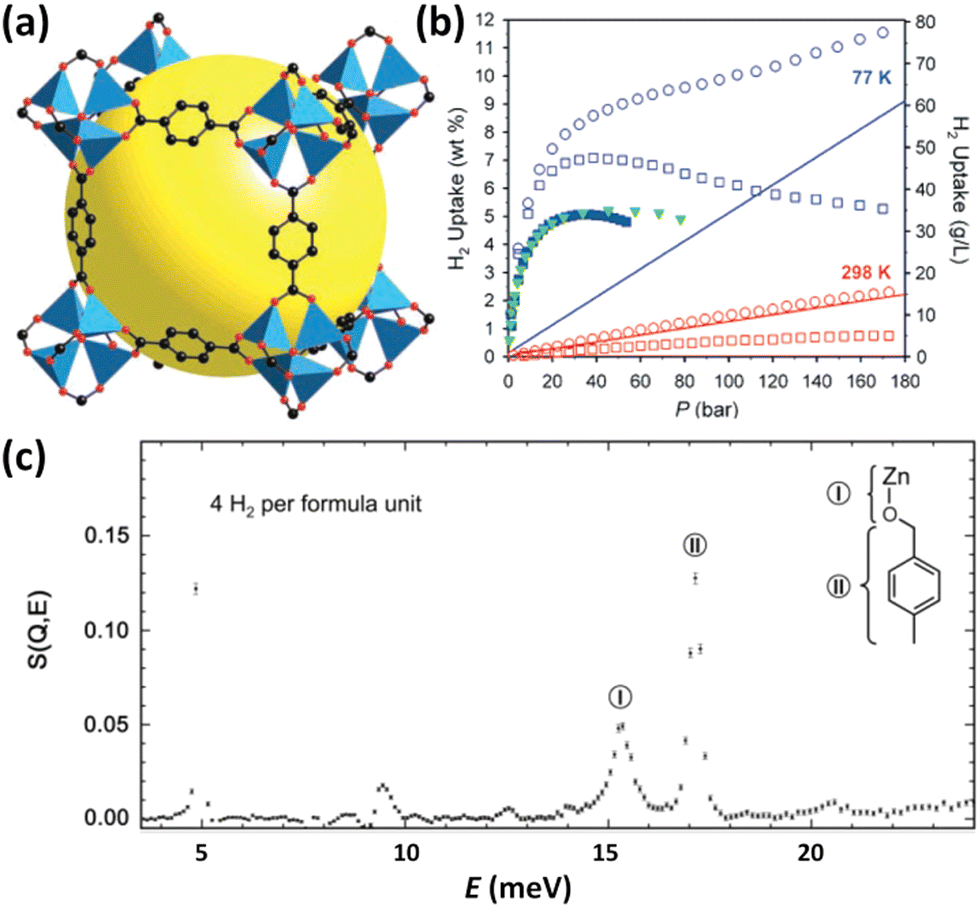 | ||
| Fig. 1 The structure, H2 adsorption properties, and H2 binding sites of MOF-5. (a) The structure is illustrated using a cube fragment with the void highlighted by a yellow sphere. (b) Comparison of the H2 isotherms of the samples with and without air exposure, which are distinguished using the filled and open symbols, respectively. The densities of pure H2 gas indicated by the solid lines were also included for a visual comparison. The square and circle symbols represent the excess and total uptake data. (c) INS spectra revealed the two binding sites corresponding to the inorganic cluster and organic ligand, which were labelled as sites I and II and schematically shown in the top right corner. Reprinted with permission from ref. 12 and 13. Copyright 2007, American Chemical Society, and Copyright 2003, Science. | ||
Apart from the IRMOF series, combination of an inorganic Zn4O-cluster with a single tritopic tricarboxylate linker or with mixed tricarboxylate/dicarboxylate linkers yielded frameworks with exceptional porosity as exemplified by MOF-180/200/205/210.19 At 77 K and 80 bar, the total gravimetric uptake capacities of MOF-200, MOF-205, and MOF-210, respectively, reached 16.3 wt%, 12.0 wt%, and 17.6 wt%, which are all higher than that of MOF-177 (11.6 wt%) under the same conditions. However, due to the low crystal density and unsuitable pore size, the volumetric uptake capacities are relatively moderate. The corresponding values, respectively, are 36, 46, and 44 g L−1, which are smaller than that of MOF-177 (50 g L−1). Other Zn4O-based ultrahigh-surface-area materials synthesized by adopting the same mixed-linker or coordination copolymerization strategy include the UMCM series. UMCM-1 derived from tritopic BTB and ditopic BDC (1,4-benzenedicarboxylate) linkers showed a high BET surface area of 4730 m2 g−1.20 Interestingly, when the BDC linker was replaced with a longer thieno[3,2]thiophene-2,5-dicarboxylate, a topologically distinct compound UMCM-2 was obtained, which is attributed to the different linker arrangement around the Zn4O cluster. UMCM-2 exhibited a BET surface area of 5200 m2 g−1 and total gravimetric and volumetric H2 uptakes of 12.4 wt% and 50 g L−1 at 77 K and 80 bar.21
2.2 Prussian blue series
Prussian blue analogues possess purely inorganic face-centred cubic framework structures. Each node is occupied by metal ions that are bridged by CN− ions to form a three-dimensional network. To satisfy the correct charge balance, framework vacancy is formed at the hexacyanometalate sites in the entire structure. Taking Fe4[Fe(CN)6]3·14H2O Prussian blue itself as an example, there is one-quarter of the [Fe(CN)6]4− sites missing, thus leading to a framework structure featuring vacancies at 25% of the [Fe(CN)6]4− sites with water molecules fulfilling the octahedral coordination of some of the Fe3+ ions. It is expected that the combination of divalent M2+ ions with [M′(CN)6]3− complexes will yield vacancies at one-third of the hexacyanometalate sites and thus Prussian blue analogues with the framework formula of M3[M′(CN)6]2 bearing larger porosity are generated. Upon dehydration, both the vacant coordination sites on the nitrogen-bound divalent M2+ cations and the polarizable CN− bridging group furnish the potential sites for binding H2. In view of the considerations mentioned above, Long et al. examined the porosities and H2 storage properties of six Prussian blue analogues M3[Co(CN)6]2 (M = Mn, Fe, Co, Ni, Cu, and Zn).22 The porosity characterization via argon sorption measurements revealed that all the compounds investigated are microporous materials, with the BET specific surface area varying from 560 to 870 m2 g−1, depending on the divalent metal ions. At 77 K and 1.2 bar, the storage capacity varies from 1.4 wt% in the Zn compound to 1.8 wt% in the Cu compound (Fig. 2a). With the exception of the Ni compound, the H2 adsorption enthalpies match the Irving–Williams stability order (Fig. 2b). The larger enthalpy of adsorption for the Ni compound is likely attributed to its smaller crystallite size. Importantly, the enthalpies of adsorption for the Prussian blue analogues are all significantly higher than the 4.7–5.2 kJ mol−1 range observed for MOF-5 without open metal sites (OMSs). Due to the high H2 adsorption capacity of Cu3[Co(CN)6]2, Hartman et al. investigated the H2 binding characteristic of this material.23 Neutron powder diffraction (NPD) studies revealed two adsorption sites for H2 molecule. One is located at the (0.25, 0.25, 0.25) crystallographic site, while the other one is situated on the exposed Cu2+ ion (Fig. 2c). Neutron vibrational spectroscopy measurements showed broad peaks (Fig. 2d), indicating a range of local binding potential for the H2 molecule adsorbed, which may be explained by the aperiodic arrangement of framework vacancies.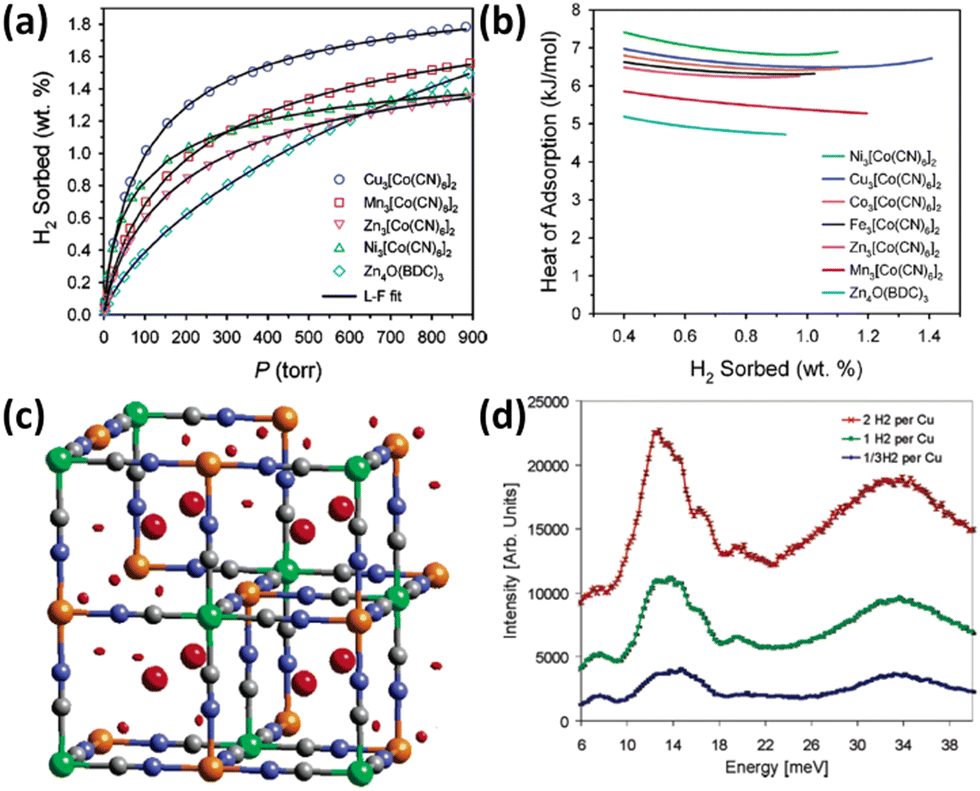 | ||
| Fig. 2 The isotherms, heat of adsorption, and binding mechanism with respect to H2 in M3[Co(CN)6]2 Prussian blue analogues (M = Mn, Fe, Co, Ni, Cu, and Zn). (a) The H2 uptake data are represented by the open symbols, and compared with the Langmuir–Freundlich fitting results. For clarity, the H2 isotherms of the Fe and Co compounds are not shown due to the isotherm similarity between the Fe and Zn compounds as well as between the Co and Mn compounds. MOF-5 was also included for comparison. (b) The isosteric heat of H2 adsorption in M3[Co(CN)6]2 compounds is plotted as a function of H2 coverages. (c) Difference Fourier analyses of NPD data revealed two sites for D2 adsorbed in the Cu compound highlighted in red colour but different shapes, which are referred to as the (1/4, 1/4.1/4) interstitial location and unsaturated copper site. (d) Neutron vibrational spectroscopy of the Cu compound loaded with different amounts of H2 displayed the broad peaks attributable to a range of local bonding potentials for the adsorbed H2 molecules. Reprinted with permission from ref. 22 and 23. Copyright 2005, American Chemical Society, and Copyright 2006, American Chemical Society. | ||
Furthermore, valence state alternation of the metal ions provided diverse Prussian blue analogues with varied concentrations of framework vacancies. The variation of framework vacancy concentration in turn affects the material properties such as permanent porosities, framework densities, and concentration of open metal sites as well as the stability of the framework against desolvation. Their interplay leads to a complicated effect on H2 adsorption performance. Eight Prussian blue analogues (Ga[Co(CN)6] (0%), Fe4[Fe(CN)6]3 (25%), Cu3[Co(CN)6]2 (33%), M2[Fe(CN)6] (M = Mn, Co, Ni, and Cu) (50%) and Co3[Co(CN)5]2; the value given in parentheses indicates framework vacancy concentration) were synthesized to compare the impact of framework vacancy concentration on the porosities and H2 sorption properties.24 The BET surface area ranges from 550 m2 g−1 for Fe4[Fe(CN)6]3 to 750 m2 g−1 for Cu3[Co(CN)6]2, while the uptake capacity at 77 K and 1.2 bar varies from 0.9 wt% in Co2[Fe(CN)6] to 2.3 wt% in Cu2[Fe(CN)6]. The maximum storage uptake capacities predicted by isotherm fitting with Langmuir–Freundlich equation are correlated with the framework vacancy concentrations, while the adsorption heats are not significantly higher for compounds incorporating framework vacancies than for fully saturated frameworks, suggesting that the H2 binding interaction in these materials is not dominated by the exposed metal coordination sites.
2.3 MOF-74 series
The MOF-74 (also referred to as CPO-27) series based on the tetraanionic dobdc (2,5-dihydroxyterephthalate/2,5-dioxido-1,4-benzenedicarboxylate) linker and dicationic metal ion represent a family of isostructural framework compounds. They feature one-dimensional honeycomb-like hexagonal channels of about 12 Å in diameter equipped with unsaturated metal coordination sites at the vertices that are accessible to the incoming guest molecules of suitable sizes. Due to the exceptionally high density of OMSs attainable by removing the axially occupied solvent molecules, they have been widely investigated for various applications including catalysis, conductivity, drug delivery, and storage and separation of various gases.25 Particularly, this structure type is compatible with the incorporation of a wide array of metal species. To date, eight different isostructural analogues with M = Mg, Mn, Fe, Co, Ni, Cu, Zn, and Cd have been fabricated. As such, they serve as an ideal platform to systematically understand the framework–adsorbate interactions.In 2008, Zhou et al. measured H2 sorption isotherms of five MOF-74 compounds based on Mg2+, Mn2+, Co2+, Ni2+, and Zn2+.29Qst analyses showed that the H2 binding energy is heavily dependent on the metal ions, following the trend of Zn < Mn < Mg < Co < Ni. Within this series, the Zn compound holds the lowest value of −8.5 kJ mol−1, while the Ni compound has the highest value of −12.9 kJ mol−1. The trend observed for the four transition metal compounds matches the Irving–Williams sequence holding true for high-spin complexes. As revealed by first-principles calculations, the major interaction between the H2 molecule and the open metal site originates from the Coulomb attraction. With regard to the Ni compound, the binding mechanism has been exposed by means of low-temperature IR spectrometry30 as well as NPD.26a During H2 adsorption, the IR absorbance bands appear in two distinct regions. The 4010–4040 cm−1 region is associated with that interacting primarily with Ni2+ ions, while the 4110–4150 cm−1 region corresponds to H2 bound to the organic ligand. On the basis of variable-temperature IR spectroscopy, the adsorption enthalpy was calculated to be −13.5 kJ mol−1, basically consistent with the studies of Zhou et al. mentioned above. NPD studies revealed that D2 was located close to the open Ni(II) ion (Fig. 3a), with the Ni(II)–D2 separation as short as 2.20 Å, which well correlates with the high initial heat of H2 adsorption. Considering that Mg2+ acts as a lightweight cation with a high charge density, the low-pressure and high-pressure H2 adsorption properties as well as the H2 binding nature of the Mg compound were further investigated in depth.26b The Mg compound exhibited a H2 uptake capacity of 2.2 wt% at 77 K and 1 bar and a zero-coverage isosteric heat of H2 adsorption at −10.3 kJ mol−1. The total adsorption amount reaches 4.9 wt% and 45 g L−1 at 77 K and 100 bar, which is significantly reduced to 0.8 wt% and 7.5 g L−1 when the temperature increases to 298 K. Analyses of variable-temperature isotherms and FTIR yielded the comparable isosteric adsorption heats of −10.3 and −12.1 kJ mol−1, respectively. NPD experiments showed that the exposed Mg2+ ion is the highest-affinity adsorption site with a Mg–D2 distance of 2.45 Å, while the oxido group is the secondary adsorption site for D2 with a D2–Ooxido distance of 3.5 Å (Fig. 3b), which is also supported by the INS studies. After this, the redox-active member of the MOF-74 series, Fe2(dobdc), was successfully synthesized,31 and the H2 storage characteristics of the Fe compound and its oxidized version were also investigated.27b The H2 uptake capacity at 77 K and 1.2 bar is 2.2 wt% and the initial H2 adsorption heat is −9.7 kJ mol−1 for the Fe(II) compound. The oxidized counterpart exhibits a higher initial H2 adsorption heat (−10 kJ mol−1) in magnitude than the un-oxidized parent solid as a result of the higher charge density and thus the stronger polarizability of Fe3+ compared to Fe2+, while it shows lower H2 storage capacity due to the reduced surface area and higher molecular weight. NPD explorations revealed that the open Fe2+ site is the primary adsorption site for D2 in the Fe(II) compound, with Fe–D2 contact of 2.47 Å (Fig. 3c). Two additional adsorption sites were also observed, in which D2 interacts with the one bound in the Fe(II) site with the intermolecular distance of 2.87–3.22 Å as well as the framework with the closest atom–D2 contacts of 3.2–3.3 Å. With respect to the oxidized counterpart Fe2(O2)(dobdc), the peroxide species was also the binding position for D2 adsorption, in addition to the three adsorption sites observed in the parent Fe(II) compound.
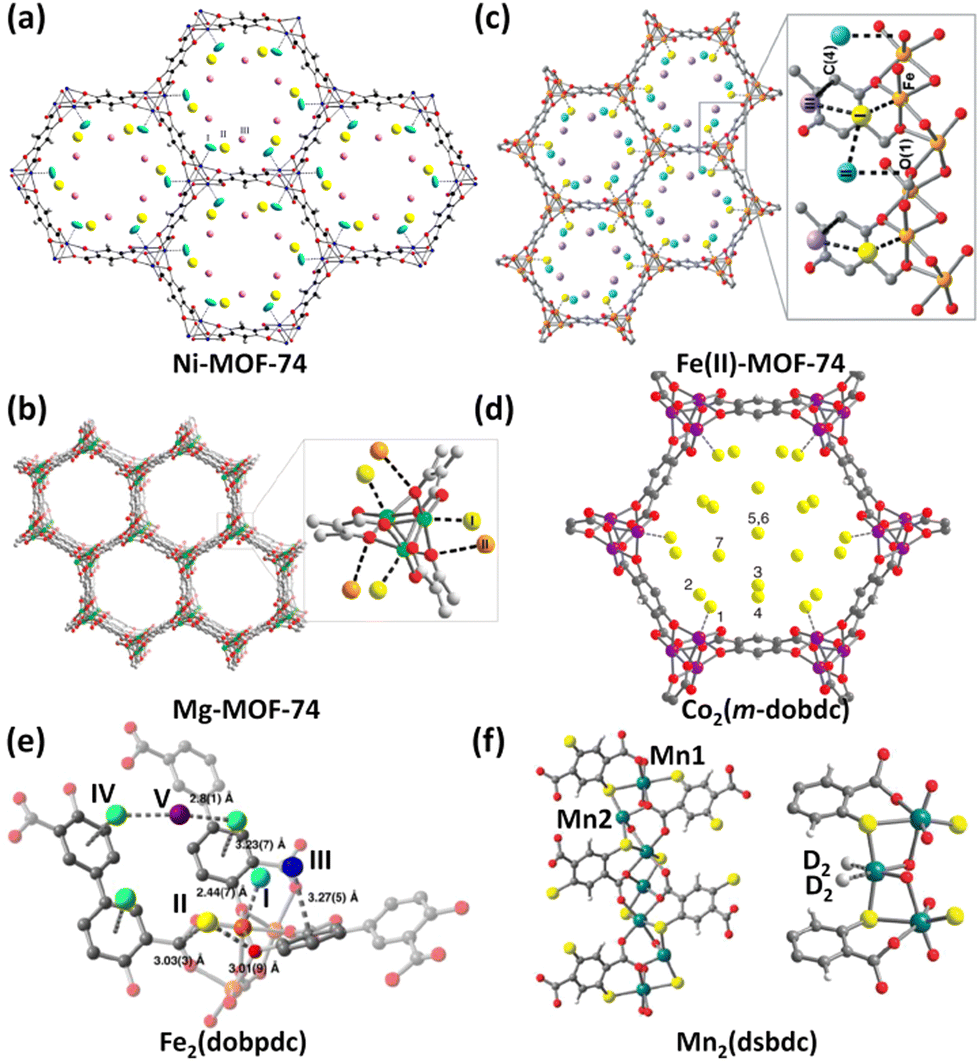 | ||
| Fig. 3 NPD experiments revealed (a) three D2 binding sites in Ni-MOF-74, (b) two D2 binding sites in Mg-MOF-74, (c) three D2 binding sites in Fe(II)-MOF-74, (d) seven D2 binding sites in the Co-MOF-74 isomer, and (e) five D2 binding sites in Fe2(dobpdc). The experimental D2 loadings are 1.5 D2 per Ni2+ ion, 0.6 D2 per Mg2+ ion, 2.25 D2 per Fe2+ ion, 3.0 D2 per Co2+ ion, and 4.5 D2 per Fe2+ ion. (f) shows two different coordination environments of Mn2+ ions and two D2 molecules bound by Mn22+ ions in the Mn2(dsbdc) compound as determined by NPD studies with a loading of 0.7 D2 per Mn22+ ion. Reprinted with permission from ref. 26–28. Copyright 2013, Elsevier; Copyright 2011–2012 and 2016, Royal Society of Chemistry; and Copyright 2014, 2016, 2018 American Chemical Society. | ||
The pore environment of MOF-74 frameworks can be further varied by linker isomerization. On using m-dobdc (4,6-dioxido-1,3-benzenedicarboxylate) as the isomeric ligand,28a the resulting framework still retains the same overall topology. Although Ni2(m-dobdc) and Ni2(dobdc) belong to a pair of ligand-originated framework isomers, the ligand field alteration results in higher charge densities of unsaturated Ni2+ centres in the isomeric version than that in the parent compound.28b As a result, Ni2(m-dobdc) exhibits a binding enthalpy of up to −13.7 kJ mol−1, which is 1.4 kJ mol−1 larger than that of Ni2(dobdc). At 298 K and 100 bar, Ni2(m-dobdc) takes up 11.9 g L−1 of H2, which is among the highest for the reported adsorbents under the same conditions. The usable capacity under the 5–100 bar pressure swing at 298 K is slightly reduced to 11.0 g L−1, which still outperforms that of compressed H2 under the conditions (7.3 g L−1). As a comparison, H2 is required to be compressed over 150 bar to obtain the same total volumetric usable capacity at 298 K. Taking Co2(m-dobdc) as an example, NPD investigations revealed seven distinct adsorption sites (Fig. 3d), among which the open Co2+ site is the strongest adsorption site (Fig. 3d). Notably, Ni2(m-dobdc) is the top-performing material with respect to the critical metric of usable volumetric H2 capacity at pressures between 5 and 100 bar and near ambient temperature.
By using extended but geometrically equivalent ligands such as 4,4-dioxido-3,3-biphenyldicarboxylate (dobpdc)28c,32 and other analogues with multiple phenylene groups,33 the pores of the MOF-74 framework can be expanded while preserving the parent framework structure. Long et al. investigated the H2 storage properties of the expanded MOF-74 family with a larger pore diameter of 18–22 Å based on the dobpdc linker and six different metal ions (Zn2+, Mn2+, Fe2+, Mg2+, Co2+, and Ni2+).28c Isosteric heat of H2 adsorption ranging from −8.4 to −12.0 kJ mol−1 followed a similar trend to the above-mentioned MOF-74 series. However, as a result of the higher surface areas and pore volumes, they exhibited enhanced gravimetric H2 uptake capacities but a lower volumetric one compared to the non-expanded MOF-74 series. Taking the Fe(II) compound as an example, NPD experiments revealed two extra adsorption sites, which were not observed for the non-expanded framework (Fig. 3e). The primary adsorption occurs in the metal centre with a Fe–D2 distance of 2.44 Å comparable to the one found for Fe2(dobdc). The secondary site is located in the middle of the two adjacent primary sites with close D2–Ccarboxyalte and D2–Oaryoxide distances of 3.03 and 3.01 Å. The third and fourth loci lie at the ligand benzene rings. The fifth site occurs in between the two sites IV. At 298 K and 100 bar, the total gravimetric capacities range from 1.3 wt% (Zn) to 1.8 wt% (Mg) and correlate reasonably well with the respective BET surface areas. Also, the drug olsalazine was employed as an extended ligand to synthesize a mesoporous series of expanded MOF-74 analogues (M = Mg, Fe, Co, Ni, and Zn) with the pore diameter of about 27 Å.34 Despite larger pore apertures, these frameworks exhibited Qst values comparable to those of M2(m-dobdc), ranging from −10.8 to −12.0 kJ mol−1. Variable-temperature H2 adsorption isotherms revealed strong adsorption at the open metal sites. Besides, these frameworks also displayed the utility for drug delivery.
In the materials mentioned above, each metal site is only capable of adsorbing a single gas molecule, thus resulting in a limited storage capacity. It is speculated that if a metal site contains more than one terminal solvent molecule, the low-coordinated metal centre generated via desolvation can accommodate more gas molecules. Based on this idea, Long et al. used a thiolated analogue (dsbdc, 2,5-disulfido-1,4-benzenedicarboxylate) of the dobdc linker to construct a Mn(II)-based framework compound.27a Different from Mn-MOF-74 in which all the metal has the same coordination environment, this compound contains two different kinds of octahedral metal centres (Fig. 3f). Mn1 is coordinated by six ligating atoms coming from the dsbdc linker, while the coordination of Mn2 is fulfilled with just four ligating atoms originating from dsbdc linkers and two cis-arranged DMF molecules. The DMF molecules can be removed upon activation as confirmed by structural analyses using synchrotron X-ray powder diffraction. NPD experiments disclosed that the exposed four-coordinated Mn2+ ion is capable of binding two gas molecules such as D2, CD4 and CO2 (Fig. 3f). The Mn2–D2 separation is 3.40 and 3.07 Å for D2 loadings of 0.7 and 1.4 per Mn2 ion, respectively, indicating relatively weak metal–D2 interactions. This is also consistent with the result of adsorption heat extracted from temperature-dependent H2 adsorption isotherms. Mn2(dsbdc) exhibited a very modest initial binding enthalpy of −5.6 kJ mol−1, significantly lower than the corresponding value of −8.8 kJ mol−1 in Mn2(dobdc). The relatively weak binding enthalpy is due to the larger radius of sulfide atom relative to that of the oxido atom.
2.4 Metal-tetrazolate frameworks
Due to the similarities between tetrazolate and carboxylate in terms of for example pKa values, the tetrazolate-based ligands have been designed and applied to construct porous MOFs.35 The use of polytetrazolate as bridging ligands is deemed to be favourable for creating rigid frameworks with exposed metal coordination sites. As a demonstration, Long et al. used 1,3,5-benzenetristetrazolate (BTT) to fabricate a Mn(II)-based compound (Mn-BTT) in which the chloride-centred square-planar [Mn4Cl] units are connected via the BTT ligands to yield an anionic sodalite-type network that is charge-balanced by [Mn(DMF)6]2+ counterions.36 Desolvation engineering enables optimization of this compound to exhibit a higher surface area (2100 m2 g−1 BET) and more accessible Mn(II) sites, and thus a better H2 storage capacity. At 77 K and 90 bar, the gravimetric and volumetric uptake capacities of Mn-BTT reached 6.9 wt% and 60 g L−1, respectively. The latter value is only 11 g L−1 lower than the liquid-H2 density. The initial adsorption heat was found to be −10.1 kJ mol−1, which is the highest value recorded for a MOF solid at that time. NPD studies with sequenced H2 loadings revealed four adsorption sites for H2 which are summarized in Fig. 4a. Both site I and site II are the two strongest adsorption sites. The D2 molecule at the site I is located on the open Mn(II) centre with a Mn(II)–D2 distance of 2.27 Å, while the one at site II displays multi-site interactions with both the chloride anion and four tetrazole rings. The D2 molecule at the third site located inside the large cuboctahedral cage has van der Waals contact with two tetrazole rings with a distance of 3.26 Å between D2 and the centroid of the tetrazole ring. The fourth H2 binding site was situated inside the small octahedral cage in which the four carbon atoms from the surrounding tetrazole rings are in the closest contact with the bound D2 molecule therein. This work provided the first neutron diffraction evidence for a metal–D2 interaction within a MOF.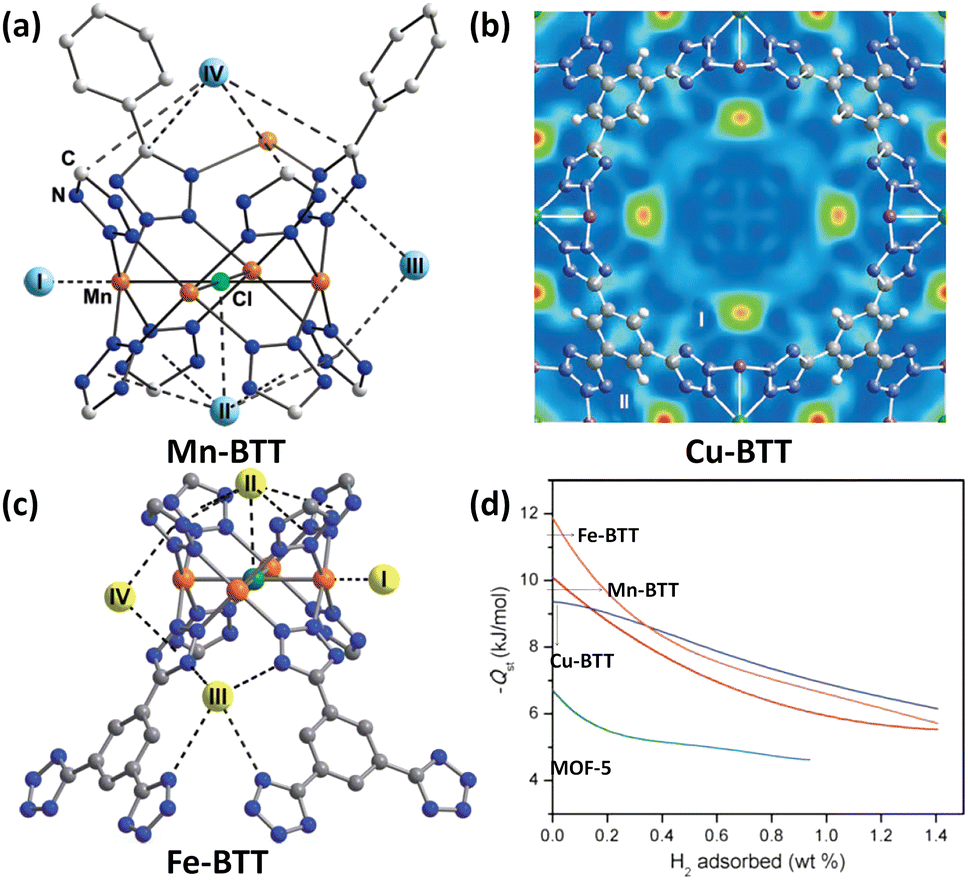 | ||
| Fig. 4 D2 binding sites in (a) Mn-BTT, (b) Cu-BTT and (c) Fe-BTT compounds as determined by the NPD experiment with the D2 loadings of 12, 30 and 20 per formula unit, respectively, and (d) comparison of their H2 adsorption heats. Reprinted with permission from ref. 36, 38 and 39. Copyright 2006, American Chemical Society; Copyright 2007, Wiley; Copyright 2010, Royal Society of Chemistry. | ||
The above experiments showed that the large adsorption enthalpy of Mn-BTT is due in part to the strong interaction between D2 molecules and unsaturated Mn2+ ions within the anionic framework skeleton. To tune the metal–D2 interaction, the same group undertook the post-synthetic ion exchange experiments and studied the H2 adsorption properties of the respective ion-exchanged frameworks.37 The results showed that the H2 adsorption enthalpies can be varied through ion exchange, and the Co(II)-exchanged compound exhibited an initial adsorption enthalpy of −10.5 kJ mol−1, which is 0.4 kJ mol−1 higher in magnitude than that of the pristine material Mn-BTT.
The inability to fully desolvate Mn-BTT limits its maximum potential for H2 storage. Replacement of Mn2+ with Cu2+ yielded a Cu-BTT compound that can however be fully activated to expose a greater number of open metal coordination sites for H2 adsorption.38 With respect to the H2 uptake, Cu-BTT exceeds Mn-BTT at 77 K and 1.2 bar, despite the lower surface area. However, the lower surface area of Cu-BTT resulted in its lower total gravimetric H2 uptake at a higher pressure of 90 bar (5.7 wt% vs. 6.9 wt%) in comparison to that of Mn-BTT. In terms of the H2 adsorption enthalpy, the initial value is lower for Cu-BTT (9.5 kJ mol−1) than for Mn-BTT (10.1 kJ mol−1) due to the Jahn–Teller effect of the copper(II) ion resulting in lower binding affinity toward H2 relative to the Mn(II) ion, which was also corroborated by NPD studies revealing that the Cu(II)–D2 distance of 2.47 Å is slighter longer than the 2.27 Å observed for the Mn(II)–D2 distance in Mn-BTT (Fig. 4b). However, with the increasing H2 loadings, the H2 adsorption heat of Cu-BTT surpassed that of Mn-BTT, which is attributed to Cu-BTT possessing more OMSs available.
Substitution of Mn(II) or Cu(II) ions in the structure with divalent cations bearing a smaller radius provides a possible approach to improve the isosteric heat of H2 adsorption because the higher charge density of the exposed metal cations on the framework surface facilitates inducing a dipole moment in H2, thus leading to stronger binding.39 To this end, they prepared an Fe(II)-based analogue via a high throughput methodology. The Fe(II) compound exhibited an initial H2 adsorption heat of −11.9 kJ mol−1, which is higher in magnitude than that of Mn-BTT (−10.1 kJ mol−1) as a result of the higher charge-to-radius ratio for Fe2+ compared to Mn2+ (Fig. 4d). Also, the initial adsorption heat of Fe-BTT is larger than that of Cu-BTT (−9.5 kJ mol−1), although Cu2+ is a smaller ion than Fe2+, which might be due to the Jahn–Teller effect of Cu(II) ions that reduces the charge at the open coordination site. However, beyond 0.4 wt% H2 uptake, the isosteric heat of H2 adsorption for Cu-BTT overtakes that of Fe-BTT. This is likely due to the greater number of desolvated M2+ ions in Cu-BTT, whereas Fe-BTT retains some bound methanol molecules. The total H2 uptake of Fe-BTT reaches 4.1 wt% and 35 g L−1 at 77 K and 95 bar. These values are below the corresponding values recorded for Mn-BTT (6.9 wt%, 60 g L−1 at 90 bar), and Cu-BTT (5.7 wt%, 53 g L−1 at 90 bar). NPD studies reveal the four D2 binding site, with the framework Fe2+ ion as the strongest-affinity site (Fig. 4c). The Fe–D2 distance is as short as 2.17 Å.
One strategy for adjusting the adsorption capacity in microporous frameworks involves ligand elongation to expand the known structure types. To this end, Long et al. designed and synthesized two expanded versions related to the aforementioned BTT ligand, namely, 1,3,5-tri-p-(tetrazol-5-yl)phenylbenzene (H3TPB-3tz) and 2,4,6-tri-p-(tetrazol-5-yl)phenyltriazine (H3TPT-3tz), and constructed the corresponding MOFs that are isotypic with that of M-BTT as revealed by single-crystal X-ray diffraction.40 Interestingly, the ligand-directed interpenetration occurs during the assembly process. Solvothermal reaction of the benzene-centred ligand with the copper ion generated the non-interpenetrated framework, while the resulting framework is doubly interpenetrated upon using the triazine-centred linker to assemble with Mn2+ or Cu2+ ion. Gas adsorption studies showed that the interpenetrated compound exhibited better framework stabilities and higher H2 storage performance. At 77 K and 80 bar, the total H2 uptake capacity of the Mn-TPT-3tz compound reached 4.5 wt% and 37 g L−1, respectively. Due to the lack of unsaturated metal centres, the volumetric storage capacity of the Mn-TPT-3tz compound is lower than that of the pristine Mn-BTT compound.
2.5 Copper-multicarboxylate frameworks
Polycarboxylate ligands are a class of the most widely used organic ligands because of their strong coordination and chelation abilities as well as flexible coordination modes. In particular, coordination of the carboxylate group with Cu2+ is prone to yield the typical Cu2(COO)4 dicopper paddlewheel unit with the terminal solvent molecules easily desorbed as a result of the Jahn–Teller effect of the copper(II) ion, thus yielding an open copper site to increase H2 adsorption. Therefore, copper-multicarboxylate frameworks have been widely explored as H2 storage materials.HKUST-1 is an iconic coordination framework compound composed of a dicopper paddlewheel bridged by a 3-connected organic linker 1,3,5-BTC (benzene-1,3,5-tricarboxilate) to form a tbo-type network. The removal of the axially bound H2O molecule makes the framework Cu(II) sites available for interaction with the H2 molecule, which is evidenced by in situ IR spectroscopy performed at 15 K showing a v(H–H) band of Cu(II)–H2 adducts.41 Also, the NPD studies established six binding sites for D2 adsorbed in HKUST-1 (Fig. 5a). The most favoured one of these sites lies in close proximity to the open Cu sites with a Cu–D2 distance of 2.39 Å.42 A triazine-centred tricarboxylate ligand, namely, 4,4′,4′′-s-triazine-2,4,6-triyltribenzoate (TATB), was used to construct a pair of framework catenation isomers (namely, PCN-6 and PCN-6′), revealing that the framework catenation is favourable for the enhancement of H2 adsorption because the interpenetration divided the large void into a smaller one that can better fit the H2 molecule.44 At 77 K and 50 bar, the total gravimetric and volumetric H2 uptake capacities of the interpenetrated compound PCN-6 are 9.5 wt% and 53 g L−1, which are systematically higher than the corresponding values of 5.8 wt% and 16.2 g L−1 for the noninterpenetrated counterpart PCN-6′. The INS studies indicated that the much stronger interactions of adsorbed H2 with the organic linker in the catenated material originate from a greater number of interacting atoms from the organic ligands, especially at high H2 loadings.
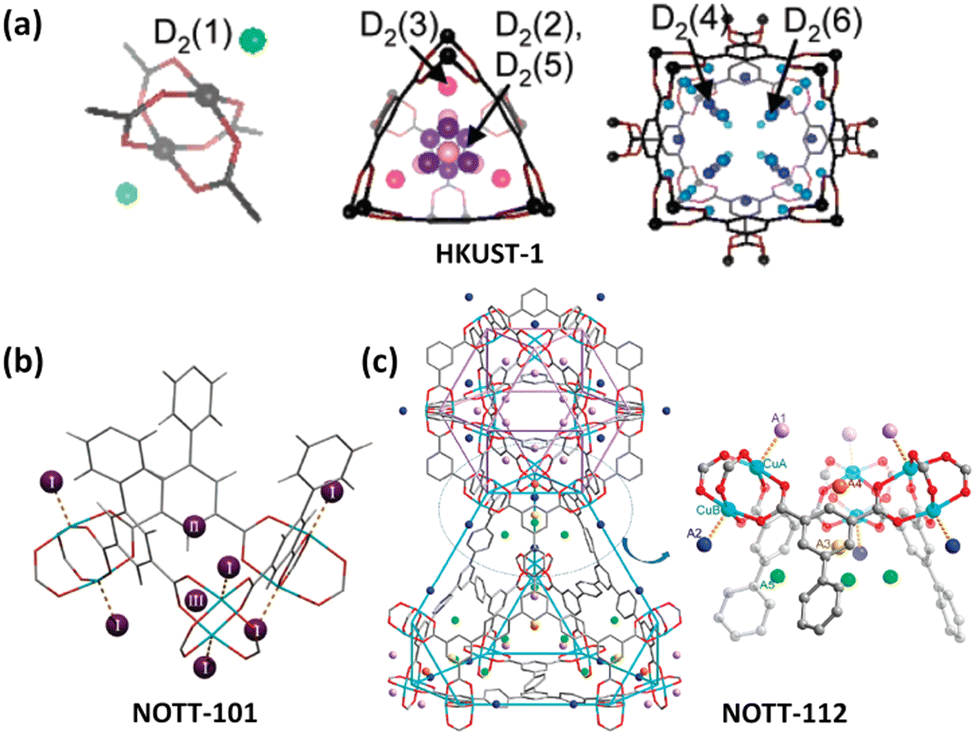 | ||
| Fig. 5 D2 binding sites in copper-multicarboxylate frameworks of (a) HKUST-1, (b) NOTT-101 and (c) NOTT-112 compounds as determined by the NPD experiment with D2 loadings of 4, 1.82 and 2.0 per copper ion, respectively. Reprinted with permission from ref. 42 and 43. Copyright 2006, and 2009–2010, American Chemical Society. | ||
Isophthalate-containing multicarboxylate ligands are in particular attractive for framework construction because of their propensity to form cage-type architectures.45 Chen et al. reported the first copper–diisophthalate framework MOF-505 based on 3,3′,5,5′-biphenyltetracarboxylic acid for H2 adsorption.46 Studies of the effect of the desolvation conditions on H2 uptake indicated the favourable role of the open copper site. The fully desolvated MOF-505 exhibited 2.47 wt% H2 uptake at 77 K and close to 1 bar. After this, a variety of diisophthalate ligands incorporating distinct central spacers between two terminal isophthalate units were developed, and the corresponding copper(II) frameworks were constructed for H2 adsorption studies. For example, Schröder et al. studied the H2 adsorption properties of a series of copper–diisopthalate frameworks based on tetracarboxylic acids bearing a range of polyaromatic backbones,43b,47 revealing that the low-pressure H2 uptake is mainly dominated by the adsorbate–adsorbent interaction that is closely related to the pore size and pore surface chemistry, while the available pore volume controls the high-pressure H2 adsorption capacities. In particular, there exists an optimal pore size to obtain a significant H2 storage density. With respect to NOTT-101, NPD studies revealed that the exposed copper site, the triangular window and the cusp formed by three phenyl rings are three main adsorption sites for H2 (Fig. 5b). The Cu–D2 distance is 2.50 Å, which is slightly longer than that observed in HKUST-1 (2.39 Å),42 but clearly not of the “Kubas”-type binding.48 Replacement of biphenyl in NOTT-102 with phenanthrene and 9,10-dihydrophenanthrene yielded the two frameworks NOTT-110 and NOTT-111. Compared to the parent compound, they exhibited a similar surface area and slightly lower pore sizes but significant enhancement of H2 uptake at the low to medium pressure region at 77 K, indicating that the ligand curvature enables enhancement of H2 adsorption.47a Zhou et al. used a methylene-bridged diisophthalate ligand to construct a pair of copper-based framework isomers (PCN-12 and PCN-12′). Comparison of their uptake properties revealed that the rational arrangement of the open copper site has a significant effect on H2 uptake.49 At 77 and 1.01 bar, the H2 uptake capacity of 3.05 wt% for PCN-12 is higher than that of 2.4 wt% for its isomer PCN-12′.
The organic linkers bearing more than two isophthalate subunits were also involved in MOF construction. A rht-type topological platform based on the dicopper paddlewheel unit and triisophthalate linkers was developed by several well-known research groups of Eddaoudi,50 Schröder,43a,51 Farha,52 and Zhou,50 and explored for high-pressure H2 adsorption studies. The entire structure of this type can be described as the packing of three types of polyhedral cages of cuboctahedron, truncated tetrahedron, and truncated octahedron in the ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1, and has the advantage of prohibiting network interpenetration. Due to ultrahigh porosity, hierarchical cage, and OMS incorporation, they exhibited impressive H2 uptake capacities. The MOFs of this structure type investigated include PCN-68/610,50 NU-100/110/111,52d and NOTT-112/113/114/115/116/119/122 series.51 At 77 K and 100 bar, the total H2 uptake of PCN-68 reaches 13 wt%.50 NPD studies on NOTT-112 have revealed five different binding sites for H2 molecules (Fig. 5c).43a Interestingly, the chemically un-equivalent copper(II) ions displayed different affinity towards H2 molecules. The H2 binding first occurred at the vacant copper site (CuA) within the cuboctahedral cage followed by the outside one (CuB) as the secondary binding site. The distances are 2.23 and 2.41 Å for CuA–D2 and CuB–D2, respectively. The D2 molecules sited at the third and fourth sites were populated nearby the triangular windows formed by three dicopper paddlewheel units and three isophthalate moieties connecting the cuboctahedral cage and the truncated tetrahedral cage. The fifth binding site is positioned with the truncated tetrahedral cage around the three-fold axis of the triangular window. Besides, a few copper-tetraisophthalate frameworks have been designed for H2 storage investigation.53 For example, three aromatic-rich binaphthalene-based octacarboxylates were employed as 8-connected organic linkers to construct porous copper-tetraisophthalate frameworks. They displayed high stability and impressive H2 adsorption. Furthermore, H2 uptake capacities can be tuned by altering side groups, ranging from 1.8 to 2.5 wt% at 77 K and 1.01 bar.53b Also, NOTT-140 with tetrahedrally-branched tetraisophthalate as a ligand was reported by Schröder's group to exhibit the total H2 uptake capacity of 6 wt% at 77 K and 20 bar.53a
:2:1, and has the advantage of prohibiting network interpenetration. Due to ultrahigh porosity, hierarchical cage, and OMS incorporation, they exhibited impressive H2 uptake capacities. The MOFs of this structure type investigated include PCN-68/610,50 NU-100/110/111,52d and NOTT-112/113/114/115/116/119/122 series.51 At 77 K and 100 bar, the total H2 uptake of PCN-68 reaches 13 wt%.50 NPD studies on NOTT-112 have revealed five different binding sites for H2 molecules (Fig. 5c).43a Interestingly, the chemically un-equivalent copper(II) ions displayed different affinity towards H2 molecules. The H2 binding first occurred at the vacant copper site (CuA) within the cuboctahedral cage followed by the outside one (CuB) as the secondary binding site. The distances are 2.23 and 2.41 Å for CuA–D2 and CuB–D2, respectively. The D2 molecules sited at the third and fourth sites were populated nearby the triangular windows formed by three dicopper paddlewheel units and three isophthalate moieties connecting the cuboctahedral cage and the truncated tetrahedral cage. The fifth binding site is positioned with the truncated tetrahedral cage around the three-fold axis of the triangular window. Besides, a few copper-tetraisophthalate frameworks have been designed for H2 storage investigation.53 For example, three aromatic-rich binaphthalene-based octacarboxylates were employed as 8-connected organic linkers to construct porous copper-tetraisophthalate frameworks. They displayed high stability and impressive H2 adsorption. Furthermore, H2 uptake capacities can be tuned by altering side groups, ranging from 1.8 to 2.5 wt% at 77 K and 1.01 bar.53b Also, NOTT-140 with tetrahedrally-branched tetraisophthalate as a ligand was reported by Schröder's group to exhibit the total H2 uptake capacity of 6 wt% at 77 K and 20 bar.53a
3. Regulation and optimization of H2 adsorption performance
As a new entry to the field of H2 storage materials, MOFs can be engineered in terms of pore dimension and shape as well as pore surface environment via the chemistry with respect to their compositions of inorganic secondary building unit (SBU), organic linkers and guest species for the purpose of modifying and improving the H2 adsorption performance. In the following section, we discussed the strategies used for increasing the H2–framework interaction and ambient-temperature H2 storage performance, balancing the gravimetric and volumetric capacities as well as improving the H2 working capacities.3.1 Increasing the H2–framework interactions and ambient-temperature H2 storage performance
| MOFs | OMS | M–D2 (centroid) distance (Å) | −Q st (kJ mol−1) | Ref. |
|---|---|---|---|---|
| a Determined by DFT calculations. | ||||
| Cu(I)-MFU-4l | Cu(I) | 1.6, 1.66a | 32 | 55 |
| V2Cl2.8(btdd) | V(II) | 1.966(8) | 20.9 | 56 |
| MFM-132 | Cu(II) | CuA–D2: 2.07(2) | 6.8 | 57 |
| CuB–D2: 2.329(15) | ||||
| Fe-BTT | Fe(II) | 2.17(5) | 11.9 | 39 |
| Ni-MOF-74 | Ni(II) | 2.201(1) | 11.9 | 26a |
| Co2(m-dobdc) | Co(II) | 2.23(5) | 11.5 | 28a |
| NOTT-112 | Cu(II) | CuA–D2: 2.23(1) | 5.64 | 43a and 51 |
| CuB–D2: 2.41(1) | ||||
| Ni2(m-dobdc) | Ni(II) | 2.25(7), 2.18(4) | 12.3 | 28a and b |
| Mn-BTT | Mn(II) | 2.27 | 10.1 | 36 |
| Co-MOF-74 | Co(II) | 2.32(2) | 10.8 | 28a |
| HKUST-1 | Cu(II) | 2.39(1) | 6.8 | 42 |
| Fe2(dobpdc) | Fe(II) | 2.44(7) | 10.0 | 28c |
| Mg-MOF-74 | Mg(II) | 2.45(4) | 10.3 | 26b |
| Cu-BTT | Cu(II) | 2.47 | 9.5 | 38 |
| Fe-MOF-74 | Fe(II) | 2.47(3) | 9.7 | 27b |
| NOTT-101 | Cu(II) | 2.50(3) | 5.38 | 43b |
| Cr-BTC | Cr(II) | 2.63(2) | 7.4 | 58 |
| Mn2(dsbdc) | Mn(II) | 3.40(4) | 5.6 | 27a |
One effective approach to fabricate OMS is to remove the terminal solvent molecules attached to the metallic node during solvothermal assembly. The degree of solvent removal is highly dependent on the activation conditions including the activation solvents, activation temperature, and ramping rate. With respect to the activation solvent, low-boiling solvents such as methanol and acetone were frequently used to exchange with the guest molecules encapsulated in the pore space and even replace the terminally coordinated amide solvent molecules such as DMF (N,N-dimethylformamide). In terms of activation temperature, a too low one does not liberate all the terminal solvent molecules, while a too high one might result in the risk of thermal collapse of the framework. In order to maximize the pore volume and OMS density determining the saturated amount of H2 adsorption and H2 binding energetics, full activation should be established. Therefore, the activation conditions need to be carefully optimized prior to H2 adsorption measurement. Long's work on the metal–tetrazolate frameworks for H2 storage has clearly highlighted the significance of desolvation on H2 adsorption performance.
As exemplified by several classes of important MOFs mentioned above, metal–H2 interaction heavily depends on the nature of the metal ion and its surrounding coordination environment. The studies on the impact of divalent metal ions on the H2 adsorption heat in MOF-74, expanded MOF-74 and M-BTT series showed that the isosteric heat of H2 adsorption usually correlates with the ionic radii of the metal ions. As the ionic radius of the metal ion decreases, the metal ion displays a higher charge density and thus a stronger polarizing ability, resulting in the enhanced interaction between metal and dihydrogen molecule. It should be mentioned that the copper ion is frequently an exception as a result of the Jahn–Teller effect originating from its d9 electronic configuration. In particular, comparison of the D2 binding behaviour of Cr-BTC58 and HKUST-142 also highlighted the importance of electronic configuration of the metal ion on H2 binding. There are two chemically un-equivalent copper ions in NOTT-112 and MFM-132, and they displayed different binding energies towards H2, indicating that the surrounding of copper ions is capable of yielding a certain effect on H2 binding. Therefore, the charge, radius, and electronic configuration as well as its arrangement and coordination environment should be carefully considered upon the design and construction of OMSs for H2 affinity improvement.
In the above examples, the Coulomb force was mainly involved in the metal–H2 interaction. Inspired by a high binding enthalpy of −80 kJ mol−1 in the metal–dihydrogen compound W(CO)3(iPr3P)2(η2-H2) discovered by Kubas and co-workers,59 Long et al. reported the first framework compound of V2Cl2.8(btdd) (H2btdd = bis(1H-1,2,3-triazolo[4,5-b],[4′,5′-i])dibenzo[1,4]dioxin) exhibiting H2 adsorption enthalpy within the optimal range for ambient temperature H2 storage by using a less-reducing, weaker π-basic vanadium site capable of backbonding interactions with H2 (Fig. 6a and b).56 In this material, vanadium is of mixed valences with the V(III):V(II) ratio of 2:3. The empty dσ orbital of V(II) in this material accepts the electron from the HOMO of the H2 molecule, and at the same time the dπ orbital offers the electron to the LUMO of the H2 molecule, thus forming a “σ + π” synergistic effect to enhance the metal–H2 interaction. This binding nature was collaborated by a series of isotherm measurements, spectroscopy characterization studies and theoretical calculations. The Clausius–Clapeyron analyses of the temperature-dependent H2 isotherms yielded an isosteric heat of H2 adsorption of −20.9 kJ mol−1, which falls in the optimal range required for ambient-temperature H2 storage. Infrared spectroscopy studies showed the appearance of two new peaks at 3919 and 4112 cm−1 relative to the bare framework spectrum upon dosing with H2 at 97 K, which are, respectively, attributed to H2 strongly bound to the open V(II) sites, and H2 physisorbed at the secondary non-metallic site within the materials. Red shift occurs for H2 bound in the metal sites in V2Cl2.8(tbdd) compared to Ni2(m-dobdc), indicating the stronger backbonding from dπ to LUMO, but the degree in red shift is much smaller than that typically characterized in molecular metal–dihydrogen complexes, indicating the attenuated dihydrogen activation. The weaker activation present in V2Cl2.8btdd is likely a result of the greater ionic charge at the metal centre as well as the weaker surrounding ligand field environment. Analyses of the variable-temperature IR spectrum using van’t Hoff's equation give a H2 binding enthalpy of −21 kJ mol−1, which is consistent with the results from isotherm measurement. Furthermore, the V–D2 interaction was directly observed through NPD studies, revealing that D2 binds at the V(II) centre with a distance of 1.966(8) Å accompanied with a slight change with respect to the V–Cl equatorial bonds and the Cl–V–Cl bond angle that contract and decrease, respectively, upon D2 binding (Fig. 6c). High-pressure H2 adsorption measurements revealed that V2Cl2.8(tbdd) achieved a total volumetric H2 uptake of 10.7 g L−1 at 298 K and 100 bar (Fig. 3d). The data demonstrated that stronger orbital-mediated interactions with π-basic metal sites provide a novel means of optimizing the thermodynamics of H2 adsorption in porous materials for storage application.
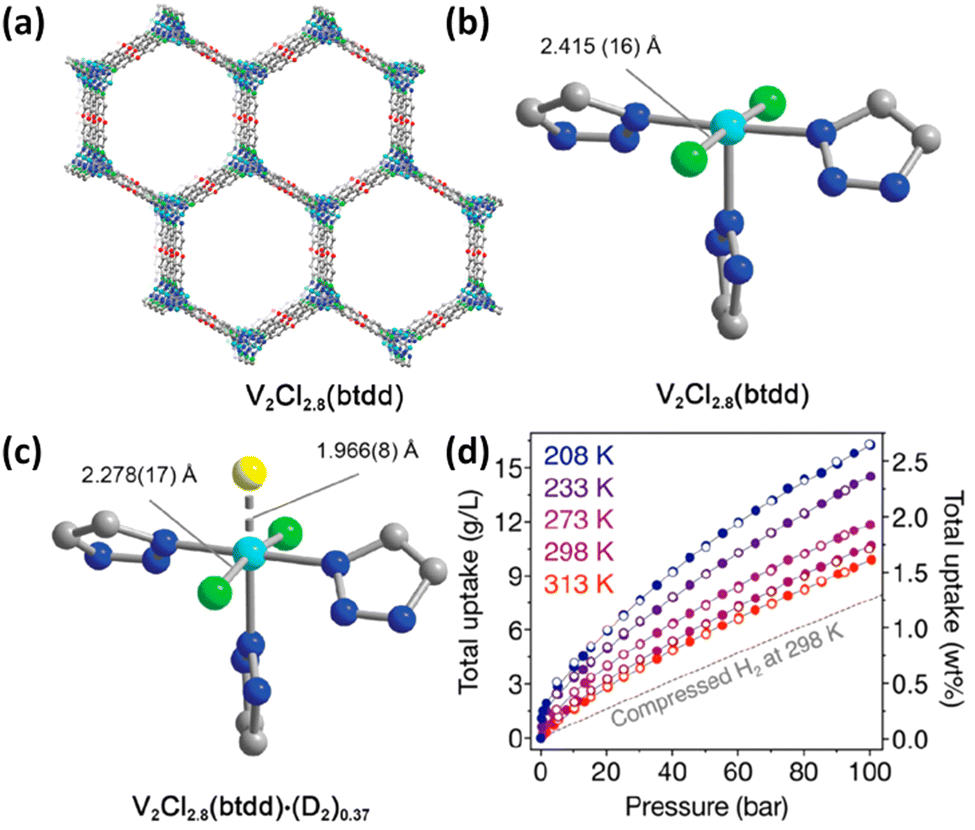 | ||
| Fig. 6 (a) The 3D structure of V2Cl2.8(btdd) and the coordination environment of the V(II) centre in (b) the activated framework and (c) in the framework dosed with 0.75 equiv. of D2 molecules. (d) The high-pressure H2 isotherm in the temperature range of 208–313 K. Reprinted with permission from ref. 56. Copyright 2021, American Chemical Society. | ||
Apart from OMS, the bridging group within the inorganic SBU was identified as the primary binding site for H2. For example, the NPD study confirms that the free bridging hydroxyl group within the pore of MFM-300(In) is the primary binding site for adsorbed D2 molecules.60 D2 forms a direct binding interaction with the bridging hydroxyl group in the pore, and the binding distance is slightly longer than that observed in MOFs with OMSs, consistent with the nature of the different binding mechanisms.
Asides from the design and selection of metal ions, cluster post-modification is recognized as one of the effective means to fine-tune the framework structure and thus the adsorption performance. Volkmer et al. reported an elegant example of post-synthetic cluster chemistry performed in the MFU-4l framework constructed from the bistriazolate BTDD2− ligand and [Zn5Cl4]6− building unit for significantly enhanced H2 binding enthalpy (Fig. 7a).55b The peripheral Cl− moieties can be exchanged with formate ions, and subsequent thermal decomposition generated the corresponding framework with the retained structure containing metal hydride complexes that can react with electrophiles such as PhCOCl (Fig. 7b). Besides, metathesis of the terminal Zn2+ with Cu2+ followed by the Cl− to HCOO− ligand exchange and subsequent thermal activation afforded the corresponding framework material [Cu(I)-MFU-4l] bearing threefold-coordinated unsaturated Cu(I) sites that showed remarkably strong but fully reversible binding of small molecules under ambient conditions (Fig. 7b and c). The H2 adsorption heat was estimated to be up to −32.3 kJ mol−1. This work highlighted the promising potential of copper(I)-containing materials bearing a higher OMS density for H2 storage. NPD studies performed by Long et al. revealed that the D2 binding in Cu-MFU-4l followed a precursor-mediated adsorption path (Fig. 7d).55a At the D2 dosing temperature of 40 K, the unsaturated copper centre serves as a very strong binding site (site I) but with very low occupancy, while the pentanuclear tetrahedral node acts as the highest occupancy site (site II). The additional site (site I*) located above site I represents a metastable physisorbed state that serves as a precursor to chemisorption. When the dosing temperature is increased to 77 K and much higher, the sites I and I* were occupied exclusively, and the occupancy of site I becomes much populated. More recently, starting from the existing framework compound MFU-4l,61 Farha et al. adopted post-synthetic ionic exchange to obtain a Li+-decorated isorecticular compound MFU-4l-Li, exhibiting a better performance than its parent compound with respect to H2 storage and release, which is mainly attributed to the increase in the pore volume after transmetalation.62 At 77 K and 100 bar, the gravimetric and volumetric H2 uptake capacities of MFY-4l-Li are 9.9 wt% and 52.4 g L−1, which are 28.6% and 12.7%% higher than the corresponding values of the parent compound (7.7 wt% and 46.5%). Under combined temperature and pressure swing conditions (77K/100 bar → 160 K/5 bar), the gravimetric and volumetric working capacities of MFU-4l-Li are 9.4 wt% and 50.2 g L−1, which also significantly exceed those examined for the parent compound (7.3 wt% and 44.3 g L−1). The H2 isosteric heat of adsorption varies from −5.4 to −3 KJ mol−1 during the entire adsorption process. The relatively modest adsorption heat might be responsible for its high working capacity under the operational conditions. The above-mentioned adsorption data place MFU-4l-Li among the best MOF-based materials for H2 storage application.
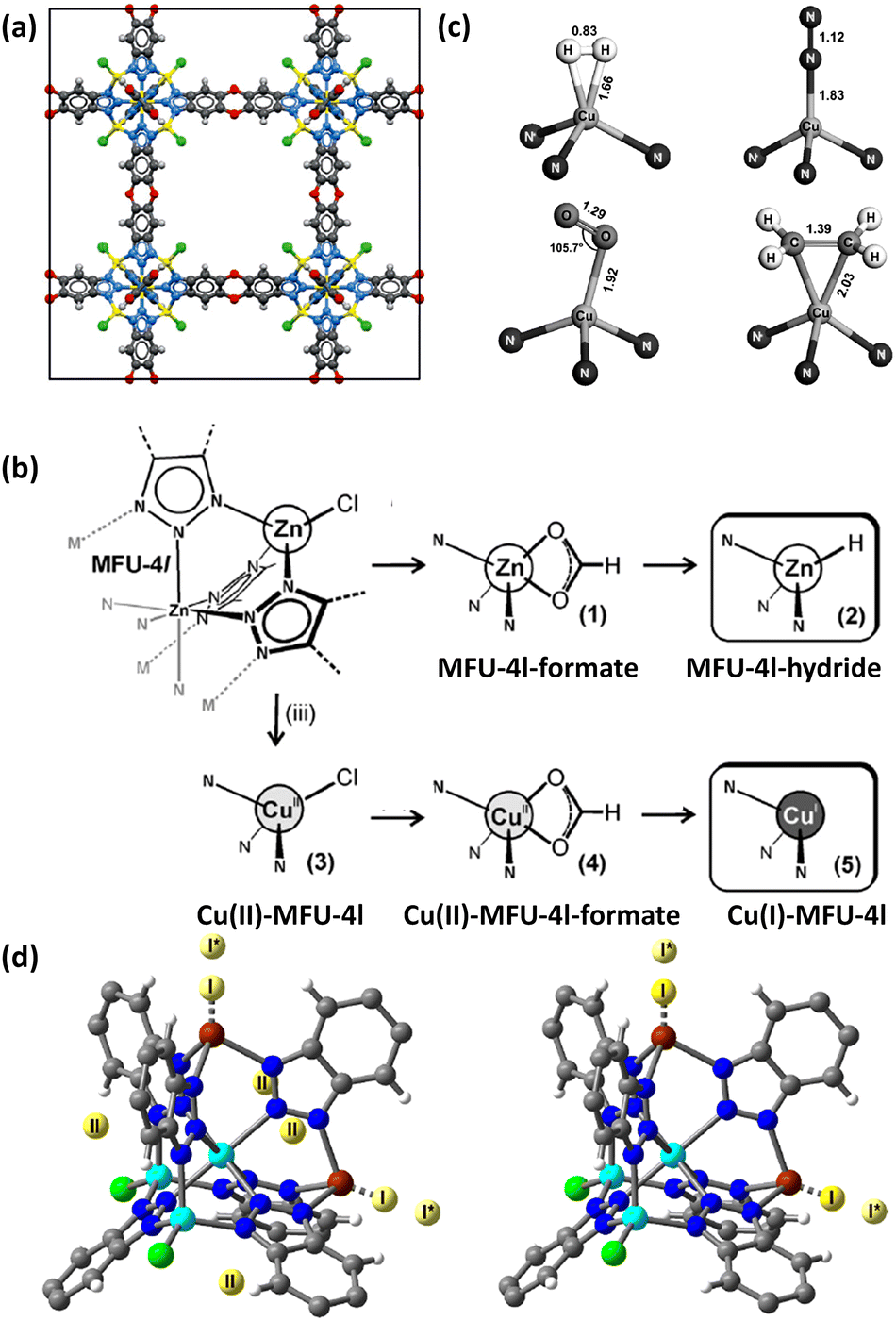 | ||
| Fig. 7 (a) The structure of MFU-4l, (b) schematic representation of the conversion of MFU-4l into its corresponding hydride-containing and Cu(I)-containing compounds, and (c) binding geometries for H2, N2, O2 and C2H4 at the Cu(I) site as determined by DFT calculations. (d) D2 binding sites for Cu(I)-MFU-4l determined at 7 K by NPD experiments at a 0.75 D2/Cu at the dosing temperatures of 40 K (left) and 77 K (right). Reprinted with permission from ref. 55a and b. Copyright 2014, Wiley. Copyright 2021, American Chemical Society. | ||
With respect to the ambient temperature H2 storage, Ni2(m-dobdc) is one of the best H2 storage materials.28b At 298 K and 100 bar, the total volumetric H2 uptake reaches 11.9 g L−1, which only slightly drops to 11.0 g L−1 upon considering the 5–100 bar working capacities. The total volumetric capacity represents a 39% increase over compressed H2 under the same conditions (7.7 g L−1). Usable H2 capacities achieved with this material are greater than those of compressed H2 under a range of conditions.
The organic ligands can be modified to induce stronger interactions with H2. Pre-synthetic incorporation of polar functional groups, electronegative heteroatoms, and crown ether as specific metal-recognizing sites into the ligand skeleton has the potential to produce the enhanced H2-binding sites.63 For example, Schröder's work showed that the pre-synthetic incorporation of functional groups such as methyl and fluorine into copper–diisophthalate frameworks endowed the resulting materials with enhanced heat of H2 adsorption.43b A Zr-MOF containing sulphur heteroatom displayed higher H2 sorption capacities than its heteroatom-free counterpart, which is attributed to the enhanced framework electronegativity arising from sulphur doping.63 SNU-200 bearing the 18-crown-6 moiety in the strut is capable of selective inclusion of cations such as K+, NH4+, and MV2+ (methyl viologen). In particular, the K+-bound compound exhibited the highest Qst value of −9.92 kJ mol−1 which is attributed to K+ ion providing accessible vacant coordination sites.64 Also, post-synthetic covalent ligand modification is also presented as a means to modify the pore structures and thus tune the H2 adsorption properties. For example, three framework compounds (IRMOF-3, UMCM-1-NH2 and DMOF-1-NH2) containing the amino group can be modified in a controlled manner with benzoic anhydride and phenylisocyanate. The resulting modified frameworks displayed enhancement in the sorption affinity of MOFs with H2 for the entire coverage range.65
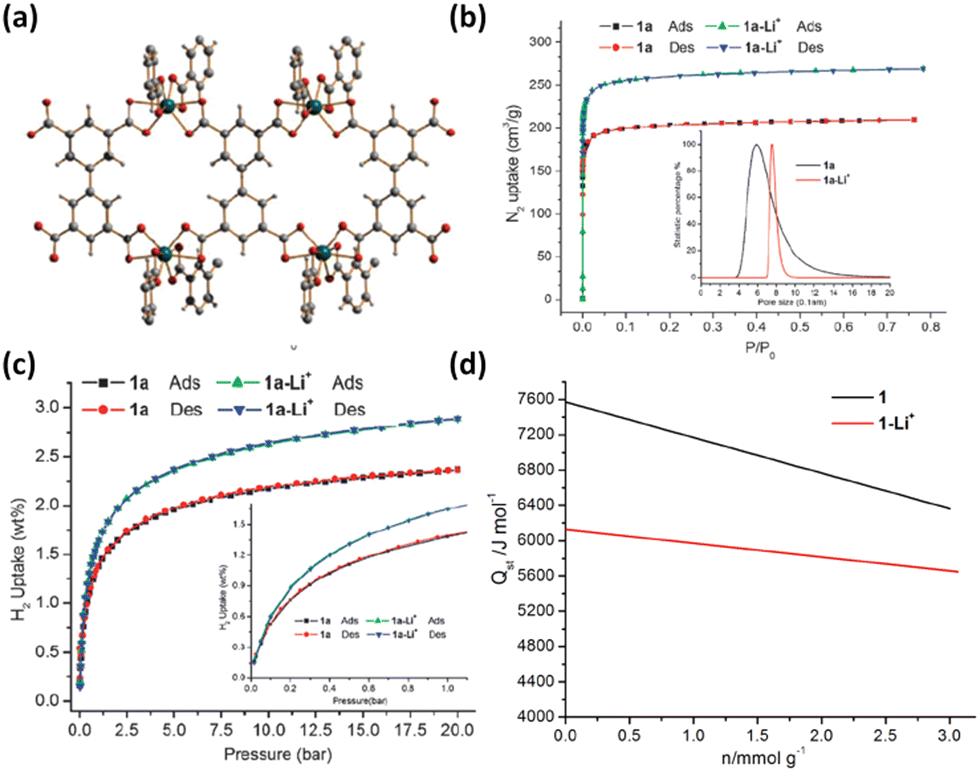
Reduction of redox-active ligands with lithium metal provides an avenue to doping the framework with Li+ ions. For example, the interpenetrated framework of Zn2(NDC)2(diPyNI) can be directly reduced with lithium metal in DMF, yielding the Li+-doped compound exhibiting a higher H2 adsorption heat over the entire loading range and a larger H2 uptake capacity at 77 K and 1 atm compared to the pristine compound (Fig. 8). The possible reasons for the significant improvement include enhanced ligand polarizability, introduction of charge-balanced Li+ ions and increased surface area induced by framework displacement.66 Goddard III et al. also employed the computational methods to confirm that such lithium-doped MOFs displayed significantly improved H2 uptake capacities at ambient temperature relative to the undoped parent compound. The Li-MOF-C300 binds 4.56 wt% H2 at 50 bar pressure, which is an order of magnitude higher than that of pure MOF-C300.67
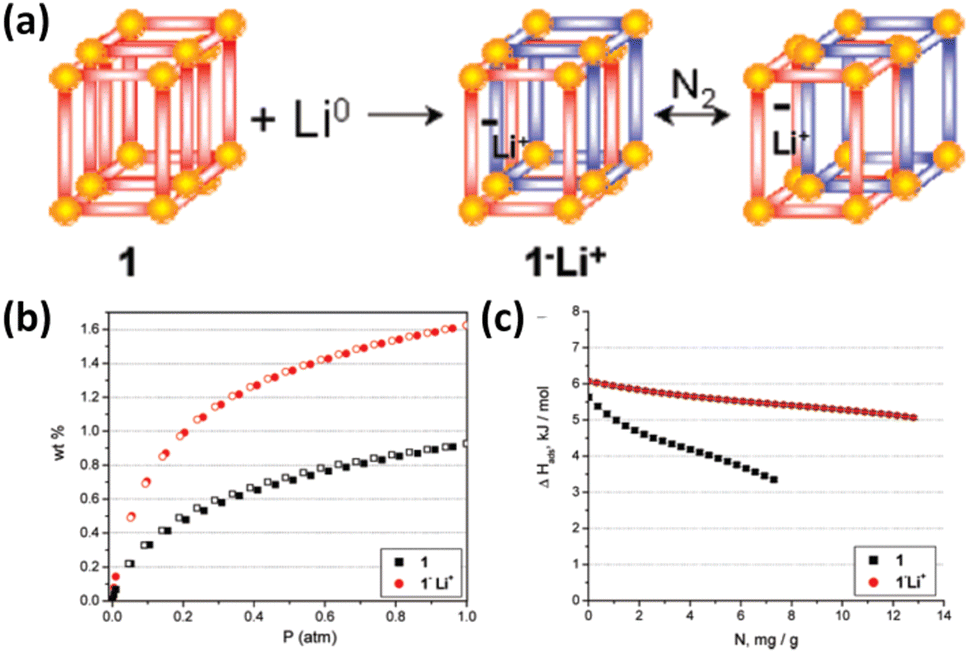 | ||
| Fig. 8 (a) Illustration of the chemical conversion of Zn2(NDC)2(diPyNI) into Zn2(NDC)2(diPyNI)·Li+via framework reduction and framework displacement leading to the enhanced (b) H2 uptake capacity and (c) isosteric heat of H2 adsorption. Reprinted with permission from ref. 66. Copyright 2007, American Chemical Society. | ||
The acid–base reaction is also utilized for linker modification. For example, the addition of a lithium alkoxide group to an organic linker is proposed as a protocol to establish Li+ doping of MOFs. A hydroxyl-modified MIL-53(Al) analogue was synthesized by substituting terephthalic acid with 2-hydroxyterephthalic acid, and subsequent deprotonation of the hydroxyl group including the bridging OH groups and the pendent OH group with lithium diisopropylamide (LDA) afforded the Li+-doping material with the preservation of the framework structure. Compared to the un-doped solid, the resulting material exhibited a slightly decreased surface area but significant enhancement in H2 uptake capacity and adsorption heat, highlighting the usefulness of Li+ doping.68
Apart from experimental exploration, theoretical investigation was performed. Froudakis et al. studied the effect of the ligand modification with lithium atoms on H2 storage capacities of MOFs via combined quantum mechanics and GCMC (grand canonical Monte Carlo) simulations.69 IRMOF-8 (naphthalene dicarboxylate as linker) and IRMOF-14 (pyrenedicarboxylate as linker) were chosen as two model MOFs in which the hydrogen atoms of the organic linkers were substituted with lithium alkoxide groups without changing the material frameworks. Compared to the unmodified MOFs, the functionalized version showed a significant improvement in terms of the interaction energies and gravimetrical H2 storage capacities at cryogenic (77 K) and ambient (300 K) temperatures.
 | ||
| Fig. 9 (a) The partial structure of (Me2NH2)[In(L)] and comparison of (b) N2 and (c) H2 isotherms at 77 K and (d) H2 adsorption heat for (Me2NH2)[In(L)] before and after Li+ exchange. Reprinted with permission from ref. 70. Copyright 2008, Royal Society of Chemistry. | ||
A strong electrostatic field can be introduced via post-synthetic ionic exchange. The ion exchange and its effect on H2 adsorption energy and uptake were investigated by utilizing a zeolite-like anionic framework compound of [Me2N]48[In48(HImDC)96] (H3ImDC = 4,5-imidazoledicarboxylic acid) as the staring compound.73 The extra-framework cation of Me2NH2+ can be fully exchanged with Li+ and Mg2+ ions. They existed in the form of aqua complexes in the resulting MOFs so that the adsorbed H2 molecules cannot directly bind with the extra-framework cations, thus resulting in similar H2 adsorption properties. However, the electrostatic field formed between the anionic framework and counterion afforded the enhanced adsorption heat of the exchanged materials compared to that of the pristine MOF because of the higher charge/size ratio of Mg2+ and Li+ ions relative to the Me2NH2+ ion.
Apart from the above-mentioned impregnation method, a direct synthesis method involving the introduction of LiOH into the solvothermal reaction system was developed for Li-doping.74 The Li-included samples displayed increased pore size, surface area, and H2 uptake capacity.
The importance of the optimized pore size for H2 adsorption was also demonstrated by NPD studies of H2 binding in a rare-earth MOF Y(BTC).78 The desolvated framework features open Y sites and tetragonal channels of about 6 Å in diameter. It was found that the strongest adsorbed position is not the OMS but is associated with the aromatic BTC linker, with a distance of 3.7 Å between the D2 and the benzene rings of the BTC linker. The results in turn indicated that small pores with an optimal pore diameter of just slightly over twice the kinetic diameter of the H2 molecule strengthen the interactions between H2 molecules and pore walls and thus increase the heat of H2 adsorption.
Apart from the pore dimension, the pore shape also plays an important effect on H2 adsorption. For example, compared to the common rectangle channels in IRMOFs, the honeycomb-like channels featuring a rolling surface in M(HBTC)(4,4′-bipy) (M = Ni and Co) strengthen the interaction of the adsorbent and H2 and thus increase the H2 adsorption properties at room temperature.79 The Ni compound shows a high H2 storage capacity of 1.20 wt% at room temperature and 72 bar.
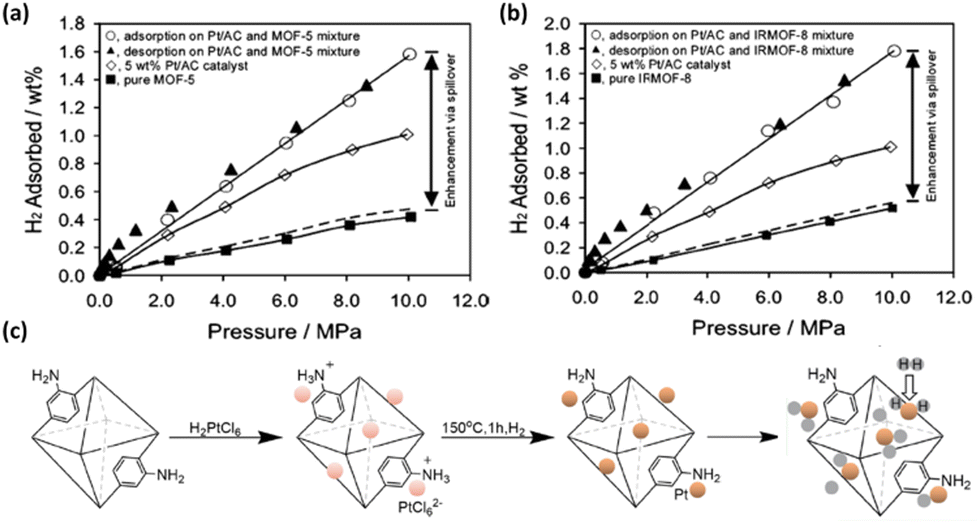 | ||
| Fig. 10 Comparison of H2 isotherms of (a) MOF-5 and (b) IRMOF-8 before and after Pt doping exhibiting significantly enhanced H2 storage capacities via the spillover effect. (c) Schematic representation of the synthetic route of Pt/aUiO that can dissociate dihydrogen to hydrogenate the organic linker. Reprinted with permission from ref. 80 and 81. Copyright 2006 and 2021, American Chemical Society. | ||
Another strategy involves metal nanoparticle fabrication inside the pore of MOFs. The preparation of Mg nanocrystals in a MOF SNU-90 based on the Zn4O cluster and the ATB (aniline-2,4,6-tribenzoate) linker was achieved by thermal decomposition of air-sensitive bis-cyclopentadienyl magnesium (Cp2Mg) vapour.82 The resulting composite material exhibited enhanced isosteric heats of H2 adsorption as well as H2 uptake at ambient temperature compared to the pristine compound, despite the lower surface area. The synergetic physisorption and chemisorption effects were responsible for the increased adsorption heat for H2 physisorption and decreased desorption temperature for H2 chemisorption.
3.2 Improving gravimetric/volumetric capacity and their balance
The gravimetric and volumetric uptake capacities are two critical indicators of H2 storage material performance, and therefore it is very important to develop effective strategies to improve them. Because the experimental and theoretical studies have revealed that the gravimetrical uptake capacity is positively correlated to the gravimetric surface area, construction of high-surface-area MOFs is a route to attaining high gravimetrical uptake capacity. This can be rationalized as follows. The critical temperature of H2 of 33 K is lower than the usual measurement temperature of 77 K. As a result, the multilayer adsorption can hardly be attained at the supercritical temperature. Monolayer adsorption dominated in the entire adsorption process in which the amount adsorbed is basically associated with the available surface area. In fact, “Chahines rule” predicts that the maximum H2 storage capacity increases linearly by 2 wt%, when the surface area increases by 1000 m2 g−1, which is decreased to 0.23 wt% when the temperature increases to ambient temperature. However, it is quite difficult to create a MOF with an extremely large surface area because of Aristotle's observation that nature abhors a vacuum. Aided by isoreticular chemistry and topological guidance, some ultrahigh-surface-area MOFs such as MOF-210,19 NU-1501,83 NOTT-11243a and PCN-68,50etc. have been synthesized. Indeed, they exhibited ultrahigh gravimetric H2 uptake capacities. Besides, utilization of a lightweight metal ion to construct metal organic frameworks is believed to be a way to improve gravimetric uptake capacity. The metal ions evaluated mainly included Mg2+,8c Be2+,84 and Al3+,83 of the main group.For the on-board transportation application, volumetric uptake capacity might be more important than the gravimetric one for a given MOF used as the H2 storage adsorbent because of the limited vehicular space. Despite its importance, MOF volumetric storage has not been studied as much as gravimetric storage. Generally, MOFs with the highest gravimetric performance exhibit modest volumetric capacities. Because the volumetric uptake capacity is a product of the gravimetric uptake and the framework/packing density, a single increase in the gravimetric surface area cannot secure the high volumetric uptake capacity. To obtain high volumetric uptake capacity, MOFs should possess a high volumetric surface area and a suitable pore size. The work from Schröder's group on engineering ligand chemistry to regulate H2 adsorption properties revealed that there exists an optimal pore size for a H2 storage material in order to realize high H2 storage densities.43b A too small pore size cannot provide enough space for H2 accommodation, while a too large pore size leads to a weak affinity to the H2 molecule. In fact, the theoretical explorations on idealized homogenous solids predicted that the pore size optimized for maximal volumetric H2 uptake capacities at 100 bar is about 7 and 10 Å at ambient temperature and cryogenic temperature, respectively.86
Asides from the structural regulation, improving the packing density of the MOF powder is also taken into account as one efficient method to optimize the volumetric storage density, which is however relatively less explored in the open literature. Indeed, inefficient material packing can lead to up to 60% loss of volumetric density on a single-crystal basis. Recently, Matzger et al. reported the significant improvement of packing density and thus volumetric uptake of the benchmark framework compound MOF-5 via engineering its morphology, size, and size distribution.85 The crystal size of MOF-5 can be regulated via changing the synthetic parameters such as feedstock ratio, reaction temperature and duration, while the introduction of carboxylate additives into the reaction system can control the relative growth rates of different crystal faces and thus tune its morphologies varying from cubic to noncubic shapes such as cuboctahedron and octahedron (Fig. 11a). For a mixture consisting of MOF-5(2349) and MOF-5(808) with different average crystal sizes shown in the brackets in a 7:1 weight ratio, the packing density can be improved by up to 33% without a significant loss of gravimetric capacity compared to the commercial MOF-5 sample. It was demonstrated by system model projections that the volumetric capacity of a typical 700 bar compressed storage system (25 g L−1) and the DOE target 2020 volumetric capacity (30 g L−1) can be surpassed via engineering crystal morphology/size or use of a bimodal distribution of cubic crystal sizes coupled with system optimization.
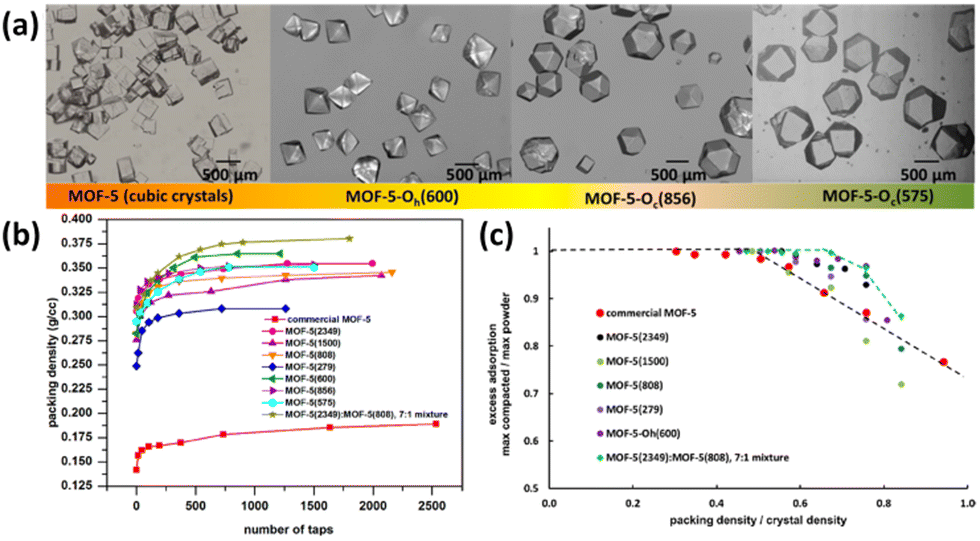 | ||
| Fig. 11 (a) Different morphologies and sizes of MOF-5 achieved through varying the solvothermal conditions and introduction of a morphology modifier. The number in the parentheses indicates the mean crystal size in microns. Comparison of (b) packing density and (c) H2 uptake indicated the importance of crystal morphology and size on optimizing volumetric H2 storage performance in MOFs. Reprinted with permission from ref. 85. Copyright 2021, American Chemical Society. | ||
There usually exists a trade-off between gravimetric and volumetric capacities reported for H2 storage materials. Because the mass and size requirement for the on-board tank must be met to make the storage system feasible, an ideal H2 adsorbent should simultaneously exhibit high gravimetric and volumetric densities. Therefore, it is crucial to ponder over the optimization of volumetric and gravimetric deliverable capacities in MOFs as concurrent objectives rather than separate ones. However, there remains a great challenge in providing satisfactory volumetric and gravimetric capacities within a single material. To date, few H2 adsorbents balance high volumetric and gravimetric capacities.83,89,90,92,93
One key step toward a satisfactory trade-off between volumetric and gravimetric capacities would be to impart a single material with both high volumetric and gravimetric surface areas. In this regard, the computation method has been of great value in accelerating this search and is an effective method to identify the optimal materials. Farha and coworkers reported the simulation-motivated synthesis of an ultraporous MOF NU-1501-M based on metal trinuclear clusters exhibiting concurrently high gravimetric and volumetric BET surface areas of 7310 m2 g−1 and 2060 m2 cm−3, which imparts the material with impressive gravimetric and volumetric storage performance for H2 and methane.83 Under the combined temperature and pressure swing conditions of 77 K/100 bar (adsorption) to 160/5 bar (desorption), agreeing with the tank design conditions proposed by DOE, the deliverable capacities reach 14.0 wt% and 46.2 g L−1, which is among the highest reported for MOFs.
3.3 Improving H2 working capacities
The H2 working capacity is defined as the amount of H2 gas released via pressure reduction and typically evaluated assuming a pressure swing between 5 bar and 100 bar under the isothermal conditions. Considering that to empty a H2 tank below the atmospheric pressure is not energetically economic, 5 bar has been taken as the lower limit of the working pressure. However, due to different structural factors influencing the H2 adsorption performance in the high and low pressure regions, how to design the MOF materials with high H2 working capacities has become a long-standing challenge that has triggered tremendous studies to improve the H2 working capacity.With respect to the isothermal pressure-swing operation (100–5 bar & 77 K), the studies of the H2 Storage Engineering Center of Excellence (HSECoE) have positioned MOF-5, one of the most widely studied MOFs, as an important benchmark material due to its uncommon balance of both working capacities of 4.5 wt% and 31.1 g L−1 at 77 K. After this, the computational screening and experimental validation have identified IRMOF-20 based on thieno[3,2-b]thiophene-2,5-dicarboxylic acid as a candidate exceeding the MOF-5 baseline in terms of gravimetric and volumetric working capacities, which reach 5.7 wt% and 33.4 g L−1.90 Recently, further computational studies led to the finding of three MOFs with capacities surpassing that of IRMOF-20. They are SNU-70 (7.8 wt%, and 34.3 g L−1), UMCM-9 (7.3 wt%, and 34.1 g L−1), and PCN-610/NU-100 (10.1 wt%, 35.5 g L−1), establishing a new high-water mark for usable H2 capacities.92
Optimization of the conditions triggering the gas release is cited as an alternative approach to H2 working capacity improvement. Given that the HSECoE has proposed designing tanks for cryo-adsorption storage that operate with charge at 77 K and 100 bar and discharge at 160 K and 5 bar, a combined temperature and pressure swing is recommended to load and deliver H2 from these systems. As such, the quantity of H2 remaining in the tank after use is minimized, and therefore the deliverable H2 is maximized. However, H2 storage in MOFs using the pressure and temperature swing methods is yet to be systematically studied under these conditions experimentally. For this purpose, Snurr et al. used computational tools to construct 13512 potential MOFs on the basis of 41 different topological structures and screened/predicted their H2 adsorption properties, revealing the feasibility of using MOFs for H2 storage under the suggested cryo-adsorption operating conditions.93 The best MOF operating in 100 bar/77 K → 5 bar/160 K adsorption/desorption cycles achieves a H2 deliverable capacity of 57 g L−1, surpassing that of current CHG (compressed H2 gas) technologies (35 g L−1, 5–700 bar). Also, computation simulations can delineate useful structure–performance relationship, which are shown in Fig. 12. A MOF material with the highest volumetric storage capacities at 77 K and 100 bar should have a void fraction of 0.85 (Fig. 12a). The largest volumetric deliverable capacity (77 K/100–160 K/5 bar) corresponds to a heat of adsorption around 4 kJ mol−1 (Fig. 12b), which is translated to a pore size of 8 Å and a low void fraction (Fig. 12c). With such an optimal heat of H2 adsorption, the deliverable capacities can vary from 36 to 57 g L−1, depending on the volumetric surface areas (Fig. 12b). The trade-off effect between gravimetric and volumetric deliverable capacities is topologically dependent. Namely, distinct topologies reach a maximum in volumetric deliverable capacity at different linker sizes (Fig. 12d). For the MOFs with the volumetric capacities over 50 g L−1, the gravimetric deliverable capacities vary from 6 to 21 wt%. For further experimental demonstration, a new isoreticular series of (4,6)-connected MOFs (she-MOF-x, x = 1–4) together with NU-1103 were synthesized and their H2 adsorption properties were measured. NU-1103, more stable than the she-MOF-X series, exhibited volumetric and gravimetric deliverable capacities of 43.2 g L−1 and 12.6 wt%, respectively. More recently, Farha and co-workers experimentally examined the gravimetric and volumetric working capacities of 14 MOFs under the new temperature and pressure swing tank design conditions.87 A reasonably linear relationship between gravimetric working capacity and pore volume as well as a relatively constant trend for volumetric working capacities was observed for all of the 14 MOFs studied. Remarkably, the MOFs tested volumetrically exceed the DOE 2020 target of 30 g L−1, which is still achievable with the exception of rht-MOF-7 even though taking into account a 25% loss in capacity upon packing. In particular, NU-125 presents the highest volumetric deliverable capacity of all MOFs studied at 49 g L−1, owing partially to the favourable integration of volumetric surface area and void fraction. Also, this MOF displays a large gravimetric deliverable capacity of 7.7% overtaking the 6.5% ultimate system target.
 | ||
| Fig. 12 Computational simulations revealing the relationship of (a) H2 loading vs. void faction, (b) volumetric H2 working capacities (ΔH2) vs. heats of H2 adsorption, with colour showing MOF volumetric surface areas (VSA), and (c) heats of H2 adsorption and MOF void fractions vs. MOF largest cavity diameters. Highlighted points denote MOFs with ΔH2 higher than 53 g L−1 and (d) volumetric vs. gravimetric H2 working capacities, with colour showing selected MOF topologies. NOTT-1103 was identified as one of the best performers in terms of balancing gravimetric and volumetric H2 working capacities with the structure and H2 isotherm shown in (e and f), respectively. Reprinted with permission from ref. 93. Copyright 2016, Royal Society of Chemistry. | ||
4. Conclusions and outlook
Zero-carbon emission and high energy density place H2 as an attractive energy carrier to replace fossil oil in the future. The key to the establishment of clean H2 energy systems lies in H2 storage. One of the promising ways is the physisorption of H2 in porous solids due to fast kinetics, reversibility, and favorable thermodynamics. As reviewed above, the past two decades have witnessed the rapid development of porous MOFs for H2 storage after a tentative testament to the potential feasibility of MOF-5 as a H2 storage material in 2003. Asides from extremely high internal porosity, the rich structure and pore chemistry as well as diverse host–guest interaction, a feature less readily achieved in zeolites and activated carbon, afford MOFs with huge potential to tune the pore metrics and chemistry and thus regulate and optimize H2 storage performance. In particular, the capacity to construct the isoreticular series makes MOFs an excellent platform for elucidating the effect of various structural parameters such as OMS, organic linker, pore size, and surface area on the H2 adsorption properties, as demonstrated in several important classes of MOFs afore-discussed, thus providing the designing principles for future H2 storage materials with better performance. To date, some benchmark materials have been established. In terms of adsorption heat, the highest value of −32 kJ mol−1 holds for Cu(I)-MFU-4l. With respect to the balanced working capacities, MOF-5, IRMOF-20, SNU-7, and NU-100 were successively identified as the best MOFs under the isothermal pressure-swing operation. Upon considering the new tank design conditions (77 K/100 bar → 160 K/5 bar), several MOFs, in particular water-stable NU-1501-Al and NU-1103, have become the best material performers (for details, see Table 2). In fact, their performance has exceeded that of compressed H2 gas techniques.| MOFs | S BET (m2 g−1) | V p (cm3 g−1) | D c (g cm−3) | Total capacities (100 K and 77 K) | Working capacities (77 K, 5–100 bar) | Working capacities (77 K/100 bar, 160 K/5 bar) | Q st (kJ mol−1) | Ref. | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gravimetric basis (wt%) | Volumetric basis (g L−1) | Gravimetric basis (wt%) | Volumetric basis (g L−1) | Gravimetric basis (wt%) | Volumetric basis (g L−1) | ||||||
| S BET: BET specific surface area; Vp: pore volume; Dc: framework density; Qst: isosteric heat of H2 adsorption at zero coverage; NA: not available. | |||||||||||
| Cu-MOF-74 | 1270 | 0.47 | 1.323 | 3.1 | 43.0 | 0.9 | 13.0 | 2.8 | 39.0 | 5.6 | 87 |
| PCN-250 | 1780 | 0.71 | 0.896 | 5.4 | 50.8 | 1.6 | 15.7 | 4.9 | 46.4 | 6.6 | 87 |
| Rht-MOF-7 | 1950 | 0.79 | 0.789 | 4.9 | 40.5 | 1.7 | 14.2 | 4.5 | 37.2 | 5.9 | 87 |
| HKUST-1 | 1980 | 0.75 | 0.879 | 5.3 | 49.3 | 1.8 | 17.1 | 4.9 | 45.4 | 6.5 | 87 |
| Zn2(BDC)2(DABCO) | 2020 | 0.76 | 0.873 | 4.9 | 44.9 | 1.4 | 13.6 | 4.6 | 42.0 | 4.9 | 87 |
| NU-1000 | 2200 | 1.48 | 0.571 | 8.0 | 49.5 | 4.6 | 29.6 | 7.6 | 47.1 | 5 | 87 |
| UiO-67 | 2360 | 0.91 | 0.688 | 5.9 | 42.8 | 2.6 | 19.9 | 5.5 | 40.3 | 5.8 | 87 |
| CYCU-3-Al | 2450 | 1.56 | 0.477 | 8.2 | 42.9 | 4.9 | 26.2 | 7.9 | 41.0 | 4.5 | 87 |
| UiO-68-Ant | 3030 | 1.17 | 0.607 | 7.6 | 49.8 | 3.8 | 25.7 | 7.1 | 46.9 | 6 | 87 |
| MFU-4l | 3160 | 1.30 | 0.559 | 7.7 | 46.5 | 4.5 | 27.5 | 7.3 | 44.3 | 5.5 | 62 and 88 |
| NU-125 | 3230 | 1.33 | 0.578 | 8.2 | 51.6 | 3.6 | 23.8 | 7.7 | 48.6 | 5.1 | 87 |
| NOTT-112 | 3440 | 1.44 | 0.446 | 8.7 | 42.7 | 4.6 | 23.5 | 8.2 | 40.4 | 5.1 | 87 |
| MOF-5 | 3512 | 1.47 | 0.61 | 8.0 | 53.3 | 4.5 | 31.1 | 7.8 | 51.9 | NA | 12 |
| NU-1500-Al | 3560 | 1.46 | 0.498 | 8.4 | 46.6 | 4.4 | 26.6 | 8.2 | 44.6 | 4.9 | 83 |
| NU-1102 | 3720 | 1.65 | 0.403 | 9.9 | 45.3 | 6.9 | 30.5 | 9.6 | 43.7 | 4.5 | 89 |
| MFU-4l-Li | 4070 | 1.66 | 0.479 | 9.9 | 52.4 | 5.9 | 32.4 | 9.4 | 50.2 | 5.4 | 62 |
| IRMOF-20 | 4073 | 1.56 | 0.51 | 9.3 | 52.7 | 5.7 | 33.4 | 9.1 | 51.0 | NA | 90 |
| NU-1101 | 4340 | 1.72 | 0.459 | 9.5 | 48.7 | 6.1 | 29.9 | 9.1 | 46.6 | 5.5 | 89 |
| BUT-22 | 4380 | 2.01 | 0.381 | 12.0 | 45.8 | 7.8 | 30.3 | 11.6 | 44.1 | 4.7 | 91 |
| SNU-70 | 4944 | 2.14 | 0.411 | 10.7 | 48.9 | 7.8 | 34.3 | 10.6 | 47.9 | NA | 92 |
| UMCM-9 | 5039 | 2.31 | 0.372 | 11.5 | 48.5 | 7.3 | 34.1 | 11.3 | 47.4 | NA | 92 |
| NU-100 | 6050 | 3.17 | 0.29 | 14.1 | 48.1 | 10.1 | 35.5 | 13.9 | 47.6 | NA | 92 |
| NU-1103 | 6246 | 2.72 | 0.298 | 12.9 | 44.9 | 10.1 | 33.3 | 12.6 | 43.2 | 3.8 | 89 and 93 |
| NU-1501-Fe | 7140 | 2.90 | 0.299 | 13.6 | 46.9 | 9.4 | 33.9 | 13.2 | 45.4 | 4 | 83 |
| NU-1501-Al | 7310 | 2.91 | 0.283 | 14.5 | 47.9 | 10.3 | 35.5 | 14.0 | 46.2 | 4 | 83 |
Given that H2 is a nonpolar diatomic molecule, the H2–framework interaction is usually quite weak that cannot meet the practical storage requirements. In fact, significant H2 uptakes are only observed at cryogenic temperatures and high pressure. The improvement of ambient temperature H2 storage capacities calls for enhancing the H2-binding energies. Theoretical calculations have revealed that the optimal H2 adsorption heat for ambient-temperature storage should fall in the range of −15 to −20 kJ mol−1. To date, various chemistries related to inorganic SBUs, organic linkers, and guest species have been intensively carried out to increase the H2 adsorption heat. The SBU chemistry includes the introduction of open metal sites and post-synthetic metal/terminal ligand exchange. The unsaturated metal centre is capable of polarizing the H2 molecule through induced dipole moment, thus yielding the dipole-induced dipole interactions. In particular, incorporation of special metal ions such as the early transition metal ion of V(II) for the formation of the Kubas-type metal–H2 interaction has been initiated since the discovery of Kubas-type metal–H2 molecular complexes. In addition to the nature of metal–H2 interaction, the concentration of open metal sites and their rational spatial arrangement were also considered in designing MOFs with OMSs for H2 storage. As demonstrated in NOTT-112, specific geometrical arrangement of open copper sites within the cuboctahedral cage strengthened the interaction of H2 and open copper sites. By means of post-synthetic metal and terminal ligand exchange, the monovalent copper(I) chemistry at SBU for example was also explored for binding H2. The utilization of an aromatic linker to construct a π-rich pore surface, chemical reduction of redox-active ligand with elemental Li(0), the acid–base reaction between the acidic functional group grafted at the organic linker and lithium-containing base for lithium doping, and post/pre-synthetic covalent modification of the ligands with polar functional groups represent notable synthetic strategies involving the linker chemistry. Regarding guest chemistry, a variety of guest species, in particular Li+ ion, can be incorporated in the MOF pores via post-synthetic ion exchange and pre-synthetic introduction of LiOH into the reaction system. In addition, metal nanoparticles can be embedded in the pore exhibiting the synergistic effect of physisorption and chemisorption. For example, the doping of Pt nanoparticles to achieve H2 spillover can improve the room-temperature H2-storage performance of MOFs.
For the development of H2-adsorbing MOFs, a high H2 sorption capacity is an important criterion. To this end, the corresponding strategies have been established. The strategies to increase the gravimetric capacities include the construction of ultrahigh surface area MOFs and employment of the lightweight alkaline and alkaline earth ions for example as metal ions. The volumetric uptake capacities can be optimized by structural tuning with a suitable pore size and volumetric surface areas, which is more difficult to achieve than the gravimetric one. Besides, engineering the crystal morphology is an effective method to improve the packing density and thus boost volumetric performance. However, the mutual restriction of structural parameters makes it quite challenging to simultaneously obtain both high capabilities. An ideal MOF material should exhibit good gravimetric and volumetric H2 uptake capacities. The computational simulation plays an important role in accelerating the identification of the optimized materials with balanced gravimetrical and volumetric capacities. Regarding working capacities, deployment of the combined temperature and pressure swing also afforded a technical route to enhance the deliverable amount of H2 fuel.
A detailed understanding of the H2 sorption mechanism including the position, occupancy, and orientation of the adsorbed H2 in the framework as well as H2–framework interactions is very crucial in H2 adsorption studies. Such information can be directly and/or indirectly acquired and extracted using advanced spectroscopic characterization techniques such as NPD, INS, in situ synchrotron powder diffraction,94 and in situ low-temperature IR spectroscopy in combination with the corresponding DFT theoretical calculations. The low X-ray scattering factor of an H atom limits the single-crystal X-ray diffraction technique, being seldom applied to structurally characterize H2 adsorption sites. Depending on the MOF structure investigated, the primary H2 adsorption sites might be located on different positions, including the open metal sites, the bridging OH groups, the organic linker, small windows and so on, which can be considered as H2 binding sites being incorporated into the framework upon the design and construction of future H2 storage materials. These investigations would afford key guidance for the rational design and construction of porous MOFs with enhanced performance.
Although significant progress in development of MOFs as H2 storage media has been made, there are many concerns that are not fully solved, including but not limited to the common issues associated with the framework stability, production cost, material machinability/shaping, and intrinsic heat management during use, to name a few. In particular, due to the weak interaction between MOFs and H2, the development of MOFs that can operate under ambient conditions has so far been unsuccessful. Further efforts should be devoted to simulating the Kubas-type metal–H2 interaction in the MOF compounds to improve H2 binding energies. Besides, the optimization and balancing of gravimetric and volumetric uptake capacities to address the trade-off effect as well as deep understanding of H2 binding mechanism via spectral techniques and structure–performance correlation is still the future research direction. Since the mechanism investigation usually involves very low temperature, the H2 binding behaviour under practical storage conditions is still unclear and requires continued investigation. We firmly believe that further collaboration between experimental and theoretical researchers and industrial partners around the world will accelerate the commercialization and industrialization of some promising MOFs applied in our daily lives for H2 fuel storage in the near future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the Natural Science Foundation of China (no. 21771162 and 51802284) and Zhejiang Provincial Natural Science Foundation of China (LY20E020007).Notes and references
- A. C. Dillon, K. M. Jones, T. A. Bekkedahl, C. H. Kiang, D. S. Bethune and M. J. Heben, Nature, 1997, 386, 377–379 CrossRef CAS.
- (a) Z. Chen, K. O. Kirlikovali, K. B. Idrees, M. C. Wasson and O. K. Farha, Chem, 2022, 8, 1–24 CrossRef; (b) Y. Yan, S. Yang, A. J. Blake and M. Schroder, Acc. Chem. Res., 2014, 47, 296–307 CrossRef CAS PubMed; (c) M. P. Suh, H. J. Park, T. K. Prasad and D.-W. Lim, Chem. Rev., 2012, 112, 782–835 CrossRef CAS PubMed; (d) D. Zhao, D. Yuan and H.-C. Zhou, Energy Environ. Sci., 2008, 1, 222–235 RSC; (e) Y. He, F. Chen, B. Li, G. Qian, W. Zhou and B. Chen, Coord. Chem. Rev., 2018, 373, 167–198 CrossRef CAS; (f) X. Lin, J. Jia, P. Hubberstey, M. Schröder and N. R. Champness, CrystEngComm, 2007, 9, 438–448 RSC; (g) J. L. C. Rowsell and O. M. Yaghi, Angew. Chem., Int. Ed., 2005, 44, 4670–4679 CrossRef CAS; (h) Y. He, W. Zhou, R. Krishna and B. Chen, Chem. Commun., 2012, 48, 11813–11831 RSC.
- (a) P. Zhou, L. Yue, X. Wang, L. Fan, D.-L. Chen and Y. He, ACS Appl. Mater. Interfaces, 2021, 13, 54059–54068 CrossRef CAS PubMed; (b) L. Fan, L. Yue, W. Sun, X. Wang, P. Zhou, Y. Zhang and Y. He, ACS Appl. Mater. Interfaces, 2021, 13, 40788–40797 CrossRef PubMed; (c) L. Fan, P. Zhou, X. Wang, L. Yue, L. Li and Y. He, Inorg. Chem., 2021, 60, 10819–10829 CrossRef CAS PubMed; (d) T. Lan, L. Li, Y. Chen, X. Wang, J. Yang and J. Li, Mater. Chem. Front., 2020, 4, 1954–1984 RSC; (e) B. Li, H. Wang and B. Chen, Chem. – Asian J., 2014, 9, 1474–1498 CrossRef CAS PubMed; (f) J.-R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS.
- (a) Y. Cui, Y. Yue, G. Qian and B. Chen, Chem. Rev., 2012, 112, 1126–1162 CrossRef CAS PubMed; (b) B. Chen, S. Xiang and G. Qian, Acc. Chem. Res., 2010, 43, 1115–1124 CrossRef CAS PubMed; (c) M. D. Allendorf, C. A. Bauer, R. K. Bhakta and R. J. T. Houk, Chem. Soc. Rev., 2009, 38, 1330–1352 RSC.
- C. Wang, T. Zhang and W. Lin, Chem. Rev., 2012, 112, 1084–1104 CrossRef CAS PubMed.
- (a) M. Zhao, S. Ou and C.-D. Wu, Acc. Chem. Res., 2014, 47, 1199–1207 CrossRef CAS PubMed; (b) A. Corma, H. García and F. X. L. I. Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS PubMed.
- (a) D. Zhao, K. Yu, X. Han, Y. He and B. Chen, Chem. Commun., 2022, 58, 747–770 RSC; (b) Y. He, W. Zhou, G. Qian and B. Chen, Chem. Soc. Rev., 2014, 43, 5657–5678 RSC; (c) P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS PubMed.
- (a) Y. E. Cheon, J. Park and M. P. Suh, Chem. Commun., 2009, 5436–5438 RSC; (b) Y. E. Cheon and M. P. Suh, Chem. Commun., 2009, 2296–2298 RSC; (c) M. Dincă and J. R. Long, J. Am. Chem. Soc., 2005, 127, 9376–9377 CrossRef PubMed.
- X. Zhang, Z. Chen, X. Liu, S. L. Hanna, X. Wang, R. Taheri-Ledari, A. Maleki, P. Li and O. K. Farha, Chem. Soc. Rev., 2020, 49, 7406–7427 RSC.
- L. Ma, A. Jin, Z. Xie and W. Lin, Angew. Chem., Int. Ed., 2009, 48, 9905–9908 CrossRef CAS PubMed.
- (a) D. P. Broom, C. J. Webb, K. E. Hurst, P. A. Parilla, T. Gennett, C. M. Brown, R. Zacharia, E. Tylianakis, E. Klontzas, G. E. Froudakis, T. A. Steriotis, P. N. Trikalitis, D. L. Anton, B. Hardy, D. Tamburello, C. Corgnale, B. A. V. Hassel, D. Cossement, R. Chahine and M. Hirscher, Appl. Phys. A: Mater. Sci. Process., 2016, 122, 151 CrossRef; (b) Y. Basdogana and S. Keskin, CrystEngComm, 2015, 17, 261–275 RSC; (c) M. Hirscher, B. Panella and B. Schmitz, Microporous Mesoporous Mater., 2010, 129, 335–339 CrossRef CAS; (d) L. J. Murray, M. Dincă and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294–1314 RSC; (e) A. W. C. V. D. Berg and C. O. Areán, Chem. Commun., 2008, 668–681 RSC.
- S. S. Kaye, A. Dailly, O. M. Yaghi and J. R. Long, J. Am. Chem. Soc., 2007, 129, 14176–14177 CrossRef CAS PubMed.
- N. L. Rosi, J. Eckert, M. Eddaoudi, D. T. Vodak, J. Kim, M. O'Keeffe and O. M. Yaghi, Science, 2003, 300, 1127–1129 CrossRef CAS PubMed.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef CAS PubMed.
- J. L. C. Rowsell, A. R. Millward, K. S. Park and O. M. Yaghi, J. Am. Chem. Soc., 2004, 126, 5666–5667 CrossRef CAS PubMed.
- H. K. Chae, D. Y. Siberio-Pérez, J. Kim, Y. Go, M. Eddaoudi, A. J. Matzger, M. O'Keeffe and O. M. Yaghi, Nature, 2004, 427, 523–527 CrossRef CAS PubMed.
- M. G. Nijkamp, J. E. M. J. Raaymakers, A. J. V. Dillen and K. P. D. Jong, Appl. Phys. A, 2001, 72, 619–623 CrossRef CAS.
- J. I. Feldblyum, A. G. Wong-Foy and A. J. Matzger, Chem. Commun., 2012, 48, 9828–9830 RSC.
- H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. Ö. Yazaydin, R. Q. Snurr, M. O'Keeffe, J. Kim and O. M. Yaghi, Science, 2010, 329, 424–428 CrossRef CAS PubMed.
- K. Koh, A. G. Wong-Foy and A. J. Matzger, Angew. Chem., Int. Ed., 2008, 47, 677–680 CrossRef CAS PubMed.
- K. Koh, A. G. Wong-Foy and A. J. Matzger, J. Am. Chem. Soc., 2009, 131, 4184–4185 CrossRef CAS PubMed.
- S. S. Kaye and J. R. Long, J. Am. Chem. Soc., 2005, 127, 6506–6507 CrossRef CAS PubMed.
- M. R. Hartman, V. K. Peterson and Y. Liu, Chem. Mater., 2006, 18, 3221–3224 CrossRef CAS.
- S. S. Kaye and J. R. Long, Catal. Today, 2007, 312, 311–316 CrossRef.
- (a) B. R. Barnett, S. T. Parker, M. V. Paley, M. I. Gonzalez, N. Biggins, J. Oktawiec and J. R. Long, J. Am. Chem. Soc., 2019, 141, 18325–18333 CrossRef CAS; (b) H. Kim and Y. Jung, J. Phys. Chem. Lett., 2014, 5, 440–446 CrossRef CAS PubMed; (c) Y. He, R. Krishna and B. Chen, Energy Environ. Sci., 2012, 5, 9107–9120 RSC; (d) E. D. Bloch, W. L. Queen, R. Krishna, J. M. Zadrozny, C. M. Brown and J. R. Long, Science, 2012, 335, 1606–1610 CrossRef CAS PubMed.
- (a) C. M. Brown, A. J. Ramirez-Cuesta, J.-H. Her, P. S. Wheatley and R. E. Morris, Chem. Phys., 2013, 427, 3–8 CrossRef CAS; (b) K. Sumida, C. M. Brown, Z. R. Herm, S. Chavan, S. Bordiga and J. R. Long, Chem. Commun., 2011, 47, 1157–1159 RSC.
- (a) T. Runčevski, M. T. Kapelewski, R. M. Torres-Gavosto, J. D. Tarver, C. M. Brownce and J. R. Long, Chem. Commun., 2016, 52, 8251–8254 RSC; (b) W. L. Queen, E. D. Bloch, C. M. Brown, M. R. Hudson, J. A. Mason, L. J. Murray, A. J. Ramirez-Cuesta, V. K. Peterson and J. R. Long, Dalton Trans., 2012, 41, 4180–4187 RSC.
- (a) M. T. Kapelewski, S. J. Geier, M. R. Hudson, D. Stück, J. A. Mason, J. N. Nelson, D. J. Xiao, Z. Hulvey, E. Gilmour, S. A. FitzGerald, M. Head-Gordon, C. M. Brown and J. R. Long, J. Am. Chem. Soc., 2014, 136, 12119–12129 CrossRef CAS PubMed; (b) M. T. Kapelewski, T. e Runčevski, J. D. Tarver, H. Z. H. Jiang, K. E. Hurst, P. A. Parilla, A. Ayala, T. Gennett, S. A. FitzGerald, C. M. Brown and J. R. Long, Chem. Mater., 2018, 30, 8179–8189 CrossRef CAS; (c) D. Gygi, E. D. Bloch, J. A. Mason, M. R. Hudson, M. I. Gonzalez, R. L. Siegelman, T. A. Darwish, W. L. Queen, C. M. Brown and J. R. Long, Chem. Mater., 2016, 28, 1128–1138 CrossRef CAS.
- W. Zhou, H. Wu and T. Yildirim, J. Am. Chem. Soc., 2008, 130, 15268–15269 CrossRef CAS.
- J. G. Vitillo, L. Regli, S. Chavan, G. Ricchiardi, G. Spoto, P. D. C. Dietzel, S. Bordiga and A. Zecchina, J. Am. Chem. Soc., 2008, 130, 8386–8396 CrossRef CAS PubMed.
- E. D. Bloch, L. J. Murray, W. L. Queen, S. Chavan, S. N. Maximoff, J. P. Bigi, R. Krishna, V. K. Peterson, F. Grandjean, O. G. J. Long, B. Smit, S. Bordiga, C. M. Brown and J. R. Long, J. Am. Chem. Soc., 2011, 133, 14814–14822 CrossRef CAS.
- (a) L.-C. Lin, J. Kim, X. Kong, E. Scott, T. M. McDonald, J. R. Long, J. A. Reimer and B. Smit, Angew. Chem., Int. Ed., 2013, 52, 4410–4413 CrossRef CAS PubMed; (b) T. M. McDonald, W. R. Lee, J. A. Mason, B. M. Wiers, C. S. Hong and J. R. Long, J. Am. Chem. Soc., 2012, 134, 7056–7065 CrossRef CAS PubMed.
- H. Deng, S. Grunder, K. E. Cordova, C. Valente, H. Furukawa, M. Hmadeh, F. Gándara, A. C. Whalley, Z. Liu, S. Asahina, H. Kazumori, M. O'Keeffe, O. Terasaki, J. F. Stoddart and O. M. Yaghi, Science, 2012, 336, 1018–1023 CrossRef CAS PubMed.
- D. J. Levine, T. e Runčevski, M. T. Kapelewski, B. K. Keitz, J. Oktawiec, D. A. Reed, J. A. Mason, H. Z. H. Jiang, K. A. Colwell, C. M. Legendre, S. A. FitzGerald and J. R. Long, J. Am. Chem. Soc., 2016, 138, 10143–10150 CrossRef CAS.
- M. Dincă, A. F. Yu and J. R. Long, J. Am. Chem. Soc., 2006, 128, 8904–8913 CrossRef.
- M. Dincă, A. Dailly, Y. Liu, C. M. Brown, D. A. Neumann and J. R. Long, J. Am. Chem. Soc., 2006, 128, 16876–16883 CrossRef.
- M. Dincă and J. R. Long, J. Am. Chem. Soc., 2007, 129, 11172–11176 CrossRef.
- M. Dincă, W. S. Han, Y. Liu, A. Dailly, C. M. Brown and J. R. Long, Angew. Chem., Int. Ed., 2007, 46, 1419–1422 CrossRef PubMed.
- K. Sumida, S. Horike, S. S. Kaye, Z. R. Herm, W. L. Queen, C. M. Brown, F. Grandjean, G. J. Long, A. Dailly and J. R. Long, Chem. Sci., 2010, 1, 184–191 RSC.
- M. Dincă, A. Dailly, C. Tsay and J. R. Long, Inorg. Chem., 2008, 47, 11–13 CrossRef.
- C. Prestipino, L. Regli, J. G. Vitillo, F. Bonino, A. Damin, C. Lamberti, A. Zecchina, P. L. Solari, K. O. Kongshaug and S. Bordiga, Chem. Mater., 2006, 18, 1337–1346 CrossRef CAS.
- V. K. Peterson, Y. Liu, C. M. Brown and C. J. Kepert, J. Am. Chem. Soc., 2006, 128, 15578–15579 CrossRef CAS PubMed.
- (a) Y. Yan, I. Telepeni, S. Yang, X. Lin, W. Kockelmann, A. Dailly, A. J. Blake, W. Lewis, G. S. Walker, D. R. Allan, S. A. Barnett, N. R. Champness and M. Schröder, J. Am. Chem. Soc., 2010, 132, 4092–4094 CrossRef CAS PubMed; (b) X. Lin, I. Telepeni, A. J. Blake, A. Dailly, C. M. Brown, J. M. Simmons, M. Zoppi, G. S. Walker, K. M. Thomas, T. J. Mays, P. Hubberstey, N. R. Champness and M. Schröder, J. Am. Chem. Soc., 2009, 131, 2159–2171 CrossRef CAS.
- (a) S. Ma, J. Eckert, P. M. Forster, J. W. Yoon, Y. K. Hwang, J.-S. Chang, C. D. Collier, J. B. Parise and H.-C. Zhou, J. Am. Chem. Soc., 2008, 130, 15896–15902 CrossRef CAS PubMed; (b) S. Ma, D. Sun, M. Ambrogio, J. A. Fillinger, S. Parkin and H.-C. Zhou, J. Am. Chem. Soc., 2007, 129, 1858–1859 CrossRef CAS PubMed; (c) D. Sun, S. Ma, Y. Ke, D. J. Collins and H.-C. Zhou, J. Am. Chem. Soc., 2006, 128, 3896–3897 CrossRef CAS PubMed.
- Y. He, B. Li, M. O'Keeffe and B. Chen, Chem. Soc. Rev., 2014, 43, 5618–5656 RSC.
- B. Chen, N. W. Ockwig, A. R. Millward, D. S. Contreras and O. M. Yaghi, Angew. Chem., Int. Ed., 2005, 44, 4745–4749 CrossRef CAS PubMed.
- (a) S. Yang, X. Lin, A. Dailly, A. J. Blake, P. Hubberstey, N. R. Champness and M. Schroder, Chem. – Eur. J., 2009, 15, 4829–4835 CrossRef CAS PubMed; (b) X. Lin, J. Jia, X. Zhao, K. M. Thomas, A. J. Blake, G. S. Walker, N. R. Champness, P. Hubberstey and M. Schröder, Angew. Chem., Int. Ed., 2006, 45, 7358–7364 CrossRef CAS PubMed.
- G. J. Kubas, Acc. Chem. Res., 1988, 21, 120–128 CrossRef.
- X.-S. Wang, S. Ma, P. M. Forster, D. Yuan, J. Eckert, J. J. Lopez, B. J. Murphy, J. B. Parise and H.-C. Zhou, Angew. Chem., Int. Ed., 2008, 47, 7263–7266 CrossRef CAS PubMed.
- D. Yuan, D. Zhao, D. Sun and H.-C. Zhou, Angew. Chem., Int. Ed., 2010, 49, 5357–5361 CrossRef CAS.
- Y. Yan, X. Lin, S. Yang, A. J. Blake, A. Dailly, N. R. Champness, P. Hubberstey and M. Schroder, Chem. Commun., 2009, 1025–1027 RSC.
- (a) G. Barin, V. Krungleviciute, D. A. Gomez-Gualdron, A. A. Sarjeant, R. Q. Snurr, J. T. Hupp, T. Yildirim and O. K. Farha, Chem. Mater., 2014, 26, 1912–1917 CrossRef CAS; (b) O. K. Farha, I. Eryazici, N. C. Jeong, B. G. Hauser, C. E. Wilmer, A. A. Sarjeant, R. Q. Snurr, S. T. Nguyen, A. Ö. Yazaydın and J. T. Hupp, J. Am. Chem. Soc., 2012, 134, 15016–15021 CrossRef CAS PubMed; (c) O. K. Farha, C. E. Wilmer, I. Eryazici, B. G. Hauser, P. A. Parilla, K. O'Neill, A. A. Sarjeant, S. T. Nguyen, R. Q. Snurr and J. T. Hupp, J. Am. Chem. Soc., 2012, 134, 9860–9863 CrossRef CAS PubMed; (d) O. K. Farha, A. Ö. Yazaydın, I. Eryazici, C. D. Malliakas, B. G. Hauser, M. G. Kanatzidis, S. T. Nguyen, R. Q. Snurr and J. T. Hupp, Nat. Chem., 2010, 2, 944–948 CrossRef CAS PubMed.
- (a) C. Tan, S. Yang, N. R. Champness, X. Lin, A. J. Blake, W. Lewis and M. Schröder, Chem. Commun., 2011, 47, 4487–4489 RSC; (b) L. Ma, D. J. Mihalcik and W. Lin, J. Am. Chem. Soc., 2009, 131, 4610–4612 CrossRef CAS.
- (a) H. Frost and R. Q. Snurr, J. Phys. Chem. C, 2007, 111, 18794–18803 CrossRef CAS; (b) S. K. Bhatia and A. L. Myers, Langmuir, 2006, 22, 1688–1700 CrossRef CAS.
- (a) B. R. Barnett, H. A. Evans, G. M. Su, H. Z. H. Jiang, R. Chakraborty, D. Banyeretse, T. J. Hartman, M. B. Martinez, B. A. Trump, J. D. Tarver, M. N. Dods, L. M. Funke, J. Börgel, J. A. Reimer, W. S. Drisdell, K. E. Hurst, T. Gennett, S. A. FitzGerald, C. M. Brown, M. Head-Gordon and J. R. Long, J. Am. Chem. Soc., 2021, 143, 14884–14894 CrossRef CAS PubMed; (b) D. Denysenko, M. Grzywa, J. Jelic, K. Reuter and D. Volkmer, Angew. Chem., Int. Ed., 2014, 53, 5832–5836 CrossRef CAS PubMed.
- D. E. Jaramillo, H. Z. H. Jiang, H. A. Evans, R. Chakraborty, H. Furukawa, C. M. Brown, M. Head-Gordon and J. R. Long, J. Am. Chem. Soc., 2021, 143, 6248–6256 CrossRef CAS.
- Y. Yan, I. d Silva, A. J. Blake, A. Dailly, P. Manuel, S. Yang and M. Schröder, Inorg. Chem., 2018, 57, 12050–12055 CrossRef CAS PubMed.
- K. Sumida, J.-H. Her, M. Dinca, L. J. Murray, O. J. M. Schloss, C. J. Pierce, B. A. Thompson, S. A. FitzGerald, C. M. Brown and J. R. Long, J. Phys. Chem. C, 2011, 115, 8414–8421 CrossRef CAS.
- G. J. Kubas, R. R. Ryan, B. I. Swanson, P. J. Vergamini and H. J. Wasserman, J. Am. Chem. Soc., 1984, 106, 451–452 CrossRef CAS.
- M. Savage, I. D. Silva, M. Johnson, J. H. Carter, R. Newby, M. Suyetin, E. Besley, P. Manuel, S. Rudić, A. N. Fitch, C. Murray, W. I. F. David, S. Yang and M. Schröder, J. Am. Chem. Soc., 2016, 138, 9119–9127 CrossRef CAS PubMed.
- D. Denysenko, M. Grzywa, M. Tonigold, B. Streppel, I. Krkljus, M. Hirscher, E. Mugnaioli, U. Kolb, J. Hanss and D. Volkmer, Chem. – Eur. J., 2011, 17, 1837–1848 CrossRef CAS PubMed.
- Z. Chen, M. R. Mian, S.-J. Lee, H. Chen, X. Zhang, K. O. Kirlikovali, S. Shulda, P. Melix, A. S. Rosen, P. A. Parilla, T. Gennett, R. Q. Snurr, T. Islamoglu, T. Yildirim and O. K. Farha, J. Am. Chem. Soc., 2021, 143, 18838–18843 CrossRef CAS PubMed.
- M. Yoon and D. Moon, Microporous Mesoporous Mater., 2015, 215, 116–122 CrossRef CAS.
- D.-W. Lim, S. A. Chyun and M. P. Suh, Angew. Chem., Int. Ed., 2014, 53, 7819–7822 CrossRef CAS.
- Z. Wang, K. K. Tanabe and S. M. Cohen, Chem. – Eur. J., 2010, 16, 212–217 CrossRef CAS.
- K. L. Mulfort and J. T. Hupp, J. Am. Chem. Soc., 2007, 129, 9604–9605 CrossRef CAS PubMed.
- S. S. Han and W. A. Goddard III, J. Am. Chem. Soc., 2007, 129, 8422–8423 CrossRef CAS.
- D. Himsl, D. Wallacher and M. Hartmann, Angew. Chem., Int. Ed., 2009, 48, 4639–4642 CrossRef CAS PubMed.
- E. Klontzas, A. Mavrandonakis, E. Tylianakis and G. E. Froudakis, Nano Lett., 2008, 8, 1572–1576 CrossRef.
- S. Yang, X. Lin, A. J. Blake, K. M. Thomas, P. Hubberstey, N. R. Champness and M. Schroder, Chem. Commun., 2008, 6108–6110 RSC.
- (a) T. Stergiannakos, E. Tylianakis, E. Klontzas, P. N. Trikalitis and G. E. Froudakis, J. Phys. Chem. C, 2012, 116, 8359–8363 CrossRef CAS; (b) P. Dalach, H. Frost, R. Q. Snurr and D. E. Ellis, J. Phys. Chem. C, 2008, 112, 9278–9284 CrossRef CAS; (c) A. Blomqvist, C. M. Araújo, P. Srepusharawoot and R. Ahuja, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20173–20176 CrossRef CAS PubMed.
- S. Yang, G. S. B. Martin, J. J. Titman, A. J. Blake, D. R. Allan, N. R. Champness and M. Schröder, Inorg. Chem., 2011, 50, 9374–9384 CrossRef CAS PubMed.
- F. Nouar, J. Eckert, J. F. Eubank, P. Forster and M. Eddaoudi, J. Am. Chem. Soc., 2009, 131, 2864–2870 CrossRef CAS PubMed.
- M. Bosch, M. Zhang, D. Feng, S. Yuan, X. Wang, Y.-P. Chen and H.-C. Zhou, APL Mater., 2014, 2, 124103 CrossRef.
- B. Chen, X. Zhao, A. Putkham, K. Hong, E. B. Lobkovsky, E. J. Hurtado, A. J. Fletcher and K. M. Thomas, J. Am. Chem. Soc., 2008, 130, 6411–6423 CrossRef CAS PubMed.
- L. Pan, M. B. Sander, X. Huang, J. Li, M. Smith, E. Bittner, B. Bockrath and J. K. Johnson, J. Am. Chem. Soc., 2004, 126, 1308–1309 CrossRef CAS.
- B. Kesanli, Y. Cui, M. R. Smith, E. W. Bittner, B. C. Bockrath and W. Lin, Angew. Chem., Int. Ed., 2005, 44, 72–75 CrossRef CAS.
- J. Luo, H. Xu, Y. Liu, Y. Zhao, L. L. Daemen, C. Brown, T. V. Timofeeva, S. Ma and H.-C. Zhou, J. Am. Chem. Soc., 2008, 130, 9626–9627 CrossRef CAS PubMed.
- Y. Li, L. Xie, Y. Liu and R. Y. X. Li, Inorg. Chem., 2008, 47, 10372–10377 CrossRef CAS PubMed.
- Y. Li and R. T. Yang, J. Am. Chem. Soc., 2006, 128, 726–727 CrossRef CAS.
- P.-C. Kang, Y.-S. Ou, G.-L. Li, J.-K. Chang and C.-Y. Wang, ACS Appl. Nano Mater., 2021, 4, 11269–11280 CrossRef CAS.
- D.-W. Lim, J. W. Yoon, K. Y. Ryu and M. P. Suh, Angew. Chem., Int. Ed., 2012, 51, 9814–9817 CrossRef CAS PubMed.
- Z. Chen, P. Li, R. Anderson, X. Wang, X. Zhang, L. Robison, L. R. Redfern, S. Moribe, T. Islamoglu, D. A. Gómez-Gualdrón, T. Yildirim, J. F. Stoddart and O. K. Farha, Science, 2020, 368, 297–303 CrossRef CAS PubMed.
- K. Sumida, M. R. Hill, S. Horike, A. Dailly and J. R. Long, J. Am. Chem. Soc., 2009, 131, 15120–15121 CrossRef CAS.
- K. Suresh, D. Aulakh, J. Purewal, D. J. Siegel, M. Veenstra and A. J. Matzger, J. Am. Chem. Soc., 2021, 143, 10727–10734 CrossRef CAS PubMed.
- M. Rzepka, P. Lamp and M. A. de la Casa-Lillo, J. Phys. Chem. B, 1998, 102, 10894–10898 CrossRef CAS.
- P. García-Holley, B. Schweitzer, T. Islamoglu, Y. Liu, L. Lin, S. Rodriguez, M. H. Weston, J. T. Hupp, D. A. Gómez-Gualdrón, T. Yildirim and O. K. Farha, ACS Energy Lett., 2018, 3, 748–754 CrossRef.
- B. J. Bucior, N. S. Bobbitt, T. Islamoglu, S. Goswami, A. Gopalan, T. Yildirim, O. K. Farha, N. Bagheri and R. Q. Snurr, Mol. Syst. Des. Eng., 2019, 4, 162–174 RSC.
- D. A. Gómez-Gualdrón, T. C. Wang, P. García-Holley, R. M. Sawelewa, E. Argueta, R. Q. Snurr, J. T. Hupp, T. Yildirim and O. K. Farha, ACS Appl. Mater. Interfaces, 2017, 9, 33419–33428 CrossRef.
- A. Ahmed, Y. Liu, J. Purewal, L. D. Tran, A. G. Wong-Foy, M. Veenstra, A. J. Matzger and D. J. Siegel, Energy Environ. Sci., 2017, 10, 2459–2471 RSC.
- B. Wang, X. Zhang, H. Huang, Z. Zhang, T. Yildirim, W. Zhou, S. Xiang and B. Chen, Nano Res., 2021, 14, 507–511 CrossRef CAS.
- A. Ahmed, S. Seth, J. Purewal, A. G. Wong-Foy, M. Veenstra, A. J. Matzger and D. J. Siegel, Nat. Commun., 2019, 10, 1568 CrossRef.
- D. A. Gómez-Gualdrón, Y. J. Colón, X. Zhang, T. C. Wang, Y.-S. Chen, J. T. Hupp, T. Yildirim, O. K. Farha, J. Zhang and R. Q. Snurr, Energy Environ. Sci., 2016, 9, 3279–3289 RSC.
- Y. Kubota, M. Takata, R. Matsuda, R. Kitaura, S. Kitagawa, K. Kato, M. Sakata and T. C. Kobayashi, Angew. Chem., Int. Ed., 2005, 44, 920–923 CrossRef CAS PubMed.
Footnote |
| † Crystallographic densities were used to calculate volumetric capacities in the main text unless specified otherwise. |
| This journal is © The Royal Society of Chemistry 2022 |