DOI:
10.1039/D0QO01453B
(Review Article)
Org. Chem. Front., 2021,
8, 1345-1363
Recent advances in cascade radical cyclization of radical acceptors for the synthesis of carbo- and heterocycles
Received
20th November 2020
, Accepted 5th January 2021
First published on 8th January 2021
Abstract
Cascade radical cyclization is an attractive synthetic method due to its high atom- and step-economy to construct carbo- and heterocycles, which have wide applications in chemical and life science industries. Radical acceptors with unsaturated bonds play a key role in realizing highly rapid, concise, and efficient cascade radical cyclization. This Review is devoted to highlighting the latest achievements in the development of cascade radical cyclization methodologies for the synthesis of carbo- and heterocycles in this rapidly growing research field. Based on the hybridization of starting materials, this Review includes C(sp)–C(sp) bond, C(sp)–C(sp2) bond, C(sp2)–C(sp2) bond, C(sp2)–C(sp3), and carbon–heteroatom bond coupling cyclization. Representative examples mainly published in last three years will be discussed for each highlighted reaction design and mechanism, providing readers with common tools for future research in cascade radical cyclization.
 Jianhua Liao | Dr Jianhua Liao was born in Jiangxi Province, People's Republic of China. In 2017, she received her PhD degree from South China University of Technology (SCUT) under the supervision of Prof. Huanfeng Jiang. During 2019 and 2020, she studied as a visiting scholar with Prof. Weiping Tang at University of Wisconsin-Madison (USA). She worked in the school of Pharmaceutical Sciences at Gannan Medical University, China. Her current research interests are focused on the synthesis of biologically active molecules and novel radical chemistry. |
 Xiao Yang | Xiao Yang was born in Shanxi Province, People's Republic of China, in 1997. In 2020, she obtained her Bachelor's degree in Pharmacy from Gannan Medical University. After graduation, she joined the group of associate professor Jianhua Liao at Gannan Medical University to carry out her Master's degree studies. Her current research interests are focused on the development of new cascade radical cyclization reactions. |
 Lu Ouyang | Dr Lu Ouyang was born in Jiangxi Province, People's Republic of China. She obtained her PhD degree from South China University of Technology (SCUT) under the supervision of Prof. Huanfeng Jiang in 2018. After graduation, she joined the school of Pharmaceutical Sciences at Gannan Medical University, China. She is interested in the development of transition-metal catalyzed synthesis of biologically active molecules. |
 Renshi Luo | Dr Renshi Luo was born in 1981 in Jiangxi Province, China. He received his B.S. degree in 2004 from Jiangxi Normal University, his M.S. degree in 2007 from South China Normal University, and his Ph.D. degree in 2011 from Sun Yat-Sen University. He finished his postdoctoral fellowship in Prof. Wenjun Tang's laboratory at Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, in 2013. He had a one-year academic visit at the University of Wisconsin-Madison in Prof. Weiping Tang's laboratory from 2016 to 2017. His research interests are in the areas of transition-metal catalysis and organocatalysis. |
1. Introduction
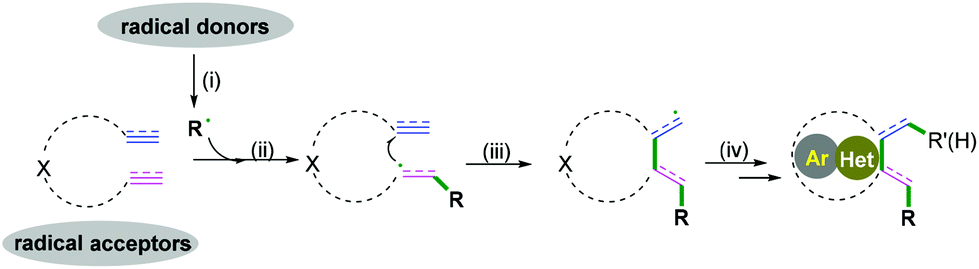
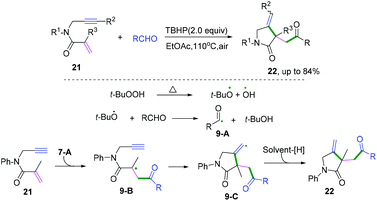
Carbo- and heterocycles are extremely important skeletons in organic synthesis; they are widely found in natural products and bioactive compounds.1 As a result, a variety of synthetic methods have been performed in the development of carbo- and heterocyclic molecules, among which cascade radical cyclization arguably constitutes the most valuable tool to date.2 Because of the sustainability of synthetic processes, cascade reactions are highly recognized as powerful strategies for the construction of high-value and often complex cyclic compounds. The processes are performed in a single preparation step with a high synthetic economy to build up the formation of more than two bonds.3 In past decades, excellent results have been achieved in this research area, and efficient protocols for the construction of carbo- and heterocycles have been disclosed. As pioneering work in radical cascade reactions, Porter and coauthors developed a systematic study of unsaturated peroxy radical cyclizations to synthesize prostaglandin analogs.3a,b In the 1980s, Stork3c and Curran groups3d elegantly reported fascinating research in the field of radical chemistry. Transient radical species were generated from halides with tri-n-butyltin hydride, followed by hex-5-enyl-like radical cyclizations and finally a hydrogen atom abstraction to deliver the final products. Later, an excellent research of the cascade radical cyclization of electron-deficient alkenes with carbon species generated from the irradiation of the N-hydroxy-2-thiopyridone esters of suitable γ,δ-unsaturated carboxylic acids to construct complex carbocycles was developed by Zard and co-workers.3e Despite these very impressive and significant advances, the synthetic potential of available chemical feedstocks as radical acceptors to construct carbo- and heterocycles with high chemo- and regioselectivity is still an imposing challenge for chemists. Therefore, many attempts have been made to create new and efficient strategies that are capable of accessing complex molecular frameworks. Different radical acceptors, such as 1,n-diynes, 1,n-enynes, alkynyl (hetero) arenes, o-alkenylarylisonitriles, 1,n-dienes, alkenyl (hetero) arenes, alkyne-tethered ketoximes, alkene-tethered ketoximes, 2-substituted phenyl isocyanides, and 2-isocyanoaryl thioethers have been elegantly applied in combination with radical species in the formation of carbo- and heterocycles. Recent cascade radical cyclization generally involves four key stages: (i) a radical formation-radical species is generated from a radical donor by a single-electron transfer (SET) process; (ii) radical addition-subsequent radical addition to an unsaturated bond leads to the formation of a carbon radical intermediate; (iii) a radical cyclization-carbon–carbon/heteroatom bond is formed; (iv) the radical quenching-carbon radical intermediate is quenched by hydrogen abstraction or another radical donor coupling to construct a carbo- and heterocyclic molecule (Scheme 1).
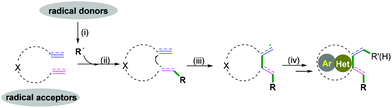 |
| | Scheme 1 General pathway of the cascade radical cyclization reaction. | |
Recently, a variety of radical cascade cyclization reactions have been developed. Although radical C–H activation/radical cross-coupling,2a catalytic cascade reactions by radical relay,2d cascade cyclization of 1,n-enynes and diynes,2b and radical cascade reaction in the synthesis of 1-indenones2e have been the subject of excellent reviews, we herein aim to examine a disparate region of radical chemistry in a more general review of radical cascade cyclization by highlighting the properties and reactivities of radical acceptors with different types of hybridization of the carbon–carbon/heteroatoms. According to the hybridization of the starting materials, this Review will embrace the classification of the carbon–carbon bond (e.g., C(sp)–C(sp), C(sp)–C(sp2), C(sp2)–C(sp2), and C(sp2)–C(sp3) bonds, section 2) and carbon–heteroatom bond coupling cyclization (e.g., C–O, C–N, and C–S bonds, section 3), with an emphasis on the properties and reactivities of the radical acceptor. This is important to enable the reader to directly compare and contrast reactivity patterns in the additions of diverse radicals to unsaturated substrates. Additionally, representative examples of radical cascade cyclization are selected for further discussion. Moreover, we highlight the reaction design and reaction mechanisms, providing readers with common strategies for new work on radical cascade cyclization in the future.
2. Carbon–carbon bond coupling cyclization
Radical cascade cyclization is widely applied to carbo- and heterocycle synthesis in the applications of medicines, agrochemicals, and other functional materials; this cyclization proceeds through a single electron transfer pathway to construct new carbon–carbon (C–C) bonds.
2.1 C(sp)–C(sp) bond coupling cyclization
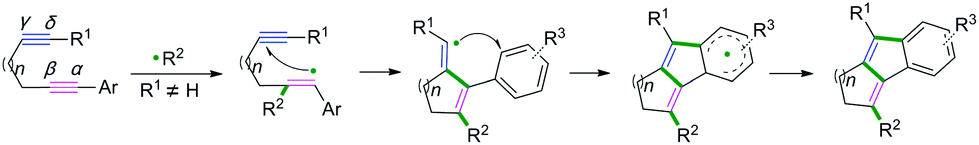
C(sp)–C(sp) bond coupling cyclizations are usually performed with the starting radical acceptors of 1,n-diynes. The general reaction process for the C(sp)–C(sp) bond coupling cyclization of 1,n-diynes is shown in Scheme 2. A single-electron transfer proceeds, resulting in the formation of a radical species under the proper conditions. The regio- and chemoselectivity for the subsequent radical attack at an unsaturated group depends on the type of substituent group of the 1,n-diynes. If R1 is not equal to H, the free radical addition attack occurs at the β position of the 1,n-diyne. Further radical cyclization to the γ position of another triple bond of the 1,n-diyne leads to a new alkenyl carbon radical intermediate, followed by further intramolecular radical cyclization with aromatics linked at the α position. Finally, deprotonation of the aryl radical intermediate with base or oxidants delivers the desired product.
 |
| | Scheme 2 Plausible mechanism of radical cyclization to 1,n-diynes. | |
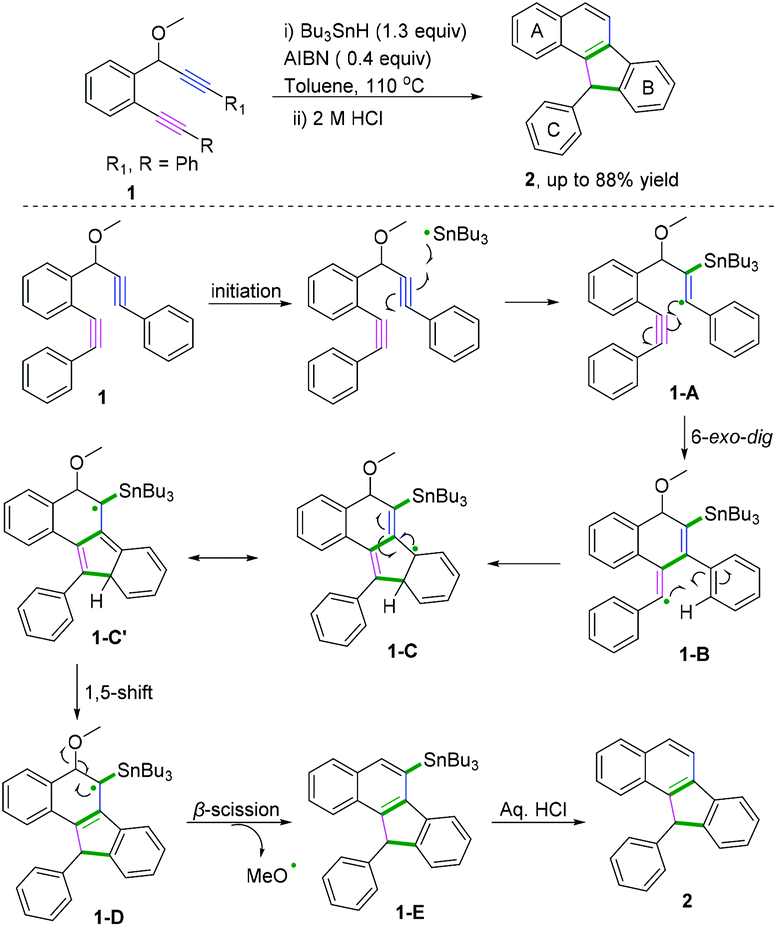
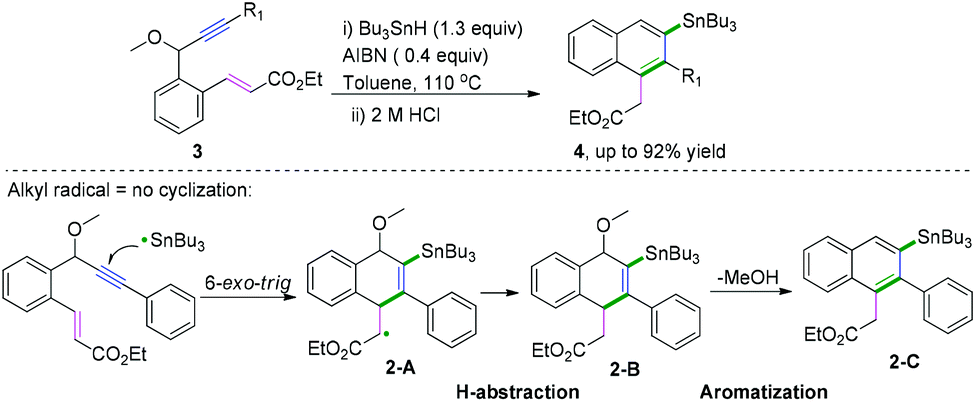
Polycyclic aromatic hydrocarbons (PAHs) are fascinating compounds that have wide utility in material sciences.4 In this regard, Alabugin and co-workers developed pioneering tin-mediated radical cascade processes that employed a tethered initiator or a traceless directing group to perform the initial radical attack on the polyalkyne substrates.5 As demonstrated in Scheme 3,5e the first example of a traceless directing group in the radical cascade transformation of enynes to efficiently construct polycyclic ribbons of tunable dimensions was reported. In this transformation, high chemo- and regioselectivity of the initial attack in skipped oligoalkynes was realized by the propargyl OR moiety. It was proposed that the reaction involved an initial formation of a radical intermediate 1-A, generated via the attack of the Bu3Sn radical at the central alkyne. The subsequent 6-exo-dig cyclization process afforded the second vinyl radical intermediate 1-B. Further attack of the neighboring phenyl π-system delivered a delocalized radical intermediate 1-C, leading to rearomatization via a 1,5-hydrogen shift. Subsequent β-scission of the OMe afforded the intermediate 1-D. The final fragmentation of intermediate 1-E led to the aromatization of the top ring into a naphthalene moiety. Interestingly, when analogous 1,6-enynes were engaged in this cascade radical reaction by the same group, the process did not form the last five-membered ring.5d As shown in Scheme 4, the final product consisted only of six-membered rings. The DFT calculations suggested that the difference between the radical cascades of oligoalkynes and enynes was the greater reactivity of vinyl radicals and the more stabilizing group of the vinyl moiety in the product in comparison with the alkyl radicals.
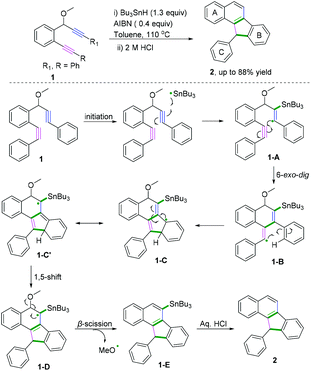 |
| | Scheme 3 Traceless directing groups in radical cascade transformations of oligoalkynes. | |
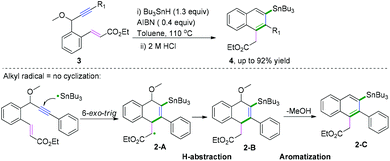 |
| | Scheme 4 Traceless directing groups in radical cascade transformations of 1,6-enynes. | |
In 2017, Xu and coauthors also demonstrated a remarkable electrochemical synthesis of nitrogen-doped PAHs through intramolecular cascade radical cyclization of diynes in a one-step process with a high step-economy (Scheme 5).6 The reaction proceeded in the presence of ferrocene, a mild and selective redox catalyst, to access varieties of electron-rich PAHs without overoxidation. The process involved formation of an unprecedented amidyl radical, 3-A, to deliver a cascade cyclization process with three rings in a single operation.
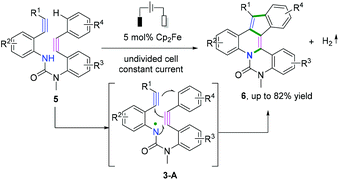 |
| | Scheme 5 Electrochemical cascade radical cyclization of diynes. | |
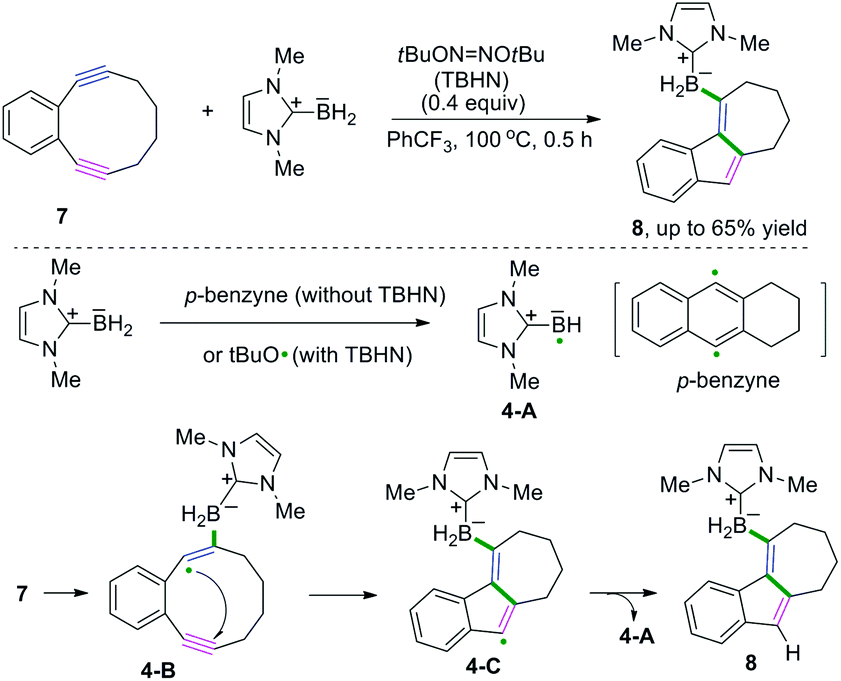
Excellent research of borylative radical cascade cyclization of 1,5-diynes to generate borylated carbocycles has been developed by Curran, Taniguchi, and coauthors (Scheme 6).7 The proposed mechanism suggested that boryl radical species 4-A from N-heterocyclic carbene-boranes (NHC-boranes) were formed firstly, initiated by di-tert-butyl hyponitrite (TBHN), followed by intermolecular addition of NHC-boryl radicals 4-A to the alkyne of the diyne. Subsequent 5-endo-dig cyclization of 1,2-diethynylbenzenes generated the alkenyl radical intermediates 4-C, whereas intermediates 4-C could abstract a hydrogen atom from NHC-borane to deliver the final borylated products. As demonstrated in Scheme 6, stabilization of the alkenyl radical intermediates 4-B by the aromatic ring and the strain of the alkynes may benefit the addition of the NHC-boryl radical species 4-A to the alkynes in this transformation.
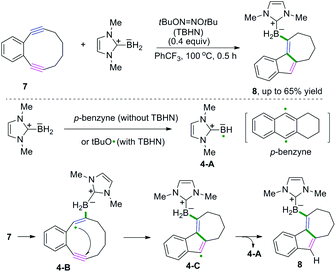 |
| | Scheme 6 Borylative radical cascade cyclization of diynes and N-heterocyclic carbene-boranes. | |
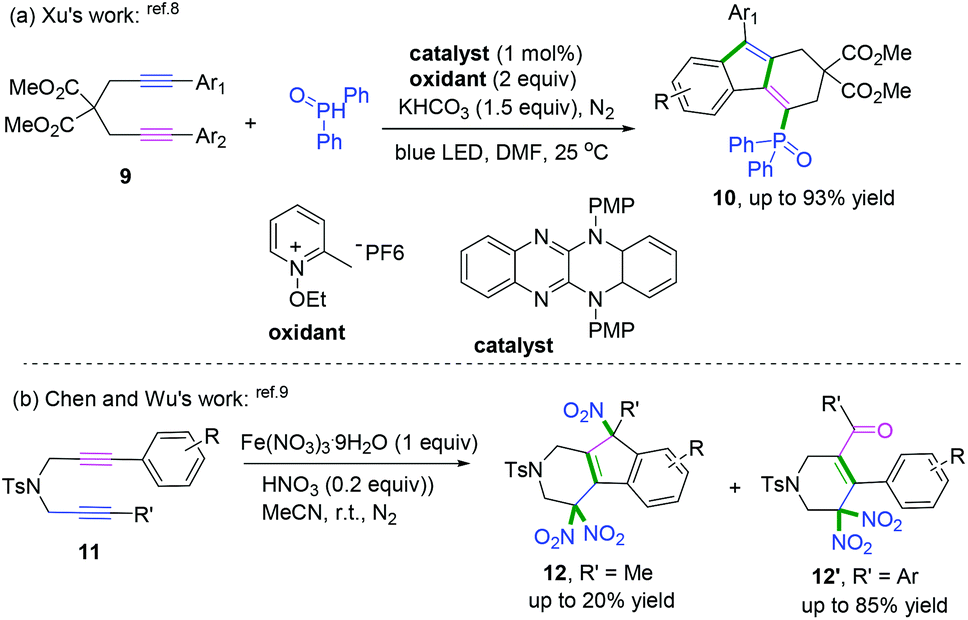
The interception of 1,6-diynes captured with radical donors, such as phosphoryl radical8 and nitroso radical (˙NO2),9 delivered fluorenes bearing different functional groups. In 2018, a series of reducing organic visible-light photocatalysts with strong absorption was designed and synthesized by Xu and co-workers and further used for the radical cascade cyclization of 1,6-diynes with phosphoryl radicals (Scheme 7a).8 The experimental observations have been corroborated through time-dependent density functional theory calculations.
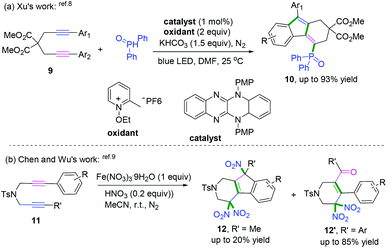 |
| | Scheme 7 Radical cascade cyclization of 1,6-diynols. | |
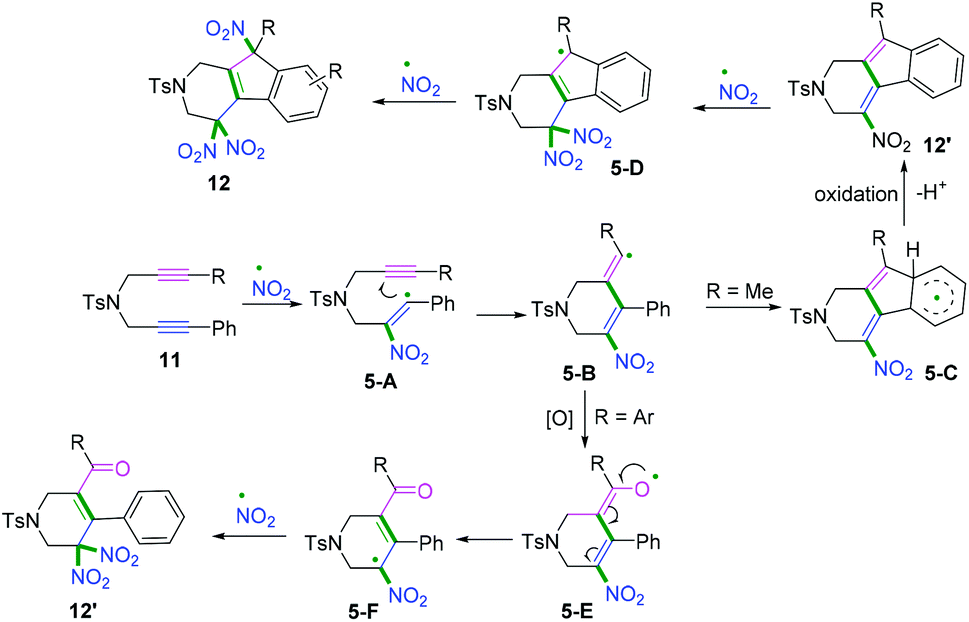
Another radical attacking addition at the β position of 1,n-diynes was demonstrated by Chen, Wu, and co-workers to synthesize cyclic gem-dinitro skeletons via cascade radical nitration of 1,6-diynes (Scheme 7b).9 The transformation proceeded with Fe(NO3)3·9H2O as an inexpensive nitrating agent, providing a mild nitrating source for the preparation of cyclic gem-dinitro compounds. The transformation was proposed to involve the initial generation of ˙NO2 radicals, followed by a sequence of cascade radical addition/cyclization/oxidation/deprotonation/further nitration processes. As demonstrated in Scheme 8, ˙NO2 was firstly generated via decomposition of Fe (NO3)3·9H2O, followed by radical addition to the triple bond. Subsequently, addition of alkenyl radical 5-A to the second triple bond afforded the cyclomethylene radical intermediates 5-B. For the alkyl-substituted 1,6-diyne substrates, the alkenyl radical intermediates 5-B would undergo an intramolecular radical addition process to afford the aromatic intermediates 5-C. A further oxidation/deprotonation/nitration sequence yielded the tri-nitro products. Meanwhile, for the aromatic 1,6-diynes, the cyclomethylene radical intermediates 5-B tended to be oxidized by iron oxo species (FevO), leading to the formation of oxygen radicals 5-E, followed by rapid tautomerization to form intermediates 5-F. Finally, the radical cross-coupling of intermediates 5-F with ˙NO2 delivered the gem-di-nitro cyclization products. It was proposed that the two different reaction processes may be affected by the stabilities and activities of the alkenyl radical intermediates.
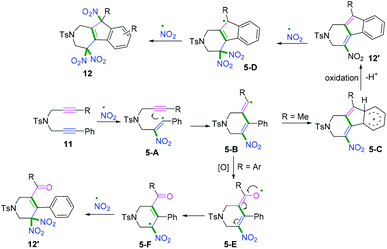 |
| | Scheme 8 Proposed mechanisms for the formation of 12 and 12′. | |
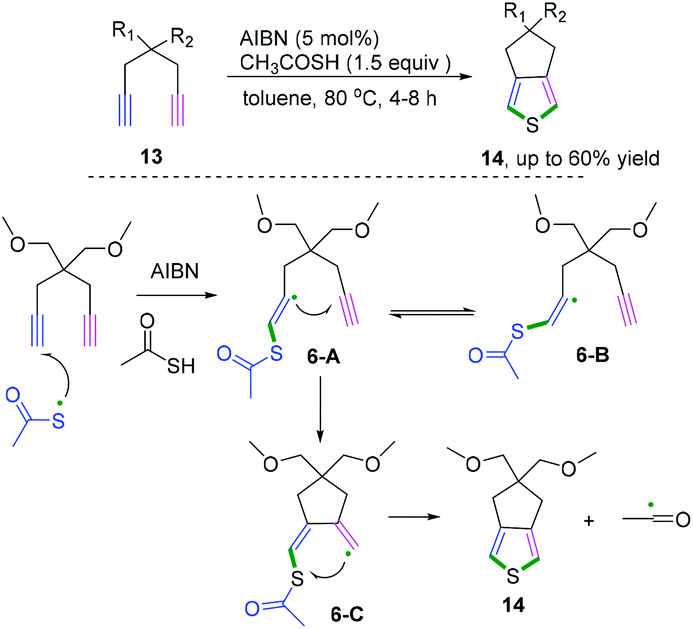
When the 1,6-diynes contain terminal alkynes, the radical addition generally occurs at a terminal position. Representative research on radical cascade cyclization of terminal alkynes for the synthesis of 3,4-fused-ring-substituted thiophenes was reported by Zade and co-workers (Scheme 9).10 The transformation proceeded in the presence of a catalytic amount of azobis-(isobutyronitrile) as a radical initiator. According to the proposed mechanism, the radical cascade initiated thiophene ring 6-C was formed by the radical cyclization of thiyl radical at the terminal position of the diyne. The subsequent cyclization underwent intramolecular homolytic substitution at the sulfur to liberate the acyl radical to furnish the desired products. Notably, it was found that only the E-vinyl radical intermediates 6-A can generate the formation of the desired fused-ring-substituted thiophenes.
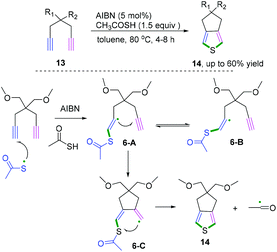 |
| | Scheme 9 Radical cascade cyclization of terminal alkynes. | |
2.2 C(sp)–C(sp2) bond coupling cyclization
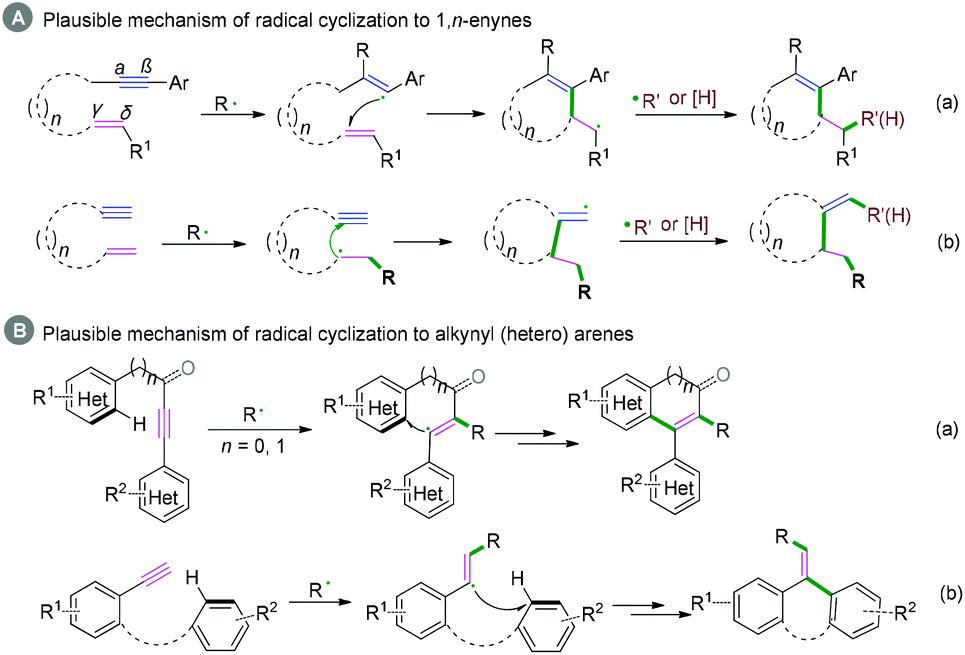
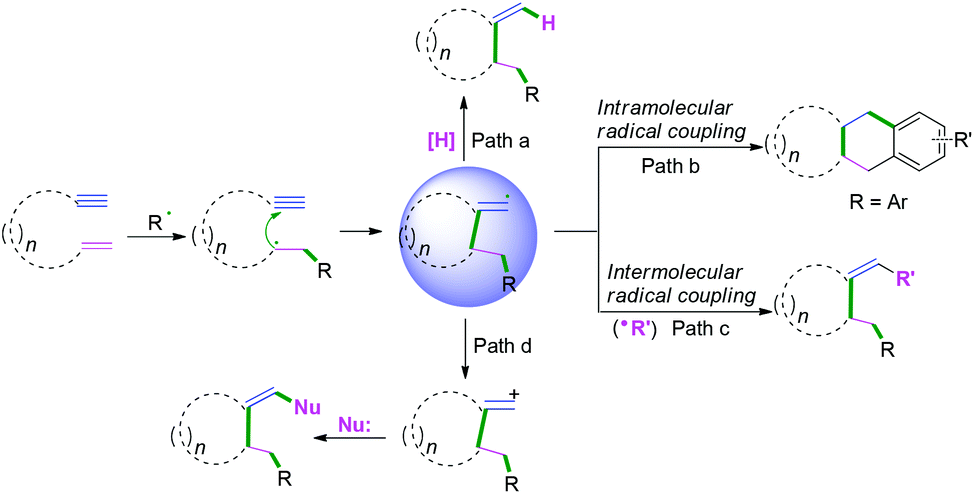
Generally, C(sp)–C(sp2) bond coupling cyclization is generated from 1,n-enynes or alkynyl (hetero) arenes, which are commonly used radical acceptors. For the radical acceptors of 1,n-enynes, there are two functionalities that participate in the cascade reactions, which have four possible directions for radical attack. According to previous studies, the order of priority of attacking unsaturated bonds usually depends on the structure of the radical acceptors and the reaction conditions (Scheme 10A). For the radical acceptors with non-terminal double bonds, although alkyne π-bonds are stronger and less reactive than the π-bonds of alkenes, selective radical attacking at the α position of the alkyne can be achieved, which may be due to the self-sorted kinetic process via the subsequent favored 5-exo-trig radical cyclization (Scheme 10A, a).11 Meanwhile, for the terminal alkenes, regioselective addition of radical species across the double bond in the 1,n-enyne occurs firstly. The regioselectivity is proposed by the fact that the pi-bond of the alkyne is stronger than alkene, leading to the lower kinetic barrier of radical addition to the double bond.12 Subsequently, the same intramolecular 5-exo-cyclization with better orbital overlap for the stereoelectronic effects finishes the cyclization. Finally, the hydrogen abstraction or further radical cross-coupling takes place to generate the observed products (Scheme 10A, b).
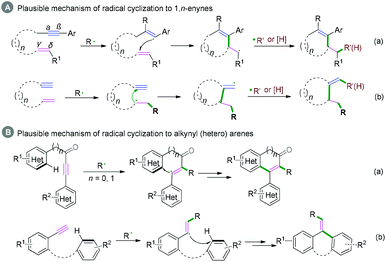 |
| | Scheme 10 Plausible mechanism of radical cyclization to 1,n-enynes or alkynyl (hetero) arenes. | |
Alkynyl (hetero) arenes are also efficient radical acceptors and are widely used in the chemical and pharmaceutical science industries. As demonstrated in Scheme 10B, there are generally two pathway strategies to realize intramolecular radical cyclization of alkynyl (hetero) arenes. One is via radical cyclization of the ortho pi-bonds of (hetero) arenes (Scheme 10B, a) while another strategy is to cyclize via the pi-bonds of other (hetero) arenes (Scheme 10B, b). The key success of this strategy lies in the formation of (hetero) aryl vinyl radical intermediates. The final rearomatization process affords the desired product.
2.2.1 C(sp)–alkenyl C(sp2) bond cyclization.
Cascade radical cyclization of 1,5-enynes.
In pioneering research published by Alabugin and co-workers, they developed a series of reversible Bu3Sn-mediated radical additions to alkynes and alkenes, yielding the formation of Sn-substituted indenes and naphthalenes from 1,5-enynes (Scheme 11).11a–c The radical pool mixtures are kinetically self-sorted via the fastest cyclization of attacking of the more reactive radical (vinyl > alkyl) at the more reactive π-bond (alkene > alkyne). The reactions were found to be highly R2-dependent, providing different products in the cascade radical cyclization of 1,5-enynes. Additionally, alkenes could be used as equivalents of alkynes in this cascade radical reaction when the cascade reaction includes a radical-ring expansion. Along the same lines, the same group developed a formal 6-endo-dig cyclization for the synthesis of quinolines from isonitriles using alkenes as synthetic equivalents of alkynes via coupling homoallylic ring expansion. The proposed mechanism revealed that a path sequence of 5-exo/3-exo/retro-3-exo afforded the formal 6-endo products.11c–e
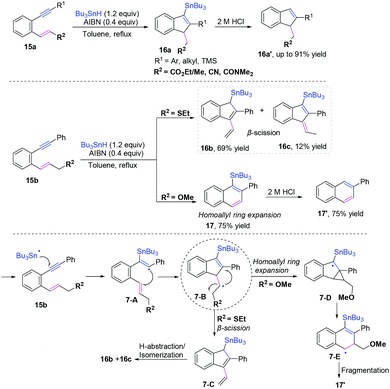 |
| | Scheme 11 Cascade radical cyclization of 1,5-enynes. | |
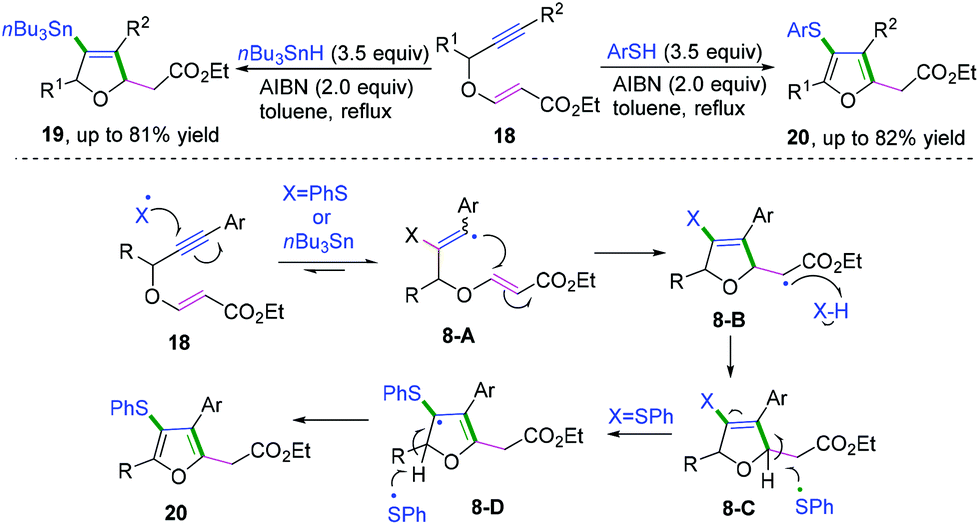
In 2019, Shelke and co-workers reported a cascade radical cyclization of 1,5-enynes to provide an efficient method for the synthesis of a variety of tetrasubstituted furans and dihydrofurans (Scheme 12).11f The proposed mechanism showed that the first selective radical addition of an initial thiyl or stannyl radical to the triple bond generated the aryl vinyl radical intermediate 8-A, followed by favored 5-exo-trig radical cyclization to afford alkyl carbon radical intermediate 8-B. Subsequently, hydrogen abstraction of intermediate 8-B from n-Bu3SnH or PhSH formed the dihydrofuran derivative 8-C. A further radical process was initiated in the presence of excess PhSH and AIBN, leading to the formation of resonance-stabilized allylic radical 8-D. Finally, hydrogen abstraction of dihydrofuran intermediates 8-D afforded the highly substituted furan products. It was supposed that because the bond energy of PhS-H is greater than that of n-Bu3Sn-H, further oxidation of dihydrofurans to the corresponding furans could be performed by thiyl radical.11g
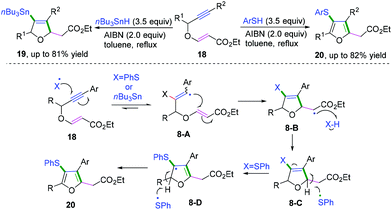 |
| | Scheme 12 Cascade radical cyclization of 1,5-enynes. | |
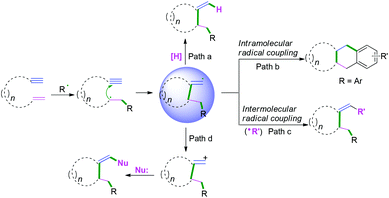 |
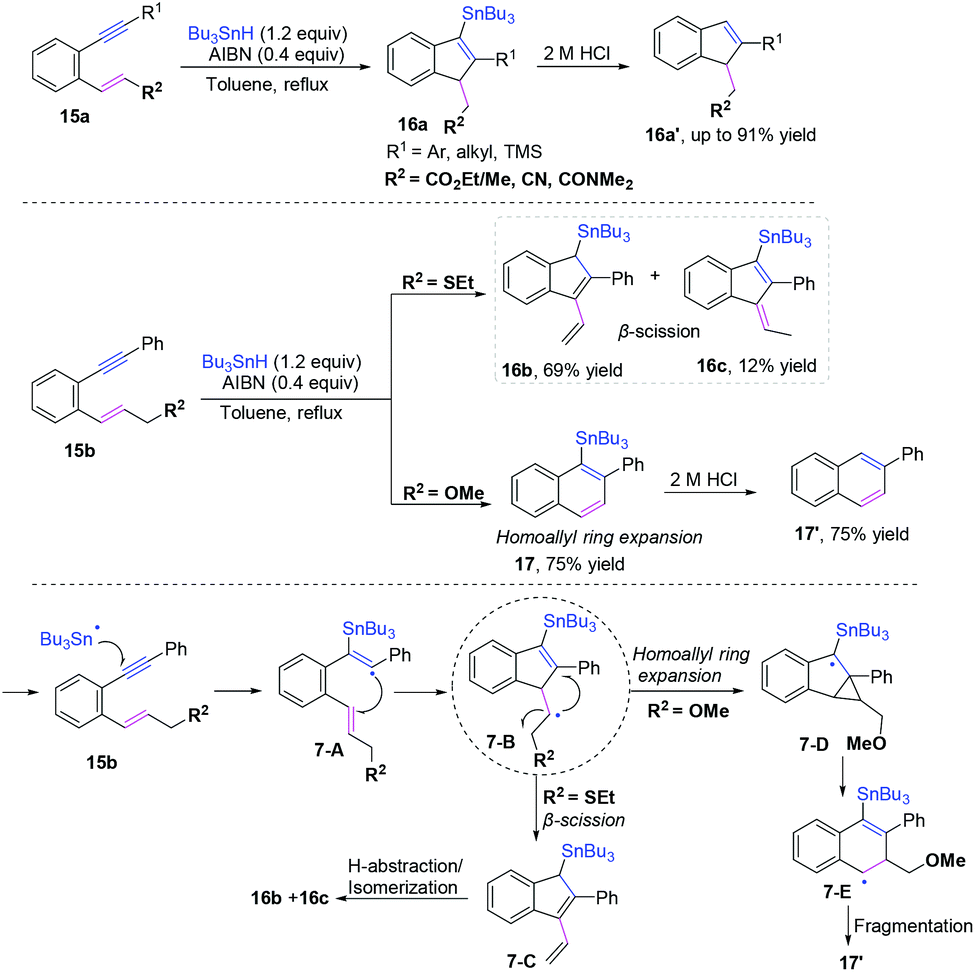
| | Scheme 13 Plausible mechanism of radical cyclization to 1,n-enynes. | |
 |
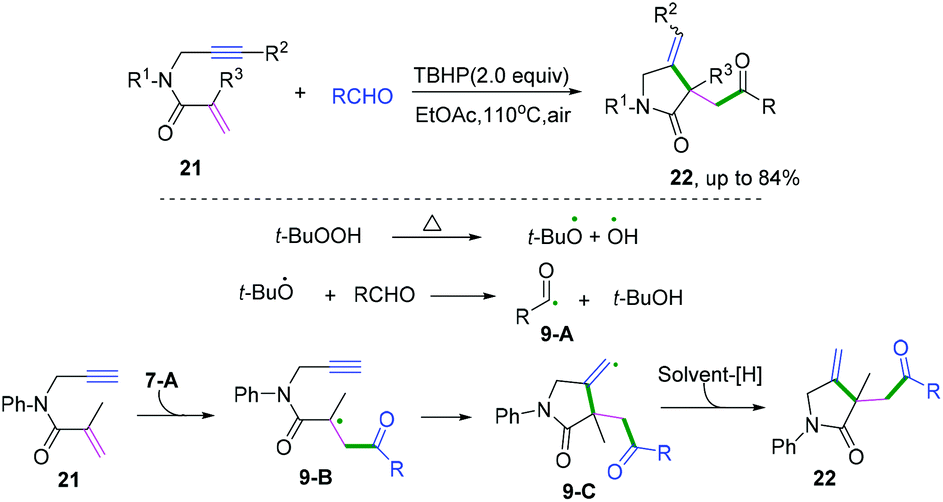
| | Scheme 14 Cascade radical cyclization of 1,6-enynes terminated with hydrogen abstraction. | |
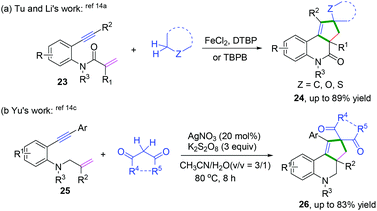 |
| | Scheme 15 Cascade radical cyclization of 1,7-enynes terminated with intramolecular C(sp3)–H coupling. | |
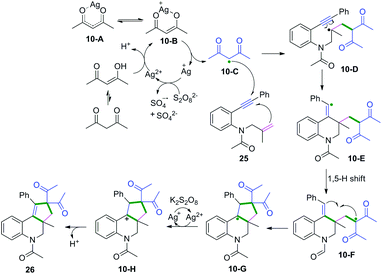 |
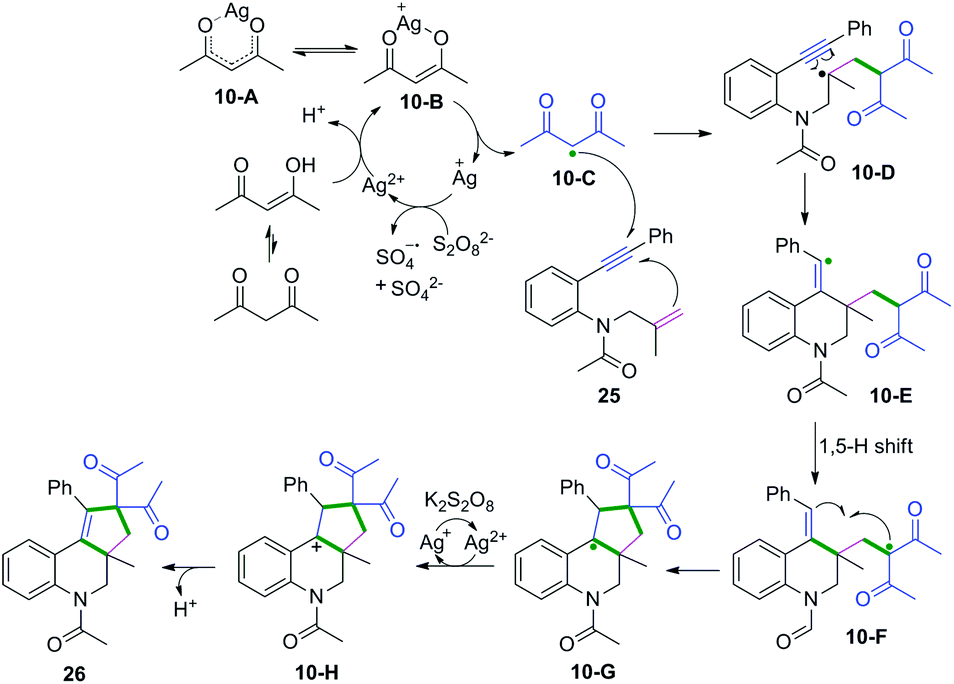
| | Scheme 16 Proposed mechanisms for the formation of 26. | |
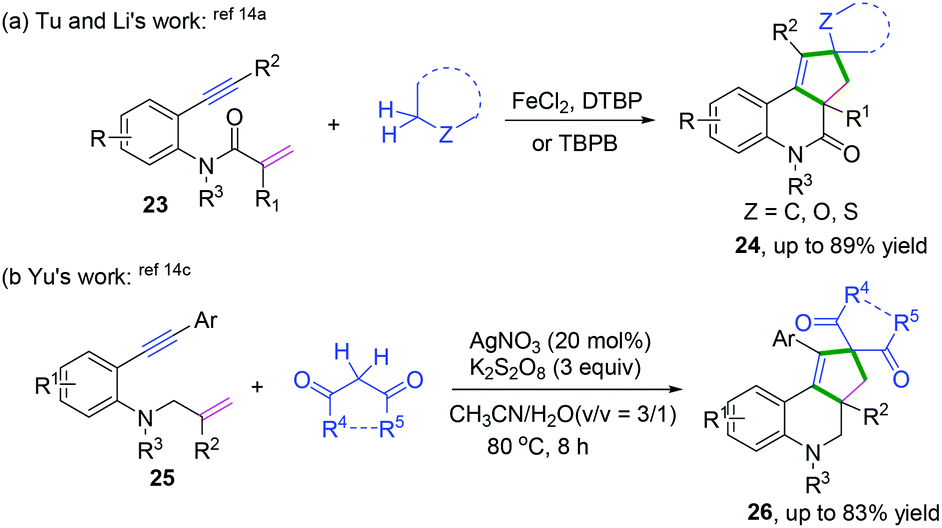
Metal-catalyzed cascade radical cyclization of 1,n-enynes (n = 6, 7) with azides or aldehydes are important methods for the synthesis of a variety of fascinating fused compounds.15 Copper-catalyzed radical [2 + 2 + 1] annulation of benzene-linked 1,n-enynes (n = 6, 7) with azides was developed by Song, Li, and co-workers (Scheme 17a).15a This protocol was successfully applied for the construction of a series of fused pyrroline compounds, which proceeded via a cascade radical addition/cyclization/annulation/azidation sequence. Later, Li and coauthors reported an iron-catalyzed cascade radical [2 + 2 + 2] annulation of benzene-linked 1,7-enynes with aldehydes to access a variety of fused pyran compounds (Scheme 17b).15b Vinyl radical intermediates 11-C generated from aldehydic radical cyclization were terminated with carbonyl compounds, reacting in a dual role to realize the domino cyclization.
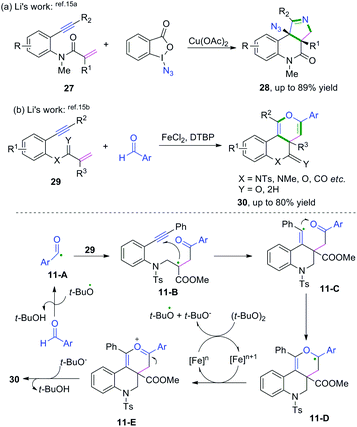 |
| | Scheme 17 Cascade radical cyclization of 1,7-enynes terminated with intramolecular radical coupling. | |
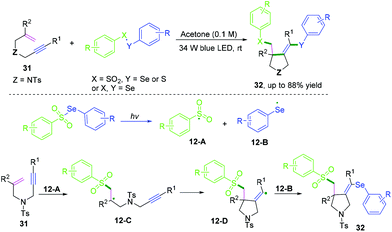 |
| | Scheme 18 Cascade radical cyclization of 1,6-enynes terminated with intermolecular radical cross-coupling. | |
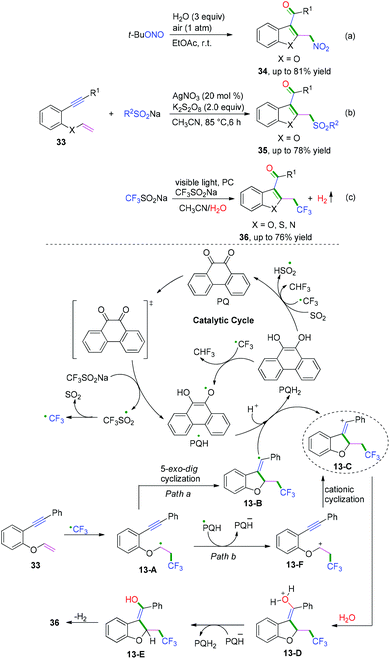 |
| | Scheme 19 Cascade radical cyclization of 1,6-enynes terminated with nucleophiles. | |
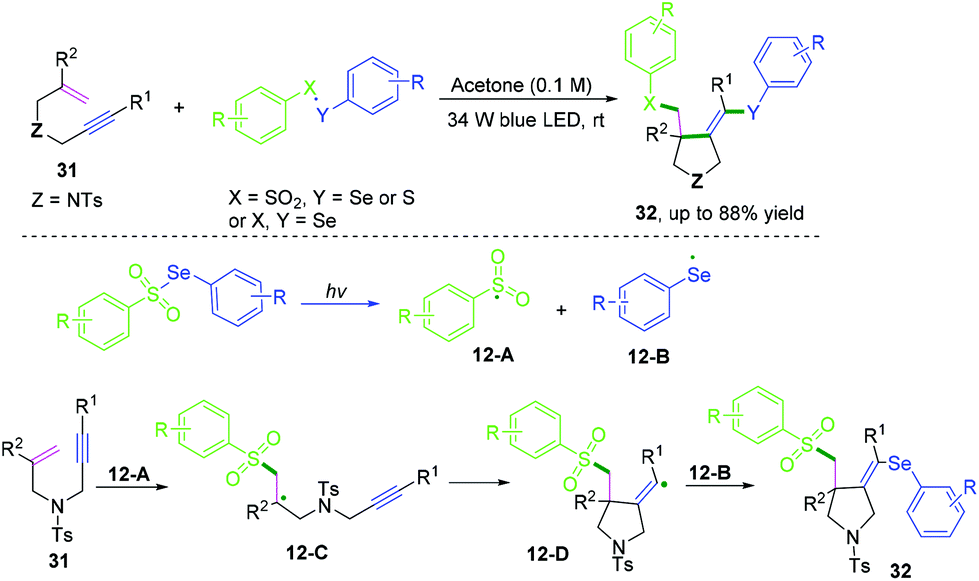
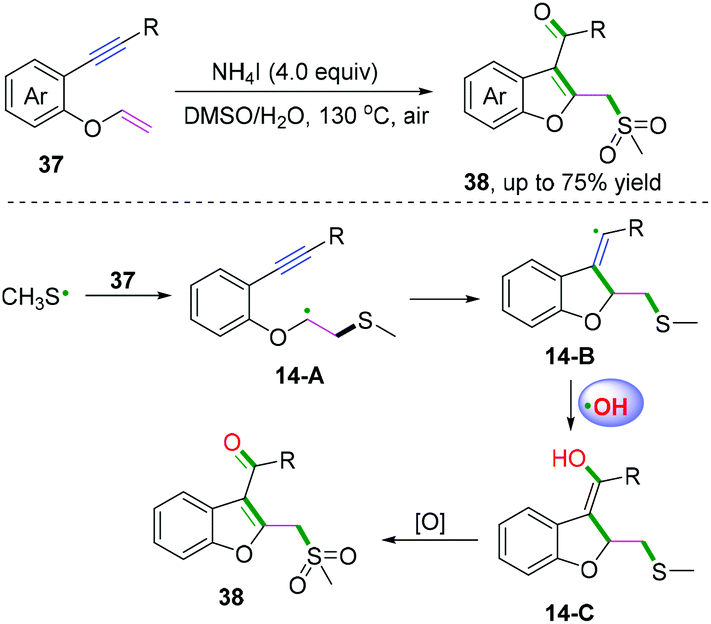
DMSO is one of the simplest starting materials used in organic chemistry. Liu, Sun, and co-workers extended the scope of cascade radical cyclization of oxygen-linked 1,6-enynes with DMSO to synthesize methylsulfonylated and carbonylated benzofurans (Scheme 20).17d In this reaction, vinyl radical intermediates 14-B were terminated by a hydroxyl radical coupling process to afford intermediates 14-C, followed by further keto-enol tautomerism and thioether oxidization to furnish the final products. However, oxidation of 14-A or 14-B into the corresponding carbocations and trapping of the vinyl cation with a water molecule to afford 14-C could not be ruled out.17b,c
 |
| | Scheme 20 Cascade radical cyclization of 1,6-enynes terminated with intermolecular radical cross-coupling. | |
2.2.2 C(sp)–(hetero) aryl C(sp2) bond cyclization.
N-Propargylindoles can act as efficient radical acceptors to capture free radicals, such as S,18 P,19 sulfonyl-,20 and acyl-containing21 radicals, which proceed to cascade cyclization to access 2-functionalized pyrrolo[1,2-a]indoles (Scheme 21).
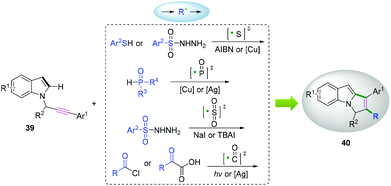 |
| | Scheme 21 Cascade radical cyclization of N-propargylindoles. | |
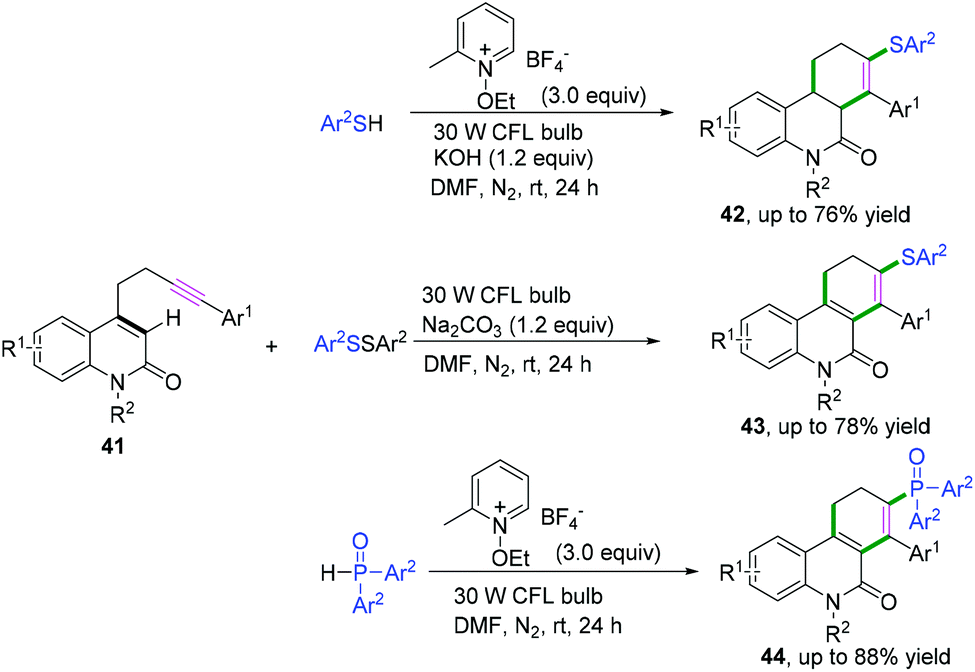
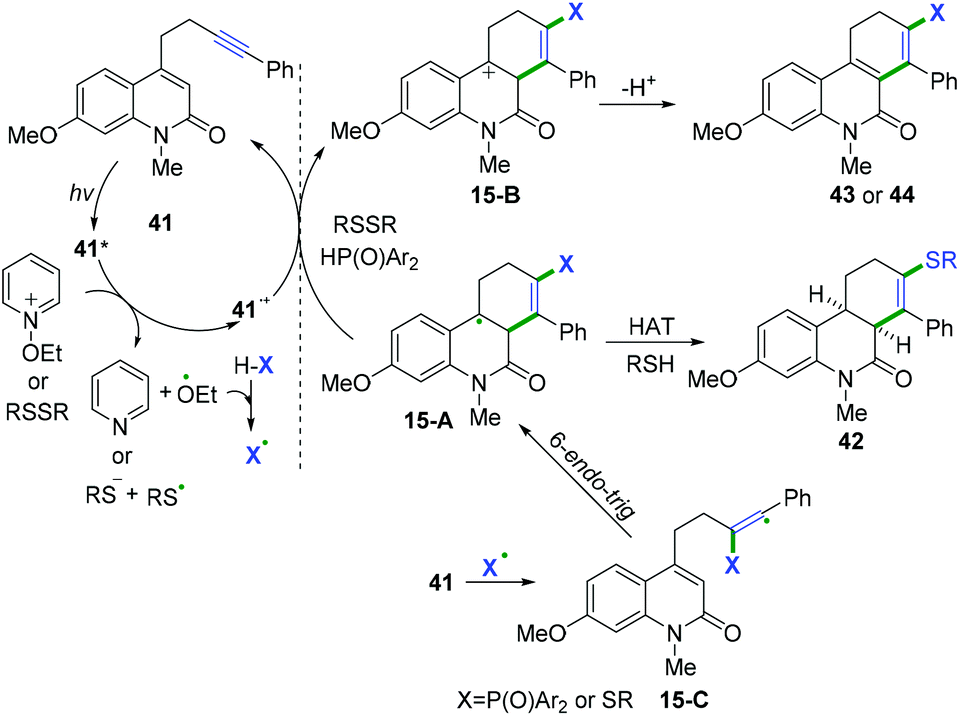
In 2019, Hong and co-workers achieved visible-light-induced radical cyclization of quinolinone-containing radical acceptors for the synthesis of a variety of dihydro- or tetrahydrophenanthridin-6(5H)-ones (Scheme 22).22 It was proposed that the reaction involved the initial generation of S- or P-centered radicals and a subsequent cascade radical addition/6-endo-trig cyclization/hydrogen atom transfer/electron transfer sequence (Scheme 23).
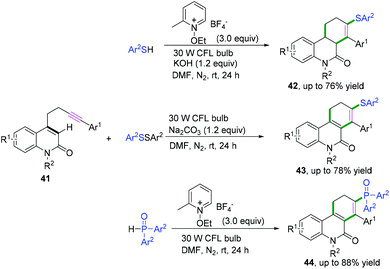 |
| | Scheme 22 Cascade radical cyclization of quinolinone-containing substrates. | |
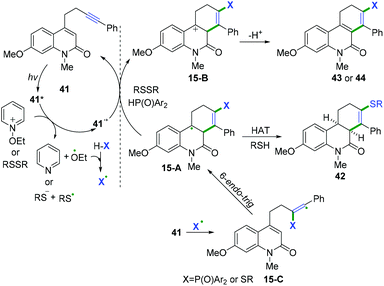 |
| | Scheme 23 Proposed mechanisms for the formation of 42–44. | |
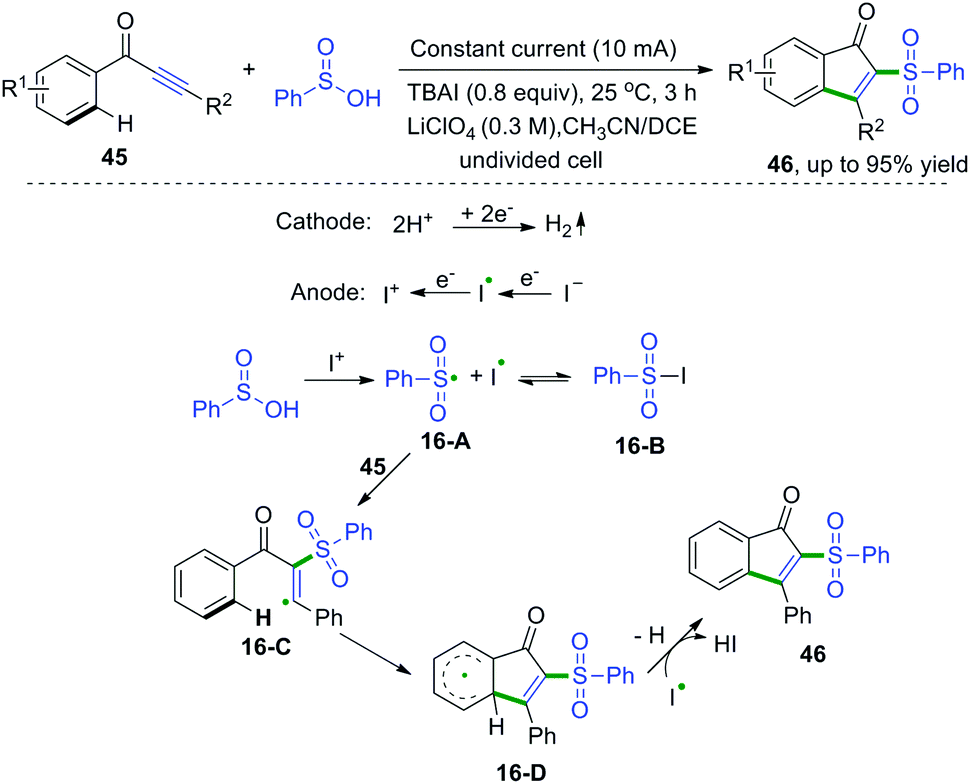
1,3-Diarylprop-2-yn-1-ones are fascinating scaffolds that have broad utility in organic chemistry. In 2017, electrooxidative cascade cyclization of 1,3-diarylprop-2-yn-1-ones with sulfinic acids was developed by Lei and co-workers (Scheme 24).23 This synthetic method was successfully applied for the construction of a series of sulfonated indenones. The proposed mechanism showed that the I+1 species was formed firstly via anodic oxidation of the iodide ion, followed by reaction with sulfinic acids to access sulfonyl radical species and iodine radicals. Subsequent radical addition of sulfonyl radicals to 1,3-diarylprop-2-yn-1-one afforded vinyl radical intermediates 16-C. Then, intramolecular cyclization of intermediates 16-C generated the radical intermediates 16-D. The final oxidation of 16-D finished the desired products.
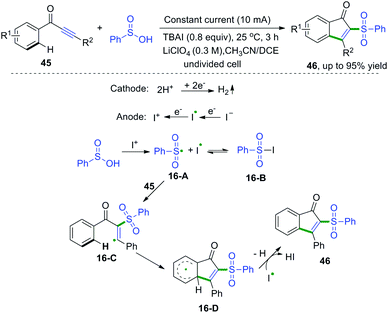 |
| | Scheme 24 Cascade radical cyclization of 1,3-diarylprop-2-yn-1-ones with sulfinic acids. | |
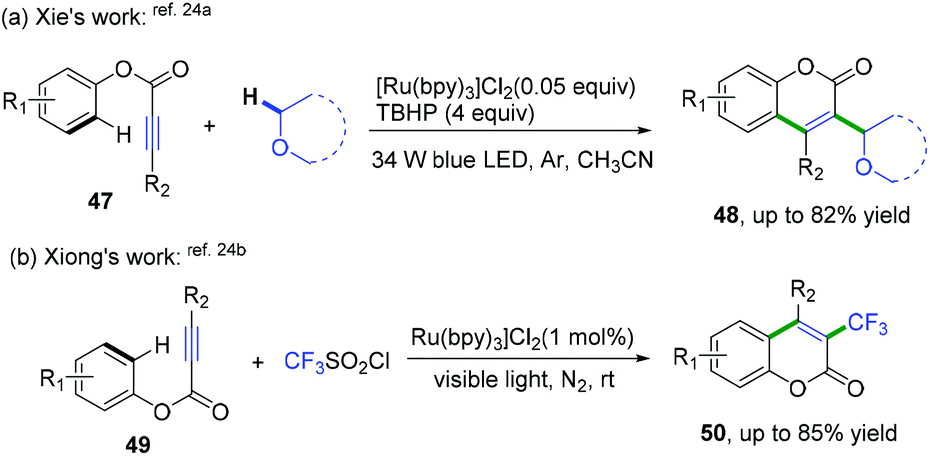
Later, She, Xiong, and co-workers also realized domino radical cyclization of ester arylpropiolates to synthesize 3-substituted coumarin derivatives (Scheme 25).24 As shown in Scheme 25a, a highly regioselective visible-light-promoted difunctionalization of ester arylpropiolates with an α-Csp3–H bond in ether derivatives to construct coumarins was achieved.24a The key intermediate of this reaction is the formation of α-oxo radical species generated by photoredox-mediated oxidation of ethers. The cascade radical cyclization of ester 3-arylpropiolates with CF3SO2Cl as the trifluoromethyl radical source was also developed (Scheme 25b).24b This reaction proceeded via a similar photo-catalytic mechanism of a cascade addition/dearomative cyclization/oxidation/aromatization process to afford a variety of 3-trifluoromethyl coumarins.
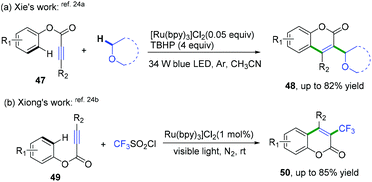 |
| | Scheme 25 Cascade radical cyclization of ester 3-arylpropiolates. | |
In 2018, Xu and co-workers developed an unprecedented electrochemical difluoromethylarylation of alkynes to construct fluorinated dibenzazepines (Scheme 26).25 This protocol was performed with a new air-stable difluoromethylation reagent, CF2HSO2NHNHBoc, pre-synthesized from commercially available reagents CF2HSO2Cl and NH2NHBoc in one step. Based on their mechanistic research, they proposed a detailed catalytic cycle, as depicted in Scheme 26. Firstly, electrolytic anodic oxidation of Cp2Fe would afford the Cp2Fe+ species, along with the reduction of MeOH to afford H2 and MeO− at the cathode. Subsequent oxidation of intermediates 17-A and the conjugate base of CF2HSO2NHNHBoc by Cp2Fe+ species generated diazene intermediates 17-Cvia the intermediacy of N-radical intermediates 17-B, followed by decomposition to generate CF2H radical, which then reacted with the triple bonds in the alkynes to furnish the vinyl radical intermediates 17-D. C-Radical intermediates 17-D underwent the preferred 7-ortho cyclization with the N-benzyl group to afford the radical intermediates 17-E in a regio- and stereoselective manner. The N-benzyl group attacked the vinyl radical from the side away from the difluoromethyl substituent to avoid steric repulsion, leading to the Z-configuration of the trisubstituted alkenes in intermediates 17-E. Final rearomatization of intermediates 17-E through electron and proton elimination afforded the desired dibenzazepine products.
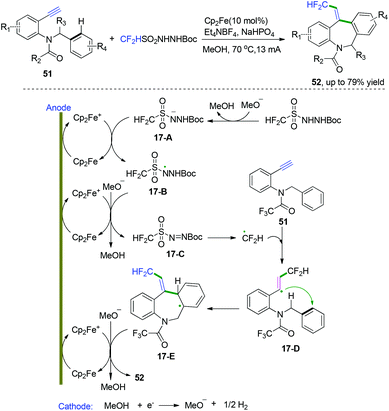 |
| | Scheme 26 Electrochemical cascade radical cyclization of alkynes. | |
2.3 C(sp2)–C(sp2) bond coupling cyclization
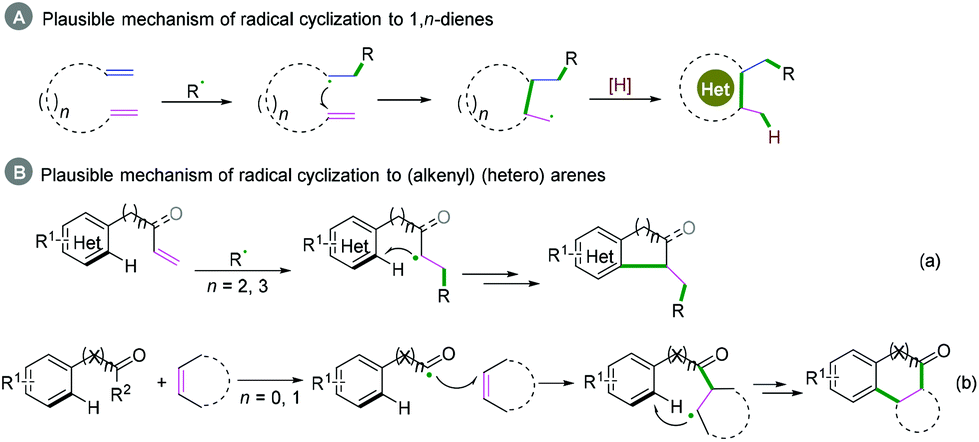
1,n-Dienes or alkenyl (hetero) arenes are also usual radical acceptors, which would benefit the construction of complex cycle structures via coupling cyclization reactions. In the past decades, many excellent xanthate-based cascade radical reactions to construct complex structures, as reported by Zard and coauthors, have been realized.26 Due to the instability of radical intermediates, coupling cyclization of 1,n-dienes is difficult to realize efficiently (Scheme 27A), while for the radical acceptors of alkenyl (hetero) arenes, the efficiency of the coupling cyclization reaction depends on whether the cyclization is intramolecular or intermolecular (Scheme 27B). Alkenyl (hetero) arenes usually act as radical acceptors with an activated terminal carbon–carbon double bond, which contributes to highly efficient intramolecular cyclization (Scheme 27B, a), while intermolecular cyclization is rarely realized efficiently owing to the higher energy barrier (Scheme 27B, b).
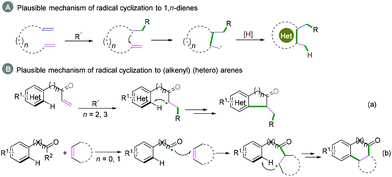 |
| | Scheme 27 Plausible mechanism of radical cyclization of 1,n-dienes or (alkenyl) (hetero) arenes. | |
2.3.1 Alkenyl C(sp2)–alkenyl C(sp2) bond coupling cyclization.
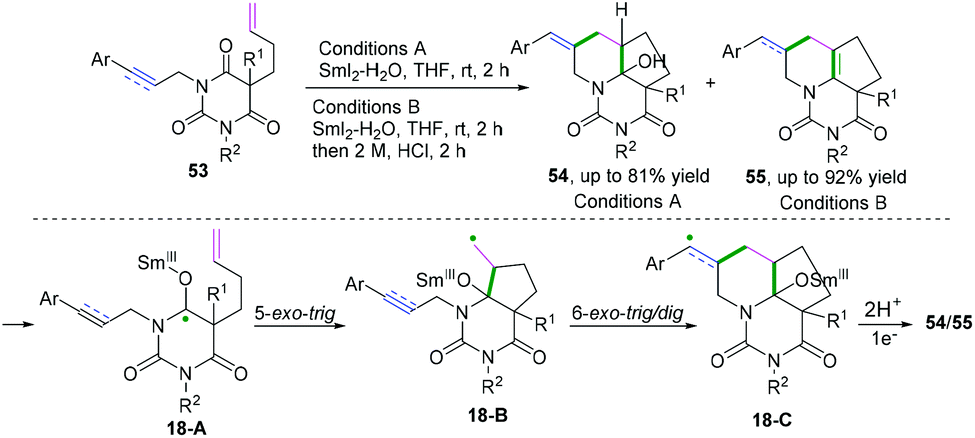
In 2016, Procter and co-workers achieved radical–radical cyclization cascades for the synthesis of hemiaminal- or enamine-containing tricyclic compounds with up to five contiguous stereo-centers (Scheme 28).27 The transformation underwent an electron-transfer reduction of amide-type carbonyls by SmI2-H2O. The proposed mechanism suggested that LiBr had a beneficial effect on achieving highly efficient radical–radical cyclization cascades. According to the reaction mechanism, enyne 53 proceeded through 5-exo-trig radical cyclization to afford radical intermediates 18-B, followed by 6-exo-trig/dig cyclization to generate intermediates 18-C. Further reduction and protonation delivered tricyclic hemiaminals 54 or the corresponding enamines 55 after a dehydration process.
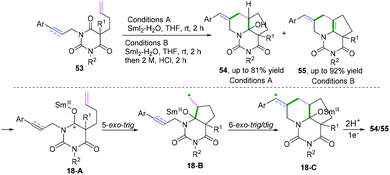 |
| | Scheme 28 Cascade radical cyclization of dienes/enynes. | |
2.3.2 Alkenyl C(sp2)–(hetero) aryl C(sp2) bond coupling cyclization.
Intramolecular cyclization.
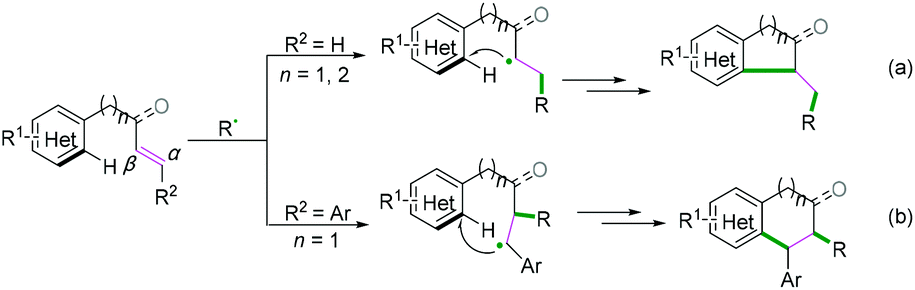
As demonstrated in Scheme 29, for radical acceptors of alkenyl (hetero) arenes, the order of priority of the attacking position of the carbon–carbon double bond depends on the type of R2 substituent group (Scheme 29). When R2 is H, the priority attacking position of the carbon–carbon double bond occurs at the α position (Scheme 29a), while it reacts at the β position for the aryl substitute; this may be due to the steric hindrance effect (Scheme 29b).
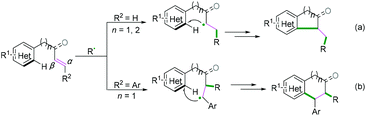 |
| | Scheme 29 Intramolecular alkenyl C(sp2)–(hetero) aryl C(sp2) bond coupling cyclization. | |
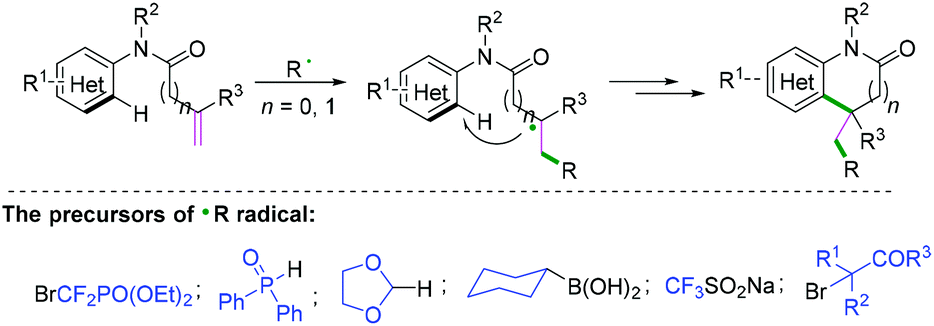
Functionalization of alkenyl N-arylamides with radical donors has become a more efficient and straightforward way to generate a variety of nitrogen heterocyclics. Radical cyclization of alkenyl N-arylamides with free radical precursors,28–34 such as BrCF2PO(OEt)2,28 diphenylphosphine oxide,29 1,3-dioxolane,30 cyclohexylboronic acid,31 CF3SO2Na,32 and alkyl bromides,33 finished the final alkenyl C(sp2)–(hetero) aryl C(sp2) bond construction (Scheme 30).
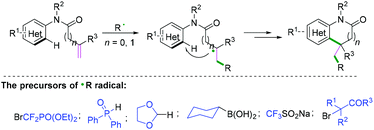 |
| | Scheme 30 Cascade radical cyclization of alkenyl N-arylamides with radical donors. | |
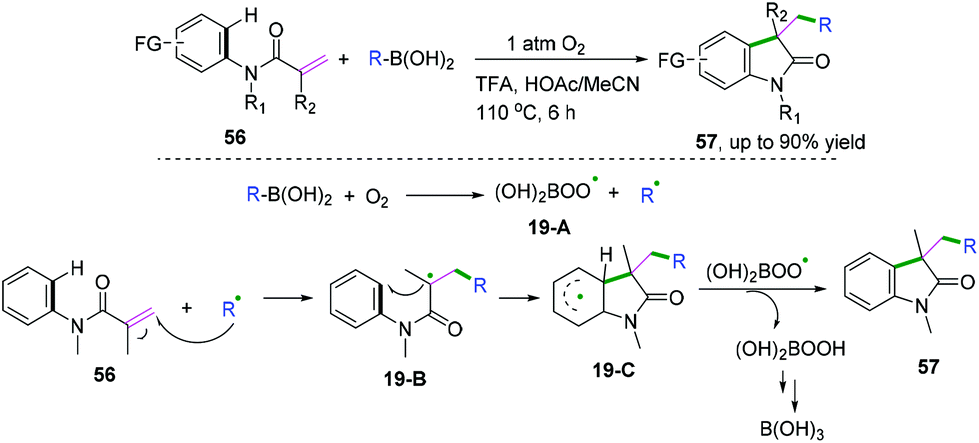
In 2018, Liu and co-workers demonstrated an efficient metal-free alkylation of N-arylacrylamides with organoboronic acid to access a wide range of medical and natural privileged scaffolds (Scheme 31).31 The control experimental results indicated that molecular oxygen was used as the terminal oxidant and that site-specificity was achieved. The proposed mechanism showed that radical 19-A and alkyl radicals were generated initially from the reaction of boronic acid and molecular oxygen. Subsequent addition of the alkyl radical to the alkene followed by cyclization would afford radical intermediate 19-C. Then, a hydrogen-atom transfer process from intermediate 19-C to 19-A furnished the desired product and peroxyboronic acid, leading finally to boric acid.
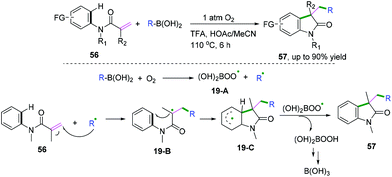 |
| | Scheme 31 Molecular oxygen-promoted radical alkylation with organoboronic acid. | |
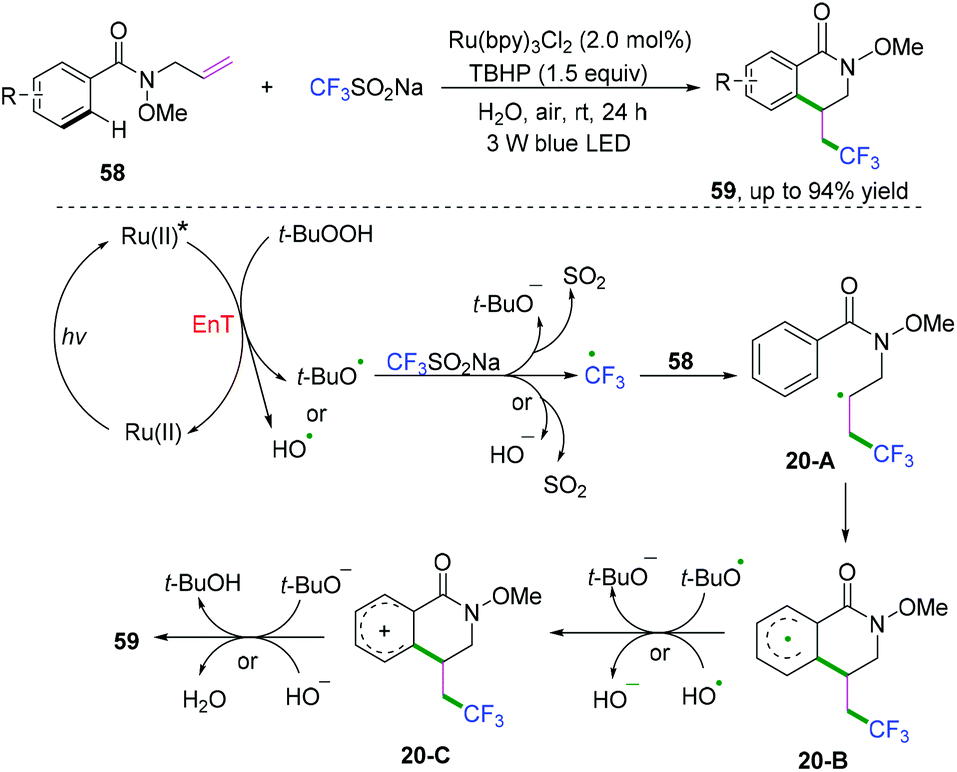
Later, Li, Wang, and coauthors reported a visible-light-induced method for cyclization of N-allylbenzamides with CF3SO2Na to synthesize trifluoromethylated dihydroisoquinolinone derivatives under mild and green reaction conditions (Scheme 32).32 Radical cyclization transformation was realized efficiently to access the desired products in high yields in water at room temperature.
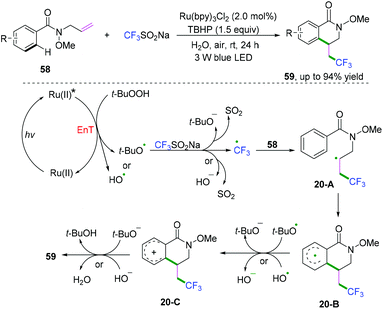 |
| | Scheme 32 Visible-light-induced radical cyclization of N-allylbenzamides with CF3SO2Na. | |
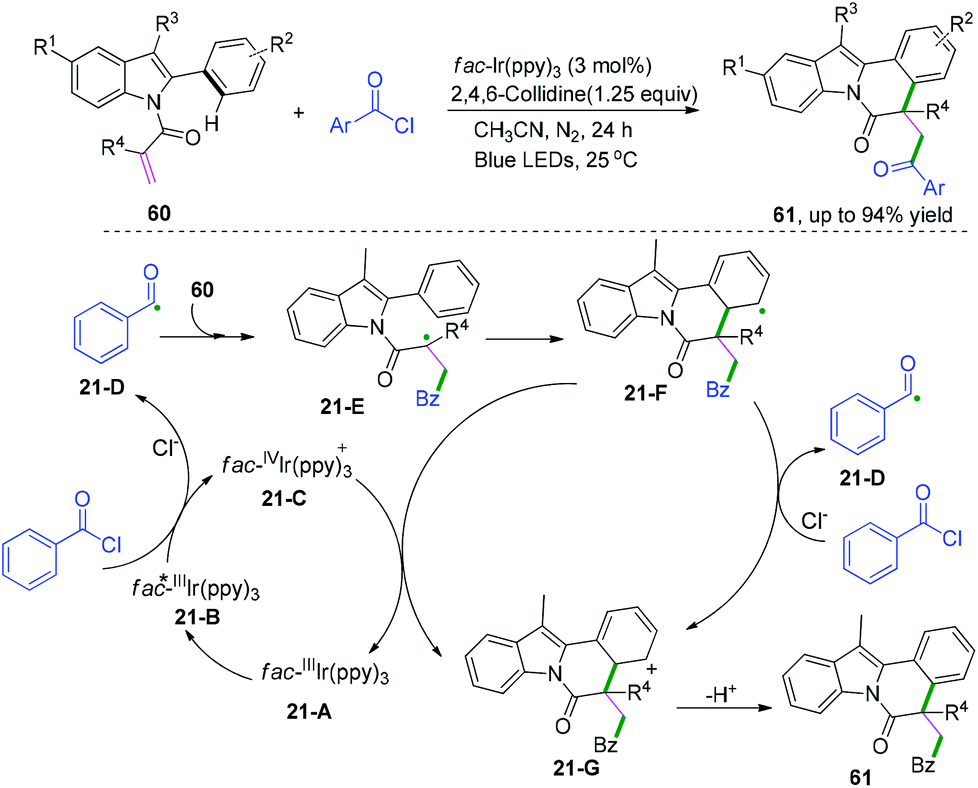
Indolo[2,1-a]isoquinoline derivatives are fascinating scaffolds that are widely found in natural products and pharmaceuticals;35 thus, they are attracting a wide range of attention in organic synthesis.36,37 Recently, visible-light-induced radical cascade transformation of 2-aryl indoles with acyl chlorides has been developed by Xu and co-workers (Scheme 33).37 This redox-neutral reaction was successfully applied for the construction of a series of functionalized valuable indolo[2,1-a]isoquinoline derivatives. The transformation proceeded initially with the formation of acyl radical species 21-D under the photo-catalytic cycle, followed by the addition reaction with 2-aryl indoles to afford a new α-acyl radical intermediate, 21-E. Subsequently, the radical intermediate 21-F was delivered via cyclization reaction of species 21-E, and the further oxidation of species 21-F afforded the cation intermediate 21-G through a SET process with the oxidized photocatalyst fac-IVIr(ppy)3, regenerating the ground state photocatalyst fac-Ir(ppy)3 for the next new photocatalytic cycle; meanwhile, alternative oxidation of species 21-F by benzoyl chloride to generate the cation 21-G cannot be ruled out. The final rapid deprotonation of cation species 21-G proceeded to regain its aromaticity and furnished the desired product.
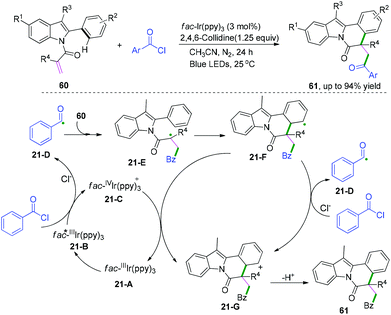 |
| | Scheme 33 Visible-light-induced radical cascade cyclization of 2-aryl indoles with acyl chlorides. | |
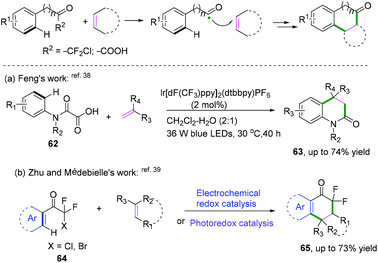 |
| | Scheme 34 Intermolecular cyclization of alkenes with aryl-substituted radical donors. | |
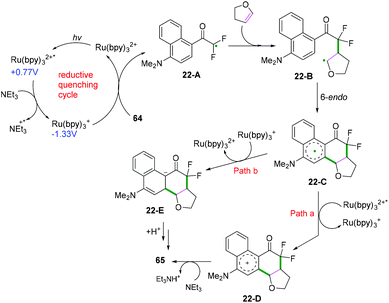 |
| | Scheme 35 Proposed mechanism for the formation of 65. | |
2.4 C(sp2)–C(sp3) bond coupling cyclization
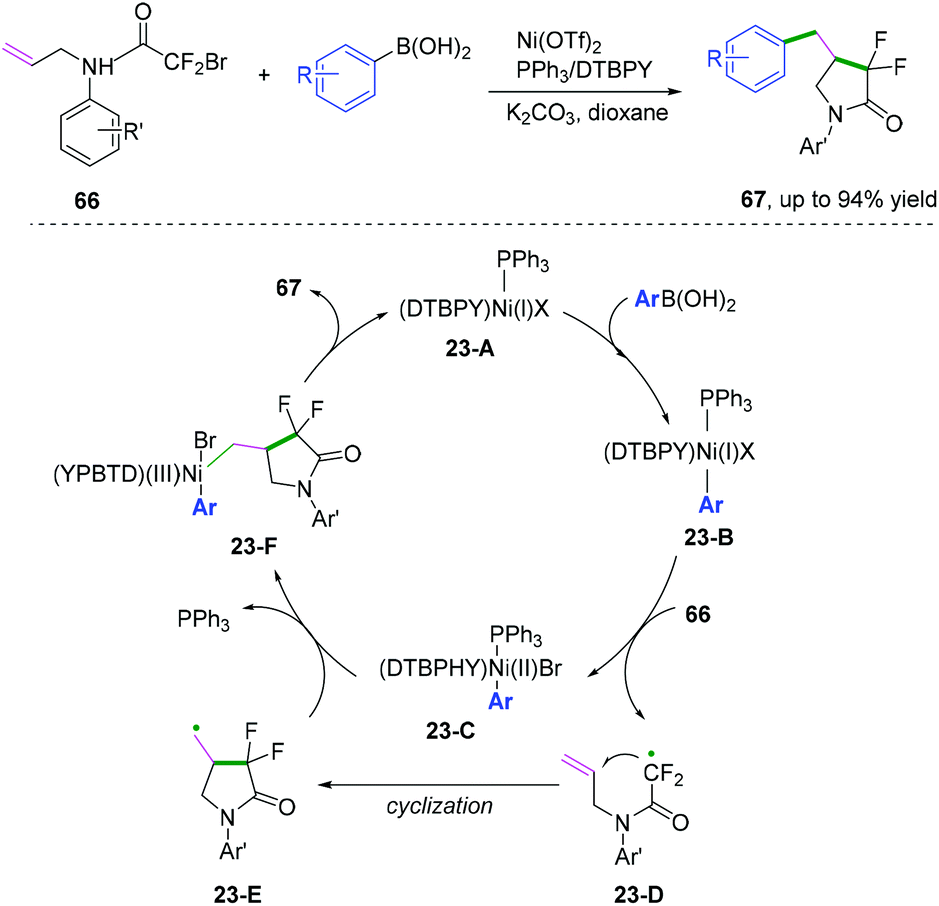
Compared with C(sp)–C(sp2) and C(sp2)–C(sp2) bond coupling cyclization, C(sp2)–C(sp3) cyclization is more challenging because the alkyl radical version to afford coupling cyclization has a higher energy barrier. In 2018, Mai, Lu, and co-workers developed a nickel-catalyzed radical cascade cyclization/arylation to access a series of 4-benzyl-3,3-difluoro-γ-lactam derivatives (Scheme 36).40 The reaction was supposed to proceed through the formation of a primary alkyl radical generated from Ni(I) complex 23-B initiated via a single electron transfer process. Further intermolecular Suzuki-type coupling cyclization afforded the desired product.
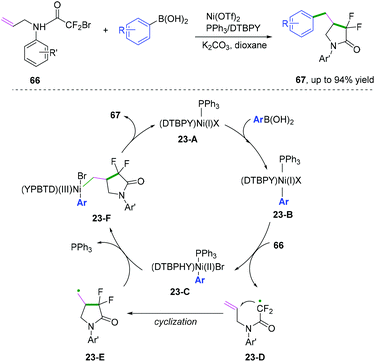 |
| | Scheme 36 Nickel-catalysed C(sp2)–C(sp3) bond coupling cyclization. | |
3. Carbon–heteroatom bond coupling cyclization
In addition to the aforementioned studies, carbon–heteroatom bond coupling cyclization initiated by heteroatom radicals is an efficient synthetic method to generate heterocycles.41
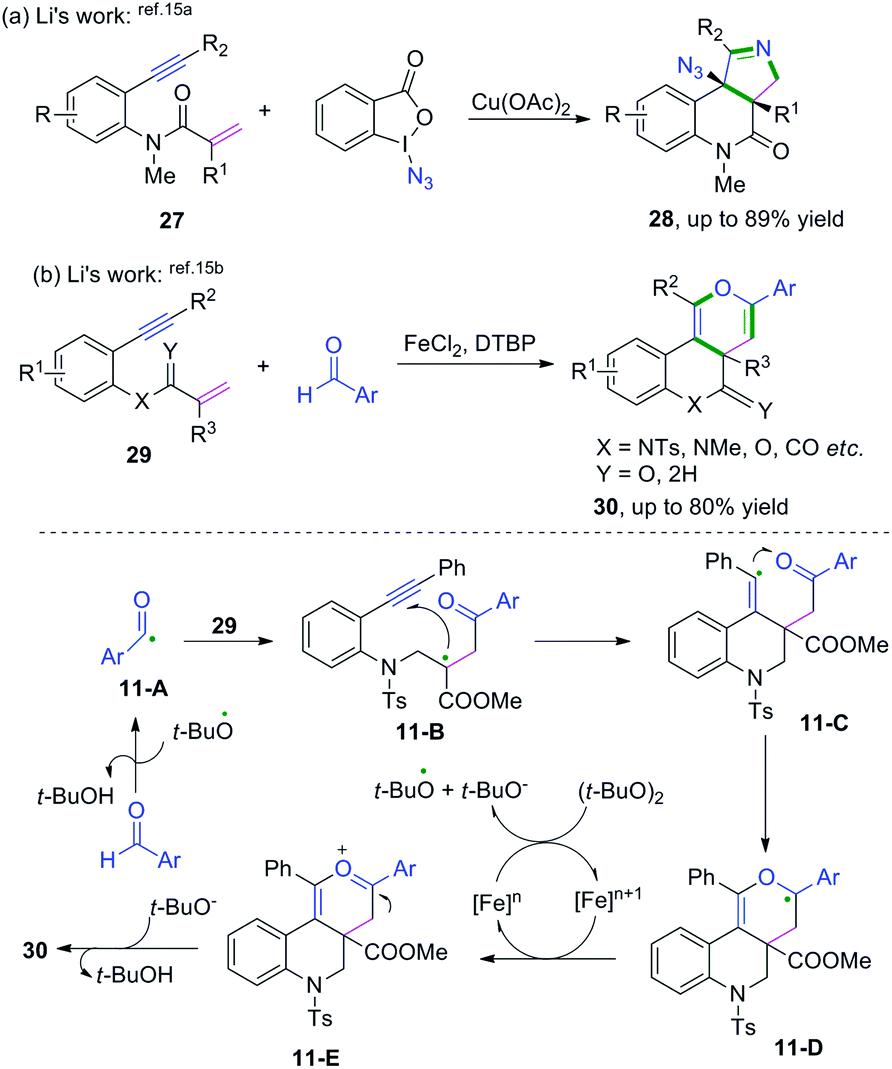
3.1 C(sp)–Heteroatom bond
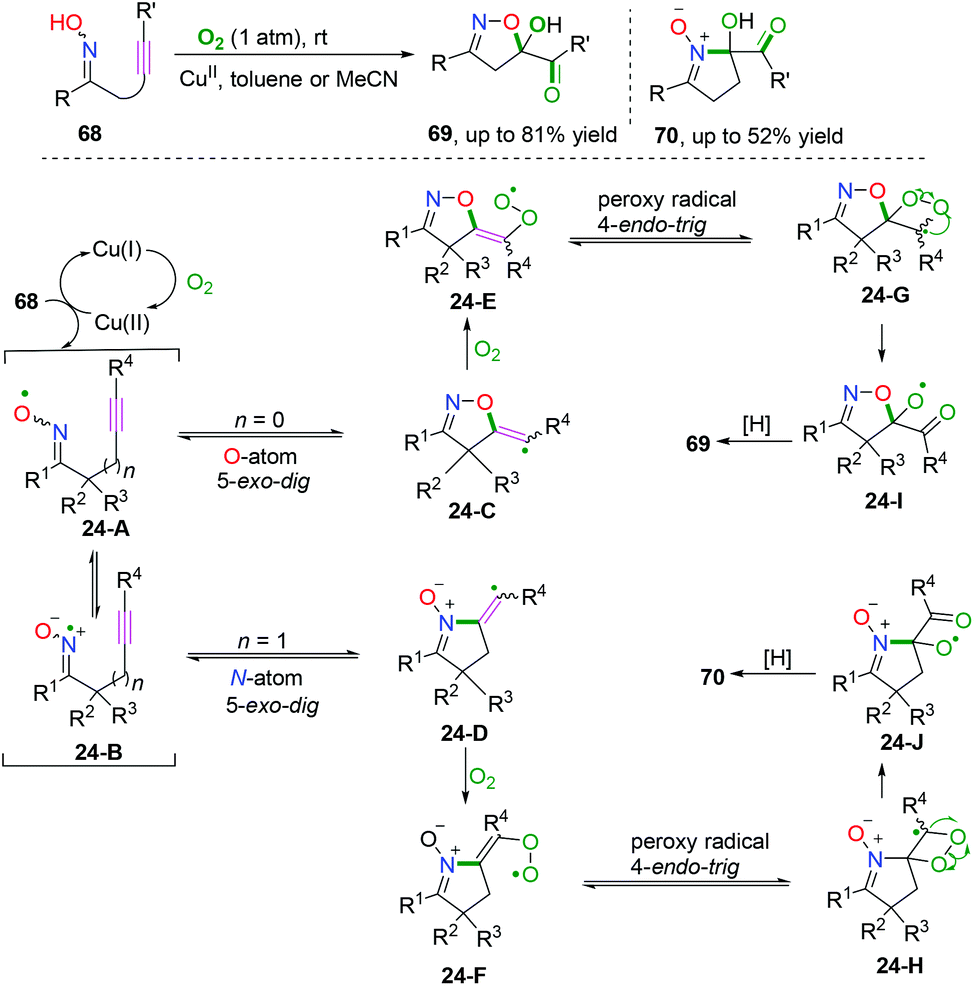
Alkyne-tethered ketoxime compounds can also serve as highly efficient radical acceptors to perform radical coupling cyclization, as reflected in the Cu-catalyzed dioxygen-activated cascade radical coupling reaction developed by Han and co-workers (Scheme 37).42 This transformation successfully accessed a range of isoxazoline/cyclic nitrone-featured α-ketols under mild conditions. A cascade iminoxyl radical dichotomous 5-exo-dig cyclization/oxygen activation/peroxy radical 4-endo-trig cyclization process was proposed by 18O isotope tracing experiments and DFT calculations.
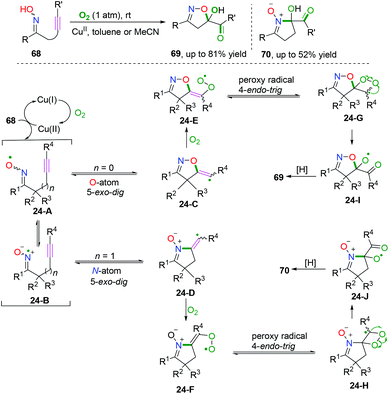 |
| | Scheme 37 Cu-catalyzed cascade radical reaction of alkyne-tethered ketoximes. | |
3.2 C(sp2)–Heteroatom bond
Recently, transition-metal catalyzed iminoxyl radical-initiated cyclization has been applied in realizing C(sp2)–heteroatom bond coupling cyclization.43–46 The general transition-metal catalyzed mechanism involves capturing the carbon-centered radical intermediates by TMn+Nu to form high-valent transient TM(n+1)+Nu species, followed by reductive elimination to afford the final products (Scheme 38A).
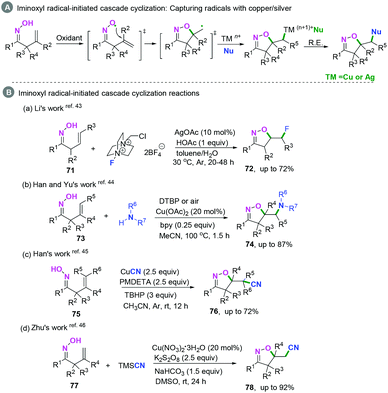 |
| | Scheme 38 Transition-metal catalyzed iminoxyl radical-initiated cyclization. | |
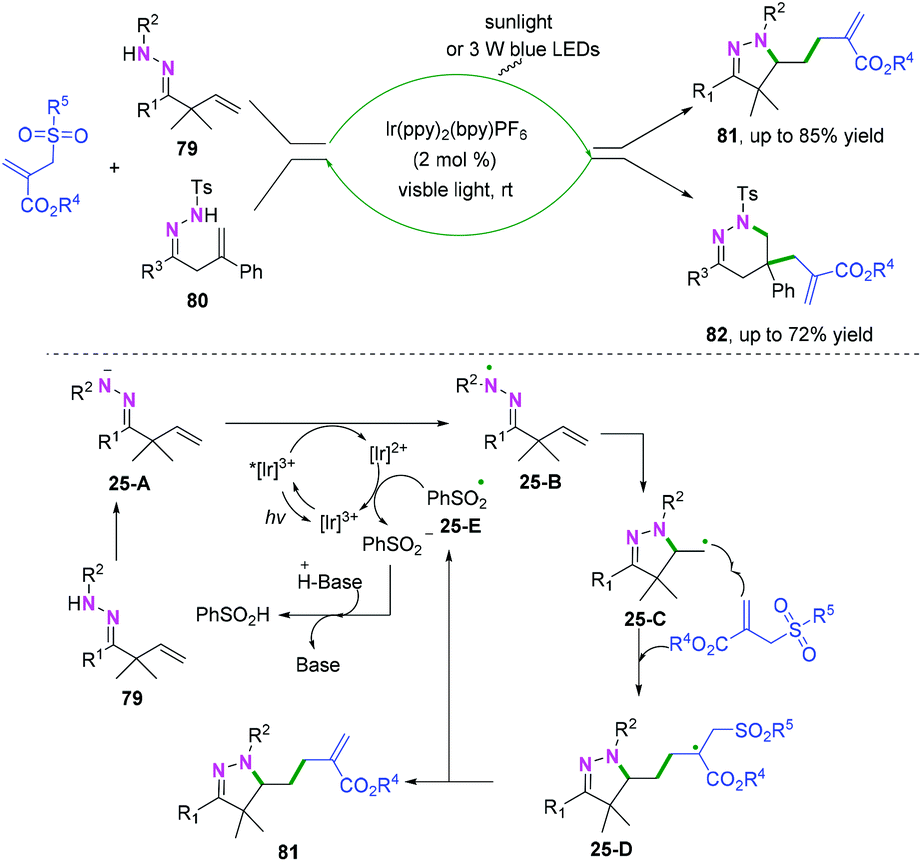
Additionally, photo-catalyzed hydrazonyl radical-mediated cyclization is an efficient strategy to realize C(sp2)–heteroatom bond coupling cyclization, as reported by Chen and coauthors (Scheme 39).47 As demonstrated in Scheme 39, carbon-centered radical intermediates 25-C generated by hydrazonyl radical 25-B-initiated cyclization are selectively trapped by allyl sulfones to afford intermediate 25-D, followed by hemolytic fragmentation to access the final desired compounds with release of the sulfonyl radical 25-E. Finally, the sulfonyl radical intermediate 25-E undergoes further single-electron transfer reduction by the reducing [Ir(ppy)2bpy]2+ species to afford the sulfonate anion and regenerate the ground state photocatalyst for the new photocatalytic cycle.
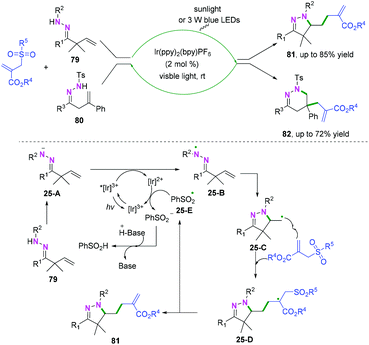 |
| | Scheme 39 Proposed mechanism for the formation of 81. | |
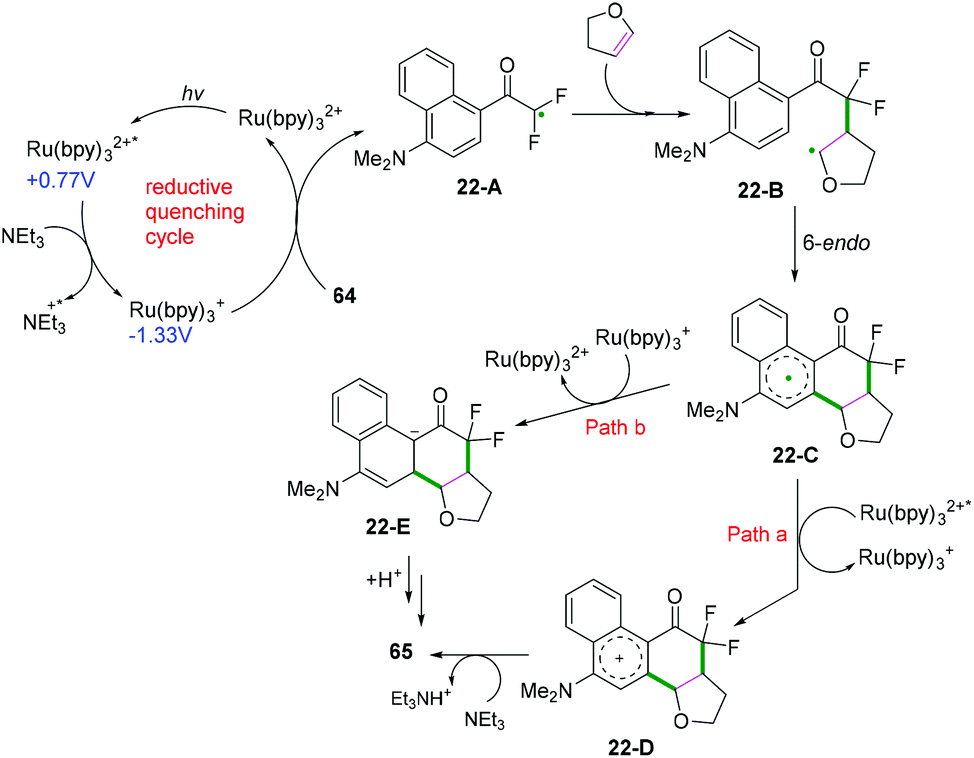
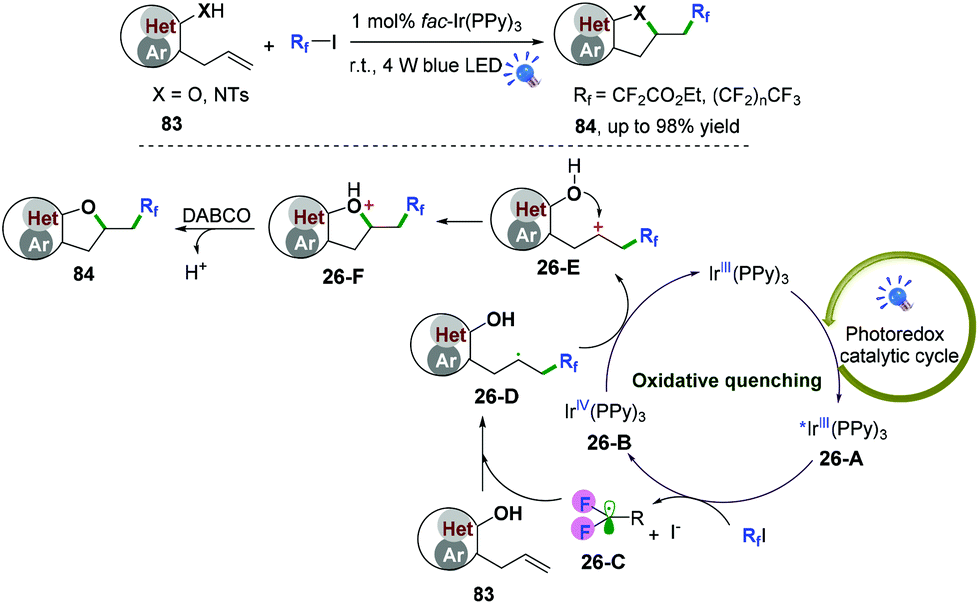
Our group also described palladium and photoredox-catalyzed oxy-/aminofluoroalkylative cascade radical cyclization of alkenes with available Rf-I reagents to generate fluoroalkylated 2,3-dihydrobenzofuran and indolin derivatives in good to excellent yields under mild conditions (Scheme 40).48,49 As shown in Scheme 40,49 first, irradiation of the photocatalyst would lead to an excited-state adduct *fac-Ir(PPy)326-A, which would then engage in electron transfer with fluoroalkyl iodide reagents (RfI) to afford the fluoroalkyl radical species 26-C and intermediates fac-IrIV(PPy)326-B. Subsequent radical addition of 26-C to alkenes would generate the alkyl radicals 26-D. Further oxidation of radical intermediates 26-D with fac-IrIV(PPy)3via a second SET process afforded the carbocation intermediates 26-E and recycled the ground state fac-IrIII(PPy)3 for the new catalytic cycle. A new C–O bond was then constructed via intramolecular 5-exo-trig cyclization to afford intermediates 26-F. Final deprotonation with DABCO would deliver the desired cyclization products. Additionally, reaction of the intermediates 26-D with iodofluoroalkane to form the corresponding secondary iodides followed by nucleophilic substitution cannot be ruled out for this reaction.
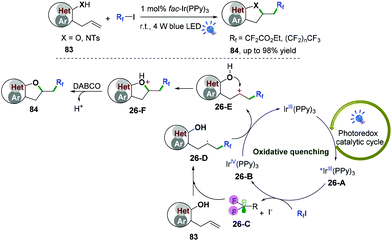 |
| | Scheme 40 Photoredox-catalyzed oxy-/aminofluoroalkylative cyclization of alkenes. | |
4. Miscellaneous examples
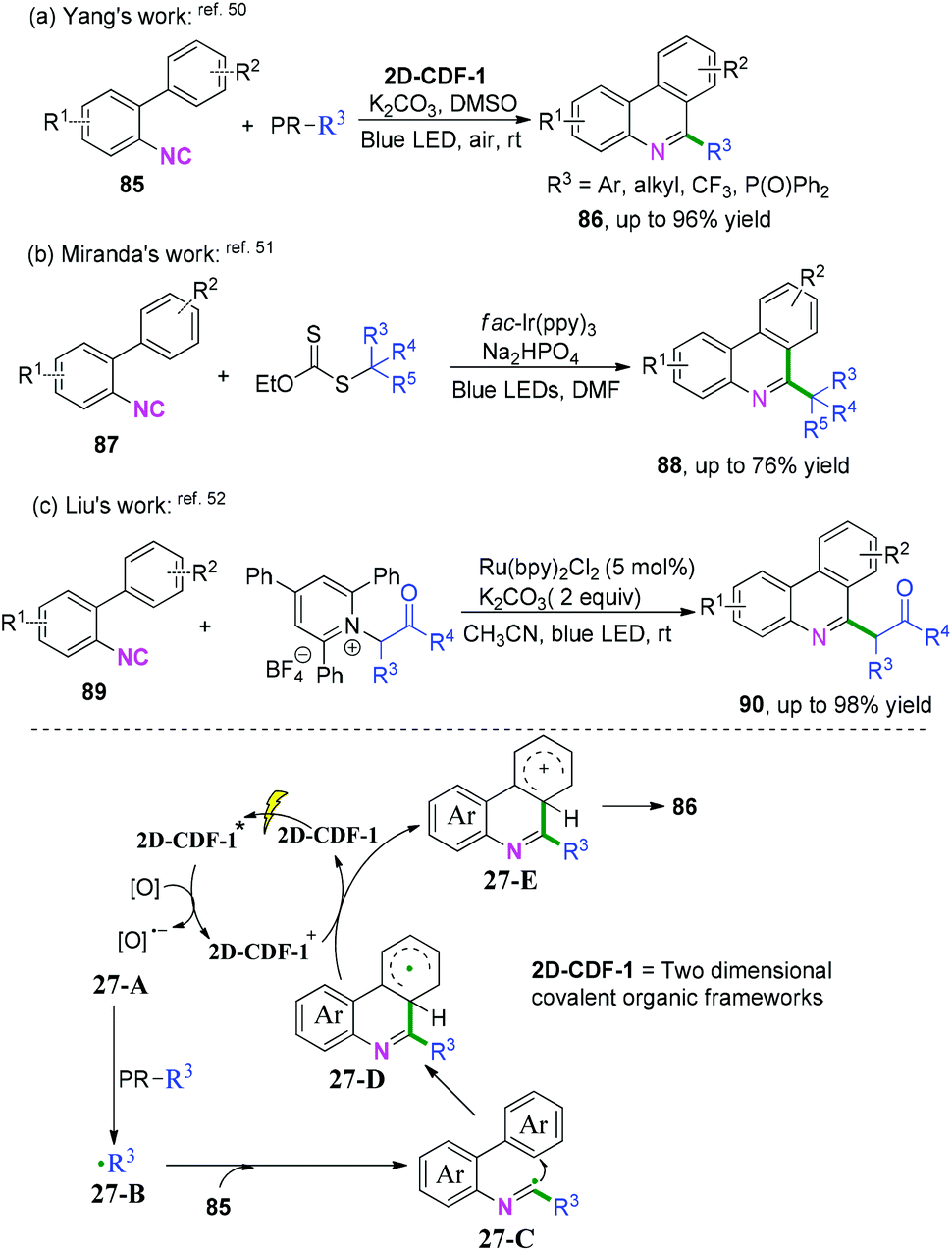
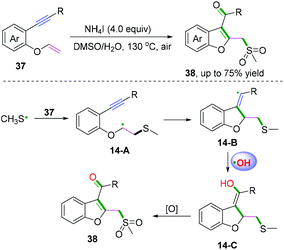
2-Substituted phenyl isocyanides have wide and extensive applications in nitrogen-containing synthesis.50–53 In a pioneering study published in 2019, Yang and co-workers developed a visible-light-induced cascade radical cyclization of 2-aryl phenyl isocyanides to construct varieties of 6-substituted phenanthridines (Scheme 41a).50 This synthetic transformation proceeded through recyclable covalent organic frameworks (2D-COFs) reacting as photocatalysts. In light of the authors’ recycling experiments, excellent recyclability of this organic framework photocatalyst was demonstrated. Later, Miranda and coauthors demonstrated a photocatalytic xanthate-based tandem radical cyclization of 2-biphenyl isocyanides for the construction of 6-alkylated phenanthridines (Scheme 41b).51 This transformation proceeded without a stoichiometric amount of oxidant in moderate yield under mild conditions. Liu and co-workers also reported an efficient visible-light-induced radical alkylation of isocyanides for the synthesis of 6-alkylated phenanthridines. In this transformation, available Katritzky pyridinium salts derived from amino acids/peptides were used as radical precursors (Scheme 41c).52
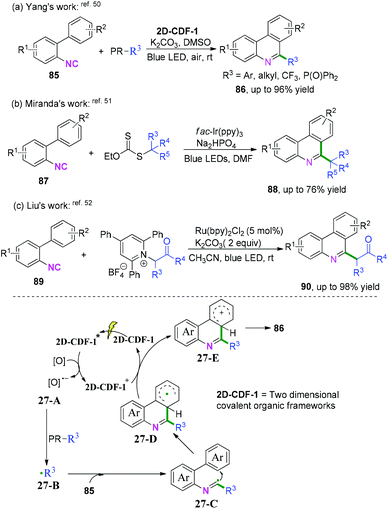 |
| | Scheme 41 Visible-light-induced cascade radical cyclization of 2-substituted phenyl isocyanides. | |
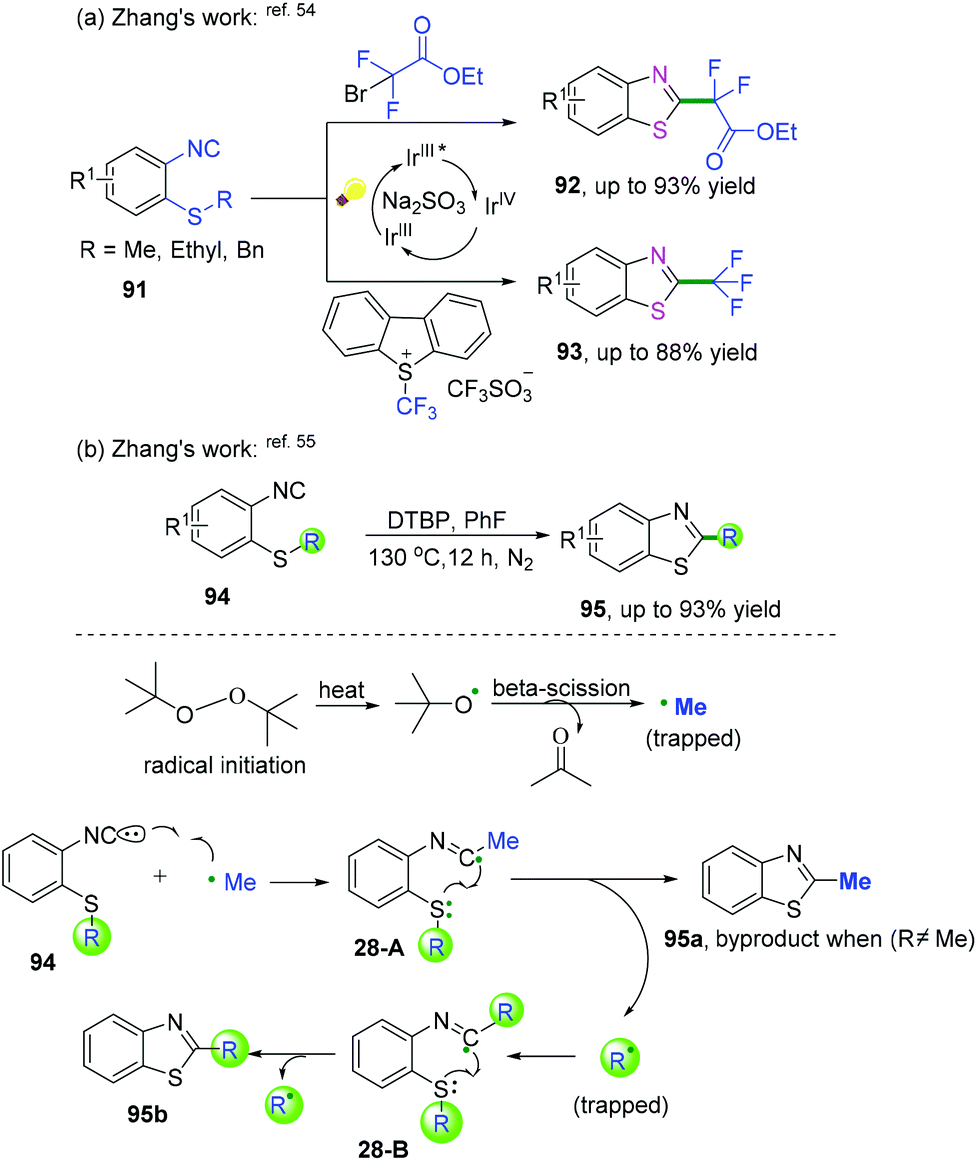
2-Isocyanoaryl thioether derivatives are highly valuable starting materials in organic chemistry. Zhang, Wu, and co-workers have further extended the radical cyclization of 2-isocyanoaryl thioethers to synthesize benzothiazole derivatives (Scheme 42).54,55 The proposed mechanism showed that sodium sulfite participated in the photocatalytic cycle as a reductant, which benefited the transformation of Ir4+ into Ir3+ to perform the fluoroalkylation under mild reaction conditions (Scheme 42a).54 Meanwhile, in the presence of di-tert-butyl peroxide, heat-induced homolytic dissociation of DTBP initially afforded a tert-butyl oxide radical, which then underwent β-scission to afford a methyl radical species. Further radical attack of the obtained methyl at the terminal carbon of the isocyano moiety delivered an imidoyl radical intermediate 28-A, with subsequent cyclization to instantaneously afford 2-methylbenzothiazole 95a with the release of R radical. The preferred R-substituted product was obtained via a similar pathway through intermediates 28-B when the R group was not equal to the methyl group (Scheme 42b).55
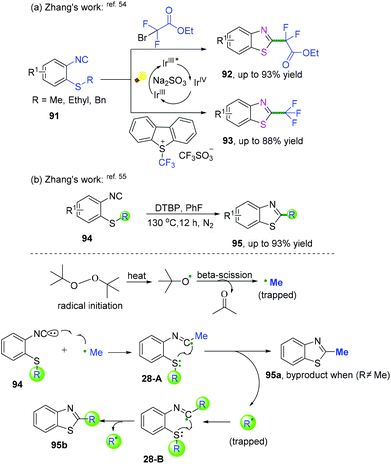 |
| | Scheme 42 Cascade radical cyclization of 2-isocyanoaryl thioethers. | |
5. Conclusions and outlook
Cascade radical cyclization has emerged as a versatile and efficient tool for the rapid synthesis of high-value and complex carbo- and heterocycles, which has attracted considerable attention in synthetic organic chemistry. This Review highlights the latest advances in cascade radical cyclization of radical acceptors. A wide range of recent achievements in the development of cascade radical cyclization for the synthesis of carbo- and heterocycles have been investigated along with these areas. Different types of hybridization of the starting materials by coupling cyclization were categorized, including C(sp)–C(sp), C(sp)–C(sp2), C(sp2)–C(sp2), C(sp2)–C(sp3) bond coupling cyclization and carbon–heteroatom bond (e.g., C–O, C–N, C–S bond) coupling cyclization. For the C(sp)–C(sp) and C(sp)–C(sp2) bond coupling cyclization, 1,5-diynes (enynes) or 1,6-diynes (enynes) react as preferred efficient radical acceptors, which benefits the construction of five- or six-membered cycles with low energy barriers as well as the C(sp)–(hetero) aryl C(sp2) and alkenyl C(sp2)–(hetero) aryl C(sp2) coupling cyclization. Additionally, for the reactions of C(sp)–alkenyl C(sp2), C(sp)–(hetero) aryl C(sp2), alkenyl C(sp2)–alkenyl C(sp2), and intramolecular alkenyl C(sp2)–(hetero) aryl C(sp2) bond cyclization, a radical acceptor with an activated terminal carbon–carbon double bond delivers high efficiency in cascade radical cyclization; this may be due to the high stability of the alkyl radical intermediate generated from radical addition to the double bond.
Despite the significant progress and extensive efforts that have been made in the past few years, several challenges remain to be addressed: (i) simple, available synthetic chemical feedstocks reacted as radical acceptors to construct carbo- and heterocycles instead of highly activated unsaturated substrates; (ii) controlled high chemo- and regioselectivity of radical species to realize excellent asymmetric synthesis; (iii) further exploitation of detailed and exact cascade radical cyclization mechanisms. We hope this Review will benefit readers who wish to exploit new protocols to construct various biological carbo- and heterocycles in the future.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors thank the National Natural Science Foundation of China (21961002, 21962004), Jiangxi provincial department of science and technology (20192BAB203004), Fundamental Research Funds for Gannan Medical University (QD202019, QD201810), and the COVID-19 emergency project of Gannan Medical University (YJ202027) for financial support.
Notes and references
-
(a)
E. C. T. aylor and J. E. Saxton, The Chemistry of Heterocyclic Compounds, Wiley-Interscience, New York, 1983, p. 1994 Search PubMed
 ;
(b)
J. A. Joule and K. Mills, Heterocyclic Chemistry, Blackwell Science, Oxford, 2000 Search PubMed ;
(c)
T. Eicher, S. Hauptmann and A. Speicher, The Chemistry of Heterocycles, Wiley-VCH Verlag GmbH & Co, Weinheim, 2nd edn, 2003 CrossRef ;
(d) A. R. Katritzky, Introduction: Heterocycles, Chem. Rev., 2004, 104, 2125 CrossRef CAS ;
(e) N. Saracoglu, Antioxidant activities of synthetic indole derivatives and possible activity mechanisms, Top. Heterocycl. Chem., 2007, 11, 145 Search PubMed .
;
(b)
J. A. Joule and K. Mills, Heterocyclic Chemistry, Blackwell Science, Oxford, 2000 Search PubMed ;
(c)
T. Eicher, S. Hauptmann and A. Speicher, The Chemistry of Heterocycles, Wiley-VCH Verlag GmbH & Co, Weinheim, 2nd edn, 2003 CrossRef ;
(d) A. R. Katritzky, Introduction: Heterocycles, Chem. Rev., 2004, 104, 2125 CrossRef CAS ;
(e) N. Saracoglu, Antioxidant activities of synthetic indole derivatives and possible activity mechanisms, Top. Heterocycl. Chem., 2007, 11, 145 Search PubMed .
- For selected reviews, see.
(a) H. Yi, G. Zhang, H. Wang, Z. Huang, J. Wang, A. K. Singh and A. Lei, Recent advances in radical C–H activation/radical cross-coupling, Chem. Rev., 2017, 117, 9016 CrossRef CAS ;
(b) J. Xuan and A. Studer, Radical cascade cyclization of 1,n-enynes and diynes for the synthesis of carbocycles and heterocycles, Chem. Soc. Rev., 2017, 46, 4329 RSC ;
(c) K. Hung, X. Hu and T. J. Maimone, Total synthesis of complex terpenoids employing radical cascade processes, Nat. Prod. Rep., 2018, 35, 174 RSC ;
(d) H.-M. Huang, M. H. Garduño-Castro, C. Morrill and D. J. Procter, Catalytic cascade reactions by radical relay, Chem. Soc. Rev., 2019, 48, 4626 RSC ;
(e) Y. Zhang, K. Sun, Q. Lv, X. Chen, L. Qu and B. Yu, Recent applications of radical cascade reaction in the synthesis of functionalized 1-indenones, Chin. Chem. Lett., 2019, 30, 1361 CrossRef CAS .
- For selected works, see:
(a) M. O. Funk, R. Isaac and N. A. Porter, Free radical cyclization of unsaturated hydroperoxides, J. Am. Chem. Soc., 1975, 97, 12814 CrossRef ;
(b) N. A. Porter and M. O. Funk, Peroxy radical cyclization as a model for prostaglandin biosynthesis, J. Org. Chem., 1975, 40(24), 3614 CrossRef CAS ;
(c) G. Stork, R. Mook Jr., S. A. Biller and S. D. Rychnovsky, Free-radical cyclization of bromo acetals. Use in the construction of bicyclic acetals and lactones, J. Am. Chem. Soc., 1983, 105, 3741 CrossRef CAS ;
(d) D. P. Curran and D. M. Rakiewicz, Tandem radical approach to linear condensed cyclopentanoids. Total synthesis of (±)-hirsutene, J. Am. Chem. Soc., 1985, 107, 1448 CrossRef CAS ;
(e) D. H. R. Barton, E. Silva and S. Z. Zard, Multiple radical additions: an expedient entry into complex frameworks, J. Chem. Soc., Chem. Commun., 1988, 4, 285 RSC ;
(f) K. C. Nicolaou, D. J. Edmonds and P. G. Bulger, Cascade reactions in total synthesis, Angew. Chem., Int. Ed., 2006, 45, 7134 CrossRef CAS ;
(g) C. Grondal, M. Jeanty and D. Enders, Organocatalytic cascade reactions as a new tool in total synthesis, Nat. Chem., 2010, 2, 167 CrossRef CAS ;
(h) L.-Q. Lu, J.-R. Chen and W.-J. Xiao, Development of cascade reactions for the concise construction of diverse heterocyclic architectures, Acc. Chem. Res., 2012, 45, 1278 CrossRef CAS ;
(i) C. M. R. Volla, I. Atodiresei and M. Rueping, Catalytic C–C bond-forming multi-component cascade or domino reactions: Pushing the boundaries of complexity in asymmetric organocatalysis, Chem. Rev., 2014, 114, 2390 CrossRef CAS ;
(j) Y. Wang, H. Lu and P.-F. Xu, Asymmetric catalytic cascade reactions for constructing diverse scaffolds and complex molecules, Acc. Chem. Res., 2015, 48, 1832 CrossRef CAS .
- For selected reviews, see:
(a) J. E. Anthony, Functionalized acenes and heteroacenes for organic electronics, Chem. Rev., 2006, 106, 5028 CrossRef CAS ;
(b) X. Feng, W. Pisula and K. Müllen, Large polycyclic aromatic hydrocarbons: Synthesis and discotic organization, Pure Appl. Chem., 2009, 81, 2203 CAS ;
(c) A. Narita, X.-Y. Wang, X. Feng and K. Müllen, New advances in nanographene chemistry, Chem. Soc. Rev., 2015, 44, 6616 RSC .
-
(a) K. Pati, A. M. Hughes, H. Phan and I. V. Alabugin,
Exo-dig radical cascades of skipped enediynes: Building a naphthalene moiety within a polycyclic framework, Chem. – Eur. J., 2014, 20, 390 CrossRef CAS ;
(b) P. M. Byers and I. V. Alabugin, Polyaromatic ribbons from oligo-alkynes via selective radical cascade: Stitching aromatic rings with polyacetylene bridges, J. Am. Chem. Soc., 2012, 134, 9609 CrossRef CAS ;
(c) I. V. Alabugin and B. Gold, “Two functional groups in one package”: Using both alkyne π-bonds in cascade transformations, J. Org. Chem., 2013, 78, 7777 CrossRef CAS ;
(d) K. Pati, G. dos Passos Gomes and I. V. Alabugin, Combining traceless directing groups with hybridization control of radical reactivity: From skipped enynes to defect-free hexagonal frameworks, Angew. Chem., Int. Ed., 2016, 55, 11633 CrossRef CAS ;
(e) K. Pati, G. dos Passos Gomes, T. Harris, A. Hughes, H. Phan, T. Banerjee, K. Hanson and I. V. Alabugin, Traceless directing groups in radical cascades: From oligoalkynes to fused helicenes without tethered initiators, J. Am. Chem. Soc., 2015, 137, 1165 CrossRef CAS .
- Z.-W. Hou, Z.-Y. Mao, J. Song and H.-C. Xu, Electrochemical synthesis of polycyclic N-heteroaromatics through cascade radical cyclization of diynes, ACS Catal., 2017, 7, 5810 CrossRef CAS .
- T. Watanabe, D. Hirose, D. P. Curran and T. Taniguchi, Borylative radical cyclizations of benzo[3,4]cyclodec-3-ene-1,5-diynes and N-heterocyclic carbene-boranes, Chem. – Eur. J., 2017, 23, 5404 CrossRef CAS .
- D. Liu, M.-J. Jiao, Z.-T. Feng, X.-Z. Wang, G.-Q. Xu and P.-F. Xu, Design, synthesis, and application of highly reducing organic visible-light photocatalysts, Org. Lett., 2018, 20, 5700 CrossRef CAS .
- X. Yi, K. Chen, W. Chen, W. Chen, M. Liu and H. Wu, Synthesis of cyclic gem-dinitro compounds via radical nitration of 1,6-diynes with Fe(NO3)3·9H2O, Org. Biomol. Chem., 2019, 17, 4725 RSC .
- A. R. Agrawal, N. R. Kumar, S. Debnath, S. Das, C. Kumar and S. S. Zade, Radical-cascade avenue for 3,4-fused-ring-substituted thiophenes, Org. Lett., 2018, 20, 4728 CrossRef CAS .
-
(a) S. Mondal, R. K. Mohamed, M. Manoharan, H. Phan and I. V. Alabugin, Drawing from a pool of radicals for the design of selective enyne cyclizations, Org. Lett., 2013, 15, 5650 CrossRef CAS ;
(b) S. Mondal, B. Gold, R. K. Mohamed and I. V. Alabugin, Design of leaving groups in radical C-C fragmentations: Through-bond 2c–3e interactions in self-terminating radical cascades, Chem. – Eur. J., 2014, 20, 8664 CrossRef CAS ;
(c) S. Mondal, B. Gold, R. K. Mohamed, H. Phan and I. V. Alabugin, Rerouting radical cascades: Intercepting the homoallyl ring expansion in enyne cyclizations via C−S scission, J. Org. Chem., 2014, 79, 7491 CrossRef CAS ;
(d) C. J. Evoniuk, M. Ly and I. V. Alabugin, Coupling cyclizations with fragmentations for the preparation of heteroaromatics: quinolines from o-alkenyl arylisocyanides and boronic acids, Chem. Commun., 2015, 51, 12831 RSC ;
(e) C. J. Evoniuk, G. P. Gomes, M. Ly, F. D. White and I. V. Alabugin, J. Org. Chem., 2017, 82, 4265 CrossRef CAS ;
(f) S. J. Gharpure, Padmaja, V. Prasath and Y. G. Shelke, Cascade radical cyclization on alkynyl vinylogous carbonates for the divergent synthesis of tetrasubstituted furans and dihydrofurans, Org. Lett., 2019, 21, 223 CrossRef CAS ;
(g) D. Crich, D. Grant, V. Krishnamurthy and M. Patel, Catalysis of stannane-mediated radical chain reactions by benzeneselenol, Acc. Chem. Res., 2007, 40, 453 CrossRef CAS .
-
(a) R. K. Mohamed, S. Mondal, B. Gold, C. J. Evoniuk, T. Banerjee, K. Hanson and I. V. Alabugin, Alkenes as alkyne equivalents in radical cascades terminated by fragmentations: Overcoming stereoelectronic restrictions on ring expansions for the preparation of expanded polyaromatics, J. Am. Chem. Soc., 2015, 137, 6335 CrossRef CAS ;
(b) I. V. Alabugin and E. Gonzalez-Rodriguez, Alkyne origami: Folding oligoalkynes into polyaromatics, Acc. Chem. Res., 2018, 51, 1206 CrossRef CAS .
- X.-D. Xu, T.-T. Cao, Y.-N. Meng, G. Zhou, Z. Guo, Q. Li and W.-T. Wei, Metal-free regioselective radical cyclization of 1,6-enynes with carbonyl compounds, ACS Sustainable Chem. Eng., 2019, 7, 13491 CrossRef CAS .
-
(a) J.-K. Qiu, B. Jiang, Y.-L. Zhu, W.-J. Hao, D.-C. Wang, J. Sun, P. Wei, S.-J. Tu and G. Li, Catalytic dual 1,1-H-abstraction/insertion for domino spirocyclizations, J. Am. Chem. Soc., 2015, 137, 8928 CrossRef CAS ;
(b) M. Hu, J.-H. Fan, Y. Liu, X.-H. Ouyang, R.-J. Song and J.-H. Li, Metal-free radical [2 + 2 + 1] carbocyclization of benzene-linked 1,n-enynes: Dual C(sp3)-H functionalization adjacent to a heteroatom, Angew. Chem., Int. Ed., 2015, 54, 9577 CrossRef CAS ;
(c) K. Sun, Y.-F. Si, X.-L. Chen, Q.-Y. Lv, N. Jiang, S.-S. Wang, Y.-Y. Peng, L.-B. Qu and B. Yu, Silver-catalyzed radical cascade cyclization of unactivated alkenes towards cyclopenta[c]quinolones, Adv. Synth. Catal., 2019, 361, 4483 CrossRef CAS .
-
(a) X.-H. Ouyang, R.-J. Song, Y. Liu, M. Hu and J.-H. Li, Copper-catalyzed radical [2 + 2 + 1] annulation of benzene-linked 1,n-enynes with azide: Fused pyrroline compounds, Org. Lett., 2015, 17, 6038 CrossRef CAS ;
(b) L. Lv and Z. Li, Iron-catalyzed radical [2 + 2 + 2] annulation of benzene-linked 1,7-enynes with aldehydes: Fused pyran compounds, Org. Lett., 2016, 18, 2264 CrossRef CAS .
- M. R. Mutra, V. S. Kudale, J. Li, W.-H. Tsai and J.-J. Wang, Alkene versus alkyne reactivity in unactivated 1,6-enynes: regio- and chemoselective radical cyclization with chalcogens under metal- and oxidant-free conditions, Green Chem., 2020, 22, 2288 RSC .
-
(a) M. Hu, B. Liu, X.-H. Ouyang, R.-J. Song and J.-H. Li, Nitrative cyclization of 1-ethynyl-2-(vinyloxy)benzenes to access 1-[2-(nitromethyl)benzofuran-3-yl] ketones through dioxygen activation, Adv. Synth. Catal., 2015, 357, 3332 CrossRef CAS ;
(b) W. Wu, S. Yi, W. Huang, D. Luo and H. Jiang, Ag-catalyzed oxidative cyclization reaction of 1,6-enynes and sodium sulfinate: Access to sulfonylated benzofurans, Org. Lett., 2017, 19, 2825 CrossRef CAS ;
(c) S. Jana, A. Verma, R. Kadu and S. Kumar, Visible-light-induced oxidant and metal-free dehydrogenative cascade trifluoromethylation and oxidation of 1,6-enynes with water, Chem. Sci., 2017, 8, 6633 RSC ;
(d) J. Zhang, S. Cheng, Z. Cai, P. Liu and P. Sun, Radical addition cascade cyclization of 1,6-enynes with DMSO to access methylsulfonylated and carbonylated benzofurans under transition-metal-free conditions, J. Org. Chem., 2018, 83, 9344 CrossRef CAS .
-
(a) S. J. Gharpure and Y. G. Shelke, Cascade radical cyclization of N-propargylindoles: Substituents dictate stereoselective formation of N-fused indolines versus indoles, Org. Lett., 2017, 19, 5022 CrossRef CAS ;
(b) J. Zhu, S. Sun, M. Xia, N. Gu and J. Cheng, Copper-catalyzed radical 1,2-cyclization of indoles with arylsulfonyl hydrazides: access to 2-thiolated 3H-pyrrolo[1,2-a]indoles, Org. Chem. Front., 2017, 4, 2153 RSC .
-
(a) S. Chen, P. Zhang, W. Shu, Y. Gao, G. Tang and Y. Zhao, Cascade phosphinoylation/cyclization/isomerization process for the synthesis of 2-phosphinoyl-9H-pyrrolo[1,2-a]indoles, Org. Lett., 2016, 18, 5712 CrossRef CAS ;
(b) H. Zhang, W. Li and C. Zhu, Copper-catalyzed cascade phosphorylation initiated radical cyclization: Access to 2-phosphorylated pyrrolo[1,2-a]indole, J. Org. Chem., 2017, 82, 2199 CrossRef CAS .
-
(a) P. Zhang, Y. Gao, S. Chen, G. Tang and Y. Zhao, Direct synthesis of 2-sulfonated 9H-pyrrolo[1,2-a]indoles via NaI-catalyzed cascade radical addition/cyclization/isomerization, Org. Chem. Front., 2017, 4, 1350 RSC ;
(b) K. Sun, X.-L. Chen, Y.-L. Zhang, K. Li, X.-Q. Huang, Y.-Y. Peng, L.-B. Qu and B. Yu, Metal-free sulfonyl radical-initiated cascade cyclization to access sulfonated indolo[1,2-a]quinolones, Chem. Commun., 2019, 55, 12615 RSC .
-
(a) Y. Liu, Z. Chen, Q.-L. Wang, P. Chen, J. Xie, B.-Q. Xiong, P.-L. Zhang and K.-W. Tang, Visible light-catalyzed cascade radical cyclization of N-propargylindoles with acyl chlorides for the synthesis of 2-acyl-9H-pyrrolo[1,2-a]indoles, J. Org. Chem., 2020, 85, 2385 CrossRef CAS ;
(b) C. R. Reddy, R. C. Kajareab and N. Punna, Silver-catalyzed acylative annulation of N-propargylated indoles with α-keto acids: Access to acylated pyrrolo[1,2-a]indoles, Chem. Commun., 2020, 56, 3445 RSC .
- K. Kim, H. Choi, D. Kang and S. Hong, Visible-light excitation of quinolinone-containing substrates enables divergent radical cyclizations, Org. Lett., 2019, 21, 3417 CrossRef CAS .
- J. Wen, W. Shi, F. Zhang, D. Liu, S. Tang, H. Wang, X.-M. Lin and A. Lei, Electrooxidative tandem cyclization of activated alkynes with sulfinic acids to access sulfonated indenones, Org. Lett., 2017, 19, 3131 CrossRef CAS .
-
(a) S. Feng, X. Xie, W. Zhang, L. Liu, Z. Zhong, D. Xu and X. She, Visible-light-promoted dual C–C bond formations of alkynoates via a domino radical addition/cyclization reaction: A synthesis of coumarins, Org. Lett., 2016, 18, 3846 CrossRef CAS ;
(b) L. Chen, L. Wu, W. Duan, T. Wang, L. Li, K. Zhang, J. Zhu, Z. Peng and F. Xiong, Photoredox-catalyzed cascade radical cyclization of ester arylpropiolates with CF3SO2Cl to construct 3-trifluoromethyl coumarin derivatives, J. Org. Chem., 2018, 83, 8607 CrossRef CAS .
- P. Xiong, H.-H. Xu, J. Song and H.-C. Xu, Electrochemical difluoromethylarylation of alkynes, J. Am. Chem. Soc., 2018, 140, 2460 CrossRef CAS .
- B. Quiclet-Sire and S. Z. Zard, Some aspects of radical cascade and relay reactions, Proc. R. Soc. A, 2017, 473, 20160859 CrossRef .
- H.-M. Huang and D. J. Procter, Radical–radical cyclization cascades of barbiturates triggered by electron-transfer reduction of amide-type carbonyls, J. Am. Chem. Soc., 2016, 138, 7770 CrossRef CAS .
- G. Yin, M. Zhu, G. Yang, X. Wang and W. Fu, Synthesis of difluoromethylenephosphonated oxindoles through visible-vight-induced radical cyclization of N-arylacrylamides, J. Fluorine Chem., 2016, 191, 63 CrossRef CAS .
- C.-X. Li, D.-S. Tu, R. Yao, H. Yan and C.-S. Lu, Visible-light-induced cascade reaction of isocyanides and N-arylacrylamides with diphenylphosphine oxide via radical C–P and C–C bond formation, Org. Lett., 2016, 18, 4928 CrossRef CAS .
- W. Ou, G. Zhang, J. Wu and C. Su, Photocatalytic cascade radical cyclization approach to bioactive indoline-alkaloids over donor–acceptor type conjugated microporous polymer, ACS Catal., 2019, 9, 5178 CrossRef CAS .
- A. Ling, L. Zhang, R. X. Tan and Z.-Q. Liu, Molecular oxygen-promoted general and site-specific alkylation with organoboronic acid, J. Org. Chem., 2018, 83, 14489 CrossRef CAS .
- L. Zou, P. Li, B. Wang and L. Wang, Visible-light-induced radical cyclization of N-allylbenzamides with CF3SO2Na to trifluoromethylated dihydroisoquinolinones in water at room temperature, Green Chem., 2019, 21, 3362 RSC .
- Y. Yamane, K. Yoshinaga, M. Sumimoto and T. Nishikata, Iron-enhanced reactivity of radicals enables C–H tertiary alkylations for construction of functionalized quaternary carbons, ACS Catal., 2019, 9, 1757 CrossRef CAS .
- Y. Mei, L. Zhao, Q. Liu, S. Ruan, L. Wang and P. Li, Synthesis of sulfone-functionalized chroman-4-ones and chromans through visible-light-induced cascade radical cyclization under transition-metal-free conditions, Green Chem., 2020, 22, 2270 RSC .
-
(a) A. J. Kochanowska-Karamyan and M. T. Hamann, Marine indole alkaloids: Potential new drug leads for the control of depression and anxiety, Chem. Rev., 2010, 110, 4489 CrossRef CAS ;
(b) M. Somei and F. Yamada, Simple indole alkaloids and those with a nonrearranged monoterpenoid unit, Nat. Prod. Rep., 2004, 21, 278 RSC ;
(c) D. A. Horton, G. T. Bourne and M. L. Smythe, The combinatorial synthesis of bicyclic privileged structures or privileged substructures, Chem. Rev., 2003, 103, 893 CrossRef CAS ;
(d) J. Ewing, G. Hughes, E. Ritchie and W. C. Taylor, Radiochemical studies on free radicals, Nature, 1952, 169, 618 CrossRef CAS ;
(e) T. Polossek, R. Ambros, S. Angerer, G. Brandi, A. Mannschreck and E. Angerer, 6-Alkyl-12-formylindolo[2,1-a]isoquinolines. Syntheses, estrogen receptor binding affinities, and stereospecific cytostatic activity, J. Med. Chem., 1992, 35, 3537 CrossRef CAS ;
(f) R. Faust, P. J. Garratt, R. Jones and L.-K. Yeh, Mapping the Melatonin Receptor. 6. Melatonin agonists and antagonists derived from 6H-Isoindolo[2,1-a]indoles, 5,6-dihydroindolo[2,1-a]isoquinolines, and 6,7-dihydro-5H-benzo[c]azepino[2,1-a]indoles, J. Med. Chem., 2000, 43, 1050 CrossRef CAS ;
(g) G. A. Kraus, V. Gupta, M. Kohut and N. Sing, A direct synthesis of 5,6-dihydroindolo[2,1-a]isoquinolines that exhibit immunosuppressive activity, Bioorg. Med. Chem. Lett., 2009, 19, 5539 CrossRef CAS .
-
(a) S. J. Mahoney and E. Fillion,
N-Fused indolines through non-carbonyl-stabilized rhodium carbenoid C-H insertion of N-aziridinyl imines, Chem. – Eur. J., 2012, 18, 68 CrossRef CAS ;
(b) K. Morimoto, K. Hirano, T. Satoh and M. Miura, Rhodium-catalyzed oxidative coupling/cyclization of 2-phenylindoles with alkynes via C−H and N−H bond cleavages with air as the oxidant, Org. Lett., 2010, 12, 2068 CrossRef CAS ;
(c) Y.-J. Li, J.-T. Zhu, H.-B. Xie, S. Li, D.-J. Peng, Z.-K. Li, Y.-M. Wu and Y.-F. Gong, Rh-catalyzed intramolecular sp2 C–H bond difluoromethylenation, Chem. Commun., 2012, 48, 3136 RSC ;
(d) M.-H. Shen, Y.-P. Pan, Z.-H. Jia, X.-T. Ren, P. Zhang and H.-D. Xu, An efficient approach to 1,2,3-trisubstituted indole via rhodium catalyzed carbene Csp3−H bond insertion, Org. Biomol. Chem., 2015, 13, 4851 RSC ;
(e) C. Bressy, D. Alberico and M. Lautens, A route to annulated indoles via a palladium-catalyzed tandem alkylation/direct arylation reaction, J. Am. Chem. Soc., 2005, 127, 13148 CrossRef CAS ;
(f) W.-T. Wei, X.-J. Dong, S.-Z. Nie, Y.-Y. Chen, X.-J. Zhang and M. Yan, Intramolecular dehydrative coupling of tertiary amines and ketones promoted by KO-t-Bu/DMF: A new synthesis of indole derivatives, Org. Lett., 2013, 15, 6018 CrossRef CAS ;
(g) N. Fuentes, W.-Q. Kong, L. Fernάndez-Sάnchez, E. Merino and C. Nevado, Cyclization cascades via N-amidyl radicals toward highly functionalized heterocyclic scaffolds, J. Am. Chem. Soc., 2015, 137, 964 CrossRef CAS .
- Y.-L. Wei, J.-Q. Chen, B. Sun and P.-F. Xu, Synthesis of indolo[2,1-a]isoquinoline derivatives via visible-light-induced radical cascade cyclization reactions, Chem. Commun., 2019, 55, 5922 RSC .
- Q.-F. Bai, C. Jin, J.-Y. He and G. Feng, Carbamoyl radicals via photoredox decarboxylation of oxamic acids in aqueous media: Access to 3,4-dihydroquinolin-2(1H)-ones, Org. Lett., 2018, 20, 2172 CrossRef CAS .
-
(a) C. Qu, P. Xu, W. Ma, Y. Cheng and C. Zhu, A novel visible light mediated radical cyclization of enol lactones: a concise method for fluorinated polycyclic lactone scaffolds, Chem. Commun., 2015, 51, 13508 RSC ;
(b) C. Adouama, R. Keyrouz, G. Pilet, C. Monnereau, D. Gueyrard, T. Noël and M. Médebielle, Access to cyclic gem-difluoroacyl scaffolds via electrochemical and visible light photocatalytic radical tandem cyclization of heteroaryl chlorodifluoromethyl ketones, Chem. Commun., 2017, 53, 5653 RSC .
- W.-P. Mai, F. Wang, X.-F. Zhang, S.-M. Wang, Q.-P. Duan and K. Lu, Nickel-catalysed radical tandem cyclisation/arylation: practical synthesis of 4-benzyl-3,3-difluoro-γ-lactams, Org. Biomol. Chem., 2018, 16, 6491 RSC .
- J. Liao, L. Ouyang, Q. Jin, J. Zhang and R. Luo, Recent advances in the oxime-participating synthesis of isoxazolines, Org. Biomol. Chem., 2020, 18, 4709 RSC .
- X.-X. Peng, D. Wei, W.-J. Han, F. Chen, W. Yu and B. Han, Dioxygen activation via Cu-catalyzed cascade radical reaction: An approach to isoxazoline/cyclic nitrone-featured α-ketols, ACS Catal., 2017, 7, 7830 CrossRef CAS .
- Y.-Y. Liu, J. Yang, R.-J. Song and J.-H. Li, Synthesis of 5-(fluoromethyl)-4,5-dihydroisoxazoles by silver-catalyzed oxyfluorination of unactivated alkenes, Adv. Synth. Catal., 2014, 356, 2913 CrossRef CAS .
- R.-H. Liu, D. Wei, B. Han and W. Yu, Copper-catalyzed oxidative oxyamination/diamination of internal alkenes of unsaturated oximes with simple amines, ACS Catal., 2016, 6, 6525 CrossRef CAS .
- F. Chen, F.-F. Zhu, M. Zhang, R.-H. Liu, W. Yu and B. Han, Iminoxyl radical-promoted oxycyanation and aminocyanation of unactivated alkenes: Synthesis of cyano-featured isoxazolines and cyclic nitrones, Org. Lett., 2017, 19, 3255 CrossRef CAS .
- F. Meng, H. Zhang, K. Guo, J. Dong, A.-M. Lu and Y. Zhu, Access to cyano-containing isoxazolines via copper-catalyzed domino cyclization/cyanation of alkenyl oximes, J. Org. Chem., 2017, 82, 10742 CrossRef CAS .
- Q.-Q. Zhao, J. Chen, D.-M. Yan, J.-R. Chen and W.-J. Xiao, Photocatalytic hydrazonyl radical-mediated radical cyclization/allylation cascade: Synthesis of dihydropyrazoles and tetrahydropyridazines, Org. Lett., 2017, 19, 3620 CrossRef CAS .
- J. Liao, L. Fan, W. Guo, Z. Zhang, J. Li, C. Zhu, Y. Ren, W. Wu and H. Jiang, Palladium-catalyzed fluoroalkylative cyclization of olefins, Org. Lett., 2017, 19, 1008 CrossRef CAS .
- J. Liao, L. Ouyang, Y. Lai and R. Luo, Photoredox-catalyzed oxy-/aminofluoroalkylative cyclization of alkenes, J. Org. Chem., 2020, 85, 5590 CrossRef CAS .
- S. Liu, W. Pan, S. Wu, X. Bu, S. Xin, J. Yu, H. Xu and X. Yang, Visible-light-induced tandem radical addition–cyclization of 2-aryl phenyl isocyanides catalysed by recyclable covalent organic frameworks, Green Chem., 2019, 21, 2905 RSC .
- P. López-Mendoza and L. D. Miranda, Photocatalytic xanthate-based radical addition/cyclization reaction sequence toward 2-biphenyl isocyanides: synthesis of 6-alkylated phenanthridines, Org. Biomol. Chem., 2020, 18, 3487 RSC .
- Z.-F. Zhu, M.-M. Zhang and F. Liu, Radical alkylation of isocyanides with amino acid-/peptide-derived Katritzky salts via photoredox catalysis, Org. Biomol. Chem., 2019, 17, 1531 RSC .
- H. Jiang, Y. Cheng, R. Wang, M. Zheng, Y. Zhang and S. Yu, Synthesis of 6-alkylated phenanthridine derivatives using photoredox neutral somophilic isocyanide insertion, Angew. Chem., Int. Ed., 2013, 52, 13289 CrossRef CAS .
- Y. Yuan, W. Dong, X. Gao, X. Xie and Z. Zhang, Sodium sulfite-involved photocatalytic radical cascade cyclization of 2-isocyanoaryl thioethers: Access to 2-CF2/CF3-containing benzothiazoles, Org. Lett., 2019, 21, 469 CrossRef CAS .
- K. Luo, W.-C. Yang, K. Wei, Y. Liu, J.-K. Wang and L. Wu, Di-tert-butyl peroxide-mediated radical C(sp2/sp3)–S bond cleavage and group-transfer cyclization, Org. Lett., 2019, 21, 7851 CrossRef CAS .
|
| This journal is © the Partner Organisations 2021 |

 *a,
Xiao
Yang
a,
Lu
Ouyang
a,
Yinlong
Lai
b,
Jiuzhong
Huang
*a,
Xiao
Yang
a,
Lu
Ouyang
a,
Yinlong
Lai
b,
Jiuzhong
Huang