
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOvercoming transport limitations in miniaturized electrophoretic delivery devices†
Maria
Seitanidou
,
Klas
Tybrandt
 ,
Magnus
Berggren
* and
Daniel T.
Simon
,
Magnus
Berggren
* and
Daniel T.
Simon
Laboratory of Organic Electronics, Department of Science and Technology, Linköping University, 60174 Norrköping, Sweden. E-mail: magnus.berggren@liu.se
First published on 11th March 2019
Abstract
Organic electronic ion pumps (OEIPs) have been used for delivery of biological signaling compounds, at high spatiotemporal resolution, to a variety of biological targets. The miniaturization of this technology provides several advantages, ranging from better spatiotemporal control of delivery to reduced invasiveness for implanted OEIPs. One route to miniaturization is to develop OEIPs based on glass capillary fibers that are filled with a polyelectrolyte (cation exchange membrane, CEM). These devices can be easily inserted and brought into close proximity to targeted cells and tissues and could be considered as a starting point for other fiber-based OEIP and “iontronic” technologies enabling favorable implantable device geometries. While characterizing capillary OEIPs we observed deviations from the typical linear current–voltage behavior. Here we report a systematic investigation of these irregularities by performing experimental characterizations in combination with computational modelling. The cause of the observed irregularities is due to concentration polarization established at the OEIP inlet, which in turn causes electric field-enhanced water dissociation at the inlet. Water dissociation generates protons and is typically problematic for many applications. By adding an ion-selective cap that separates the inlet from the source reservoir this effect is then, to a large extent, suppressed. By increasing the surface area of the inlet with the addition of the cap, the concentration polarization is reduced which thereby allows for significantly higher delivery rates. These results demonstrate a useful approach to optimize transport and delivery of therapeutic substances at low concentrations via miniaturized electrophoretic delivery devices, thus considerably broadening the opportunities for implantable OEIP applications.
Introduction
Organic bioelectronics is unique as the translator of signals between biology and technology, and is a promising technological solution to a variety of diagnostic and therapeutic challanges.1–4 Biosensing,5–7 electrophysiological recording8 and drug delivery systems9–11 based on organic electronic materials have been demonstrated to record and regulate biological functions that previously have been very difficult, or even impossible, to achieve with pharmaceutical or traditional (bio)electronic techniques.12,13 “Iontronics” is the subfield of organic bioelectronics that combines electronic and ionic properties of organic electronics.14–16 It is therefore uniquely suited for biological applications where ionic (biochemical) signals can be triggered, released, and addressed electronically. The fundamental component of iontronics is the organic electronic ion pump (OEIP), an electrophoretic delivery device that can be considered an iontronic resistor.17–19 OEIPs and other similar iontronic devices are based on transporting, or “pumping”, charged species through cation- or anion-exchange membranes (CEMs or AEMs), resulting in high spatiotemporal delivery resolution, high dosage precision (ideally one electron per delivered monovalent ion), and, unlike analogous microfluidics-based techniques, there is ideally no liquid flow accompanied with the actual compound delivery.14 OEIP technology has been used to trigger cell signaling in vitro,1,20 to control epileptiform activity in brain slice models,21–23 to effect sensory function in vivo,19 as e-therapy for pain in awake animals,17 and even to modulate plant growth via phytohormone delivery.24 Traditionally, the bioelectronics demonstrations of OEIP technology have required photolithographic techniques on planar substrates to sequentially pattern active and passive areas. However, for implantation scenarios, this design faces some scale and rigidity limitations along with issues related to invasiveness (planar substrates/geometry and flexibility). To address the need for OEIP technology which overcomes the previous limitations, we demonstrate OEIPs devices based on glass capillary fibers that are filled with an anionic polyelectrolyte (CEM) (Fig. 1A and B). Capillary fibers offer several design advantages for use with implantable OEIP devices. The ion exchange membrane (IEM) channel of a planar OEIP, made via thin film processing, can be tailored based on an intrinsic ionic resistivity, leading to a geometry-dependent (length/width ratio) ionic resistance value.14 Large ions tend to present a relatively higher resistance for more narrow channels given the same length/width ratio. This is due to less swelling occurring perpendicular to the substrate for narrower channels, as compared to wider ones, thus providing less water uptake inside the IEM which is necessary to promote high ionic conductivity (Fig. 1C).17 Fiber capillary OEIPs provide a larger ion-transport cross-section, as compared to a thin narrow channel manufactured using thin film processing, thus the capillary OEIPs facilitates transport of larger ions such as drugs and neurotransmitters. In addition, the capillary pipe simultaneously serves as both encapsulation and substrate for the OEIP channel (Fig. 1C). Such devices, assuming a narrow outer diameter, can easily be inserted and brought into close proximity to targeted cells, tissues, and organs – particularly in vivo – and could be considered as a starting point for other fiber-based OEIP technologies enabling favorable implantable device geometries. | ||
| Fig. 1 A) Experimental setup and schematic illustration of CEM and geometries. B) Photograph of OEIP fiber capillary device. C) Schematic illustration of planar and fiber OEIP and the channel swelling respectively. D) Counterion concentration at the fiber inlet versus current density (left). Typical current–voltage curve and transition between regions i and iii to determine the limiting current: (i) non limiting current linear region, (ii) limiting current region, (iii) over-limiting current region with potential water splitting (right); and schematic illustration of the three regions in relation to the OEIP device. | ||
OEIPs are based on the selective transport of charged species through CEMs (i.e., anion selective membranes). CEMs are characterized by a high concentration of fixed negative charges and the permselectivity holds if the ionic concentrations in the adjacent electrolytes are lower than the fixed charge concentration of the CEM (Donnan exclusion).25 The ionic current through the membrane is represented by the combination of migration controlled by the electric field and diffusion along concentration gradients, with diffusion most noticeable when no potential is applied. The current through the OEIP is driven by electrochemical electrode reactions in the source and target reservoirs. In this work Ag/AgCl paste electrodes were used which operate according to eqn (1).
| AgCl(s) + e− ↔ Ag(s) + Cl− | (1) |
The capacity of the Ag/AgCl electrodes was measured in a three-electrode setup by chronopotentiometry and calculated from C = IΔt, where I is the constant charging current (100 μA) and Δt is the time during which the applied voltage is stable (Fig. S1†). The Ag/AgCl electrode capacity was ≈200 mC, which was much higher than the transported charge in the OEIP experiments (≈3 mC).
During delivery, high concentrations of ions are accumulated at the release site resulting in a steep concentration gradient. Concentration polarization refers to the emergence of concentration gradients at the interface between the CEM and an electrolyte as an electric current pass through the system. The transport rate of ions depends on the value of electric current flowing through the membrane. For CEMs, the electrolyte concentration decreases in the close vicinity to the membrane on the side where cations enter into the channel, i.e. the feeding or source side, since the electric field, close to the inlet, forces cations into and anions away from the membrane inlet. At the other side of the membrane, cations escape the membrane outlet, and anions migrate toward the membrane, thus the electrolyte concentration is increased at this side of the OEIP. The resulting electric (ionic) current (I) typically increases linearly with voltage (V) at low voltages according to Ohm's law, until the concentration drops to zero at the inlet side due to concentration polarization, and the point of limiting current of the system is reached.26,27 According to the classic theory of concentration polarization, as the current density increases, the concentration of the ions on the feeding side (fiber inlet) of the membrane drops and approaches zero at the very interface, and then forms a space-charge region (SCR) (Fig. 1D). Meanwhile, the concentration increases on the opposite side (fiber outlet) (Fig. 1D) due to the membrane selectivity. For current levels above the limiting current, an additional potential loss is introduced.28 The electric field at the inlet increases sharply as a result of ion depletion and causes electric field-enhanced water splitting, which then generates protons and hydroxide ions along the inlet interface.29–32
In Fig. 1D an archetypical ionic current–voltage (I–V) curve is given for a CEM channel with the three distinct regions in the I–V regions clearly displayed. The first linear region (i) follows Ohm's law for given electrolyte and membrane resistivity values. The second region (ii) is characterized by a plateau resulting from the reduced counterion concentration at the fiber inlet. In the third region (iii), the linear increase in the I–V characteristics, above the limiting current, is referred to as the over-limiting current.26,33–35 This over-limiting current occurs due to electrically-induced convection, electroconvection, electroosmotic convection and/or water splitting at the inlet of the membrane.36,37 According to this theory, inhomogeneities of the membrane cause an irregular electric field that interacts with the extended space charge, which is built up due to an excess of counterions adjacent to the membrane which is established above the limiting current density.38 The limiting current density is here determined as the intersection of the curve tangents of regions (i) and (ii) (Fig. 1D). In this work, we address the limiting current issues by presenting a systematic investigation of the I–V curves for CEMs under different experimental conditions (different charged molecules at different concentrations). The polyanion 2-acrylamido-2-methylpropane sulfonic acid (AMPSA) cross-linked with the polyalcohol polyethylene glycol (PEG) was chosen as the model CEM material system (Fig. 1A). The experimental values of the limiting current, thus obtained, have been compared with those acquired from finite-element modeling. To obtain effective ion transport, we realized OEIPs with increased area at the CEM inlet by introducing an ion selective cap to prevent the limiting current and associated water splitting.
Methods
OEIP fiber capillaries fabrication
Glass fiber capillaries (25 μm and 50 μm inner diameter and 125 μm outer diameter, Polymicro Technologies, CM Scientific), coated with polyimide to provide abrasion resistance and to maintain mechanical strength were used for OEIP fabrication. The capillary was immersed into a beaker that contained sulfuric acid (concentrated) and was soaked for a controlled removal of the polyimide coating. When heated to 100 °C, the sulfuric acid removes the polyimide under a slow stirring for 20 min. The glass capillaries were then rinsed with DI water. The capillary was glued to a needle adapter using a glue and heat gun and connected vertically, via 5 ml disposable polypropylene syringe, to a nitrogen line fitting for flushing. The syringe reservoir was filled with different solvents (1 ml) and the valve, connected to the nitrogen supply line, was opened to give the desired flow rate. The procedure was performed as follows: the capillary was flushed with nitrogen at 5 bar for 5 min. Then it was flushed with 2 M KOH(aq) and the left for 2 hours to allow for etching of the surface. This etching step increases the hydrophilicity of the capillary as KOH solution increases the surface silanol concentration as hydroxide ions react with the silanol groups of the silica surface to produce silicate ions.39 Next, the capillary was flushed with DI water for 10 min and dried by nitrogen flushing for 5 min. After etching the capillary, the silanization step was introduced. The capillary was flushed with 3-(trimethoxysilyl) propyl methacrylate (10 wt% in toluene) for 1 hour followed by drying with nitrogen flushing for 5 min and ethanol flushing for 10 min. The silanization step introduces vinylic groups on the surface of the fused silica capillary and ensures that the polymer is attached covalently to the capillary wall. The silanizing agent reacts with the silanol group on the glass surface and methacrylic groups are expressed on the surface providing hydrophobic characteristics.39 Finally, the capillary was flushed with the acrylate monomer 2-acrylamido-2-methylpropane sulfonic acid (AMPSA, Sigma-Aldrich, MW 2000, 50 wt%) using a dark syringe tube for 20 min. AMPSA was mixed with polyethylene glycol diacrylate (PEG-DA, MW 575, 2 wt%) and a photoinitiator (0.5 wt% 2-hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone, Sigma Aldrich) that promotes cross-linking in DI water. Next, the capillary was exposed to UV light (UVS-28 EL Series 8 Watt UV lamp, 254 nm). The polymerization was completed after 10 min. After the polymerization, the capillary was cut into desired device lengths (15 mm) by a fiber cleaver and these were then mounted directly onto heat shrink tubes/reservoirs. To hydrate the polyanion channel, OEIP capillary devices were soaked and stored in DI water before use. To introduce the ion selective cap, the outer surface of the end of the capillaries was functionalized. The capillaries were soaked in 2 M KOH(aq) solution for 2 hours and then in silane for 1 hour followed by ethanol soaking for 30 min. The last step was to introduce the ion selective cap at the fiber inlet, which is defined by the polyanion AMPSA material mixed with (3-glycidyloxypropyl) trimethoxysilane (GOPs), a photoinitiator (2-hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone, Sigma Aldrich) for cross-linking in DI water and an aliphatic adhesion promoter. The capillary was exposed to UV light for 10 min. The capillaries were flushed with nitrogen during the polymerization step.Electrical characterization
To investigate the limiting current for the fiber capillaries, electrical characterization was performed using a Keithley 2602 SourceMeter with custom designed LabVIEW software. These were used to source voltage in the range 0–5 V simultaneously recording the current at a scan rate of 5 mV s−1. The limiting current was studied and evaluated for HCl, KCl and acetylcholine chloride (AChCl) at different concentrations (100 mM, 10 mM, 1 mM, 0.1 mM) with and without the ion selectivity cap at the inlet of the fiber. Immediately before the main experiments, the channels were loaded with the desired ions. The devices were operated in a target solution of 100 mM KCl(aq) at a constant voltage of 1 V. The source reservoir was loaded with 100 mM HCl(aq), KCl(aq), AChCl(aq) respectively.Computational simulations
The device was modelled by the Nernst–Planck–Poisson equations (2)–(4), which were solved with the COMSOL Multiphysics 5.3a software, according to the procedure in ref. 40.| ji = −Di(∇ci + zifci∇V) | (2) |
| ∇ji = −dci/dt | (3) |
 | (4) |
The model assumes a homogeneous membrane with a fixed charge concentration cfix. A 2D axisymmetric geometry was used for the model, effectively making it a 2D computational problem (Fig. S2†). Special care was taken in meshing the CEM-electrolyte interface, with mesh sizes in the order of 0.1 nm perpendicular to the interface, as quantities changes very rapidly at this surface. The inner and outer diameter of the capillary were set to 25 μm and 130 μm, respectively. The fixed charge concentration was set to 1.8 M (estimated experimentally). The diffusion coefficients in water were set according to literature to DK+ = 19.6 × 10−10, DCl− = 20.3 × 10−10 and DACh+ = 5.5 × 10−10 m2 s−1.41 The diffusion coefficients are lower within the membrane and were decreased by a factor of 0.154 to match the experimental data. The diameter of the selective cap was set to 100 μm. No contribution from convection or water splitting was included in the model. The concentration boundary conditions for the source reservoir were set according to the corresponding experimental conditions and the potential was set to the applied potential VP. The target reservoir was electrically grounded and comprised 100 mM KCl in all experiments. The OEIP outlet was implemented according to the membrane-electrolyte Donnan equilibrium (c+ = 1809.9 mM, c− = 9.9 mM, V = −0.19 V) to create a constant interface which does not affect the source side of the device. The limiting current was defined as the current for which the concentration next to the membrane was 1/1000 of the bulk electrolyte concentration and it was determined by ramping the driving voltage from 0 V to 4 V and picking the current value for which the concentration criteria was fulfilled.
Results and discussion
Fiber capillary OEIPs with 15 mm long/25 μm and 50 μm inner diameter and 7 mm long/25 μm inner diameter CEM channels were fabricated (Fig. 1A and B) to investigate the effect of geometrical characteristics of the capillary pump at the limiting current and the water splitting regime. To investigate the limiting current for the fiber CEMs, the source reservoir was loaded with a 100 mM HCl(aq) solution. The voltage was then sourced within the range of 0–5 V and the currents were recorded at a scan rate of 5 mV s−1 for different HCl concentrations (10 mM, 1 mM, 0.1 mM). The limiting current and threshold voltage for capillaries with 50 μm inner diameter matched the measured data for capillaries with 25 μm inner diameter reasonably well (Fig. S3B†). The geometrical characteristics of the capillary pump affect its resistance, thus affecting the limiting current and the water splitting regime. As the channel is an ionic resistor, a shorter channel results in a lower resistance, meaning that the limiting current is reached at a lower voltage. The length of the channel doesn't affect the limiting current density but does affect the threshold voltage (Fig. S3C†). The experiment was repeated for the 25 μm inner diameter capillary by switching the source electrolyte to KCl and AChCl to investigate ions of different sizes and of different biological relevance. The limiting current of the CEM membrane was measured in the same manner as for HCl, at different electrolyte concentrations. The obtained I–V curves clearly show the three characteristic regions, and they are most pronounced for the relatively lower electrolyte concentrations (1 and 0.1 mM). From the results given in Fig. 2A–C, the limiting current depends on the concentration, but also on the diffusion coefficient of the ions as Ilim,H+ > Ilim,K+ > Ilim,ACh+ as expected. From a device perspective it is important to understand the nature of the over-limiting current. During water splitting, H+ and OH− ions are produced at the inlet, with the H+ being transported through the OEIP into the target with potentially undesirable effects.42,43 To determine if water splitting occurred, the fiber inlet and outlet were monitored using a horizontal spectral microscope system during KCl (1 mM) delivery in real time (Fig. 2D and E). A phenol red pH indicator was added to the KCl source electrolyte and a methyl red pH indicator was added to KCl target electrolyte. The OEIP was operated continuously in the range 0–3.5 V and microscope images were taken every 1.5 min. The red channel images given in Fig. 2D was generated by first subtracting an image prior to the experiment from the time series, then amplifying the remaining red channel of the image by a factor of 5. The solution, including the phenol red indicator, exhibits a gradual transition from yellow to red over the pH range from 6.8 to 8.2. Above pH 8.2, phenol red turns into a bright red color. The cations (H+) flow leftward into the fiber inlet and the anions (OH−) flow out from the inlet, causing a color change to occur. At the outlet side a KCl (1 mM) solution, including the methyl red indicator, exhibited a gradual transition from yellow to red below the pH range from 6.2 to 4.4. Below pH 6.2, methyl exhibits a bright red color. The cations (H+) flow leftward (Fig. 2E) into the fiber inlet toward the outlet, causing a color change to occur at the fiber outlet. The limiting current was reached at 0.5 V and OH− and H+ started to appear at the fiber inlet and outlet, respectively, at higher voltages, confirming the presence of water splitting. For the voltages and currents beyond this point, the major contribution to an increase of ion current through the CEM is then related to H+ transport. A corresponding change of the pH was recorded at the fiber inlet and outlet, and was due to accumulation and transport of remaining OH− and H+, respectively. | ||
| Fig. 2 Experimental I–V curves with the three characteristic regions for three electrolytes A) HCl, B) KCl, C) AChCl at different concentrations (100 mM, 10 mM, 1 mM, 0.1 mM) in the range 0–5 V scanned at a rate of 5 mV s−1. The dashed lines show the calculated limiting currents from the computational model for the three electrolytes at different concentrations. D) Microscope images (enhanced red channel) of pH changes at the inlet during 1 mM KCl delivery. E) Microscope images of pH changes at the outlet during KCl (1 mM) delivery. | ||
To better understand the ion transport processes at the inlet, the fiber OEIP inlet was modelled with the Nernst–Planck–Poisson equations,40 which previously have been applied to similar iontronic devices.44 The inlet was modelled for various ionic species, concentrations, and applied voltages in order to determine the limiting currents, which here is defined as the current for which the concentration at the inlet has decreased by a factor 1000 with respect to the electrolyte bulk concentration. The calculated limiting currents are shown as dashed lines in Fig. 2A–C, and the calculated values matches the measured data reasonably well. Fig. 3A shows the concentration and voltage profiles at the inlet (x = 0 μm) for c = 10 mM KCl and V = 0–4 V. Because of the spherical geometry at the inlet, the concentration gradient has a nonlinear profile, in contrast to the semi-linear concentration gradient often calculated for flat CEM membranes. Before the limiting current is reached, the concentration profile becomes steeper with increasing voltage. Once the limiting current is reached, most sections of the concentration profile become more or less independent of the applied voltage and the concentration profiles scales then linearly with the bulk electrolyte concentration. At this point any increase in applied voltage results in a greater loss in the electric potential at the electrolyte–CEM interface as shown in Fig. 3B. The resulting high electric field at the interface is the cause of local convection effects and water splitting in real devices.
 | ||
| Fig. 3 Concentration and voltage at the inlet for 10 mM KCl. A) The K+ concentration profiles at the inlet for V = 0–4 V. The curves for 3 V and 4 V are nearly identical. B) Voltage profiles for V = 0–4 V. For lower voltages the voltage drop at the CEM is quite small, while at limiting current conditions (V = 3 V and 4 V) additionally applied voltage is dropped at the interface (x: distance from fiber inlet). | ||
Based on the analysis above we conclude that the cause of the irregularities in current–voltage characteristics is generated by concentration polarization at the inlet. Also, we found that this generates water splitting at the inlet, which leads to transport and delivery of H+, an undesirable consequence for most devices and applications. The measurements and simulations also show that this issue is especially severe for larger molecules (lower diffusion coefficient) at lower concentrations in the source electrolyte, a common scenario when delivering biologically relevant molecules. To resolve this issue, we therefore re-engineered the inlet geometry to allow for higher currents without reaching the limiting current. We achieved this by designing an ion selective cap at the fiber inlet (Fig. 4A) to increase the area of the electrolyte–CEM interface. The OEIP fiber inlet was functionalized and immersed for 20 min into the AMPSA solution. The fiber inlet with the ion selective drop was exposed to UV radiation for 10 min. Fig. 4B and C shows the I–V curves obtained, with the addition of the ion selective cap for the CEM at different HCl and AChCl concentrations. Indeed, the measured I–V curves for OEIPs with the cap do not show the typical signs of limiting currents which are characteristic for devices without a cap (Fig. 2A–C). Simulations of devices with a cap of 100 μm diameter confirm the effect (Fig. 4D and E) as this cap increases the limiting current by an order of magnitude. The influence of the cap on the limiting current can be understood by considering the ion concentration that decreases from that of the bulk electrolyte to nearly zero at the electrolyte–CEM interface. The selectivity of the CEM causes the ion transport to be diffusion-dependent, thus the current density depends on the ion concentration gradient and the total current depends therefore on the interface area times the current density. For small inlets/caps, the geometry is close to spherical and the limiting current thus scales faster than linearly with respect to inlet area. For larger caps, the diffusion gradient starts to resemble that of a planar membrane due to the decreasing curvature of the surface, and the limiting current will thus scale linearly with the surface area of the inlet cap.
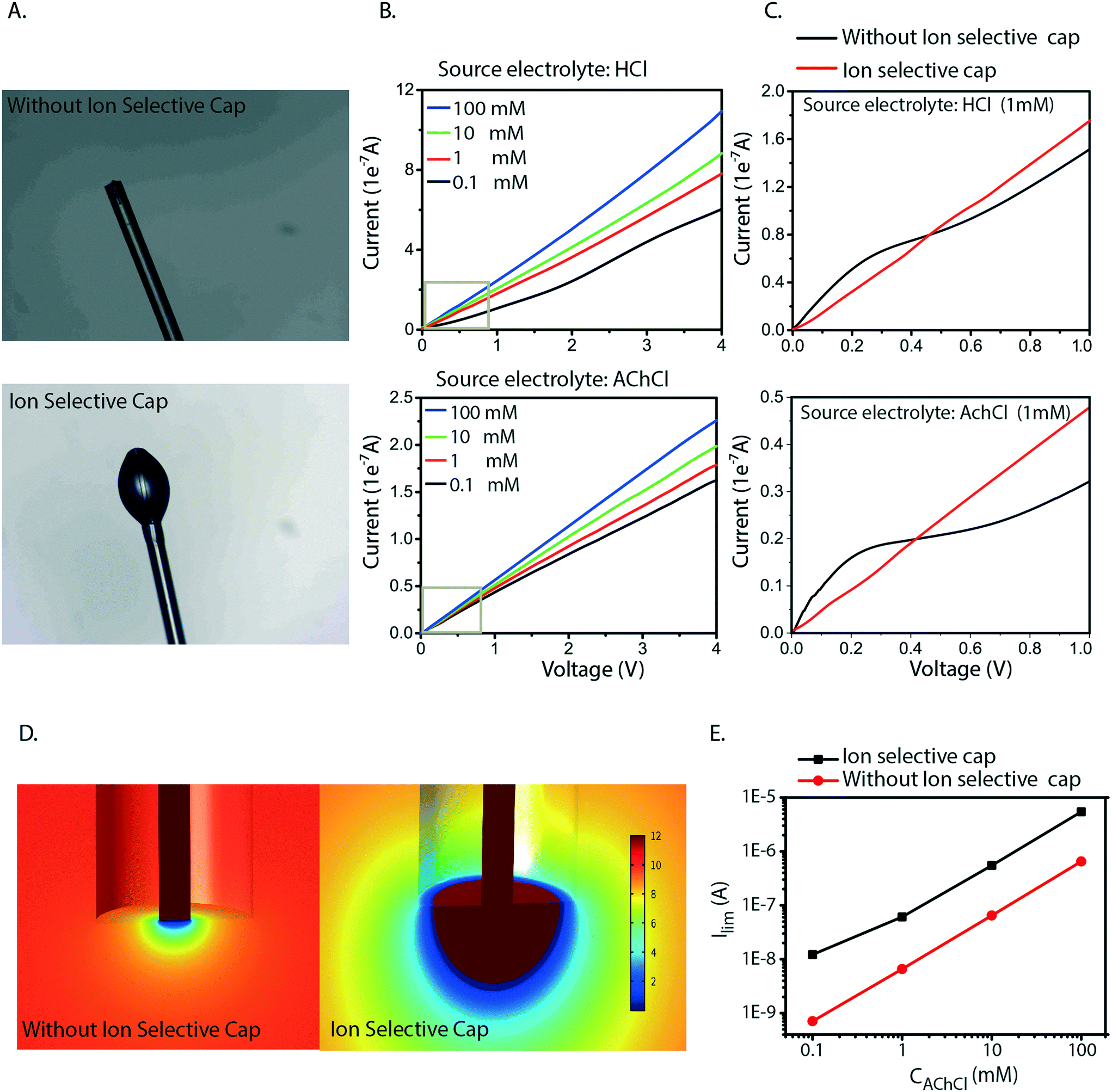 | ||
| Fig. 4 A) Microscope images of fiber with and without the ion selective cap at the inlet. B) Current–voltage curves for HCl and AChCl at different concentrations (100 mM, 10 mM, 1 mM, 0.1 mM), in the range 0–5 V scanned at a rate of 5 mV s−1, after the introduction of an ion selective cap at the inlet. C) Comparison current–voltage curves of 0.1 mM HCl and AChCl with and without the ion selective cap. D) Simulations of limiting current for different AChCl concentrations with and without a 100 μm large ion selective cap around fiber inlet. E) Calculated ACh+ gradient at fiber inlet for 10 mM source concentration at limiting current conditions with and without the ion selective cap. | ||
The selective cap resolves several of the issues related to miniaturization of OEIPs. As the OEIPs must be operated below the limiting current to avoid water splitting, it is crucial that the limiting current is high enough for the envisioned applications. The cap effectively resolves this by gathering ions from a larger surface of the electrolyte, thereby avoiding depletion-limitation of ion transport along the interface. This will then allow the OEIP to operate properly with a source electrolyte of low concentration including ionic species with low diffusion coefficient; a common combination for biologically active substances and applications. Also, the areal cap allows for the use of arbitrarily small channels as it entirely decouples the inlet from the channel dimensions.
Conclusions
We have investigated irregularities observed in the I–V curves for miniaturized capillary OEIPs. The I–V curves obtained for HCl, KCl, and AChCl source electrolytes all show three characteristic regions, which are associated with concentration polarization occurring at the inlet. Concentration polarization can lead to OEIP operation in the over-limiting current regime, which can cause water splitting. Indeed, when investigated, water splitting was found to occur at the inlet of the capillary OEIP and is typically undesired as this process can generate pH changes and loss in transport efficiency. A combined experimental and simulation study established a good correlation between the limiting ion current and the electrolyte concentration and diffusion coefficient of the ion. Our findings show that it would be problematic to transport many biologically relevant ions, as they typically come at low concentrations and often exhibit low diffusion coefficients. We solved this limitation and restriction by developing an ion selective cap manufactured at the inlet, which significantly increases the surface area of the inlet and thereby the limiting current of the OEIP. This will enable miniaturized OEIPs to transport biologically relevant ions at relatively much higher rates and thereby address a wider range of biomedically relevant applications and needs, such as in applications related to neurological disorders and diseases where a wide window of drug delivery frequencies and amplitudes is desired.Author contributions
Maria Seitanidou carried out the experiments, fabricated and characterized the devices, and wrote the manuscript. Klas Tybrandt developed and performed the theoretical computational models. Klas Tybrandt conceived the original idea. Magnus Berggren and Daniel T. Simon supervised the project. All authors discussed the results and contributed to the final manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors wish to thank Dr. Erik Gabrielsson and Dr. David Poxson for their assistance in designing and developing devices. This work was primarily supported by the Swedish Foundation for Strategic Research. Additional funding was provided by the Advanced Functional Materials SFO-center at Linköping University, the Önnesjö Foundation, and the Knut and Alice Wallenberg Foundation.References
- J. Isaksson, P. Kjäll, D. Nilsson, N. Robinson, M. Berggren and A. Richter-Dahlfors, Nat. Mater., 2007, 6, 673–679 CrossRef CAS PubMed
.
- G. G. Malliaras, Biochim. Biophys. Acta, 2013, 1830, 4286–4287 CrossRef CAS PubMed
- R. M. Owens and G. G. Malliaras, MRS Bull., 2010, 35, 449–456 CrossRef CAS
- T. Someya, Z. Bao and G. G. Malliaras, Nature, 2016, 540, 379–385 CrossRef CAS PubMed
- L. Torsi, M. Magliulo, K. Manoli and G. Palazzo, Chem. Soc. Rev., 2013, 42, 8612–8628 RSC
- J. T. Mabeck and G. G. Malliaras, Anal. Bioanal. Chem., 2005, 384, 343–353 CrossRef PubMed
- A. Dodabalapur, Mater. Today, 2006, 9, 24–30 CrossRef CAS
- N. Lago and A. Cester, Appl. Sci., 2017, 7, 1292 CrossRef
- L. Ricotti, A. Cafarelli, V. Iacovacci, L. Vannozzi and A. Menciassi, Curr. Nanosci., 2015, 11, 144–160 CrossRef CAS
- C. Boehler and M. Asplund, J. Biomed. Mater. Res., Part A, 2015, 103, 1200–1207 CrossRef PubMed
- R. Wadhwa, C. F. Lagenaur and X. T. Cui, J. Controlled Release, 2006, 110, 531–541 CrossRef CAS PubMed
- D. T. Simon, E. O. Gabrielsson, K. Tybrandt and M. Berggren, Chem. Rev., 2016, 116, 13009–13041 CrossRef CAS PubMed
- D. T. Simon, K. C. Larsson, D. Nilsson, G. Burström, D. Galter, M. Berggren and A. Richter-Dahlfors, Biosens. Bioelectron., 2015, 71, 359–364 CrossRef CAS PubMed
- T. Arbring Sjöström, M. Berggren, E. O. Gabrielsson, P. Janson, D. J. Poxson, M. Seitanidou and D. T. Simon, Adv. Mater. Technol., 2018, 3, 1700360 CrossRef
- X. Strakosas, J. Selberg, Z. Hemmatian and M. Rolandi, Adv. Sci., 2017, 4, 1600527 CrossRef PubMed
- C. Z. Y. Deng, E. Josberger, J. Jin, A. F. Rousdari, B. A. Helms, M. P. Anantram and M. Rolandi, Sci. Rep., 2013, 1–8 CAS
- A. Jonsson, Z. Song, D. Nilsson, B. A. Meyerson, D. T. Simon, B. Linderoth and M. Berggren, Sci. Adv., 2015, 1, e1500039 CrossRef PubMed
- J. Isaksson, D. Nilsson, P. Kjäll, N. D. Robinson, A. Richter-Dahlfors and M. Berggren, Org. Electron., 2008, 9, 303–309 CrossRef CAS
- D. T. Simon, S. Kurup, K. C. Larsson, R. Hori, K. Tybrandt, M. Goiny, E. W. H. Jager, M. Berggren, B. Canlon and A. Richter-Dahlfors, Nat. Mater., 2009, 8, 742–746 CrossRef CAS PubMed
- K. Tybrandt, K. C. Larsson, S. Kurup, D. T. Simon, P. Kjäll, J. Isaksson, M. Sandberg, E. W. H. Jager, A. Richter-Dahlfors and M. Berggren, Adv. Mater., 2009, 21, 4442–4446 CrossRef CAS
- A. Jonsson, S. Inal, I. Uguz, A. J. Williamson, L. Kergoat, J. Rivnay, D. Khodagholy, M. Berggren, C. Bernard, G. G. Malliaras and D. T. Simon, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 9440–9445 CrossRef CAS PubMed
- A. J. Williamson, J. Rivnay, L. Kergoat, A. Jonsson, S. Inal, I. Uguz, M. Ferro, A. Ivanov, T. A. Sjöström, D. T. Simon, M. Berggren, G. G. Malliaras and C. Bernard, Adv. Mater., 2015, 27, 3138–3144 CrossRef CAS PubMed
- C. M. Proctor, A. Slézia, A. Kaszas, A. Ghestem, I. del Agua, A.-M. Pappa, C. Bernard, A. J. Williamson and G. G. Malliaras, Sci. Adv., 2018, 4, eaau1291 CrossRef PubMed
- D. J. Poxson, M. Karady, R. Gabrielsson, A. Y. Alkattan, A. Gustavsson, S. M. Doyle, S. Robert, K. Ljung, M. Grebe, D. T. Simon and M. Berggren, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 4597–4602 CrossRef CAS PubMed
-
H. Strathmann, Ion-Exchange Membrane Separation Processes, Elsevier, 2004 Search PubMed
- V. M. Barragan and C. Ruiz-Bauza, J. Colloid Interface Sci., 1998, 205, 365–373 CrossRef CAS PubMed
- J. A. Manzanares, K. Kontturi, S. Mafe, V. M. Aguilella and J. Pellicer, Acta Chem. Scand., 1991, 45, 115–121 CrossRef CAS
- V. M. Aquilella, S. Mafe, J. A. Manzanares and J. Pellicer, J. Membr. Sci., 1991, 61, 177–190 CrossRef
- N. Káňavová and L. Machuča, Period. Polytech., Chem. Eng., 2014, 58, 125–130 CrossRef
- N. Káňavová, L. Machuča and D. Tvrzník, Chem. Pap., 2014, 68, 324–329 Search PubMed
- R. Valerdi-Pérez and J. Ibánez-Mengual, Desalination, 2001, 141, 23–37 CrossRef
- J. J. Krol, M. Wessling and H. Strathmann, J. Membr. Sci., 1999, 162, 145–154 CrossRef CAS
- T. Scarazzato, D. C. Buzzi, A. M. Bernardes and D. C. R. Espinosa, Braz. J. Chem. Eng., 2015, 32, 831–836 CrossRef
- C. Korzenowski, M. A. S. Rodrigues, L. Bresciani, A. M. Bernardes and J. Z. Ferreira, J. Hazard. Mater., 2008, 152, 960–967 CrossRef CAS PubMed
- M. A. S. Rodrigues, F. D. R. Amado, J. Z. Xavier, K. F. Streit, A. M. Bernardes and J. L. N. Ferreira, J. Cleaner Prod., 2008, 605–611 CrossRef
- J. A. Manzanares, S. Mafd and H. J. Reiss, J. Phys. Chem., 1993, 97, 8524–8530 CrossRef CAS
- V. I. Zabolotskii, K. A. Lebedev and E. G. Lovtsov, Russ. J. Electrochem., 2006, 42, 931–941 CrossRef
- I. Rubinstein, J. Chem. Soc., Faraday Trans., 1991, 87, 2079–2087 RSC
- J. Courtois, M. Szumski, E. Byström, A. Iwasiewicz, A. Shchukarev and K. Irgum, J. Sep. Sci., 2006, 29, 14–24 CrossRef CAS PubMed
- K. Tybrandt, Soft Matter, 2017, 13, 8171–8177 RSC
- E. M. Sega, C. F. Tormena, P. R. de Oliveira, R. Rittner, L. W. Tinoco, J. Figueroa-Villar and N. F. Höehr, J. Mol. Struct., 2006, 797, 44–48 CrossRef CAS
- H. Strathmann, J. Krol, H.-J. Rapp and G. Eigenberger, J. Membr. Sci., 1997, 125, 123–142 CrossRef CAS
- H. J. Lee, H. Strathmann and S. Moon, Desalination, 2006, 190, 43–50 CrossRef CAS
- N. Boon and M. O. de la Cruz, Soft Matter, 2015, 11, 4793–4798 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9lc00038k |
| This journal is © The Royal Society of Chemistry 2019 |