
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical and cellular antioxidant activity of flavone extracts of Labisia pumila before and after in vitro gastrointestinal digestion†
Hui Wu ,
Hongru Xi,
Furao Lai,
Juanjuan Ma and
Huifan Liu*
,
Hongru Xi,
Furao Lai,
Juanjuan Ma and
Huifan Liu*
College of Food Science and Engineering, South China University of Technology, 81 Wu Shan Lu, Tianhe Qu, Guangzhou Shi, Guangdong Sheng 510640, China. E-mail: 13600008265@163.com; Fax: +86-20-87112532; Tel: +86-20-87112853
First published on 28th March 2018
Abstract
Two kinds of flavone extracts were extracted and purified from Labisia pumila (LP). High-performance liquid chromatography (HPLC) analysis was used to determinate the flavones in the extracts, and catechin, glycitin, rutin, naringin, and myricetin were identified in the LP leaf extract (LPL-F) while genistin, naringin, and myricetin were found in the stem extract (LPS-F). Specific flavonol compounds mediated the satisfactory scavenging abilities. The flavone leaf extracts performed better than the stem extracts in chemical antioxidative activities but worse in cellular antioxidative capabilities. The chemical and cellular antioxidative activities were not obviously changed by gastrointestinal digestion but slightly changed at the last 2 hours of intestinal digestion because prolonged exposure to alkaline conditions could destroy the structure of flavonoids. Changes in MDA and GSH content, and enzyme activities of SOD and GSH-Px in human erythrocytes during GI digestion indicated the possible intracellular antioxidant-detoxifying mechanisms were through attenuating AAPH-induced oxidative stress by inhibiting ROS generation, in which stem extracts performed the better.
1. Introduction
Labisia pumila (LP), locally known in Malaysia as Kacip Fatimah, is one of the popular natural product-based medicines belonging to the Myrsinaceae family that is widely grown in tropical rainforests of southeast Asia.1 The health-promoting uses of LP include as a post-partum medication to maintain the female reproductive system and enhance sexual function.2 Numerous studies have reported that LP shows great benefits in the prevention and treatment of diseases related to estrogen deficiencies, which may be strongly mediated by its pleiotropic actions including phytoestrogen and antioxidant effects.3,4 Currently, various types of phytochemicals have been identified in LP, and most possess beneficial properties that are consistent with its traditional use, which has been practiced for thousands of years in Asia. The LP extracts mainly contain flavonoids, polyphenols, saponins, and organic acids.1 Total flavonoids, an important secondary metabolite of LP, mainly include flavonoids and their glycosides, isoflavones, flavonols and their glycosides, isoflavanones, and dihydroisoflavones. The parent compounds of flavonoids are quercetin, myricetin, kaempferol, rutin, protocatechuic acid, catechin, naringin, daidzin, genistein, and anthocyanin. Flavonoids exhibit strong bioactivity such as free radical scavenging, antioxidant, antitumor, antiradiation, and cardiovascular system protection.5 Therefore, studying LP flavonoids and their antioxidative abilities is of considerable significance.Several methods are used to assess the antioxidative capacity of flavones based on electron transfer (ET) including the 1,1-diphenyl-2-picrylhydrazyl (DPPH) radical scavenging activity (DRSA), ferric ion reducing antioxidant power (FRAP), and 2,2-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) (ABTS)-scavenging assays.6 The specific reaction and transfer reactions in an electron transfer (ET)-based assay may be explained as follows: probe (oxidant) + electron from antioxidant → reduced probe + oxidized antioxidant.7 In vitro methods for measuring antioxidant capacity are based on chemicals reactions that are simple and easy processes but are unable to assess toxicity, bioavailability, and metabolites of antioxidant components. A newly developed approach, the 2,2-azobis(2-amidinopropane) dihydrochloride (AAPH)-induced erythrocyte hemolysis assay, quantifies the antioxidant ability of bioactive compounds in cell culture and evaluates accessibility in an assay where AAPH acted as the free radical.8
The antioxidative activities of extracts may vary under different conditions. For example, under the human gastrointestinal (GI) conditions, the absorptivity of the intestinal epithelial cell, strong acid, high content of bile acids, and types of enzymes and their interaction may affect the activity of extracts, indicating that the actual beneficial effects of total antioxidative flavonoids on human health are affected following consumption.9,10 In vitro GI digestion models are known to simulate the processes in the human digestive tract by sequential exposure of test substances to gastric and intestinal conditions this model is a simple, cheap, and reproducible tool to assess the bio-accessibility of nutrients, digestion of substances, and dissolution of pharmaceutical dosage forms.11
The therapeutic effects of LP on estrogen deficiency-related disorders were reported to be induced by its flavone content and relevant antioxidative abilities.4 Therefore, this present study was designed to identify the optimal conditions for ethanol extraction of the total flavones of LP. In addition, we characterize the types and amounts of flavones present in LP and their antioxidative abilities using electron transfer (ET)-based and red cell models. After developing the in vitro GI digestion model, we investigated the ET-based antioxidative capacities of the flavones. The information provided by this study may contribute to further studies of the digestion fermentation parameters of flavones, which may provide useful information for studying flavones extracted from LP.
2. Materials and methods
2.1 Materials
Dried LP samples (1 kg) were purchased from Forest Research Institute Malaysia (FRIM). Human blood samples were collected from three healthy adult volunteers. Vitamin C (VC), AAPH, DPPH, and ABTS were purchased from Sigma-Aldrich (St. Louis, MO, USA). Phosphate-buffered saline (PBS, pH 7.4) was purchased from Gibco Life Technologies (Grand Island, NY, USA). Catechin, puerarin, epicatechin, daidzin, glycitin, rutin, genistin, naringin, myricetin, daidzein, luteolin, quercetin, apigenin, genistein, kaempferol, and chrysin were purchased from Sigma-Aldrich. All other chemicals and reagents used were of analytical grade and commercially available.2.2 Plant preparation
The dried LP (1 kg) was carefully separated into leaf and stem samples, which were subsequently crushed into a powder using a tissue triturator and passed through a 40-mesh screen. The collected leaf and stem samples were immediately stored at −4 °C until use. The detailed sample image is shown in the ESI (Fig. S1†).2.3 Optimal extract of total flavonoid using response surface methodology (RSM)
Ethanol was used to extract the flavone from the LP samples. Single-factor experiments were designed to determine the critical factors affecting the extraction yield of flavone from LP. Four parameters were analyzed, namely extractive temperature, ethanol concentration, extraction time, and solid-to-solvent ratio. Based on the results of the single-factor experiments, three critical factors were selected for the design of the response surface experiment using statistical software. A Box–Behnken design with three factorial points, three levels, and three central points, leading to 15 sets of experiments, was implemented to determine the optimal extraction conditions for flavone extraction from LP. The extraction efficiency of total flavonoid for each parameter was determined using the method of Mavundza.12 The optimal extraction conditions determined based on the RSM were subsequently used to obtain the flavone extracts. The collected flavone extracts of the stem (cLPS-F) and leaf (cLPL-F) were then lyophilized and stored at −20 °C until use.2.4 Sample purification
A total of 0.25 g cLPS-F and cLPL-F were dissolved in 50 mL ultrapure water, filtered through 0.22 μm microporous filters, loaded onto AB-8 macroporous resin (1.6 × 35 cm), and then eluted at a flow rate of 1 mL min−1 with 500 mL each of ultrapure water and 40% ethyl alcohol sequentially. The eluents were analyzed by the methods described above. Following elution and lyophilization, the flavonoids fractions LPS-F and LPL-F were obtained.2.5 Determination of chemical components
To comprehensively elucidate the chemical compositions of the flavone extracts, the polysaccharide, protein, polyphenol, and total flavonoid contents of the cLPL-F, cLPS-F, LPS-F, and LPL-F were evaluated. After with glucose as the standard, the phenol-sulfuric acid method was used to determine the polysaccharide content.13 Furthermore, the method required the use of coomassie brilliant blue G-250 with bovine serum albumin as the standard.13 The polyphenol content was measured using folin reagents with gallic acid as the standard. The total flavonoid content was measured using the method of Mavundza with some modification.12 In brief, 9 mg of the flavone extracts was weighed accurately, dissolved in 5.16 mL ultrapure water, and 0.3 mL sodium nitrite (NaNO2, 5%) was added to the sample solutions, which were then submerged for 6 min. Then, 0.3 mL aluminum nitrate [10%] was added to the mixtures, which were submerged for another 6 min, and then 4 mL sodium hydroxide (NaOH, 4%) was added, followed by submersion for 15 min in the dark. When the reactions were completed, the absorbance of the mixture was evaluated immediately at 510 nm. The standard curve was constructed using rutin standard solution.2.6 High-performance liquid chromatography (HPLC) analysis
The flavonoid compositions of the LPS-F and LPL-F were characterized using high-performance liquid chromatography HPLC based on the method described by Yong et al, with some modifications.14 The flavonoid standards used were catechin, puerarin, epicatechin, daidzin, glycitin, rutin, genistin, naringin, myricetin, daidzein, luteolin, quercetin, apigenin, genistein, kaempferol, and chrysin. A total of 20 μL of each flavone extract was loaded in the HPLC system, which was equipped with an analytical column (Intersil ODS-3, 5 μm 4.6 × 150 mm) The mobile phase used consisted of 0.1% methanoic acid (solvent A) and acetonitrile (solvent B). The pH of the water was adjusted to 2.5 with 1% methanoic acid. The isoflavonoid and flavonoid compounds were detected at 280 nm. The column was equilibrated with 10% solvent B for the first 5 min. Then, the ratio of solvent B was increased to 90% in the next 10 min and maintained for 5 min. Subsequently, the flow rate was set to 0.6 mL min−1, solvent B was decreased from 90% to 10% in 5 min, and then maintained at 10% solvent for the last 5 min.2.7 Simulated in vitro gastric and intestinal digestion
The simulated small intestinal electrolyte solution (SIES) consisted of 5.4 g NaCl, 0.65 g KCl, and 0.33 g calcium chloride hydrate (CaCl2H2O) dissolved in 1 L ultrapure water and was adjusted to a final pH of 7 using 0.1 M NaOH. In addition, for the pancreatin enzyme solution, 14 g pancreatin enzyme was dissolved in 200 g water and centrifuged for 10 min at 4800 × g after gently mixing for 10 min. Then, 100 g of the supernatant was added to the following solution, 200 g 4% bile salt and 100 g 7% pancreatic enzymes (w/w), which were added to 100 g SIES. Then, 13 mg trypsin was added to the adequately mixed solution, and its pH was adjusted to 7.5 with 0.1 M NaOH.
All samples and the control were dialyzed (molecular weight [MW] cut off 100 Da) against ultrapure water, which was changed every 3 hours until the ionic concentration of the dialysate was equal to that of the ultrapure water. The residual tubes of the sample and control groups were primed to simulate intestinal digestion by adding 12 mL SIES after adjusting the pH of each tube to 7 with 1 M NaHCO3. The tubes were mixed completely and digested in an incubator at 95 rpm at 37 °C in the dark under anaerobic conditions. The samples digested under simulated intestinal conditions (LPS-F-I and LPL-F-I) and the control (control-I) were also analyzed after 1, 2, 4 and 6 hours digestion using the corresponding tubes. All intestinal digestion samples and the control groups were immediately dialysis (MW cut off 100 Da) against ultrapure water, which was changed every 3 hours until the ionic concentration of the dialysate was equal to that of the ultrapure water. All digested samples were then lyophilized and stored at −20 °C until use.
2.8 Determination of chemical antioxidant properties
where, Ac and As are the absorbance values of the control and sample groups, respectively. The DRSA% of the four fractions was analyzed by comparison with the concentration of the sample that caused a 50% reduction of the DPPH radical.
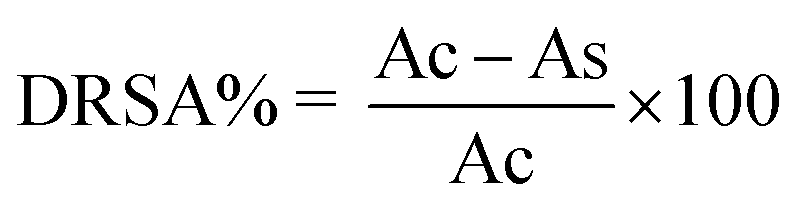
where, Ac and As are the absorbance values of the control and sample groups, respectively.
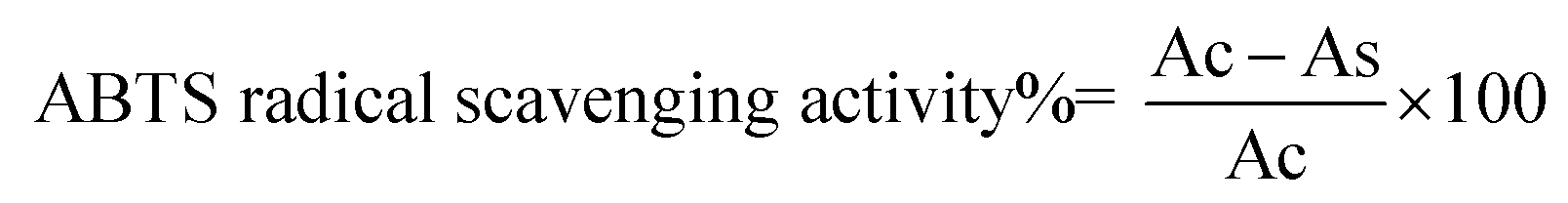
2.9 Determination of AAPH-induced erythrocyte hemolysis
According to the method of Zhang et al., with some modification, the protective effect of the before and after digestion LPS-F and LPL-F solutions against AAPH-induced erythrocyte oxidative hemolysis was analyzed.19 All experiments were performed in compliance with “China food safety standards GB151935-2014”, and approved by the ethics committee at South China University of Technology. Informed consents were obtained from human participants of this study. Erythrocytes were obtained from the blood samples by washing them three times with PBS (pH 7.4) by centrifugation (1200 × g, 10 min, 4 °C). The erythrocyte suspensions were diluted 4-fold with PBS (pH 7.4) and then 200 μL samples were added to 200 μL PBS or the LPS-F and LPL-F solutions, including the before digestion (0.375, 0.75, 1.5, 3, 6 and 12 μg mL−1) and after digestion (4 μg mL−1) sample solutions. The mixtures were incubated for 30 min at 37 °C and 400 μL 0.2 mM AAPH was added, followed by incubation for another 2 hours with gentle shaking. Subsequently, the well-incubated mixture was diluted with 3.2 mL PBS (pH 7.4) and centrifuged (1200 × g, 10 min, 4 °C). The absorbance of the supernatant was measured at 540 nm similar to the sample-treated group. Ultrapure water was added to the above suspension to obtain 100% hemolysis. The hemolysis inhibition rate was calculated as follows: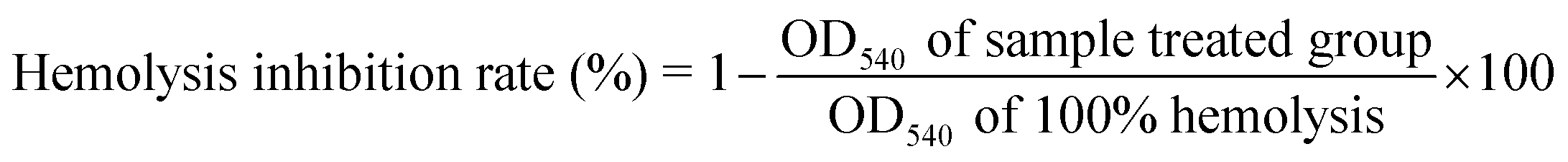
2.10 Estimation of intracellular ROS level
The erythrocytes were obtained as described by Zhang et al., with minor modifications, but the supernatant was discarded after 3 washes with PBS (pH 7.4) and erythrocytes were resuspended after dilution in a 5-fold volume of PBS (pH 7.4). Further, 100 μL of the erythrocyte resuspension was used to determine the intracellular ROS levels. After centrifugation (1200 × g, 10 min, 4 °C), 200 μL of 10 μM DCFH-DA was added, followed by incubation for 25 min at 37 °C in the dark. The solution was washed and resuspended in 600 μL PBS (pH 7.4). The fluorescence intensity of erythrocytes was measured with a Fluoroskan Ascent microplate fluorometer (ex/em, 488/525 nm) (Thermo Scientific).2.11 Determination of MDA, GPX, SOD and GSH
The erythrocytes were obtained as described by Zhang et al., with minor modifications, but the supernatant was discarded after 3 washes with PBS (pH 7.4) and erythrocytes were added to double ultrapure water in an ice-water bath for 10 min and then centrifuged (1200 × g, 10 min, 4 °C) to obtain the celllysis buffer. The content of GSH and MDA were determined using GSH and microscale MDA kits, respectively. A cellular glutathione peroxidase kit and total superoxide dismutase assay kit were used to determine the activities of GSH-Px and SOD, respectively.2.12 Statistical analysis
The data are expressed as the mean ± standard deviation (SD) of three replicates. Significant differences between the means of different parameters were calculated using Duncan's multiple-range test using the statistical package for the social sciences (SPSS) 17.0 software (SPSS Inc., Chicago, IL, USA). Differences were considered statistically significant at P < 0.05.3. Results and discussions
3.1 Analysis of response surface
The results of the single-factor experiments (data not shown) were used to develop a Box–Behnken Design (BBD) for the RSM to optimize the extraction conditions of flavone extracts from LP. The lack of fit (0.0772) indicated that the experimental results had a good fit with the model. In this case, A, B, C, AB, AC, BC, A2, B2, and C2 were all significant to the yield. The final predictive equation was obtained by neglecting the insignificant factors as shown below:| Y = 39.69 − 0.32 × A − 0.17 × B + 0.019 × C + 0.14 × A × B + 0.19 × A × C − 0.48 × B × C − 3.29 × A2 − 1.52 × B2 − 1.53 × C2 |
According to the RSM design, the optimum conditions for flavone extraction from LP were a ratio of material to solvent of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :59.01, the temperature of 85.13 °C, and ethanol concentration of 34.40%. The predicted result was 39.7032 mg g−1. To facilitate the experiment, a ratio of material to solvent, temperature, ethanol concentration, and time of 1:60 (w/v), 85 °C, 35%, and 90 min, respectively were selected to validate the RSM model.20 The flavone extract yields were 39.41 ± 0.311 and 39.05 ± 0.667 mg g−1 from the stem and leaf, respectively, in accordance with the predicted result.
:59.01, the temperature of 85.13 °C, and ethanol concentration of 34.40%. The predicted result was 39.7032 mg g−1. To facilitate the experiment, a ratio of material to solvent, temperature, ethanol concentration, and time of 1:60 (w/v), 85 °C, 35%, and 90 min, respectively were selected to validate the RSM model.20 The flavone extract yields were 39.41 ± 0.311 and 39.05 ± 0.667 mg g−1 from the stem and leaf, respectively, in accordance with the predicted result.
3.2 Purification of flavone from LP
To remove the various water-soluble impurities and obtain the purified flavone extracts of LP, the cLPS-F and cLPL-F were first eluted with ultrapure water on an AB-8 macroporous resin. Polysaccharide, one of the water-soluble impurities, was used as the impurity indicator to determinate the time for elution in ultrapure water. As shown in Fig. 1A, more than one absorbance peak was observed at 490 nm from the tubes 10 to 23, indicating the polysaccharides in cLPL-F were eluted successfully. Therefore, the 40% ethanol elution began after elution with 30 tubes of water at the same flow rate, and the chromatogram of LPL-F is shown in Fig. 1B. At a wavelength of 510 nm, only one symmetrical peak was observed, indicating that the major flavone extracted from the leaf of LP was in the LPL-F. The same results were obtained for the characterization of the LP stem with a slight difference, shown in Fig. 1C and D. The polysaccharides in cLPS-F were different from those in the cLPL-F based on the one absorbance peak from tubes 3 to 18 at 490 nm observed during the ultrapure water elution as shown in Fig. 1C. Moreover, the ethanol elution was started after elution with 20 tubes of water and two absorbance peaks were detected at 510 nm, indicating that two flavones were extracted in cLPS-F. When considering the flavone content of the extracts and the convenience of the flavone fraction collection (Fig. 1B), only the eluent with an absorbance peak at high levels at 510 nm (tubes 10 to 15) was collected and designated as the LPS-F flavone extracts. However, since there was a low flavone content in the stem fraction of LP, we may perform unique structural characterization and relevant bioactivity analyses in the next stage of our research studies.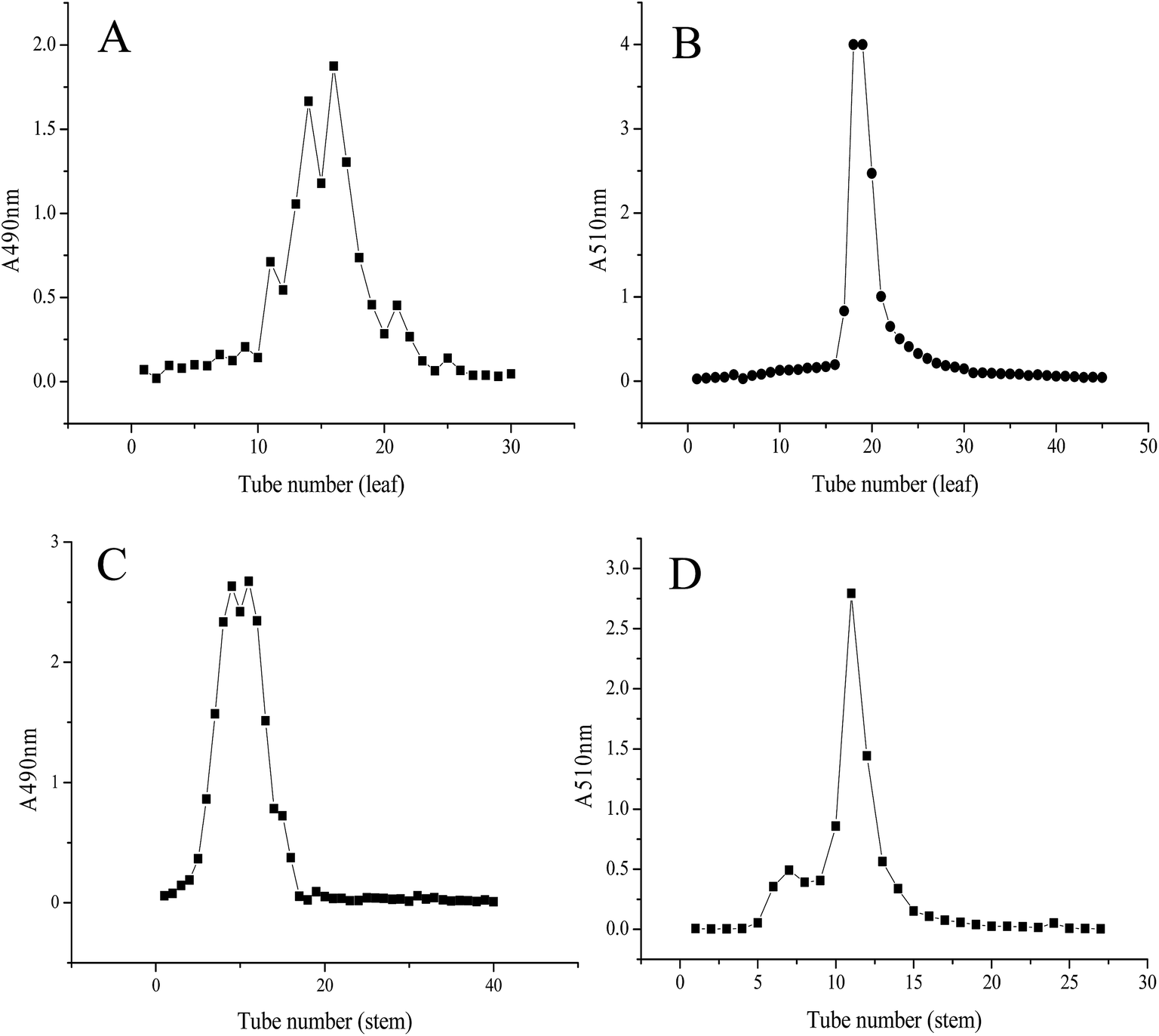 | ||
| Fig. 1 Chromatography of the Labisia pumila (LP) leaf extract at absorbance wavelength of (A) 490 nm and (B) 510 nm. LP stem extract at absorbance wavelength of (C) 490 nm and (D) 510 nm isolated from LP using AB-8 macroporous resin. | ||
3.3 Analysis of chemical components
Four major bioactive compounds consisting of total sugars, protein, total polyphenol, and flavones were analyzed in this study, and the results are shown in Table 1.| cLPL-F | cLPS-F | LPL-F | LPS-F | |
|---|---|---|---|---|
| a Data are shown as the mean ± SD (n = 3).b Not detected or lower than limit of quantification. | ||||
| Proteina (μg per mg FW) | 3.09 ± 0.15 | 2.07 ± 0.12 | NDb | ND |
| Total polyphenol, (μg gallic acid per mg FW) | 28.69 ± 0.43b | 65.82 ± 0.36 | 96.60 ± 0.44 | 118.20 ± 11.04 |
| Total flavones, (μg rutin per mg FW) | 86.17 ± 4.21 | 82.45 ± 14.16 | 381.20 ± 14.55 | 667.22 ± 19.36 |
| Neutral sugar, (μg per mg FW) | 771.34 ± 14.38 | 226.13 ± 10.25 | 309.18 ± 11.30 | 511.03 ± 12.24 |
Considering protein content variation, we observed that the protein content in cLPL-F was significantly higher than that in cLPS-F was, indicating that the types and amounts of protein in the stem and leaf of LP were different. However, after elution with 40% ethanol, no proteins were detected in LPS-F and LPL-F, indicating that the major water-soluble proteins present in LP were efficiently eluted by ultrapure water.
The total flavone content in cLPS-F and cLPL-F was very similar, but after 40% ethanol elution, the content in LPL-F was approximately two times that in LPS-F. Moreover, the total flavone content in the leaf and stem extracts after purification increased by approximately 4.5 and eight times, respectively compared to the levels before purification. Regarding the changes in total polyphenol, the content in the cLPS-F was approximately three times that in the cLPL-F and after purification, the content in the LPS-F and in LPL-F increased by 3.5 and two times compared with that in the crude leaf and stem. Flavones are reported to be polyphenolic compounds that contain a C15 flavone skeleton, which indicates that the total polyphenol content included the flavone content.12 Because the increase in total flavones was significant higher than that of total polyphenols, their variations in the stem and leaf before and after purification could have been improved by elution with 40% ethanol, which effectively purified the flavone content.
The content of neutral sugars in the cLPL-F was approximately 3.5 times that in the cLPS-F but that in the LPS-F was nearly two times that in LPL-F. As previously reported, the structure of various types of flavones such as genistin, naringin, and rutin, contain glycosidic bonds, which would be detected as neutral sugar using the phenol-sulfate method.21 Therefore, based on the changes in neutral sugars in the leaf and stem flavone extracts, we could conclude that there were polysaccharides in the crude flavone extracts and the stems contained the most. However, since the elution was performed with ultrapure water, the high content of neutral sugars in the LPL-F and LPS-F were due to the glycosidic bonds present in the flavone compounds.
3.4 HPLC analysis of LPS-F and LPL-F
In our study, 16 types of flavones standards were used in the qualitative and quantitative analyses of the flavone compositions of the LPL-F and LPS-F. The details of the appearance time, standard curve, and relevant coefficient of determination (R2) values of each flavone sample are shown in Table 2. We discovered that except for the appearance time of apigenin and genistein (27.728 and 27.863 min, respectively), which were close, the other 14 standard flavones were separated obviously in the HPLC analysis. Furthermore, the standard curves of all the flavone standards performed satisfactorily (R2 = 0.990–0.999), indicating that the HPLC analysis conditions in our study were suitable for evaluating the type and relevant concentrations of the flavones in LPL-F and LPS-F (Table 3).| Sample name | Appearance time | Standard curve | R2 |
|---|---|---|---|
| Catechin | 14.409 | y = 0.20661x − 0.55675 | 0.99488 |
| Puerarin | 15.581 | y = 0.62291x − 0.68303 | 0.99102 |
| Epicatechin | 16.642 | y = 0.30391x − 0.60681 | 0.99944 |
| Daidzin | 17.433 | y = 1.62062x − 4.9338 | 0.99071 |
| Glycitin | 17.675 | y = 0.5935x − 1.15971 | 0.99001 |
| Rutin | 18.506 | y = 0.33798x − 0.58622 | 0.99916 |
| Genistin | 19.996 | y = 0.97846x − 2.12908 | 0.9926 |
| Naringin | 20.887 | y = 0.90085x − 1.2498 | 0.99925 |
| Myricetin | 23.643 | y = 3.8276x − 74.865 | 0.9991 |
| Daidzein | 24.482 | y = 1.51482x − 1.73103 | 0.99296 |
| Luteolin | 25.287 | y = 1.20201x − 3.91584 | 0.99896 |
| Quercetin | 25.50 | y = 0.7697x − 2.82202 | 0.99821 |
| Apigenin | 27.728 | y = 1.46804x − 2.33909 | 0.99956 |
| Genistein | 27.863 | y = 1.48016x − 2.05509 | 0.99369 |
| Kaempferol | 28.347 | y = 0.98661x − 2.8085 | 0.99963 |
| Chrysin | 34.082 | y = 2.81375x − 5.19779 | 0.9993 |
| Sample name | Flavone type | Appearance time | Concentration (μg mL−1) |
|---|---|---|---|
| LPL-F | Genistin | 20.581 ± 0.587 | 4.875 ± 0.012 |
| Naringin | 21.112 ± 0.226 | 4.896 ± 0.027 | |
| Myricetin | 23.832 ± 0.191 | 20.325 ± 2.443 | |
| LPS-F | Catechin | 14.552 ± 0.147 | 18.366 ± 1.182 |
| Glycitin | 17.792 ± 0.180 | 5.108 ± 0.031 | |
| Rutin | 19.146 ± 0.649 | 5.833 ± 0.022 | |
| Naringin | 20.940 ± 0.0542 | 5.002 ± 0.017 | |
| Myricetin | 23.981 ± 0.339 | 20.313 ± 2.347 |
After comparing the appearance time of the extracted and standard flavones, we concluded that the following main flavonol compounds were detected in the LPS-F, catechin, glycitin, rutin, naringin and myricetin while the LPL-F mainly contained genistin, naringin, and myricetin. The peak area of each flavonol compound and the relevant standard curve were used to determine the concentrations of the flavonol compounds.
3.5 ET-based scavenging capabilities
The LPL-F and LPS-F flavone extracts contained various flavonols, which have all shown potential antioxidative activity. Therefore, in our study, ET-based in vitro free radical scavenging assays were performed to evaluate the antioxidative abilities, and the results are shown in Fig. 2A to C. The results of the reducing power and DPPH and ABTS free radical scavenging assay performed using graded concentrations of all the LPL-F, and LPS-F samples revealed different levels of antioxidative activities. The results of the DPPH free radical scavenging assay (Fig. 2A) shows the scavenging rate of the LPL-F at concentrations from 0.375 to 6 μg mL−1 was 33.50 ± 1.53 to 80.94 ± 5.35%, which was significantly lower than that of LPS-F (36.44 ± 1.33 to 82.35 ± 3.17%) but higher than that of VC (30.94 ± 0.97 to 63.04 ± 2.01%). Moreover, at a concentration of 12 μg mL−1, the DPPH free radical scavenging rate of LPL-F (80.56 ± 4.98%) was comparable to that of LPS-F (79.92 ± 3.66%) and significantly lower than that of VC (91.56 ± 4.33%).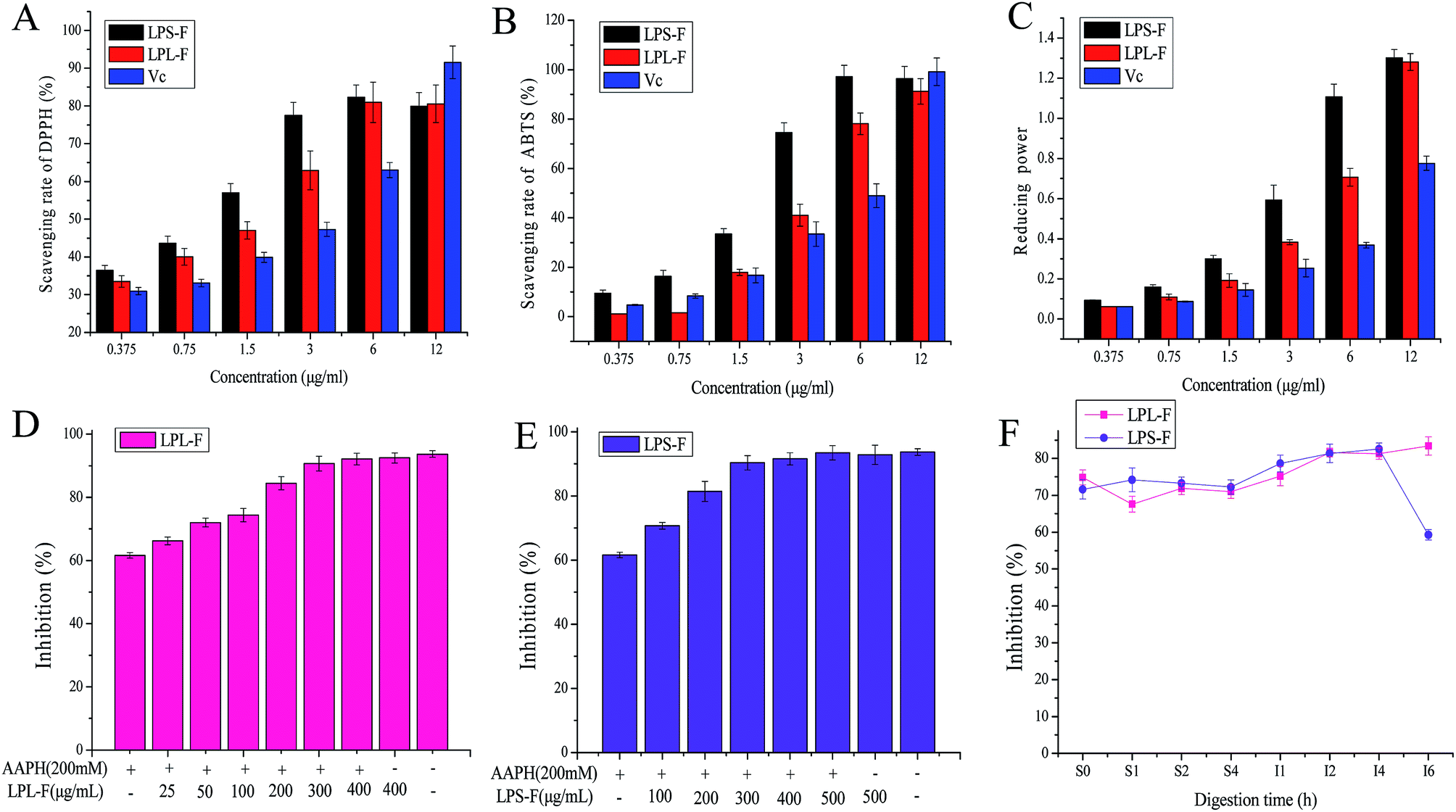 | ||
| Fig. 2 Scavenging ability of Labisia pumila leaf and stem extract fraction (LPL-F and LPS-F, respectively) on (A) 1,1-diphenyl-2-picrylhydrazyl (DPPH) and (B) 2,2-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) (ABTS) free radical. (C) Ferric ion reducing antioxidative power of both extracts. Ultrapure water and vitamin C (VC) groups were the control and positive control groups, respectively. Protective effects of (D) LPL-F and (E) LPS-F on 2,2-azobis (2-amidinopropane) dihydrochloride (AAPH)-induced erythrocyte hemolysis. (F) Protective effects of gastrointestinal (GI)-digested LPL-F and LPS-F on AAPH-induced erythrocyte hemolysis. AAPH alone (200 mM) was used as the positive control. Data are means ± standard deviations of three independent experiments. | ||
In the ABTS free radical scavenging assay, the activity of LPL-F (1.195 ± 0.012 to 1.594 ± 0.054%) was significantly lower than that of LPS-F (9.561 ± 1.221 to 16.334 ± 2.417%) and that of VC (4.780 ± 0.187 to 8.367 ± 0.871%) at concentration from 0.375 to 0.75 μg mL−1. At a concentration of 1.5 μg mL−1, the scavenging rate of LPL-F (17.928 ± 1.241%) was equal to that of VC (16.733 ± 1.998%) and significantly lowers than that of LPS-F (33.466 ± 2.21%). When the sample concentration increased from 3 to 6 μg mL−1, the scavenging rate of LPL-F (41.035 ± 4.447 to 78.087 ± 4.325%) was higher than that of VC (33.466 ± 4.996 to 49.004 ± 4.789%) but was still much lower than that of LPS-F (74.502 ± 3.987 to 97.414 ± 4.541%).
However, at a concentration of 12 μg mL−1 the ABTS free radical scavenging rate decreased in the following order: LPL-F (91.235 ± 5.214%) < LPL-F (96.414 ± 4.968) < VC (99.203 ± 5.574%). The reducing power tendencies were similar to those observed with the DPPH free radical scavenging rate with a slight difference. Specifically, the reducing power of the LPL-F sample at concentrations from 0.375 to 12 μg mL−1 (0.062 ± 0.01 to 1.281 ± 0.042%) were all significantly higher than that of VC (0.062 ± 0.02 to 0.776 ± 0.035%) but were obviously lower than that of LPS-F (0.093 ± 0.02 to 1.302 ± 0.041%).
In general, the antioxidative activities of LPS-F were higher than those of LPL-S were, and the reason might because the types and amounts of flavonols compounds in LPS-F were considerably more abundant than that in LPL-F, which was in line with the results of the HPLC analysis.22,23 Moreover, at low concentration, the in vitro antioxidative capabilities of the LPS-F and LPL-F were comparable and mostly higher than that of VC, indicating that the flavone extracts of the stem and leaf of LP exhibited very satisfactory pharmacological effects that are health-promoting and show great value of flavones extracts from L. pumila as functional food potential.
3.6 Scavenging capabilities during digestion
Although the in vitro ET-based antioxidative assays of the flavone extracts revealed strong scavenging capabilities, the actual abilities of human consumption need to be evaluated.24 Therefore, the antioxidative activities during artificial GI digestion were analyzed and are shown in Fig. 3A to C. In general, the antioxidative capabilities were not different for gastric digestion at time 0 to 4 h, and the DPPH free radical scavenging rate of LPS-F was from 36.20 ± 1.77 to 35.47 ± 0.97%, and LPL-F was from 35.32 ± 0.75 to 35.76 ± 1.81%. Furthermore, the ABTS free radical scavenging rate of LPS-F was from 86.67 ± 1.24 to 86.91 ± 1.63 and that of LPL-F was from 84.15 ± 1.22 to 85.91 ± 1.74, while the ferric reducing power of LPS-F was from 0.503 ± 0.002 to 0.636 ± 0.009 and that of LPL-F was from 0.244 ± 0.012 to 0.404 ± 0.004. The reasons for the chemical antioxidative capabilities during digestion were basically lower than of without digestion may because the electrolytes contained in digestive fluid affect the steric hindrance of free radicals therefore decreased the possibility for extracts to attack and scavenge, especially the DPPH free radicals.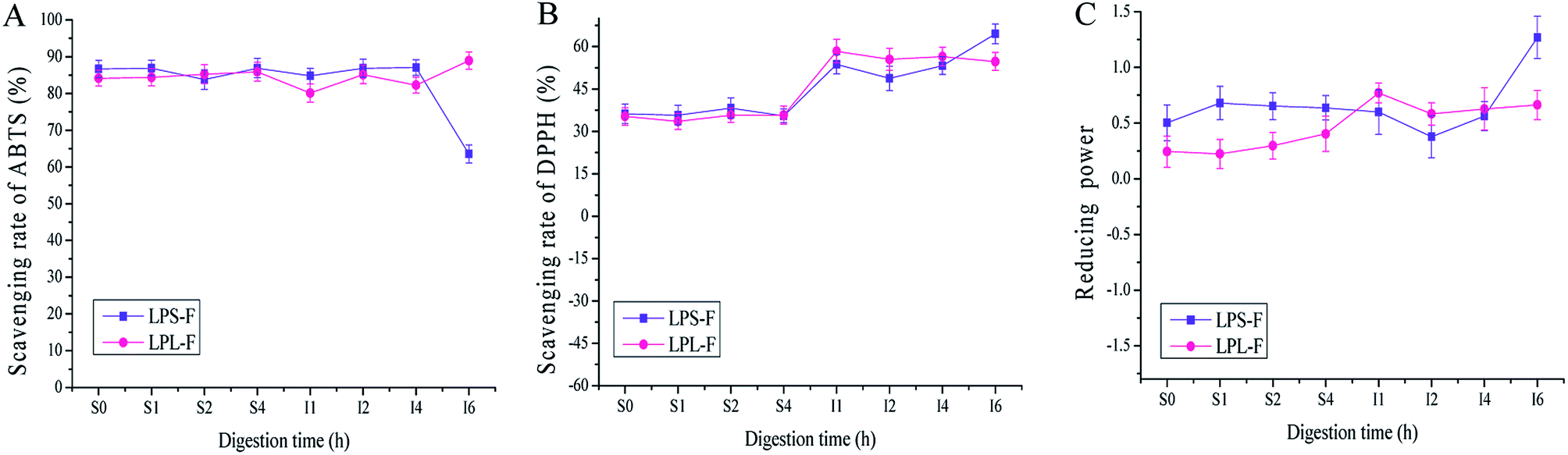 | ||
| Fig. 3 Scavenging ability of Labisia pumila leaf and stem extract fractions (LPL-F and LPS-F respectively) during gastrointestinal (GI) on 2,2-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) (ABTS) (A) and 1,1-diphenyl-2-picrylhydrazyl (DPPH) (B) free radicals. (C) Ferric ion reducing antioxidative power of both extracts. Data are means ± standard deviations of three independent experiments. | ||
Regarding the intestinal digestion, the antioxidative activities were slight stable but generally different variation at the last 2 hours digestions. As shown in Fig. 3A, the ABTS scavenging rate was stable in the first 4 hours (LPS-F, to 87.04 ± 2.01; LPL-F to 82.26 ± 2.99%), and LPS-F showed down while LPL-F shown increased from 4 to 6 hours (LPS-F, 63.52201 ± 2.44% and LPL-F, 88.93 ± 3.28%). As shown in Fig. 3B, the DPPH scavenging rate of the LPS-F stably increased at the digestive time from I1 h to I6 h, (LPS-F, from 53.72 ± 1.89% to 64.52 ± 3.05%). Furthermore, at the intestinal digestive time at I1 h, the scavenging rate of LPl-F increased at 58.39 ± 1.83%, whereas that of LPL-F decreased to 54.74 ± 0.047% at I6 h. As shown in Fig. 3C, the ferric reducing power of LPL-F and LPS-F at digestive time S4 h to I2 h also decreased slightly (LPS-F to 0.378 ± 0.013 and LPL-F to 0.582 ± 0.021). Furthermore, from I2 to I6 h the increasing tendencies of the LPS-F and LPL-F flavone extracts were similar at 0.562 ± 0.013 to 1.269 ± 0.003 and 0.628 ± 0.021 to 0.663 ± 0.007, respectively.
It has been reported that the structure of flavonoids is stable in acid conditions such as gastric fluid, but very easily damaged when exposed to alkaline environments.24,25 The parent nucleus of flavonoids is 2-phenyl-chromone, and some flavonol compounds contained glycosidic bonds. Under the alkaline condition, the glycosidic bonds of flavonoids could be broken down rapidly, and the cyclic structure could be changed, decreasing the content and relevant antioxidative capabilities. In addition, the different digestive enzymes, ionic strengths, and their reactions in gastric and intestinal environments could also affect the structure and content of flavonoid.26 However, regarding the chemical antioxidative changes during GI digestion, the structures of the two extracts were stable under gastric fluid as well as the first 4 hours when exposed to alkaline environments. Whereas the upper 4 hours continuous exposed to alkaline environment may destroy the parent nucleus and glycosidic bonds contained in flavonols extracts, and this conclusion was in line with relevant ref. 27 and 28. Therefore, the various types and amounts of flavonols compounds in stem and leaf resulting in the different chemical antioxidative capabilities. In general speaking, we could conduct that the flavonols extracts in LP performed stable chemical antioxidative activities during GI digestive environments, and may be able to reach intestinal lumens to scavenging free radicals.
3.7 Attenuation of erythrocyte hemolysis by LPS-F and LPL-F before and after GI digestion
After hemolysis induced by AAPH free radicals, the protective effects of flavone extracts on erythrocytes were evaluated, and the results are shown in Fig. 2D and E. The two flavone extracts exhibited strong inhibitory effects on hemolysis, which appeared to be concentrations-dependent. Moreover, the group treated only with 200 mM AAPH showed the lowest hemolysis inhibition at 61.62 ± 0.86%, indicating that AAPH successfully induced the hemolysis of erythrocytes. In addition, the group treated with LPL-F 400 μg mg−1 only exhibited the highest inhibition rate of 92.48 ± 1.64, and that of the LPS-F 500 μg mg−1 was 92.84 ± 3.03%, and the control group (93.71 ± 1.03) exhibited a comparable effect to that of the maximum concentration groups. This observation indicates that PBS or flavone treatment only showed no effects on the hemolysis of erythrocytes. The LPL-F, at concentrations from 25 to 400 μg mg−1, was a significantly increased inhibition rate from 66.23 ± 1.24 to 92.13 ± 1.86% and concentration of LPS-F from 100 to 500 μg mg−1 showed inhibition from 70.74 ± 1.03 to 93.50 ± 2.19%. These observations indicate that both flavone extracts effectively protected the erythrocytes against AAPH-induced hemolysis. Moreover, the LPL-F exhibited relatively better protective effects than those of the LPS-F because the inhibition induced by a low concentration (50 μg mg−1) of LPL-F was 72.01 ± 1.36%, which is comparable to that induced by the high concentration (100 μg mg−1) of LPS-F (70.74 ± 1.03%). The results of hemolysis inhibition rate were contrary to that of chemical antioxidative assays, which indicated that LPL-F was easier to cross the membrane of erythrocyte and able to scavenge AAPH free radicals inside the cells. The relatively high absorptive rate of LPL-F may contribute to the relatively low types and amounts of flavonols compounds contained in the extract.During the gastric digestion, the inhibitory effects of the flavone extracts did not vary significantly, and the rate of the LPL-F (74.86 ± 2.64 to 71.02 ± 1.94%) was higher than that of LPS-F (71.63 ± 2.03 to 72.26 ± 1.83%). However, from the intestinal digestion time 1 to 5 h, the inhibition rates all increased significantly, and that of the LPL-F was from 75.22 ± 2.31 to 81.29 ± 2.09%, and LPS-F was from 78.65 ± 2.64 to 82.57 ± 1.95%. Furthermore, these tendencies were in line with the results of the in vitro antioxidative assays.
It has been reported that the mechanism of the protective effects of the extracts on hemolysis of erythrocyte is mediated by the antioxidant content. Specifically, the antioxidants directly act as free radical scavengers or enhance the enzyme and non-enzyme intracellular system.29 Therefore protect these compounds protect erythrocytes from free radical attack and reverse the saturation of the red cell cytomembrane. The flavone extracts in our study were all low molecular weight bioactive compounds and, therefore, we inferred that their effects on AAPH-induced hemolysis were mainly attributable to the antioxidative flavonol compounds, which act as scavengers. Whereas, during intestinal digestion time 4 to 6 hour, the inhibition rate of LPL-F continuously increased but of LPS-F was decreased rapidly (LPL-F, 83.40 ± 2.46% and LPS-F, 59.27 ± 1.36%), in which the decrease in the protective effects during intestinal digestion was mainly caused by the decreased content of antioxidative flavonol compounds.
Both antioxidative mechanisms during GI digestion were evaluated and the results were shown in Fig. 4, including non-enzymatic and enzymatic antioxidant systems. Through evaluating the intensity of DCF fluorescence, erythrocyte intracellular ROS generation was prevented by extracts antioxidative protective effects, as shown in Fig. 4A. In summary, the free radical scavenger LPS-F inhibited intracellular AAPH-induced ROS generation in erythrocytes was relatively higher than that of LPL-F (LPS-F, from 222.04 ± 20.26 to 189.17 ± 19.29% of control, and LPL-F, from 82.07 ± 19.89 to 252.8 ± 19.19% of control), whereas during the last intestinal digestive 2 hours, extracts performed the best (LPS-F, 35.84578 ± 12.75% of control and LPL-F, 54.43 ± 11.17% of control). Because the excess ROS generation could cause lipid peroxidation and therefore increases MDA levels, the MDA level was measured during the digestion and the data was shown in Fig. 4C. The level of MDA was 2.29 ± 0.51 nmol mg−1 protein in LPS-F, and was lower than that of LPL-F, which was 6.13 ± 0.67 nmol mg−1 protein at I1 hour. Excess MDA negatively affects tumor promotion, cellular metabolism disruption, and cell membrane dysfunction, where extracts from LP through GI digestion still shown protective effects on erythrocytes from the negative effects from MDA.30
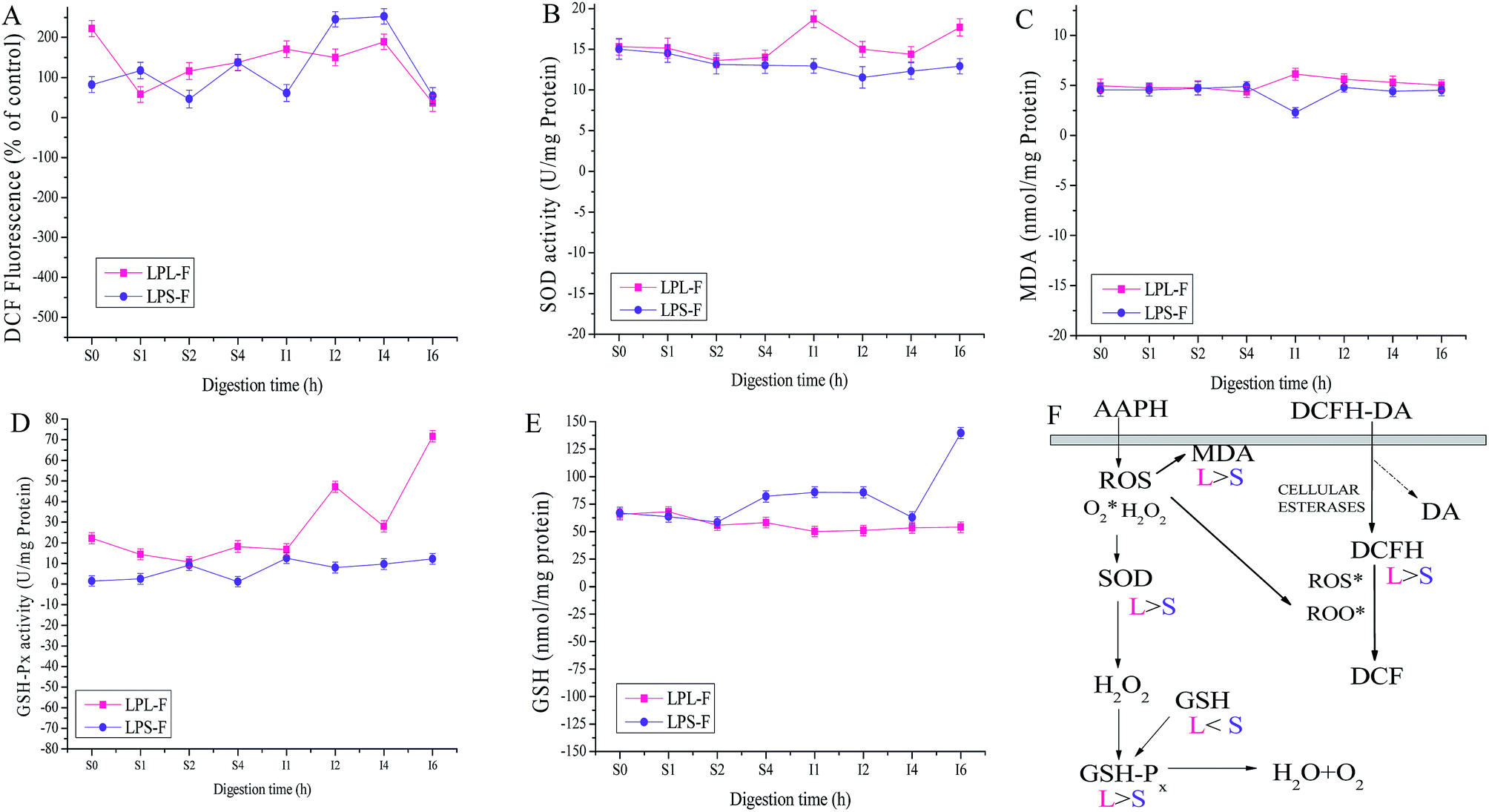 | ||
| Fig. 4 (A) Inhibitory activity of LPL-F and LPS-F during GI-digestion samples on AAPH-induced ROS overexpression in human erythrocytes. Changes in enzyme activities of SOD (B), MDA (C) content, GSH-Px (D), and GSH (E) in human erythrocytes. Erythrocytes were pretreated with LPL-F and LPS-F during GI-digestion samples for 30 min prior to AAPH (200 mM) treatment for 2 h; Data are mean ± standard deviations of three independent experiments. (F) The possible intracellular antioxidant-detoxifying mechanisms of LPL-F and LPS-F during GI-digestion samples attenuating AAPH-induced oxidative stress by inhibiting ROS generation. | ||
In order to determine whether the enzymatic antioxidative system in erythrocytes was affected by extracts during GI digestive environments, the activities of antioxidant enzymes including SOD and GPx were evaluated. As shown in Fig. 4B and D, the higher enzyme activities of SOD and GPx performed in the attenuation of erythrocyte hemolysis by LPS-F, which was 71.65 ± 2.74 U mg−1 protein of GPx and 17.68 ± 1.06 U mg−1 protein of SOD respectively at the 6 hour of intestinal digestion. The relatively high enzyme activity of GPx in LPS-F contributed to the relatively low content of GSH, which was 53.99 ± 4.92 nmol mg−1 protein at the 6 hour of LPS-F intestinal digestion, as shown in Fig. 4E. In the above pathway, excess hydrogen peroxide can be reduced by GPx to prevent hydrogen peroxide from being converted into more active species such as hydroxyl radicals and SOD was able to catalyze the conversion of highly reactive superoxide anion to less reactive oxygen and hydrogen peroxide through dismutation. In addition, GPx catalyzed the conversion of glutathione from the reduced form GSH.31 Based on the tendencies of the above enzyme activities after induced by the digestive extracts, we infer that extracts were able to pass through the erythrocyte membrane and directly inhibited ROS generation and MDA formation induced by AAPH and also regulated intracellular antioxidant enzyme activity, in which LPS-F performed the better and the details of the protective effects of extracts during oxidative erythrocyte hemolysis induced by AAPH are shown in Fig. 4F.
4. Conclusions
Various types and high contents of flavones were identified in the extracts of LP. In our study, the flavone extracts of the stem and leaf were investigated separately. First, to enhance the efficiency of the flavone extraction process, single-factor experiment and relevant RSM were used to determinate the optimal extraction parameters. We determined that the optimal conditions were a ratio of material to solvent, temperature, ethanol concentration, and time of 1:60 (w/v), 85 °C, 35% and 90 min, respectively after adjustments. During the purification of the flavone extracts of the stem and leaf, elution using ultrapure water removed various water-soluble impurities, especially polysaccharides and water-soluble proteins. Furthermore, subsequent elution with 40% ethanol increased the purity of the flavones compared with that of the crude flavone extracts of both the stem and leaf. Therefore, we determined that the 40% ethanol elution performed satisfactorily by increasing the flavone content if the LP extracts. The HPLC analysis of LPS-F and LPL-F revealed that catechin, glycitin, rutin, naringin, and myricetin were the main constituents in LPS-F, whereas genistin, naringin, and myricetin were mainly detected in LPL-F. In vitro, antioxidative assays showed that the reducing power, DPPH and ABTS free radical scavenging effects of LPL-F were lower than those of LPS-F were, which be attributable to the higher abundance and types of relevant flavonol compounds in the LPS-F. Furthermore, at sample concentrations of 0.375 to 6 μg mL−1, the antioxidative capabilities of the flavone extracts were significantly higher than that of VC. Moreover, the attenuation of erythrocyte hemolysis by the LPS-F and LPL-F was contrary to the effects observed in the ET-based antioxidative assays. This effect was satisfactory with inhibition rates of up to 90.36 and 90.702% ± 2.35 for LPS-F and LPS-F, respectively at concentrations higher than 300 μg mL−1. During the gastric digestion, the in vitro antioxidative capabilities and inhibition rate of erythrocytes hemolysis were basically stable, and no obvious changes occurred in the flavone content of the extracts. However, during the intestinal digestion, the antioxidative activities keep stable at the first 4 hours intestinal digestion and exhibited various tendencies at the end of the GI digestion. This observation indicates that although the flavones extracts exhibited satisfactory antioxidative capabilities in vitro and after gastric digestion, these activities were greatly affected by the long-term intestinal digestion and the alkaline pH may be the major factors that reduce the antioxidative activities. This study may provide some useful information on the analysis of LP and the characterized digested flavone extracts.
Conflicts of interest
There are no conflicts to declare.References
- L. S. Chua, N. A. Latiff, S. Y. Lee, C. T. Lee, M. R. Sarmidi and R. A. Aziz, Food Chem., 2011, 127, 1186–1192 CrossRef CAS PubMed
.
- M. Norhaiza, M. Maziah and M. Hakiman, J. Med. Plants Res., 2009, 3, 217–223 CAS
- L. S. Chua, S. Y. Lee, N. Abdullah and M. R. Sarmidi, Fitoterapia, 2012, 83, 1322 CrossRef CAS PubMed
- M. N. Norhayati, A. George, N. H. Hazlina, A. K. Azidah, H. I. Idiana, K. S. Law, I. S. Bahari, W. M. Zahiruddin, E. Liske and A. Azreena, J. Med. Food, 2014, 17, 929 CrossRef PubMed
- A. Q. Laghari, S. Memon, A. Nelofar and A. H. Laghari, Anal. Chem., 2011, 2, 871–878 CAS
- D. Huang, B. Ou and R. L. Prior, J. Agric. Food Chem., 2005, 53, 1841 CrossRef CAS PubMed
- L. K. Macdonaldwicks, L. G. Wood and M. L. Garg, J. Sci. Food Agric., 2006, 86, 2046–2056 CrossRef CAS
- H. Liu, J. Ma and H. Wu, RSC Adv., 2017, 7, 48913–48924 RSC
- N. Andrade, J. R. Araújo, A. Correia-Branco, J. V. Carletti and F. Martel, J. Funct. Foods, 2017, 36, 429–439 CrossRef CAS
- C. Chen, B. Zhang, X. Fu, L. J. You, A. M. Abbasi and R. H. Liu, Food Hydrocolloids, 2016, 58, 171–178 CrossRef CAS
- J. L. Hu, S. P. Nie, F. F. Min and M. Y. Xie, Carbohyd. Polym., 2013, 92, 1143 CrossRef CAS PubMed
- E. J. Mavundza, T. E. Tshikalange, N. Lall, A. A. Hussein, F. N. Mudau and J. J. M. Meyer, J. Med. Plants Res., 2010, 4, 2584–2587 CrossRef
- W. Wei, L. Feng, W. R. Bao, D. L. Ma, C. H. Leung, S. P. Nie and Q. B. Han, Planta Med., 2016, 64, 881 CAS
- F. Yong, Y. F. Zhang, B. Catron, Q. L. Chan, Q. H. Hu and J. A. Caruso, J. Anal. At. Spectrom., 2009, 24, 1657–1664 RSC
- N. L. Mkandawire, S. A. Weier, C. L. Weller, D. S. Jackson and D. J. Rose, LWT--Food Sci. Technol., 2015, 62, 662–667 CrossRef CAS
- B. Yang, H. Yang, J. Li, Z. Li and Y. Jiang, Food Chem., 2011, 124, 551–555 CrossRef CAS
- Q. L. Luo, Z. H. Tang, X. F. Zhang, Y. H. Zhong, S. Z. Yao, L. S. Wang, C. W. Lin and X. Luo, Int. J. Biol. Macromol., 2016, 89, 219 CrossRef CAS PubMed
- B. Moktan, J. Saha and P. K. Sarkar, Food Res. Int., 2008, 41, 586–593 CrossRef CAS
- M. Zhang, G. Wang, F. Lai and W. Hui, J. Agric. Food Chem., 2016, 64, 1921–1931 CrossRef CAS PubMed
- F. Jin, X. Sui, F.-S. Ma and S.-H. Wang, Filtr. Sep., 2017, 54, 38–40 CrossRef
- P. K. Jain, M. D. Kharya, A. Gajbhiye, U. V. S. Sara and V. K. Sharma, Herba Pol., 2010, 7, 1089–1099 Search PubMed
- L. Y. Cui and F. C. Department, Food Res. Development, 2014, 20 CAS
- J. Hyun, D. S. Woo YHwang, G. Jo, S. Eom, Y. Lee, J. C. Park and Y. Lim, Bioorg. Med. Chem. Lett., 2010, 20, 5510 CrossRef CAS PubMed
- L. You, M. Zhao, R. Joem and J. Ren, Food Chem., 2010, 120, 810–816 CrossRef CAS
- J. Bouayed, L. Hoffmann and T. Bohn, Food Chem., 2011, 128, 14 CrossRef CAS PubMed
- R. J. Green, A. S. Murphy, B. Schulz, B. A. Watkins and M. G. Ferruzzi, Mol. Nutr. Food Res., 2007, 51, 1152 CAS
- K. Argyri, M. Komaitis and M. Kapsokefalou, Food Chem., 2006, 96, 281–289 CrossRef CAS
- M. Bermudezsoto, F. Tomasbarberan and M. Garciaconesa, Food Chem., 2007, 102, 865–874 CrossRef CAS
- G. Wang, Z. Lei, Q. Zhong, W. Wu, H. Zhang, T. Min, H. Wu and F. Lai, Food Chem., 2017, 217, 332–341 CrossRef CAS PubMed
- W. Liao, Z. Ning, L. Chen, Q. Wei, E. Yuan, J. Yang and J. Ren, J. Agric. Food Chem., 2014, 62, 8648 CrossRef CAS PubMed
- M. Zhang, Z. Hong, H. Li, F. Lai, X. F. Li, Y. Tang, M. Tian and W. Hui, J. Agric. Food Chem., 2016, 64 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra00142a |
| This journal is © The Royal Society of Chemistry 2018 |